

This article summarizes the data supporting FDA approval of atezolizumab and pembrolizumab for the treatment of patients with advanced urothelial carcinoma ineligible for cisplatin‐based chemotherapy and the subsequent revision of the indications for both agents.
Keywords: Locally advanced or metastatic urothelial carcinoma, Bladder cancer, Platinum‐containing chemotherapy, Atezolizumab, Programmed death‐ligand 1 antibody, Pembrolizumab, Programmed death receptor‐1 antibody immunotherapy
Abstract
The U.S. Food and Drug Administration (FDA) granted accelerated approval to atezolizumab and pembrolizumab in April and May 2017, respectively, for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin‐containing chemotherapy. These approvals were based on efficacy and safety data demonstrated in the two single‐arm trials, IMvigor210 (atezolizumab) and KEYNOTE‐052 (pembrolizumab). The primary endpoint, confirmed objective response rate, was 23.5% (95% confidence interval [CI]: 16.2%–32.2%) in patients receiving atezolizumab and 28.6% (95% CI: 24.1%–33.5%) in patients receiving pembrolizumab. The median duration of response was not reached in either study and responses were seen regardless of PD‐L1 status. The safety profiles of both drugs were generally consistent with approved agents targeting PD‐1/PD‐L1. Two ongoing trials (IMvigor130 and KEYNOTE‐361) are verifying benefit of these drugs. Based on concerning preliminary reports from these trials, FDA revised the indications for both agents in cisplatin‐ineligible patients. Both drugs are now indicated for patients not eligible for any platinum‐containing chemotherapy or not eligible for cisplatin‐containing chemotherapy and whose tumors/infiltrating immune cells express a high level of PD‐L1. The indications for atezolizumab and pembrolizumab in patients who have received prior platinum‐based therapy have not been changed. This article summarizes the FDA thought process and data supporting the accelerated approval of both agents and the subsequent revision of the indications.
Implications for Practice.
The accelerated approvals of atezolizumab and pembrolizumab for cisplatin‐ineligible patients with advanced urothelial carcinoma represent the first approved therapies for this patient population. These approvals were based on single‐arm trials demonstrating reasonable objective response rates and favorable durations of response with an acceptable toxicity profile compared with available non‐cisplatin‐containing chemotherapy regimens. However, based on concerning preliminary reports from two ongoing phase III trials, the FDA revised the indication for both agents in cisplatin‐ineligible patients. Both are now indicated either for patients not eligible for any platinum‐containing chemotherapy or not eligible for cisplatin‐containing chemotherapy and whose tumors have high expression of PD‐L1.
摘要
美国食品和药品管理局 (FDA) 分别于 2017 年 4 月和 5 月同意加速批准将阿特朱单抗和帕博利珠单抗用于治疗无法接受含顺铂化疗的局部晚期或转移性尿路上皮癌患者。上述批准以在两个单臂试验中显示的有效性和安全性数据为依据:IMvigor210(阿特朱单抗)和 KEYNOTE‐052(帕博利珠单抗)。主要终点指标即确定的客观缓解率,在接受阿特朱单抗治疗的患者中为 23.5% [95% 置信区间 (CI):16.2%–32.2%],在接受帕博利珠单抗治疗的患者中为 28.6%(95% CI:24.1%–33.5%)。在这两个研究中均未达到缓解的中位持续时间,而且,无论 PD‐L1 状态如何,都可以观察到缓解。这两种药物的安全特性与以 PD‐1/PD‐L1 为靶点的批准药剂基本一致。两个正在进行的试验(IMvigor130 和 KEYNOTE‐361)正在检验这两种药物的益处。根据来自上述试验的相关初步报告,FDA 修改了这两种药剂在无法接受顺铂治疗的患者中的适应证。现在,这两种药物适用于无法接受任何含铂化疗或者无法接受含顺铂化疗且肿瘤/浸润免疫细胞表达高水平 PD‐L1 的患者。阿特朱单抗和帕博利珠单抗在既往曾接受铂类治疗的患者中的适应证没有变化。本文概述了支持加速批准这两种药剂以及随后对适应证进行修改的 FDA 思考过程和数据。
实践意义:加速批准阿特朱单抗和帕博利珠单抗用于治疗无法接受顺铂治疗的晚期尿路上皮癌患者,这提出了针对此类患者群体的首个批准疗法。上述批准以单臂试验为依据,与可用的不含顺铂化疗方案相比,此类单臂试验显示出合理的客观缓解率、良好的反应持续时间以及可接受的毒性特征。不过,根据来自两个正在进行的 III 期试验的相关初步报告,FDA 修改了这两种药剂在无法接受顺铂治疗的患者中的适应证。现在,这两种药剂适用于无法接受任何含铂化疗或者无法接受含顺铂化疗且肿瘤表达高水平 PD‐L1 的患者。
Introduction
Urothelial carcinoma (UC) is the most common malignancy in the urinary tract and accounts for approximately 16,000 deaths annually in the U.S. [1], [2]. Although most UCs are non‐muscle invasive at diagnosis and can be managed effectively with surgical resection and/or intravesical therapies, approximately 10%–15% of patients may develop invasive, locally advanced, and metastatic urothelial carcinoma [3]. Approximately 10% of patients have regionally advanced or metastatic disease at diagnosis [1]. The standard of care for patients with advanced disease is cisplatin‐containing chemotherapy [2]. However, approximately 30%–50% of patients are considered ineligible for cisplatin‐based chemotherapy because of comorbidities [4]. These patients are generally defined as those with a creatinine clearance less than 60 mL/minute, an Eastern Cooperative Oncology Group (ECOG) performance score of ≥2, or greater than or equal to grade 2 hearing impairment or peripheral neuropathy.
