

Abstract
In arsenic-endemic regions of the world, arsenic exposure correlates with diabetes mellitus. Multiple animal models of inorganic arsenic (iAs, as As3+) exposure have revealed that iAs-induced glucose intolerance manifests as a result of pancreatic β-cell dysfunction. To define the mechanisms responsible for this β-cell defect, the MIN6-K8 mouse β-cell line was exposed to environmentally relevant doses of iAs. Exposure to 0.1–1 µM iAs for 3 days significantly decreased glucose-induced insulin secretion (GIIS). Serotonin and its precursor, 5-hydroxytryptophan (5-HTP), were both decreased. Supplementation with 5-HTP, which loads the system with bioavailable 5-HTP and serotonin, rescued GIIS, suggesting that recovery of this pathway was sufficient to restore function. Exposure to iAs was accompanied by an increase in mRNA expression of UDP-glucuronosyltransferase 1 family, polypeptide a6a (Ugt1a6a), a phase-II detoxification enzyme that facilitates the disposal of cyclic amines, including serotonin, via glucuronidation. Elevated Ugt1a6a and UGT1A6 expression levels were observed in mouse and human islets, respectively, following 3 days of iAs exposure. Consistent with this finding, the enzymatic rate of serotonin glucuronidation was increased in iAs-exposed cells. Knockdown by siRNA of Ugt1a6a during iAs exposure restored GIIS in MIN6-K8 cells. This effect was prevented by blockade of serotonin biosynthesis, suggesting that the observed iAs-induced increase in Ugt1a6a affects GIIS by targeting serotonin or serotonin-related metabolites. Although it is not yet clear exactly which element(s) of the serotonin pathway is/are most responsible for iAs-induced GIIS dysfunction, this study provides evidence that UGT1A6A, acting on the serotonin pathway, regulates GIIS under both normal and pathological conditions.
Keywords: arsenic, diabetes, glucuronidation, insulin secretion, serotonin
INTRODUCTION
The total number of individuals with diabetes mellitus (DM) is expected to reach ~693 million by 2045 (8). Given the substantial comorbidities associated with DM, it is critical to define and address its causes. Although behavioral, nutritional, and genetic factors clearly account for much of DM risk, endocrine-disrupting chemicals (EDCs) have recently emerged as potential contributors (2, 41, 53, 57). One such EDC is arsenic, a toxic metalloid abundantly distributed in the earth’s crust.
Arsenic exposure is an ongoing public health problem, as it naturally contaminates the shallow groundwater underneath an estimated 140 million people worldwide (50). The situation is particularly acute in the Araihazar region of Bangladesh, where arsenic-contaminated groundwater is estimated to cause 21–24% of all mortality (2). At elevated exposure levels (>150 µg/l), arsenic likely plays a causal role in DM (39, 41). In addition, animal studies have revealed that chronic arsenic exposure induces glucose intolerance resulting from insufficient insulin secretion from pancreatic β-cells (12, 32, 37, 60). For these reasons, the relationship between arsenic exposure and β-cell function is under investigation.
Pancreatic β-cells respond to elevated blood glucose by secreting insulin in a tightly regulated multistep process (25). A rise in extracellular glucose enhances β-cell glucose uptake, glycolysis, Krebs cycle flux, and then oxidative phosphorylation, which utilizes O2 and reducing equivalents to convert ADP to ATP. The ensuing increase in the ratio of ATP:ADP closes ATP-sensitive K+ channels, causing membrane depolarization, activating voltage-gated Ca2+ channels, and allowing waves of Ca2+ influx. The rise in intracellular Ca2+ finally enables insulin granule fusion with the plasma membrane to achieve insulin secretion. In addition to this classical mechanism of ATP-dependent insulin secretion, incretins and several other endogenous compounds modulate the sensitivity of β-cells to glucose stimulation via a complex network of signaling cascades, ultimately affecting the rate of secretory vesicle fusion in response to changes in extracellular glucose levels.
A variety of pathways have been implicated in the arsenic-mediated suppression of glucose-induced insulin secretion (GIIS), including inhibition of the Krebs cycle (16, 45), mitochondrial defects (16, 34, 66), excess oxidative stress (7, 9, 21, 43), and altered levels of secretory-complex participants (14). Recently, there have been reports that arsenic activates the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcription factor, which enhances the expression of several antioxidant response element-regulated genes (21, 24, 46, 47, 62). This activation increases scavenging of free radicals, which is believed to suppress the natural oscillations in reactive oxygen species (ROS) that contribute to glucose-induced insulin secretion (GIIS), generating a tradeoff in β-cells between survival and function (15, 20). In the present study, inorganic arsenite (iAs, as As3+) was utilized to interrogate the molecular mechanisms responsible for arsenic-induced suppression of GIIS.
MATERIALS AND METHODS
Animal care.
Male C57BL/6JJcl mice at 8 wk of age were obtained from CLEA (Tokyo, Japan). Animals were group-housed under specific-pathogen-free conditions at 23 ± 2°C and 55 ± 10% relative humidity with a 12-h light-dark cycle and were provided with water and commercially obtained CE-2 diet (CLEA) at the Animal Facility of Kobe Biotechnology Research and Human Resource Development Center of Kobe University. Animals were euthanized for islet isolation by sodium pentobarbital overdose. All animal experiments were approved by the Committee on Animal Experimentation of Kobe University and carried out in accordance with the Guidelines for Animal Experimentation at Kobe University.
Cell culture.
MIN6-K8 cells (passages 19–29) were cultured at 37°C with 5% CO2 in Dulbecco’s modified eagle’s medium (no. 5796; Sigma-Aldrich, St. Louis, MO) containing 10% heat-inactivated fetal bovine serum (French Origin; Biowest, Nuaillé, France). This cell line, which was recently derived from the same IT6 mouse model utilized to generate the original MIN6 cell line, was selected because of its low basal insulin secretion, robust and dose-dependent glucose-induced insulin secretion, and unique responsiveness to incretins (23, 28).
Arsenic treatment.
Twelve- to twenty-four hours after plating in normal medium, 10% of the medium in each well was replaced with a 10× iAs (NaAsO2; Sigma-Aldrich) solution in cell culture medium. Once per day thereafter, all medium was replaced with cell culture medium containing the indicated final concentration of iAs. For 2-day exposure studies, the medium was changed one time, and for 3-day exposure studies, the medium was changed two times. Controls were subjected to the same media-change protocols without the addition of iAs, as daily media changes dramatically increased the total number of cells per well (data not shown).
Insulin secretion.
Insulin secretion experiments were performed as described previously (22). MIN6-K8 cells were preincubated for 30 min in KRBH (2.8 mM glucose ± iAs) and then incubated for 30 min in KRBH containing 2.8 to 16.7 mM glucose, 16.7 mM glucose + 0.1 to 1 nM GLP-1 (no. 4344-V; Peptide Institute, Osaka, Japan), or 11.2 mM glucose + 4–100 µM 5-hydroxytryptophan (5-HTP; no. 18918-21; Nacalai Tesque, Kyoto, Japan). Following stimulation, the buffer in each well was saved for insulin release determination. Triton solution (0.1% in KRBH) was added to each well. Plates were gently swirled for 5 min and then frozen to lyse cells. Secreted insulin and intracellular insulin contents were measured by the Cis Bio Ultra-sensitive HTRF assay (Gif sur Yvette, France). For all 2- and 3-day exposure studies, iAs was present in preincubation buffers but was absent from stimulation buffers because preliminary experiments indicated that iAs did not have an impact on GIIS in that timeframe (data not shown).
Trypan blue exclusion.
Cells were suspended in media containing 0.2% trypan blue dye for at least 3 min, and then, live/dead cells were automatically detected and quantified on the TC20 Automated Cell Counter (Bio-Rad, Hercules, CA).
Metabolomics.