No approved therapies specifically for the cisplatin‐ineligible population were available prior to April 2017. Non‐cisplatin‐based chemotherapy (e.g., carboplatin) was generally used but considered inferior to cisplatin [5]. Results from a few randomized trials of non‐cisplatin‐based regimens in this patient population along with several small single‐arm trials of the most common regimens, including gemcitabine in combination with carboplatin or oxaliplatin, demonstrated objective response rates (ORRs) of approximately 30%–45% with median response durations of approximately 5–8 months. Overall survival (OS) in these patients is poor, ranging from 7 to 10 months [6], [7], [8]. Cytotoxic therapy is also poorly tolerated in these patients, with a high incidence of hematologic toxicities including neutropenic fever [6]. Thus, there is an unmet need for effective and tolerable treatments in cisplatin‐ineligible patients with advanced urothelial carcinoma.
Monoclonal antibodies that disrupt the interaction of programmed death receptor‐1 (PD‐1) with programmed death‐ligand 1 (PD‐L1) release tumor‐mediated inhibition of the immune response and have demonstrated antitumor activity in urothelial carcinoma. On May 18, 2016, the U.S. Food and Drug Administration (FDA) granted accelerated approval to atezolizumab (TECENTRIQ; Genentech, Inc., South San Francisco, CA), a monoclonal antibody that binds PD‐L1, for use in patients with urothelial carcinoma that had progressed during or after platinum‐based chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum‐containing chemotherapy [9]. This approval was based on a similar ORR and improved duration of response (DoR) compared with historical data for available chemotherapy. Additional accelerated approvals of anti‐PD‐1 and anti‐PD‐L1 antibodies in the second‐line urothelial carcinoma setting were subsequently granted to nivolumab, durvalumab, and avelumab, and were also based on ORR and DoR [10]. On April 17, 2017, the FDA granted accelerated approval for atezolizumab in patients with urothelial carcinoma who are not eligible for cisplatin. This was based on a preplanned cohort from Study IMvigor210. IMvigor210 enrolled one cohort of patients who were cisplatin‐ineligible and one who had received prior platinum‐based therapy. Pembrolizumab (KEYTRUDA; Merck, Whitehouse Station, NJ), a PD‐1‐blocking antibody, was initially approved for urothelial carcinoma in both patients who had received platinum‐based therapy and the cisplatin‐ineligible population on May 18, 2017. The pembrolizumab regular approval in patients with locally advanced or metastatic urothelial carcinoma with disease progression during or following platinum‐containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum‐containing chemotherapy was based on an improvement in OS compared with Investigator's choice of chemotherapy (vinflunine, paclitaxel, or docetaxel) in a randomized trial. The pembrolizumab accelerated approval in cisplatin‐ineligible patients was based on the phase II single‐arm KEYNOTE‐052 trial. Herein, we summarize key review findings that supported the two approvals of atezolizumab and pembrolizumab in this novel cisplatin‐ineligible patient population, as well as FDA's rationale for revising the indications based on emerging data from two ongoing trials.
Clinical Trial Design
Cohort 1 of IMvigor210 (ClinicalTrials.gov Identifier NCT02108652) and KEYNOTE‐052 (ClinicalTrials.gov Identifier NCT02335424) were similar in design. Both studies were single arm, open‐label trials that enrolled patients with advanced urothelial carcinoma who were ineligible for cisplatin‐containing chemotherapy. Both trials enrolled patients regardless of PD‐L1 expression levels. Cisplatin ineligibility was defined as one of the following: (a) impaired renal function (glomerular filtration rate >30 but <60 mL/minute), (b) greater than or equal to grade 2 hearing loss, (c) greater than or equal to grade 2 peripheral neuropathy, or (d) ECOG performance score of 2.