MIN6-K8 cells were incubated in KRBH containing 2.8 mM glucose for 60 min followed by 5 min of stimulation with KRBH containing 11.2 mM glucose. KRBH was removed; cells were washed quickly with ice-cold distilled, deionized water; and extraction buffer (67.5% CH3OH, 7.5% CHCl3, and 25% H2O) was added. Cells were scraped and extracts were collected into vials and then snap frozen in liquid nitrogen. Samples were processed as described previously (22). Analysis was performed using MetaboAnalyst software (Research Resource Identifier: SCR_015539) according to the recommended analysis pipeline (61). Metabolite signals were normalized to the control values, and log transformation was performed. Two-dimensional principal component analysis revealed one major outlier (outside the 95% confidence interval for that exposure group) that was subsequently removed before fold-change analysis. In comparing iAs-exposed metabolite levels to control levels, the threshold for significance was set at an unadjusted P < 0.05 and a fold change >2.0.
RNA sequencing.
Total RNA was isolated using the Qiagen RNA extraction kit per the manufacturer’s instructions. Samples were processed and fragments per kilobase of transcript per million mapped reads (FPKM) values were determined by the RNA-sequencing (RNA-seq) service provider Macrogen (Seoul, Korea), who also mapped trimmed reads to the Mus musculus reference genome using TopHat and then performed transcript assembly using Cufflinks. Data were log transformed and quantile normalization was performed with Preprocess Core’s R library by Macrogen. Samples with at least one FPKM value of 0 or with a mean FPKM value <0.1 were removed from consideration. Genes for which all iAs-exposed samples were above or below control values were targeted for further analysis. This left 43 genes on which Student’s t-tests were performed, with a threshold for significance set to an unadjusted P < 0.1 and fold-change greater than ±1.4. Sequencing data were uploaded to the National Center for Biotechnology Information’s Sequence Read Archive (SRA accession no. SRP139450).
Quantitative PCR.
RNA was isolated from MIN6-K8 cells or islets of Langerhans using the Qiagen RNA extraction kit per the manufacturer’s instructions (kit no. 74106; Qiagen, Hilden, Germany). From this, cDNA was prepared using the ReverTra Ace qPCR RT kit per the manufacturer’s instructions (no. FSQ-101; Toyobo, Osaka, Japan). Taqman probes (Thermo Fisher Scientific, Waltham, MA) were used to quantify relative gene expression. For mouse samples, the reference gene used was Gapdh. For human studies, the relative expression of UGT1A6 was quantified by first finding the ΔCt value for UGT1A6 against each of three different reference genes (18S, HPRT1, and ACTB), followed by calculation of the geometric mean of the relative expression as described previously (55). Probe details are provided in Table 1.
Table 1.
Genes and corresponding Taqman probe details used for qPCR
| Target | Species | Abbreviation | Taqman Probe ID |
|---|---|---|---|
| S100 Calcium binding protein A4 | Mouse | S100a4 | Mm00803372_g1 |
| Zinc finger, DHHC-type containing 22 | Mouse | Zdhhc22 | Mm01308190_m1 |
| Nuclear protein transcription regulator 1 | Mouse | Nupr1 | Mm00498104_m1 |
| Histone cluster 1, H3e | Mouse | Hist1h3e | Mm03017763_gH |
| Insulin-like 3 | Mouse | Insl3 | Mm00502738_m1 |
| UDP glucuronosyltransferase 1 family, polypeptide A6A | Mouse | Ugt1a6a | Mm01967851_s1 |
| Annexin A13 | Mouse | Anxa13 | Mm00466133_m1 |
| Uroplakin 3A | Mouse | Upk3a | Mm00452321_m1 |
| Ribosomal protein L26 | Mouse | Rpl26 | Mm00452579_g1 |
| Glyceraldehyde phosphate dehydrogenase | Mouse | GAPDH | Mm99999915_g1 |
| Dopa-decarboxylase | Mouse | Ddc | Mm00516688_m1 |
| Aralkymine N-acetyltransferase | Mouse | Aanat | Mm01252562_g1 |
| Tryptophan hydroxylase 1 | Mouse | Tph1 | Mm01202614_m1 |
| Tryptophan hydroxylase 2 | Mouse | Tph2 | Mm00557715_m1 |
| N-acetylserotonin O-methyltransferase | Mouse | Asmt | Mm04335444_m1 |
| Ribosomal protein 18S | Human | 18S | Hs99999901_s1 |
| β-Actin | Human | ACTB | Hs99999903_m1 |
| Hypoxanthine phosphoribosyltransferase 1 | Human | HPRT1 | Hs01003267_m1 |
| UDP-glucuronosyltransferase family 1A6 | Human | UGT1A6 | Hs01592477_m1 |
Gene knockdown.
For knockdown experiments, siGENOME SMARTpool siRNA products, containing a total of four unique and specific target sequences for each gene (described in Table 2), were purchased from Dharmacon (Lafayette, CO). The siRNA was administered at the time of plating for each experiment.
Table 2.
siRNA sequences and identification numbers for gene knockdown products
| Target Gene | siRNA Product ID | Target Sequence |
|---|---|---|
| Ugt1a6a | D-057978–01 | AGGAAAAUCUUCUCAGUUA |
| Ugt1a6a | D-057978–02 | UGAAGGAGAUAGUAGAACA |
| Ugt1a6a | D-057978–03 | CAACAUGAUCUUCCUAGGA |
| Ugt1a6a | D-057978–04 | CAACAUGAUUGUCGUGGAC |
| Tph1 | D-047461–01 | GAGCAUAACUAGUGCCAUG |
| Tph1 | D-047461–02 | GCUAUGAACUACAAACAUG |
| Tph1 | D-047461–03 | CCGAUCAGCUCACUGCGAA |
| Tph1 | D-047461–04 | GGGUUAGCCUUUCGAGUCU |
Mouse islets.
Islets were harvested from 12-wk-old male mice as previously described (63). Islets were exposed to the indicated concentrations of iAs in RPMI medium (R8758; Sigma) supplemented with penicillin/streptomycin and 10% heat-inactivated FBS for 2 days. After the first 24 h of iAs exposure, 80% of the medium was replaced with fresh medium containing the indicated concentration of iAs. On the final day of exposure, GIIS was assessed by batch incubation as described previously (63). Male mice were selected for study because sex-specific differences in the expression levels of glucuronosyltransferase proteins have been reported in other tissues (58).
Human islets.
Human islets were collected by Prodo Laboratories (Aliso Viejo, CA) at their laboratory from donor tissue supplied by the United Network for Organ Sharing (Richmond, VA). Islets were cultured for a total of 5–6 days prior to the start of experiments. Donor characteristics are provided in Table 3. Islets were incubated in islet culture medium from Prodo Laboratories containing 105 mg/dl glucose for 3 days with iAs. The morning after plating, 10% of the medium was replaced with a 10× solution of iAs in culture medium. Each day for 2 consecutive days thereafter, 80% of the medium was replaced with fresh medium containing the indicated concentration of iAs.
Table 3.
Donor characteristics for human islets
| Donor No. | Prep. ID | Age, yr | Sex | BMI | HbA1C, % (mmol/mol) |
Ethnicity |
|---|---|---|---|---|---|---|
| 1 | HP-18054-01 | 40 | M | 25.3 | 5.3 (34) | Hispanic |
| 2 | HP-18063-01 | 30 | F | 18.8 | 4.4 (25) | Caucasian |
| 3 | HP-18094-01 | 39 | M | * | 4.5 (26) | Caucasian |
BMI, body mass index; M, male; F, female.
The BMI of donor 3 could not be calculated due to anatomical complications. Prep., Preparation.
O2 consumption.
O2 consumption was measured using the SeaHorse XF Mito Stress Test (no. 103015; Agilent Technologies, Santa Clara, CA) on the SeaHorse XFe24 Analyzer (Agilent Technologies) following the manufacturer’s instructions. Basal assay conditions were 1 mM sodium pyruvate, 2 mM glutamine, and 25 mM glucose in SeaHorse assay buffer (Agilent Technologies). Optimized concentrations of 2 µM oligomycin, 2–3 µM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone, and 0.5 µM rotenone A/antimycin were used. Results were analyzed using Agilent Wave software. Inorganic arsenic was absent during the 1-h preincubation and assay steps to avoid contamination of the SeaHorse Analyzer equipment. O2 consumption data were normalized to total protein per well for each exposure group as determined by BCA assay.
Mitochondrial mass quantification.