KEYNOTE‐052 additionally defined patients with New York Heart Association Class III heart failure as ineligible to receive cisplatin and thus eligible for the trial. Both trials required patients to either be previously untreated or have disease progression more than 12 months from their last dose of platinum‐containing neoadjuvant or adjuvant chemotherapy. Key exclusion criteria for both trials included a history of autoimmune disease requiring administration of systemic immunosuppressive medications.
IMvigor210 included two patient cohorts; Cohort 1 enrolled patients who were cisplatin ineligible. For the approval described herein, the FDA review focused primarily on patients enrolled in Cohort 1.
Patients received either an intravenous infusion of 1,200 mg of atezolizumab or 200 mg of pembrolizumab every 3 weeks. Patients on atezolizumab could continue treatment until unacceptable toxicity or radiographic or symptomatic progression. Patients on pembrolizumab could continue treatment until unacceptable toxicity, confirmed radiographic disease progression, investigator's decision to withdraw the subject, completion of 24 months of treatment, or development of an intercurrent illness that prevented further treatment.
Patients receiving atezolizumab were scanned every 9 weeks for the first 54 weeks and every 12 weeks thereafter. Patients receiving pembrolizumab were scanned at 9 weeks, then every 6 weeks for 48 weeks and every 12 weeks thereafter. The primary endpoint of both trials was confirmed ORR using RECIST 1.1 as determined by blinded independent central review (BICR). Duration of response was also assessed.
PD‐L1 expression status was prospectively assessed in IMvigor210 at a central laboratory using the Ventana PD‐L1 (SP142) Assay. Although PD‐L1 expression status was prospectively assessed in KEYNOTE‐052 using the Agilent PD‐L1 IHC22C3 pharmDx assay, a marketing application for the diagnostic test was not initially submitted with the pembrolizumab application.
Each drug has an ongoing randomized trial that could be used for confirmation of clinical benefit: IMvigor130 (NCT02807636) and KEYNOTE‐361 (NCT02853305). The designs of both trials were similar. Both trials enrolled patients with locally advanced or metastatic urothelial cancer who were newly diagnosed or had received neoadjuvant/adjuvant therapy more than 12 months prior to study entry. Patients were enrolled regardless of PD‐L1 status. Patients enrolled in IMvigor130 were stratified by PD‐L1 staining in tumor‐infiltrating immune cells (IC), IC0, IC1, or IC2/3, where IC2/3 corresponds to ≥5% staining. Patients enrolling in KEYNOTE‐361 were stratified by a Combined Positive Score (CPS) reflecting staining of both tumor and immune cells ≥10 or < 10. In IMvigor130, patients were randomized to atezolizumab plus chemotherapy, chemotherapy alone, or monotherapy with atezolizumab. In KEYNOTE‐361, patients were randomized to pembrolizumab plus chemotherapy, chemotherapy alone, or monotherapy with pembrolizumab. Chemotherapy consisted of gemcitabine plus either cisplatin or carboplatin in both studies. Prior to randomization, investigators determined whether the patient would receive cisplatin or carboplatin. Strict criteria for cisplatin ineligibility were not prespecified; however, investigators were required to provide the reason for cisplatin ineligibility. The coprimary endpoints of IMvigor 130 are progression‐free survival (PFS) and OS in the combination arm versus the chemotherapy‐alone arm and OS in the atezolizumab monotherapy versus chemotherapy arms, all in the intent‐to‐treat (ITT) populations, while OS in PD‐L1‐high patients will be evaluated if OS is positive in both the combination versus chemotherapy ITT populations and atezolizumab monotherapy versus chemotherapy ITT populations. KEYNOTE‐361 incorporates multiple hierarchical hypotheses with initial primary endpoints of PFS and OS in the combination versus chemotherapy arms and subsequent noninferiority and superiority endpoints of OS in the pembrolizumab versus chemotherapy arm.
Results
Atezolizumab
IMvigor210 enrolled 119 patients in Cohort 1. Key demographic and disease characteristics of these patients are summarized in Table 1. The median age of Cohort 1 was 73 years. Approximately 26% of patients had lymph node‐only disease, whereas 21% had liver involvement. The most common reason for cisplatin ineligibility was impaired renal function.
Table 1. Key baseline characteristics of patients in Cohort 1 of IMvigor210 and KEYNOTE‐052.