Cells were incubated for 30 min at 37°C with 50 nM MitoTracker Green (no. M7514; Thermo Fisher Scientific) plus 10 µM Hoechst 33342 nuclear stain (no. 62249; Thermo Fisher Scientific) in prewarmed KRBH containing 16.7 mM glucose. To quantify mitochondrial mass, the integrated mitochondrial signal per image was divided by the number of nuclei stained within that image.
Serotonin glucuronidation activity.
Lysates were prepared in 50 mM Tris·HCl buffer (pH 7.4) and 250 mM sucrose with a protease inhibitor cocktail (ref. no. 11697498001; Sigma) and sonicated briefly, and then snap frozen in liquid nitrogen and stored for later analysis. Serotonin glucuronidation was determined using lysates at a uniform protein concentration of 0.5 mg/ml in a buffer containing 5 mM serotonin as described previously (52). The rate of serotonin-glucuronide formation was normalized to total protein as determined by BCA assay. The investigator carrying out this assay was blind to the sample conditions and groupings.
Statistical analysis.
One-way ANOVA tests with Holm-Sidak multiple comparison adjustment or individual Student’s t-tests were performed using GraphPad Prism software, except where explicitly stated. The specific comparisons made are indicated in each figure. No additional statistical comparisons were done other than those indicated by a statistical comparison marker of NS (for not significant) or the indicated significance symbols. All values are represented as means ± SE. The use of “n” in the figure legends refers to the number of biological replicates per condition represented in each panel.
RESULTS
β-Cell and islet function.
We and other groups reported that mice chronically exposed to iAs became glucose intolerant and that circulating insulin was insufficient to meet the metabolic demand, accompanied by signs of pancreatic dysfunction (32, 37). This led to the conclusion that a β-cell insufficiency may underlie iAs-induced glucose intolerance. To interrogate this phenomenon, we developed a model system of iAs exposure in the MIN6-K8 mouse pancreatic β-cell line wherein the most common form of arsenic found in drinking water, iAs, at environmentally relevant concentrations, was administered (59). Three days of iAs exposure at the lowest concentration tested (0.1 µM) significantly decreased GIIS at 8.8 mM and 11.2 mM glucose stimulation (Fig. 1A). Exposure to higher concentrations up to 1 µM reduced GIIS by >50% at 11.2 mM glucose stimulation and almost completely eliminated GIIS at 8.8 mM stimulation. Despite this decrease in GIIS, neither potassium-stimulated nor basal insulin secretion was affected following iAs exposure (Fig. 1B). An inhibitory effect of 1 µM iAs on GIIS was also observed after only 2 days of exposure (Fig. 1C) but was not observed when iAs was administered only during the final 30 min of the insulin secretion assay (Fig. 1D).
Fig. 1.

Effects of inorganic arsenite (iAs) on insulin secretion. A and B: glucose-induced insulin secretion (GIIS) (n = 5–6) (A) and potassium-induced insulin secretion (n = 3–4) (B) in MIN6-K8 cells following 3 days of iAs exposure. C: GIIS following 2 days of iAs exposure (n = 5–6). D: GIIS following 30 min of iAs exposure (iAs included in stimulation buffer only) (n = 3–4). E and F: 2 days of iAs exposure in mouse islets (E) and 3 days of iAs exposure in human islets (F). A–C, E and F: 3 independent experiments were performed. D: 2 independent experiments were performed. Statistics: A, C, and F: for each glucose concentration, one-way ANOVA was performed comparing iAs exposure groups to the control group. B, D, and E: Student’s t-tests were performed for each stimulation concentration. *P < 0.05, **P < 0.01, ΩP < 0.0001.
The GIIS-inhibitory effects of long-term iAs exposure were confirmed in isolated islets of Langerhans from mice and humans. GIIS in mouse islets exposed to 2 µM iAs for 2 days was significantly decreased at both 8.8 and 11.1 mM glucose without changes to basal secretion (Fig. 1E). Human islets exposed to either 1 or 2 µM iAs for 3 days displayed a 30–40% reduction in GIIS (Fig. 1F).
Cell number, transcription factors, and antioxidant genes.
MIN6-K8 cells exposed to 1 µM iAs for 3 days showed no differences in total DNA, total protein, cell number, or viability (Fig. 2A). Additionally, expression levels of several previously identified β-cell identity-regulating transcription factors (40) were unchanged by iAs (Fig. 2E). Two antioxidant genes, superoxide dismutase 1 (Sod1) and superoxide dismutase 2 (Sod2), were increased modestly by iAs (Fig. 2E).
Fig. 2.

Markers of cell and mitochondrial health. A, left to right: cells per well (n = 12) and percentage of cells alive as determined by trypan blue staining (n = 12), total DNA (n = 4), and total protein (n = 5) per well. B: fluorescence microscopy showing total mitochondrial staining with MitoTracker Green (top) and Hoechst 33342 staining of nuclei (bottom) following 3 days of inorganic arsenite (iAs) exposure (3 images/well and 3 wells/condition were examined). C: total mitochondrial signal detected in each image, number of nuclei counted per image, and mitochondrial mass calculated as the ratio of mitochondrial signal to the number of nuclei. D: O2 consumption during mitochondrial stress test as measured by the SeaHorse Analyzer, normalized to total protein per well (n = 5). E: Taqman qPCR of β-cell identity-regulating transcription factors and antioxidant genes (n = 6–12). NS, not significant. A–D: 3 independent experiments were performed. E: 2 independent experiments were performed. Statistics: A and D: Student’s t-tests. C and E: one-way ANOVA between each iAs exposure and the control. *P < 0.05, **P < 0.01.
Mitochondrial mass and O2 consumption.
Prior studies reported that chronic arsenic exposure alters mitochondrial function, which generated the hypothesis that similar mechanisms might be at work in this model (16, 43). As a general indicator of mitochondrial status (10), mitochondrial mass was measured following 3 days of iAs exposure (Fig. 2B). Mitochondrial signal per image, number of nuclei stained, and mitochondrial signal normalized for nuclei were all unchanged from 0 to 1 µM iAs exposure (Fig. 2C). At 5 µM iAs, a significant increase in mitochondrial staining per nucleus was observed, reflecting an increase in mitochondrial mass. This concentration was five times higher than the concentration used to study insulin-secretory effects (1 µM) and was therefore not investigated further.
To assess the effects of iAs on mitochondrial function in a more comprehensive manner (6), O2 consumption under a variety of chemical stressors was measured using the SeaHorse Analyzer Mito Stress Test (Fig. 2D). O2 consumption is an indicator of oxidative phosphorylation, occurring as Krebs cycle-generated reducing equivalents are used to pump protons across the inner mitochondrial membrane for ATP production. Several direct and derived measures of O2 consumption were unchanged in this model system following 3 days of 1 µM iAs exposure, including O2 consumption associated with ATP production and maximal respiration capacity (data not shown). Collectively, these measurements suggested that the GIIS defect induced by iAs could not be explained by mitochondrial mass or respiration.
Serotonin metabolism.
An unbiased metabolomics-based approach was utilized to evaluate the effects of iAs exposure in MIN6-K8 cells. A 50% decrease in 5-HTP was observed in 1 µM iAs-exposed MIN6-K8 cells following 5 min of glucose stimulation (Fig. 3A). As 5-HTP is the rate-limiting precursor for serotonin synthesis, direct measurement of serotonin in cultured MIN6-K8 cells exposed for 3 days to 1 µM iAs was performed by ELISA, revealing that serotonin was also decreased by 1 µM iAs exposure in MIN6-K8 cells (Fig. 3B).
Fig. 3.

Metabolomics and 5-hydroxytryptophan (5-HTP) intervention following 3 days of inorganic arsenite (iAs) exposure. A: 5-HTP measured by unbiased metabolomics analysis as described in materials and methods (n = 11). B: serotonin in lysates measured by ELISA (n = 3–4). C: insulin secretion following supplementation with 5-HTP during 30 min of preincubation plus glucose stimulation. A and B: 2 independent experiments were performed. C: 3 independent experiments were performed. Statistics: A: analysis detailed in materials and methods. B: Student’s t-test. C: one-way ANOVA between each condition and each of the two 0-µM 5-HTP conditions. *P < 0.05, ***P < 0.001, ΩP < 0.0001.