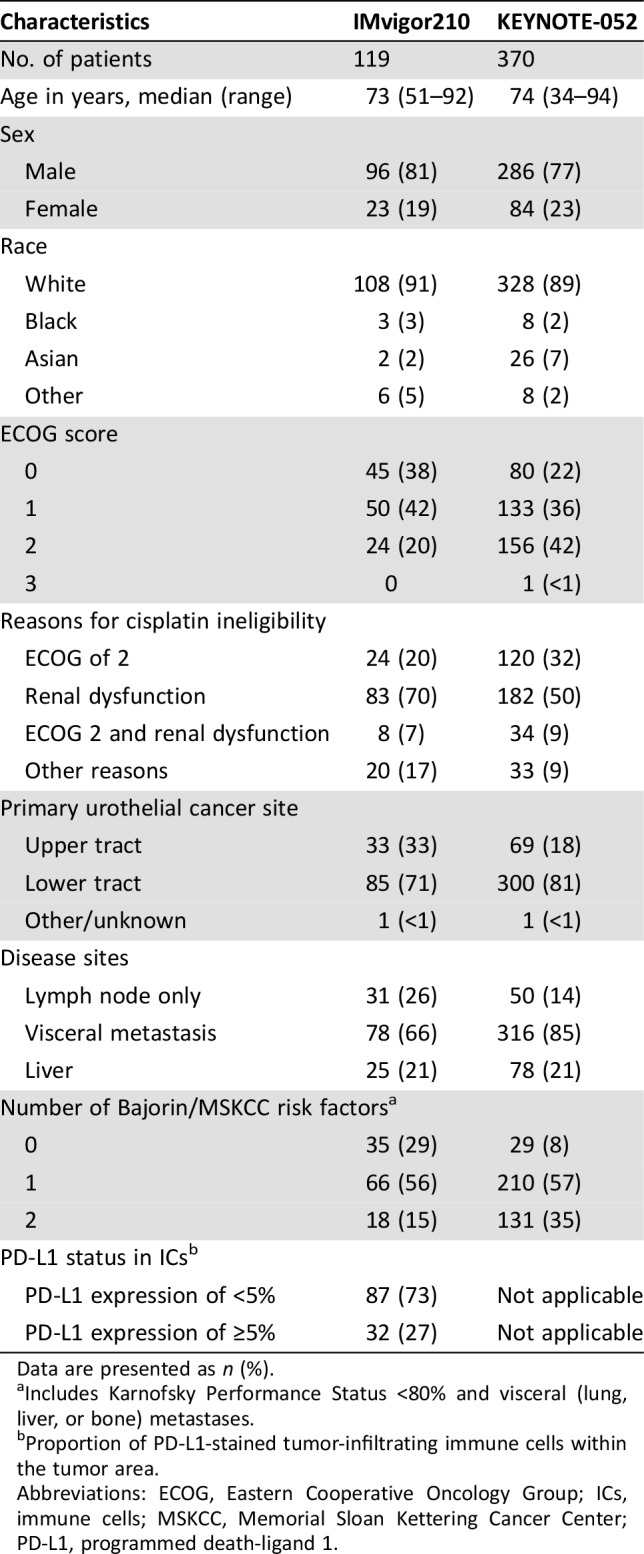
Data are presented as n (%).
Includes Karnofsky Performance Status <80% and visceral (lung, liver, or bone) metastases.
Proportion of PD‐L1‐stained tumor‐infiltrating immune cells within the tumor area.
Abbreviations: ECOG, Eastern Cooperative Oncology Group; ICs, immune cells; MSKCC, Memorial Sloan Kettering Cancer Center; PD‐L1, programmed death‐ligand 1.
As of the data cutoff, 18% of patients were on study treatment and 82% of patients discontinued study treatment. Of the discontinued patients, 77% were due to disease progression and 10% due to adverse events. The median treatment duration was 3.6 months (range: 0.02–20 months).
Efficacy.
Efficacy results are shown in Table 2. With a median follow‐up time of 14.4 months, BICR‐confirmed ORR was 23.5% (95% confidence interval [CI]: 16.2%–32.2%) in all treated patients. Both complete responses (CRs) and partial responses (PRs) were observed. Median DoR in responders was not reached as of the data cutoff date of March 14, 2016, and the observed response durations ranged from 3.7 to 16.6+ months. At the time of data cutoff, responses were ongoing for at least 6 months in 64.2% of responding patients and for at least 12 months in 21.4% of responding patients.
Table 2. Efficacy results of Cohort 1 of IMvigor210 and KEYNOTE‐052.
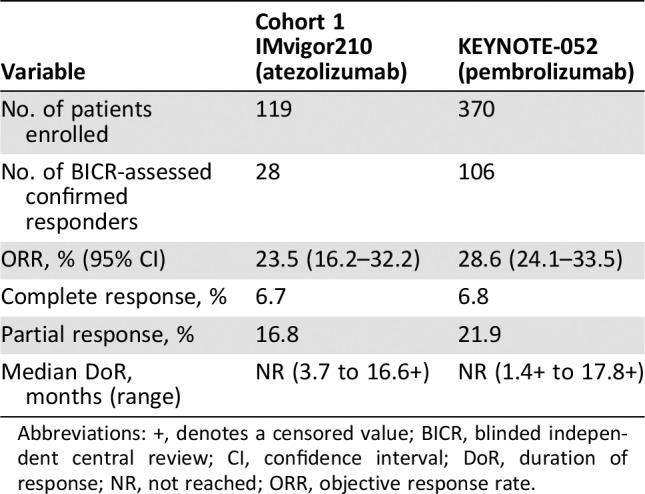
Abbreviations: +, denotes a censored value; BICR, blinded independent central review; CI, confidence interval; DoR, duration of response; NR, not reached; ORR, objective response rate.