In β-cells, exogenous 5-HTP is rapidly taken up by L-type amino acid transporters and converted to serotonin, which then accumulates in secretory granules (13, 27). To test whether repletion of this pathway could rescue the iAs-induced GIIS defect, 5-HTP was administered to iAs-exposed cells during preincubation and costimulation with glucose as part of the GIIS assay. This treatment reversed the iAs-induced reductions in GIIS without significantly affecting basal insulin secretion (Fig. 3C). These results suggested that part of the effects of iAs were mediated by changes in the serotonin pathway.
RNA sequencing.
To better understand how iAs might disrupt serotonin metabolism, RNA-seq was performed. Nine candidate genes were identified using the criteria detailed in materials and methods (Fig. 4, A and B). Expression changes were confirmed for four of the nine candidate genes: ribosomal protein 26, UDP-glucuronosyltransferase 1 family, polypeptide a6a (Ugt1a6a), uroplakin 3a, and S100 calcium binding protein a4 (Fig. 4B). Although each of these genes could have been hypothesized to mediate the effects of iAs, only one of these genes, Ugt1a6a, has a known function that overlaps with the observed disruption of serotonin metabolism. UGT1A6A and its human homolog, UGT1A6, are phase II detoxification enzymes that conjugate glucuronic acid to serotonin and other small aromatic amines for cellular export and excretion (11, 33, 35). Ugt1a6a had the highest level of expression among Ugt1 gene family members in MIN6-K8 cells as determined by RNA-seq analysis (Fig. 4C), and its expression was robustly upregulated in mouse islets following iAs exposure (Fig. 4D). Additionally, the expression of its human homolog, UGT1A6 increased in each set of donor islets following 3 days of iAs exposure (Fig. 4D), revealing that this gene and its human homolog are both robustly responsive to iAs, making it the highest priority candidate gene for further interrogation.
Fig. 4.

RNA sequencing (RNA-seq) and gene candidate confirmation following inorganic arsenite (iAs) exposure. A: volcano plot of RNA-seq results. Gray boundaries marked by dotted lines represent thresholds for candidate gene identification as described in materials and methods (n = 3). B: relative gene expression derived from (top) RNA-seq fragments per kilobase of transcript per million mapped reads (FPKM) values and (bottom) independent Taqman qPCR gene expression measurements for candidate genes following 3 days of iAs exposure (n = 4). C: original FPKM values of Ugt1a6a and other Ugt1a family member genes from the same data set used in A (n = 3). D: Taqman qPCR of Ugt1a6a following 2 days (left) or iAs exposure in mouse islets and 3 days (right) of iAs exposure in human islets. NS, not significant; ND, not detected in control and/or iAs-exposure groups. A–C: RNA-seq was performed on samples collected during 1 experiment. B and D: 3 independent experiments were performed for Taqman analyses. Statistics: A and B: analysis detailed in materials and methods. C: no separate statistical analyses were performed aside from the unbiased assessment of all RNA-seq data as described in materials and methods. D: one-way ANOVA was performed for mouse islet Ugt1a6a gene expression; however, human islet data were not subjected to statistical analysis. *P < 0.05, ΩP < 0.0001.
Although the upregulation of Ugt1a6a could help explain the observed effects of iAs, the serotonin metabolism pathway is regulated at multiple levels. For instance, arsenic in the drinking water of mice has previously been shown to decrease serotonin levels in the brain by decreasing serotonin production through changes in the expression of the rate-limiting enzyme for serotonin biosynthesis, tryptophan hydroxylase 1 (Tph1) (38). To interrogate the serotonin-metabolic state of these cells following iAs exposure, both RNA-seq and Taqman gene expression assays were used. Tph1 expression was not altered by iAs exposure (Fig. 5A). Furthermore, there were no observed differences in the expression levels of tryptophan hydroxylase 2 (Tph2), dopa decarboxylase (Ddc), and monoamine oxidase (MAO). Interestingly, gene expression of aralkymine N-acetyltransferase (Aanat), which catalyzes the conversion of serotonin to N-acetylserotonin and eventually melatonin, was barely detected by RNA-seq and undetectable by Taqman assay (Fig. 5C). Expression characteristics of Tph2 were similar to Aanat. Tryptophan as the substrate utilized by Tph1 for the production of 5-HTP and cAMP as a positive regulator of Tph1 expression (26, 65) (see Fig. 7, left) were quantified from the aforementioned metabolomics analysis. There were no changes observed in levels of these metabolites in iAs-exposed MIN6-K8 cells (Fig. 5D).
Fig. 5.
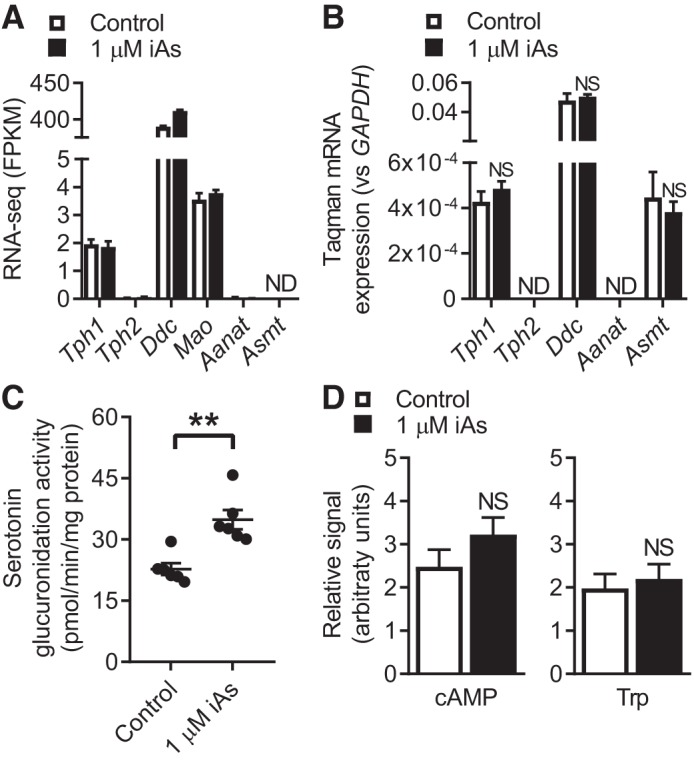
Serotonin metabolism characteristics. A and B: RNA sequencing (RNA-seq) (A) or Taqman (B) expression values for genes related to serotonin metabolism. C: the rate of serotonin glucuronidation in MIN6-K8 lysates following 3 days of inorganic arsenite (iAs) exposure, normalized to total protein. This assay is described in more detail in materials and methods. (n = 6). D: cyclic AMP (cAMP) and tryptophan (Trp) content quantified by metabolomics as described previously (n = 11). NS, not significant; ND, not detected. A: RNA-seq was performed on samples collected during 1 experiment. B: 3 independent experiments. C and D: 2 independent experiments. Statistics: A: RNA-seq data were analyzed as described in materials and methods. B and C: Student’s t-test. D: analysis detailed in materials and methods. **P < 0.01.
Fig. 7.

Model of endogenous- and inorganic arsenite (iAs)-induced serotonin metabolism. Left: UGT1A6A negatively regulates glucose-induced insulin secretion (GIIS) by glucuronidation of serotonin (and potentially its metabolites). Right: iAs exposure increases Ugt1a6a expression, raising the rate of glucuronidation, decreasing serotonin metabolites, ultimately lowering GIIS.
Serotonin glucuronidation activity.
Given that UGT1A6A catalyzes serotonin glucuronidation, and iAs increased Ugt1a6a gene expression, we hypothesized that iAs would increase the rate of serotonin glucuronidation in MIN6-K8 cells. Consistent with this rationale, the rate of serotonin glucuronidation was increased by 53% in lysates from MIN6-K8 cells exposed to 1 µM iAs for 3 days (Fig. 5C). Thus a functional consequence of iAs exposure in MIN6-K8 cells is increased serotonin glucuronidation.