Responses were observed in both PD‐L1 expression subgroups. The confirmed ORR was 21.8% (95% CI: 13.7%–32.0%) in patients with PD‐L1 expression of <5% and 28.1% (95% CI: 13.8%–46.8%) in those with PD‐L1 expression of ≥5% in ICs. There were six CRs (6.9%) in patients with PD‐L1 expression of <5% and two CRs (6.3%) in those with PD‐L1 expression of ≥5%. Median duration of response was not reached in either subgroup. Responses were ongoing for at least 6 months in 82% of responding patients and for at least 12 months in 29% of responding patients.
In exploratory subgroup analyses, responses were observed in patients with nonbladder urothelial carcinoma, visceral metastases, and prior Bacillus Calmette‐Guerin (BCG) treatment. Patients with a Bajorin risk score of 2 [11] had a lower response rate (11.1% [95% CI: 1.4%–34.7%]) compared with those with scores of 0–1 (25.7% [95% CI: 17.6%–35.4%]). Durable responses were observed in all the subgroups.
Toxicity.
All 119 patients enrolled in Cohort 1 received at least one dose of atezolizumab and were included in the safety analysis. Immune‐mediated adverse events (imAEs) were defined as events requiring the use of systemic steroids with no alternate etiology, endocrine events, and other events thought to be immune‐related. Immune‐mediated adverse events were generally managed with administration of high‐dose (1–2 mg/kg daily of prednisone or equivalent) corticosteroids followed by a taper and interruption of atezolizumab therapy. In total, 19.3% of patients experienced an imAE, including 12.6% of patients who required systemic corticosteroids and 6.7% who required only hormone replacement therapy. Five percent of patients received an oral prednisone dose equivalent to ≥40 mg daily for an immune‐mediated adverse event. The reported imAEs in this cohort included hypothyroidism, rash, hepatic injury, colitis, arthritis, adrenal insufficiency, and rhabdomyolosis. The pattern of imAEs was generally consistent with that observed with other approved PD‐1/PD‐L1‐targeted products. Thyroid function tests were routinely collected only at baseline and end of study, such that the reported incidence of hypothyroidism may underestimate the true incidence.
Five patients (4.2%) who were treated with atezolizumab experienced one of the following events, which led to death: sepsis, cardiac arrest, myocardial infarction, respiratory failure, or respiratory distress, within 30 days of last drug administration. One additional patient with herpetic meningoencephalitis was reported to have died as a result of disease progression. Four percent of patients treated with atezolizumab discontinued treatment because of an adverse event. The causes of discontinuation were diarrhea/colitis, fatigue, hypersensitivity, and dyspnea.
The most common grade 1–4 adverse events occurring in at least 20% of patients treated with atezolizumab were fatigue (52%), decreased appetite (26%), nausea (25%), urinary tract infections (22%), pyrexia (21%), and constipation (21%). The most common grade 3–4 adverse events occurring in at least 2% of patients treated with atezolizumab were fatigue (8%), urinary tract infection (5%), diarrhea (4%), intestinal obstruction (3%), decreased appetite (3%), sepsis (3%), back/neck pain (3%), renal failure (3%), and hypotension (3%). The laboratory abnormalities worsening from baseline to grade 3–4 in at least 2% of patients treated with atezolizumab included hyponatremia (15%), hyperglycemia (10%), lymphopenia (9%), anemia (7%), increased alkaline phosphatase (7%), increased creatinine (5%), hypophosphatemia (4%), increased alanine aminotransferase (4%), increased aspartate aminotransferase (4%), hyperkalemia (3%), hypermagnesemia (3%), and hyperbilirubinemia (3%).
Pembrolizumab
KEYNOTE‐052 enrolled 370 patients. Key demographic and disease characteristics of these patients are summarized in Table 1. The median age was 74 years. Approximately 14% of patients had lymph node‐only metastatic disease, whereas 21% had liver involvement. The most common reason for cisplatin ineligibility was impaired renal function. As of the data cutoff, 50% of patients were on study treatment and 50% of patients discontinued study treatment. Of the discontinued patients, 80% were due to disease progression and 10% due to adverse events. The median treatment duration was 3.4 months (range: 0.03–19.94 months).
Efficacy.
Efficacy results are shown in Table 2. With a median follow‐up time of 7.8 months, ICR‐confirmed ORR was 28.6% (95% CI: 24.1%–33.5%) in all treated patients. Both CRs and PRs were observed. Median DoR in responders was not reached as of the data cutoff date of December 19, 2016, and the observed response durations ranged from 1.4+ to 17.8+ months.