The combined gene expression, metabolomics, and enzymatic activity results suggested that iAs may increase Ugt1a6a gene expression, depleting serotonin or serotonin-related metabolites and leading to the suppression of GIIS. To determine whether the observed increase in Ugt1a6a expression might mediate the iAs-induced impairment in GIIS, Ugt1a6a expression was knocked down by siRNA in MIN6-K8 cells. Knockdown efficiency was ~70%. Knockdown in the absence of iAs significantly enhanced glucose- and incretin-induced insulin secretion without significant effects on basal- or potassium-stimulated insulin secretion (Fig. 6A), demonstrating that this gene plays a suppressive role in GIIS under normal conditions. Knockdown in the context of 2 days of iAs exposure reversed the effects of iAs on GIIS by 63% and increased basal insulin secretion in iAs-exposed cells by 70% (Fig. 6B).
Fig. 6.
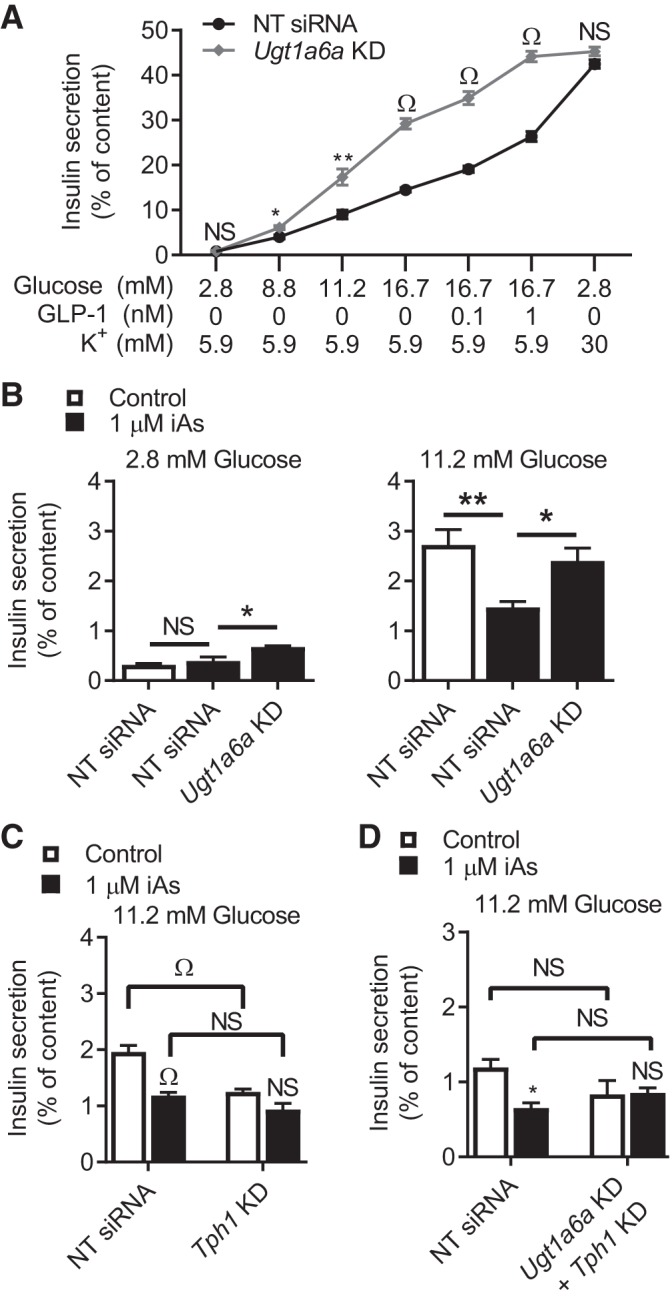
Glucose-induced insulin secretion (GIIS) following gene knockdown. A: effects of Ugt1a6a knockdown on insulin secretion in response to glucose, incretin, or potassium stimuli in the absence of arsenic exposure (n = 4). B: effects of Ugt1a6a knockdown on 2.8 and 11.2 mM glucose after 2 days of 1 µM inorganic arsenite (iAs) exposure (n = 4). C and D: 11.2 mM GIIS following 3 days of 1 µM iAs exposure and knockdown (KD) of the indicated gene(s). NS, not significant. A and B: 4 independent experiments were performed. C and D: 2 independent experiments were performed. Statistics: A: 1 Student’s t-test between knockdown and control results was performed for each stimulation condition. B: one-way ANOVA was performed between Scramble KD + 1 µM iAs and other conditions. *P < 0.05, **P < 0.01, ΩP < 0.0001. B and C: one-way ANOVA was performed comparing targeted knockdown to nontarget (NT siRNA) knockdown within each iAs exposure condition and between iAs exposure conditions within each knockdown group. All statistical comparisons performed are indicated.
Serotonin metabolism.
If the effects of iAs on GIIS were mediated by a decrease in serotonin or downstream metabolites, then predepletion of serotonin by inhibiting serotonin biosynthesis would be expected to decrease GIIS and ameliorate the effects of iAs exposure. To test this hypothesis, the serotonin pathway itself was targeted using siRNA-mediated knockdown of Tph1. In agreement with this hypothesis, depleting the serotonin pathway with Tph1 siRNA in our model decreased GIIS significantly and eliminated the effects of 3 days of 1 µM iAs exposure on GIIS (Fig. 6C). Knockdown efficiency was 53% (data not shown). This suggested that some change in serotonin metabolites at or downstream of Tph1 activity was responsible for the effects of iAs.
Finally, to examine whether effects of Ugt1a6a knockdown were dependent on intact serotonin metabolism, both Tph1 and Ugt1a6a siRNA were administered simultaneously. In this case, double knockdown inhibited GIIS similarly to individual Tph1 knockdown in the absence of iAs, and blocked the GIIS-enhancing effects of Ugt1a6a knockdown in the context of 1 µM iAs exposure. These data collectively suggest that UGT1A6A negatively regulates GIIS under normal conditions (Fig. 7, left), and iAs exposure increases Ugt1a6a expression, enhancing its repressive effects (Fig. 7, right), in both instances through the conjugation of serotonin and/or metabolites involved in serotonin metabolism.
DISCUSSION
Mice exposed to iAs for several weeks develop glucose intolerance with a combination of defects in insulin secretion (32, 37). The present study supports a central role for β-cell dysfunction in the link between iAs exposure and glucose intolerance and offers new insights into the mechanisms by which iAs may disrupt β-cell function. In light of the restorative effects of Ugt1a6a knockdown and the dependence on serotonin metabolites to achieve these results, we propose that UGT1A6A acts as a negative regulator of GIIS by facilitating the glucuronidation of serotonin or its metabolites and that the inhibitory effects of iAs exposure are largely mediated by the upregulation of Ugt1a6a gene expression.
We have not identified the specific metabolite(s) in the serotonin pathway that are most important for this model, but our findings suggest that serotonin itself may be the primary mediator of arsenic’s effects and the major target of UGT1A6A. Although historically a source of significant debate (36, 44, 48, 49), recent reports have shown that serotonin can potentiate GIIS through autocrine activation of serotonin receptor 3a following secretion (30, 31, 42) or receptor-independent intracellular protein serotonylation (44). Serotonin is also the only endogenous metabolite in the pathway that mouse UGT1A6A is known to target (4, 54). It is unlikely that the products of serotonin metabolism downstream of AANAT activity (N-acetylserotonin, melatonin, and 6-hydroxymelatonin) are involved, as Aanat expression in our model was nearly undetectable. This is consistent with other studies reporting that inbred mice on the C57BL/6 genetic background (the same strain from which the MIN6-K8 cell line was derived) possess a defect in Aanat expression (17, 51). Therefore, our model should be suitable for comparison with several published in vivo studies of arsenic exposure utilizing C57BL/6 mice (reviewed in Ref. 39). Additionally, the products of monoamine oxidase, such as 5-HIAL, 5-HTOL, or 5-HIAA, may play a role, but they are not currently recognized as major regulators of β-cell function (19).
The mechanism by which iAs exposure upregulated Ugt1a6a expression is not currently known; however, prior studies in other tissues have shown that Ugt1a6a is positively regulated by the transcription factor Nrf2 (18, 33, 56). Multiple groups have demonstrated that iAs induces Nrf2 transcriptional activity in β-cells and islets as part of the response to oxidative damage (21, 62). In these previous studies, Nrf2 regulated the expression levels of antioxidant genes Sod1, Sod2, and catalase, which in turn suppressed the natural oscillations in ROS that couple glucose metabolism to insulin secretion in β-cells. In our study, the same two Nrf2-activated genes (Sod1 and Sod2) were mildly upregulated in response to iAs exposure, providing indirect evidence of Nrf2 transcriptional activity, increasing its plausibility as a regulator of Ugt1a6a gene expression. This hypothesis must be investigated in detail, however, before concluding that a regulatory relationship exists between Nrf2 and Ugt1a6a in β-cells.