Responses were observed in both PD‐L1 expression subgroups. The confirmed ORR was 21% (95% CI: 16%–26%) in patients with CPS <10 or unknown and 47% (95% CI: 38, 57%) in those with CPS ≥10. There were 8 CRs (3%) in patients with CPS <10 or unknown and 17 CRs (15%) in those with CPS ≥10. Median duration of response was not reached in either subgroup. At the time of data cutoff, responses were ongoing for at least 6 months in 52% of responding patients and for at least 12 months in 7% of responding patients.
Responses were observed in patients with lymph node‐only metastases, visceral metastases, and prior BCG treatment, and in patients who received prior adjuvant/neoadjuvant therapy.
Toxicity.
All the 370 patients in this study received at least one dose of pembrolizumab and were included in the safety analysis. In total, 17% of patients experienced an immune‐mediated adverse event, including 8% of patients who required systemic corticosteroids and 8% who required hormone replacement therapy. Five percent of patients received an oral prednisone dose equivalent to ≥40 mg daily for an immune‐mediated adverse event. One imAE was myositis that led to multiple organ failure and death despite use of corticosteroids. The pattern of immune‐related adverse events was generally consistent with other approved agents targeting the PD‐1/PD‐L1 pathway.
Eighteen patients (5%) treated with pembrolizumab died as a result of an adverse event. Thirteen of these patients (3.5%) died within 30 days of the last dose of sepsis (5), renal failure (2), stroke, pneumonia, aspiration, respiratory failure, colonic perforation, and unknown cause. Eleven percent of patients discontinued pembrolizumab because of an adverse event. The most common cause of discontinuation were infections, including pneumonia, urinary tract infections, and sepsis.
The most common grade 1–4 adverse events occurring in at least 20% of patients treated with pembrolizumab were fatigue (38%), musculoskeletal pain (24%), decreased appetite (22%), constipation (21%), rash (21%), and diarrhea (20%). The most common grade 3–4 adverse events occurring in at least 2% of patients treated with pembrolizumab were urinary tract infection (9%), anemia (7%), fatigue (6%), musculoskeletal pain (4.9%), hyponatremia (4.1%), elevated liver function tests (3.5%), hematuria (3%), abdominal pain (2.7%), and diarrhea (2.4%). The grade 3–4 laboratory abnormalities occurring in at least 2% of patients treated with pembrolizumab included anemia (7%), elevated blood creatinine (4.3%), hyponatremia (4.1%), elevated alkaline phosphatase (2.2%), and hyperkalemia (2.2%).
IMvigor130 and KEYNOTE‐361
In the two ongoing clinical trials of atezolizumab (IMvigor130) and pembrolizumab (KEYNOTE‐361), the Data Monitoring Committee (DMC) for each study performed an early unplanned review and found that patients in the monotherapy arms of both trials with PD‐L1‐low status had decreased survival compared with patients who received cisplatin‐ or carboplatin‐based chemotherapy. There was no change in the adverse event profile of either drug. Both Merck, manufacturer of pembrolizumab, and Genentech, manufacturer of atezolizumab, stopped enrolling patients whose tumors have PD‐L1‐low status to the atezolizumab or pembrolizumab monotherapy arms per the DMCs’ recommendations. For patients already recruited in the monotherapy arms, the DMC recommended continuation in the trial without treatment modification but that they be reconsented. The monotherapy arms remain open for new enrollment only to patients whose tumors have PD‐L1‐high status; the combination arms and the chemotherapy arms of both studies also remain open to all patients regardless of PD‐L1 status.
Discussion
FDA review of the initial supplemental applications found that treatment with atezolizumab or pembrolizumab had a favorable benefit‐risk profile (Table 3) in patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin‐based chemotherapy. Treatment with either agent elicited confirmed objective antitumor responses in approximately 24%–29% of patients in the single‐arm trials. Objective response rates in these trials were similar to or greater than those seen in the treatment of patients who had received prior platinum‐based therapy. This may be due to the extent of prior therapy and to the higher proportion of patients with lymph node‐only disease in the trials of cisplatin‐ineligible patients. Although the median DoRs were not reached at the data cutoff for the response analyses in either study, the duration of response was substantially longer than that seen historically with cytotoxic chemotherapy. The majority of responding patients maintained their response for ≥6 months and some for ≥12 months, indicating durable responses with either product. There was a slight decrease in ORR in patients treated with atezolizumab or pembrolizumab who had tumors considered PD‐L1 negative by the Ventana PD‐L1 (SP142) Assay or the Agilent PD‐L1 IHC 22C3 pharmDx assay, respectively, compared with those with tumors considered PD‐L1 positive. However, in both trials, responses were observed with long durations of response in both the PD‐L1‐high and PD‐L1‐low subgroups. Responses were also observed in all demographic and disease subgroups in both trials, including patients with various primary tumor sites, prior BCG treatment, and visceral metastases. Duration of response did not appear to differ by subgroup.