Although these data support a novel model of iAs-induced β-cell dysfunction mediated by enzymatic depletion of a serotonin metabolite, the present study has some limitations. The specific molecule targeted by UGT1A6A remains unidentified. The only endogenous metabolite that UGT1A6A has been reported to conjugate is serotonin (4, 54), but the human homolog UGT1A6 is known to target several different serotonin-related metabolites (5, 35, 52), each of which may affect GIIS or β-cell resistance to oxidative damage (29, 38, 64). It is therefore warranted to evaluate whether UGT1A6A is capable of catalyzing the conjugation of any other serotonin metabolites (to our knowledge a comprehensive evaluation of this capability has not been reported). Additionally, the other three genes that were significantly altered by iAs exposure were not evaluated, although they may also contribute to the GIIS defects observed. Finally, some intermediates of iAs disposal, such as mono- and dimethylated arsenic, uniquely affect β-cells at lower concentrations (16). Therefore, future studies should utilize different physiologically-relevant species of iAs.
Despite its limitations, this report is the first to implicate glucuronidation in the control of GIIS, to describe the responsiveness of Ugt1a6a to arsenic exposure β-cells, and to identify serotonin metabolism as a β-cell-specific factor in EDC exposure. We provided evidence that disruption of serotonin metabolism is a mechanism by which environmental toxicants can impair insulin secretion, disabling β-cell normal adaptive capacity and potentiating glucose intolerance in the context of compounding diabetogenic factors. In the context of in vivo experiments, the implications for altered serotonin production may be broader, as serotonin has been demonstrated to affect glucagon secretion via paracrine action on α-cell serotonin receptors (1, 3). While associations between environmental toxicants and diabetes risk continue to mount, further delineating the mechanisms by which these agents disrupt β-cell physiology will be essential for devising strategies to reduce the burden of this devastating disease in exposed populations.
GRANTS
This study was supported by the Japan Society for the Promotion of Science Postdoctoral Fellowship for Overseas Researchers PF01 and PF02 (to C. M. Carmean); Japan Society for the Promotion of Science Grant-in-Aid for Scientific Research (S) 24229007 (to S. Seino); American Diabetes Association Grant 1-17-JDF-033; U.S. Public Health Service Grants R01-ES-028879, P30-ES-027792, and P30-DK-020595 (to R. M. Sargis); and a Honjo International Scholarship (to O. S. Oduori). This work was also funded in part by MSD K.K. Japan (to S. Seino).
DISCLOSURES
R. M. Sargis has received honoraria from CVS/Health. S. Seino has consulted for JCR Pharmaceuticals and held scientific advisory positions with Kansai Electric Power Medical Research Institute and Servier Laboratories. S. Seino has served on speaker’s bureaus for Novo Nordisk Pharma K.K. and Sumitomo Dainippon Pharma, Co., Ltd., and Novartis Pharma K.K. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
C.M.C., N.Y., and S.S. conceived and designed research; C.M.C., O.S.O., C.K., A.K., A.G.K., G.H., M.L., S.H., and M.K. performed experiments; C.M.C., N.Y., H.T., O.S.O., C.K., A.G.K., G.H., M.L., and M.K. analyzed data; C.M.C., N.Y., H.T., R.M.S., and S.S. interpreted results of experiments; C.M.C. prepared figures; C.M.C. drafted manuscript; C.M.C., N.Y., H.T., O.S.O., A.G.K., R.M.S., and S.S. edited and revised manuscript; C.M.C., N.Y., H.T., O.S.O., C.K., A.K., A.G.K., G.H., M.L., S.H., M.K., R.M.S., and S.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Graeme Bell, Matthew J. Brady, Brian Layden, Nissim Hay, Louis Philipson, Barton Wicksteed, Mustafa Omami, Jose Oberholzer, Joe I. Walker, Grace Honkawa, Junko Kamei, and Masako Hirata for support and guidance. We also thank Chihiro Seki, Ayako Kawabata, Niina Ota, Ritsuko Hoshikawa, Tomohide Hayami, Kouhei Honda, Naoya Murao, Daniel Ruiz, and Mahira Hashim for sharing technical expertise.
REFERENCES
- 1.Almaça J, Molina J, Menegaz D, Pronin AN, Tamayo A, Slepak V, Berggren PO, Caicedo A. Human beta cells produce and release serotonin to inhibit glucagon secretion from alpha cells. Cell Rep 17: 3281–3291, 2016. doi: 10.1016/j.celrep.2016.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Argos M, Kalra T, Rathouz PJ, Chen Y, Pierce B, Parvez F, Islam T, Ahmed A, Rakibuz-Zaman M, Hasan R, Sarwar G, Slavkovich V, van Geen A, Graziano J, Ahsan H. Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet 376: 252–258, 2010. doi: 10.1016/S0140-6736(10)60481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennet H, Balhuizen A, Medina A, Dekker Nitert M, Ottosson Laakso E, Essén S, Spégel P, Storm P, Krus U, Wierup N, Fex M. Altered serotonin (5-HT) 1D and 2A receptor expression may contribute to defective insulin and glucagon secretion in human type 2 diabetes. Peptides 71: 113–120, 2015. doi: 10.1016/j.peptides.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Bock KW. Roles of human UDP-glucuronosyltransferases in clearance and homeostasis of endogenous substrates, and functional implications. Biochem Pharmacol 96: 77–82, 2015. doi: 10.1016/j.bcp.2015.04.020. [DOI] [PubMed] [Google Scholar]
- 5.Bock KW, Köhle C. UDP-glucuronosyltransferase 1A6: structural, functional, and regulatory aspects. Methods Enzymol 400: 57–75, 2005. doi: 10.1016/S0076-6879(05)00004-2. [DOI] [PubMed] [Google Scholar]
- 6.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakraborty D, Mukherjee A, Sikdar S, Paul A, Ghosh S, Khuda-Bukhsh AR. [6]-Gingerol isolated from ginger attenuates sodium arsenite induced oxidative stress and plays a corrective role in improving insulin signaling in mice. Toxicol Lett 210: 34–43, 2012. doi: 10.1016/j.toxlet.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, Malanda B. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 138: 271–281, 2018. doi: 10.1016/j.diabres.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 9.Cobo JM, Castiñeira M. Oxidative stress, mitochondrial respiration, and glycemic control: clues from chronic supplementation with Cr3+ or As3+ to male Wistar rats. Nutrition 13: 965–970, 1997. doi: 10.1016/S0899-9007(97)00338-9. [DOI] [PubMed] [Google Scholar]
- 10.Cottet-Rousselle C, Ronot X, Leverve X, Mayol JF. Cytometric assessment of mitochondria using fluorescent probes. Cytometry A 79A: 405–425, 2011. doi: 10.1002/cyto.a.21061. [DOI] [PubMed] [Google Scholar]
- 11.Court MH. Isoform-selective probe substrates for in vitro studies of human UDP-glucuronosyltransferases. Methods Enzymol 400: 104–116, 2005. doi: 10.1016/S0076-6879(05)00007-8. [DOI] [PubMed] [Google Scholar]
- 12.Dávila-Esqueda ME, Morales JM, Jiménez-Capdeville ME, De la Cruz E, Falcón-Escobedo R, Chi-Ahumada E, Martin-Pérez S. Low-level subchronic arsenic exposure from prenatal developmental stages to adult life results in an impaired glucose homeostasis. Exp Clin Endocrinol Diabetes 119: 613–617, 2011. doi: 10.1055/s-0031-1287782. [DOI] [PubMed] [Google Scholar]
- 13.Di Gialleonardo V, Signore A, Scheerstra EA, Visser AK, van Waarde A, Dierckx RA, de Vries EF. 11C-hydroxytryptophan uptake and metabolism in endocrine and exocrine pancreas. J Nucl Med 53: 1755–1763, 2012. doi: 10.2967/jnumed.112.104117. [DOI] [PubMed] [Google Scholar]