Table 3. Benefit‐risk assessments of atezolizumab and pembrolizumab for first‐line use in cisplatin‐ineligible patients with advanced urothelial carcinoma at the time of initial accelerated approval.

Abbreviations: ORR, objective response rate; PD‐1, programmed death receptor‐1; PD‐L1, programmed death‐ligand 1.
Given the toxicities associated with cisplatin, carboplatin is often substituted for cisplatin in combination with other agents, including gemcitabine, as first‐line therapy. In a randomized phase III trial comparing two carboplatin‐based regimens in this population, the median survival was only 8–9 months [7]. For patients who received carboplatin and gemcitabine, the confirmed objective response rate was 36%, whereas the median response duration was only 5.4 months. The most common severe toxicities associated with chemotherapy regimens in this patient population were neutropenia (46%), thrombocytopenia (19%), and anemia (18%) [6].
Although the response rates for atezolizumab and pembrolizumab were lower relative to those for chemotherapy in the same disease setting, the responses were more durable, with durable responses of more than 12 months observed in a small number of patients. The safety profiles were largely consistent with those seen in the second‐line setting. Approximately 10%–20% of patients experienced immune‐mediated adverse events that required either use of systemic corticosteroids or hormone replacement. Apart from endocrine events, immune‐mediated toxicity was typically reversible. Although the nature of toxicity differed from that of cytotoxic chemotherapy, the incidence of high‐grade toxicity related to atezolizumab and pembrolizumab compared favorably, and only 5% of patients required high‐dose corticosteroids in either trial. These durable responses combined with different and potentially improved safety profile represent an improvement over available chemotherapies and are reasonably likely to predict clinical benefit in a patient population with a high unmet medical need.
Taken together, the evidence, as summarized in our Benefit‐Risk Assessment in Table 3, was considered sufficient for the respective accelerated approvals of atezolizumab and pembrolizumab for the intended clinical use. This provides the first nonchemotherapeutic treatments in this disease setting and addresses an unmet medical need for this unique patient population. The results from these trials should not be extrapolated to patients who may be eligible for cisplatin‐based chemotherapy.
Based on the regulatory requirements for accelerated approval, the continued approval of atezolizumab and pembrolizumab for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). Preliminary, early analysis reports generated by the respective DMCs from IMvigor130 (17% of OS events) and KEYNOTE‐361 (19% of OS events) demonstrated decreased survival in patients with PD‐L1‐low status on the monotherapy arms. Although the study stopped accrual of patients with low PD‐L1 status to the monotherapy arms, given the previous accelerated approvals for pembrolizumab and atezolizumab monotherapy for the cisplatin‐ineligible populations, FDA no longer considered the benefit‐risk profile favorable for all cisplatin‐ineligible patients. Therefore, on June 18, 2018, the indication for both agents was modified to include only patients who are not eligible for cisplatin‐containing chemotherapy and who have high expression of PD‐L1 or are not eligible for any platinum‐containing chemotherapy regardless of the level of PD‐L1 expression. As there are some patients for whom any platinum‐containing chemotherapy (cisplatin or carboplatin) is not indicated, FDA worded part of the indication for use of these drugs in patients not eligible for any platinum‐containing regimen regardless of tumor PD‐L1 status. There were substantial differences between IMvigor210 and KEYNOTE‐052 and the ongoing randomized trials. The key difference is that both IMvigor210 and KEYNOTE‐052 required patients to meet strict criteria concerning platinum eligibility, whereas the choice of cisplatin‐ or carboplatin‐based therapy was made by the Investigator on the randomized phase III trials.
This change in indication based on PD‐L1 expression was made prior to companion diagnostic approval, given the urgency in refining the product labeling for concerns over decreased survival. The companion diagnostic approvals were agreed upon as postmarketing commitments and were subsequently approved on July 2, 2018 (atezolizumab), and August 16, 2018 (pembrolizumab).
Conclusion
The efficacy and safety results described above demonstrated an acceptable benefit‐risk profile to support the initial accelerated approval of atezolizumab and pembrolizumab for the treatment of patients with locally advanced or metastatic urothelial carcinoma who are not eligible for cisplatin‐based chemotherapy. These approvals were based on confirmed ORRs comparable to available therapy in combination with improved DoRs and favorable safety profiles as well as the unmet need in this patient population. Per accelerated approval regulations, continued approval in patients with urothelial carcinoma who are not eligible for cisplatin‐containing chemotherapy may be contingent upon verification and description of clinical benefit in confirmatory trials. Based on emerging results from these ongoing, large, randomized trials, with coprimary endpoints of PFS and OS, the indication statement for both agents was revised to exclude patients eligible for platinum‐based chemotherapy with low PD‐L1 expression. This revision reflects the balance between the benefit of early availability of promising drugs to patients and the risk of accelerated approval without verification of clinical benefit [12]. The early reports of decreased survival are preliminary and the confirmatory studies are still ongoing; thus, further revisions to the indication may be warranted.