- 14.Díaz-Villaseñor A, Burns AL, Salazar AM, Sordo M, Hiriart M, Cebrián ME, Ostrosky-Wegman P. Arsenite reduces insulin secretion in rat pancreatic beta-cells by decreasing the calcium-dependent calpain-10 proteolysis of SNAP-25. Toxicol Appl Pharmacol 231: 291–299, 2008. doi: 10.1016/j.taap.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 15.Douillet C, Currier J, Saunders J, Bodnar WM, Matoušek T, Stýblo M. Methylated trivalent arsenicals are potent inhibitors of glucose stimulated insulin secretion by murine pancreatic islets. Toxicol Appl Pharmacol 267: 11–15, 2013. doi: 10.1016/j.taap.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dover EN, Beck R, Huang MC, Douillet C, Wang Z, Klett EL, Stýblo M. Arsenite and methylarsonite inhibit mitochondrial metabolism and glucose-stimulated insulin secretion in INS-1 832/13 β cells. Arch Toxicol 92: 693–704, 2018. doi: 10.1007/s00204-017-2074-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ebihara S, Marks T, Hudson DJ, Menaker M. Genetic control of melatonin synthesis in the pineal gland of the mouse. Science 231: 491–493, 1986. doi: 10.1126/science.3941912. [DOI] [PubMed] [Google Scholar]
- 18.Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, Harada T, Yamamoto M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci 59: 169–177, 2001. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 19.Feldman JM, Chapman B. Monoamine oxidase inhibitors: nature of their interaction with rabbit pancreatic islets to alter insluin secretion. Diabetologia 11: 487–494, 1975. doi: 10.1007/BF01222097. [DOI] [PubMed] [Google Scholar]
- 20.Fu J, Hou Y, Xue P, Wang H, Xu Y, Qu W, Zhang Q, Pi J. Nrf2 in Type 2 diabetes and diabetic complications: Yin and Yang. Curr Opin Toxicol 1: 9–19, 2016. doi: 10.1016/j.cotox.2016.08.001. [DOI] [Google Scholar]
- 21.Fu J, Woods CG, Yehuda-Shnaidman E, Zhang Q, Wong V, Collins S, Sun G, Andersen ME, Pi J. Low-level arsenic impairs glucose-stimulated insulin secretion in pancreatic beta cells: involvement of cellular adaptive response to oxidative stress. Environ Health Perspect 118: 864–870, 2010. doi: 10.1289/ehp.0901608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gheni G, Ogura M, Iwasaki M, Yokoi N, Minami K, Nakayama Y, Harada K, Hastoy B, Wu X, Takahashi H, Kimura K, Matsubara T, Hoshikawa R, Hatano N, Sugawara K, Shibasaki T, Inagaki N, Bamba T, Mizoguchi A, Fukusaki E, Rorsman P, Seino S. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep 9: 661–673, 2014. doi: 10.1016/j.celrep.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hashim M, Yokoi N, Takahashi H, Gheni G, Okechi OS, Hayami T, Murao N, Hidaka S, Minami K, Mizoguchi A, Seino S. Inhibition of SNAT5 induces incretin-responsive state from incretin-unresponsive state in pancreatic β-cells: study of β-cell spheroid clusters as a model. Diabetes 67: 1795–1806, 2018. doi: 10.2337/db17-1486. [DOI] [PubMed] [Google Scholar]
- 24.He X, Chen MG, Lin GX, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2 x Keap1 x Cul3 complex and recruiting Nrf2 x Maf to the antioxidant response element enhancer. J Biol Chem 281: 23620–23631, 2006. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- 25.Henquin JC. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52: 739–751, 2009. doi: 10.1007/s00125-009-1314-y. [DOI] [PubMed] [Google Scholar]
- 26.Huang Z, Liu T, Chattoraj A, Ahmed S, Wang MM, Deng J, Sun X, Borjigin J. Posttranslational regulation of TPH1 is responsible for the nightly surge of 5-HT output in the rat pineal gland. J Pineal Res 45: 506–514, 2008. doi: 10.1111/j.1600-079X.2008.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutton JC, Peshavaria M, Tooke NE. 5-Hydroxytryptamine transport in cells and secretory granules from a transplantable rat insulinoma. Biochem J 210: 803–810, 1983. doi: 10.1042/bj2100803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iwasaki M, Minami K, Shibasaki T, Miki T, Miyazaki J, Seino S. Establishment of new clonal pancreatic β-cell lines (MIN6-K) useful for study of incretin/cyclic adenosine monophosphate signaling. J Diabetes Investig 1: 137–142, 2010. doi: 10.1111/j.2040-1124.2010.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kemp DM, Ubeda M, Habener JF. Identification and functional characterization of melatonin Mel 1a receptors in pancreatic beta cells: potential role in incretin-mediated cell function by sensitization of cAMP signaling. Mol Cell Endocrinol 191: 157–166, 2002. doi: 10.1016/S0303-7207(02)00064-3. [DOI] [PubMed] [Google Scholar]
- 30.Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, Fujitani Y, Kawamori R, Miyatsuka T, Kosaka Y, Yang K, Honig G, van der Hart M, Kishimoto N, Wang J, Yagihashi S, Tecott LH, Watada H, German MS. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16: 804–808, 2010. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim K, Oh CM, Ohara-Imaizumi M, Park S, Namkung J, Yadav VK, Tamarina NA, Roe MW, Philipson LH, Karsenty G, Nagamatsu S, German MS, Kim H. Functional role of serotonin in insulin secretion in a diet-induced insulin-resistant state. Endocrinology 156: 444–452, 2015. doi: 10.1210/en.2014-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirkley AG, Carmean CM, Ruiz D, Ye H, Regnier SM, Poudel A, Hara M, Kamau W, Johnson DN, Roberts AA, Parsons PJ, Seino S, Sargis RM. Arsenic exposure induces glucose intolerance and alters global energy metabolism. Am J Physiol Regul Integr Comp Physiol, 314: R294–R303, 2018. doi: 10.1152/ajpregu.00522.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Köhle C, Badary OA, Nill K, Bock-Hennig BS, Bock KW. Serotonin glucuronidation by Ah receptor- and oxidative stress-inducible human UDP-glucuronosyltransferase (UGT) 1A6 in Caco-2 cells. Biochem Pharmacol 69: 1397–1402, 2005. doi: 10.1016/j.bcp.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Krauss S, Zhang CY, Scorrano L, Dalgaard LT, St-Pierre J, Grey ST, Lowell BB. Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest 112: 1831–1842, 2003. doi: 10.1172/JCI200319774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnaswamy S, Hao Q, Von Moltke LL, Greenblatt DJ, Court MH. Evaluation of 5-hydroxytryptophol and other endogenous serotonin (5-hydroxytryptamine) analogs as substrates for UDP-glucuronosyltransferase 1A6. Drug Metab Dispos 32: 862–869, 2004. doi: 10.1124/dmd.32.8.862. [DOI] [PubMed] [Google Scholar]
- 36.Lindström P, Sehlin J. Opposite effects of 5-hydroxytryptophan and 5-hydroxytryptamine on the function of microdissected ob/ob-mouse pancreatic islets. Diabetologia 24: 52–57, 1983. doi: 10.1007/BF00275948. [DOI] [PubMed] [Google Scholar]
- 37.Liu S, Guo X, Wu B, Yu H, Zhang X, Li M. Arsenic induces diabetic effects through beta-cell dysfunction and increased gluconeogenesis in mice. Sci Rep 4: 6894, 2014. doi: 10.1038/srep06894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, Piao F, Li Y. Protective effect of taurine on the decreased biogenic amine neurotransmitter levels in the brain of mice exposed to arsenic. Adv Exp Med Biol 776: 277–287, 2013. doi: 10.1007/978-1-4614-6093-0_26. [DOI] [PubMed] [Google Scholar]
- 39.Maull EA, Ahsan H, Edwards J, Longnecker MP, Navas-Acien A, Pi J, Silbergeld EK, Styblo M, Tseng CH, Thayer KA, Loomis D. Evaluation of the association between arsenic and diabetes: a National Toxicology Program workshop review. Environ Health Perspect 120: 1658–1670, 2012. doi: 10.1289/ehp.1104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minami K, Miyawaki K, Hara M, Yamada S, Seino S. Tracing phenotypic reversibility of pancreatic β-cells in vitro. J Diabetes Investig 1: 242–251, 2010. doi: 10.1111/j.2040-1124.2010.00051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Navas-Acien A, Silbergeld EK, Streeter RA, Clark JM, Burke TA, Guallar E. Arsenic exposure and type 2 diabetes: a systematic review of the experimental and epidemiological evidence. Environ Health Perspect 114: 641–648, 2006. doi: 10.1289/ehp.8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohara-Imaizumi M, Kim H, Yoshida M, Fujiwara T, Aoyagi K, Toyofuku Y, Nakamichi Y, Nishiwaki C, Okamura T, Uchida T, Fujitani Y, Akagawa K, Kakei M, Watada H, German MS, Nagamatsu S. Serotonin regulates glucose-stimulated insulin secretion from pancreatic β cells during pregnancy. Proc Natl Acad Sci USA 110: 19420–19425, 2013. doi: 10.1073/pnas.1310953110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pan X, Jiang L, Zhong L, Geng C, Jia L, Liu S, Guan H, Yang G, Yao X, Piao F, Sun X. Arsenic induces apoptosis by the lysosomal-mitochondrial pathway in INS-1 cells. Environ Toxicol, 31: 133–141, 2016. doi: 10.1002/tox.22027. [DOI] [PubMed] [Google Scholar]
- 44.Paulmann N, Grohmann M, Voigt JP, Bert B, Vowinckel J, Bader M, Skelin M, Jevsek M, Fink H, Rupnik M, Walther DJ. Intracellular serotonin modulates insulin secretion from pancreatic beta-cells by protein serotonylation. PLoS Biol 7: e1000229, 2009. doi: 10.1371/journal.pbio.1000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrick JS, Jagadish B, Mash EA, Aposhian HV. Monomethylarsonous acid (MMA(III)) and arsenite: LD(50) in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem Res Toxicol 14: 651–656, 2001. doi: 10.1021/tx000264z. [DOI] [PubMed] [Google Scholar]
- 46.Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56: 1783–1791, 2007. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- 47.Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, Collins S, Andersen ME. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol 244: 77–83, 2010. doi: 10.1016/j.taap.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pontiroli AE, Micossi P, Foá PP. Effects of serotonin, of its biosynthetic precursors and of the anti-serotonin agent metergoline on the release of glucagon and insulin from rat pancreas. Horm Metab Res 10: 200–203, 1978. doi: 10.1055/s-0028-1093434. [DOI] [PubMed] [Google Scholar]
- 49.Pulido OM, Bencosme SA, de Bold ML, de Bold AJ. Intracellular pancreatic B cell serotonin and the dynamics of insulin release. Diabetologia 15: 197–204, 1978. doi: 10.1007/BF00421239. [DOI] [PubMed] [Google Scholar]
- 50.Ravenscroft P, Brammer H, Richards K. Arsenic Pollution: A Global Synthesis. Chichester, UK: Wiley-Blackwell, 2009. [Google Scholar]
- 51.Roseboom PH, Namboodiri MA, Zimonjic DB, Popescu NC, Rodriguez IR, Gastel JA, Klein DC. Natural melatonin ‘knockdown’ in C57BL/6J mice: rare mechanism truncates serotonin N-acetyltransferase. Brain Res Mol Brain Res 63: 189–197, 1998. doi: 10.1016/S0169-328X(98)00273-3. [DOI] [PubMed] [Google Scholar]
- 52.Sakakibara Y, Katoh M, Kawayanagi T, Nadai M. Species and tissue differences in serotonin glucuronidation. Xenobiotica 46: 605–611, 2016. doi: 10.3109/00498254.2015.1101509. [DOI] [PubMed] [Google Scholar]
- 53.Sung TC, Huang JW, Guo HR. Association between arsenic exposure and diabetes: a meta-analysis. BioMed Res Int 2015: 368087, 2015. doi: 10.1155/2015/368087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uchihashi S, Nishikawa M, Sakaki T, Ikushiro S. Comparison of serotonin glucuronidation activity of UDP-glucuronosyltransferase 1a6a (Ugt1a6a) and Ugt1a6b: evidence for the preferential expression of Ugt1a6a in the mouse brain. Drug Metab Pharmacokinet 28: 260–264, 2013. doi: 10.2133/dmpk.DMPK-12-NT-091. [DOI] [PubMed] [Google Scholar]
- 55.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: RESEARCH0034, 2002. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh J, Jenkins RE, Wong M, Olayanju A, Powell H, Copple I, O’Neill PM, Goldring CE, Kitteringham NR, Park BK. Identification and quantification of the basal and inducible Nrf2-dependent proteomes in mouse liver: biochemical, pharmacological and toxicological implications. J Proteomics 108: 171–187, 2014. doi: 10.1016/j.jprot.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, Xie Z, Lin Y, Zhang D. Association of inorganic arsenic exposure with type 2 diabetes mellitus: a meta-analysis. J Epidemiol Community Health 68: 176–184, 2014. doi: 10.1136/jech-2013-203114. [DOI] [PubMed] [Google Scholar]
- 58.Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 76: 215–228, 2009. doi: 10.1124/mol.109.056705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu MM, Chiou HY, Ho IC, Chen CJ, Lee TC. Gene expression of inflammatory molecules in circulating lymphocytes from arsenic-exposed human subjects. Environ Health Perspect 111: 1429–1438, 2003. doi: 10.1289/ehp.6396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu W, Yao X, Jiang L, Zhang Q, Bai J, Qiu T, Yang L, Gao N, Yang G, Liu X, Chen M, Sun X. Pancreatic islet-autonomous effect of arsenic on insulin secretion through endoplasmic reticulum stress-autophagy pathway. Food Chem Toxicol 111: 19–26, 2018. doi: 10.1016/j.fct.2017.10.043. [DOI] [PubMed] [Google Scholar]
- 61.Xia J, Wishart DS. Using metaboanalyst 3.0 for comprehensive metabolomics data analysis. Curr Protoc Bioinformatics 55: 14.10.11–14.10.91, 2016. doi: 10.1002/cpbi.11. [DOI] [PubMed] [Google Scholar]
- 62.Yang B, Fu J, Zheng H, Xue P, Yarborough K, Woods CG, Hou Y, Zhang Q, Andersen ME, Pi J. Deficiency in the nuclear factor E2-related factor 2 renders pancreatic β-cells vulnerable to arsenic-induced cell damage. Toxicol Appl Pharmacol 264: 315–323, 2012. doi: 10.1016/j.taap.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yasuda T, Shibasaki T, Minami K, Takahashi H, Mizoguchi A, Uriu Y, Numata T, Mori Y, Miyazaki J, Miki T, Seino S. Rim2alpha determines docking and priming states in insulin granule exocytosis. Cell Metab 12: 117–129, 2010. doi: 10.1016/j.cmet.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 64.Yoo JM, Lee BD, Sok DE, Ma JY, Kim MR. Neuroprotective action of N-acetyl serotonin in oxidative stress-induced apoptosis through the activation of both TrkB/CREB/BDNF pathway and Akt/Nrf2/Antioxidant enzyme in neuronal cells. Redox Biol 11: 592–599, 2017. doi: 10.1016/j.redox.2016.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Deng R, Yang X, Xu W, Liu Y, Li F, Zhang J, Tang H, Ji X, Bi Y, Wang X, Zhou L, Ning G. Glucose potentiates β-cell function by inducing Tph1 expression in rat islets. FASEB J 31: 5342–5355, 2017. doi: 10.1096/fj.201700351R. [DOI] [PubMed] [Google Scholar]
- 66.Zhu XX, Yao XF, Jiang LP, Geng CY, Zhong LF, Yang G, Zheng BL, Sun XC. Sodium arsenite induces ROS-dependent autophagic cell death in pancreatic β-cells. Food Chem Toxicol 70: 144–150, 2014. doi: 10.1016/j.fct.2014.05.006. [DOI] [PubMed] [Google Scholar]