Author Contributions
Conception/design: Daniel L. Suzman, Sundeep Agrawal, Yang‐min Ning, V. Ellen Maher, Laura L. Fernandes, Stella Karuri, Shenghui Tang, Rajeshwari Sridhara, Jason Schroeder, Kirsten B. Goldberg, Amna Ibrahim, Amy E. McKee, Richard Pazdur, Julia A. Beaver
Collection and/or assembly of data: Daniel L. Suzman, Sundeep Agrawal, Yang‐min Ning, V. Ellen Maher, Laura L. Fernandes, Stella Karuri, Shenghui Tang, Rajeshwari Sridhara, Jason Schroeder, Kirsten B. Goldberg, Amna Ibrahim, Amy E. McKee, Richard Pazdur, Julia A. Beaver
Data analysis and interpretation: Daniel L. Suzman, Sundeep Agrawal, Yang‐min Ning, V. Ellen Maher, Laura L. Fernandes, Stella Karuri, Shenghui Tang, Rajeshwari Sridhara, Jason Schroeder, Kirsten B. Goldberg, Amna Ibrahim, Amy E. McKee, Richard Pazdur, Julia A. Beaver
Manuscript writing: Daniel L. Suzman, Sundeep Agrawal, Yang‐min Ning, V. Ellen Maher, Laura L. Fernandes, Stella Karuri, Shenghui Tang, Rajeshwari Sridhara, Jason Schroeder, Kirsten B. Goldberg, Amna Ibrahim, Amy E. McKee, Richard Pazdur, Julia A. Beaver
Final approval of manuscript: Daniel L. Suzman, Sundeep Agrawal, Yang‐min Ning, V. Ellen Maher, Laura L. Fernandes, Stella Karuri, Shenghui Tang, Rajeshwari Sridhara, Jason Schroeder, Kirsten B. Goldberg, Amna Ibrahim, Amy E. McKee, Richard Pazdur, Julia A. Beaver
Disclosures
The authors indicated no financial relationships.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Spiess PE, Agarwal N, Bangs R et al. Bladder cancer, version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2017;15:1240–1267. [DOI] [PubMed] [Google Scholar]
- 3.Park JC, Citrin DE, Agarwal PK et al. Multimodal management of muscle invasive bladder cancer. Curr Probl Cancer 2014;38:80–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galsky MD, Hahn NM, Rosenberg J et al. Treatment of patients with metastatic urothelial cancer “unfit” for cisplatin‐based chemotherapy. J Clin Oncol 2011;29:2432–2438. [DOI] [PubMed] [Google Scholar]
- 5.Galsky MD, Chen GJ, Oh WK et al. Comparative effectiveness of cisplatin‐based and carboplatin‐based chemotherapy for treatment of advanced urothelial carcinoma. Ann Oncol 2012;23:406–410. [DOI] [PubMed] [Google Scholar]
- 6.Necchi A, Pond GR, Raggi D et al. Efficacy and safety of gemcitabine plus either taxane or carboplatin in the first‐line setting of metastatic urothelial carcinoma: A systematic review and meta‐analysis. Clin Genitourin Cancer 2017;15:23–30.e22. [DOI] [PubMed] [Google Scholar]
- 7.De Santis M, Bellmunt J, Mead G et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin‐based chemotherapy: EORTC study 30986. J Clin Oncol 2012;30:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Santis M, Wiechno P, Bellmunt J et al. Vinflunine‐gemcitabine versus vinflunine‐carboplatin as first‐line chemotherapy in cisplatin‐unfit patients with advanced urothelial carcinoma: Results of an international randomized phase II trial (JASINT1). Ann Oncol 2016;27:449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ning YM, Suzman D, Maher VE et al. FDA approval summary: Atezolizumab for the treatment of patients with progressive advanced urothelial carcinoma after platinum‐containing chemotherapy. The Oncologist 2017;22:743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ning YM, Maher VE, Beaver JA et al. Accelerating early access to immunotherapies for advanced urothelial carcinoma. The Oncologist 2018;23:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bajorin DF, Dodd PM, Mazumdar M et al. Long‐term survival in metastatic transitional‐cell carcinoma and prognostic factors predicting outcome of therapy. J Clin Oncol 1999;17:3173–3181. [DOI] [PubMed] [Google Scholar]
- 12.Beaver JA, Howie LJ, Pelosof L et al. A 25‐year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: A review. JAMA Oncol 2018;4:849–856. [DOI] [PubMed] [Google Scholar]