

Abstract
Photoelectrochemical cells are widely studied for solar energy conversion. However, they have rarely been used for the synthesis of high added-value organic molecules. Here we describe a strategy to use hematite, an abundant and robust photoanode, for non-directed arene C-H amination. Under illumination the photo generated holes in hematite oxidizes electron-rich arenes to radical cations which further react with azoles to give nitrogen heterocycles of medicinal interest. Unusual ortho-selectivity has been achieved probably due to a hydrogen bonding interaction between the substrates and the hexafluoroisopropanol co-solvent. The method exhibits broad scope and is successfully applied for the late-stage functionalization of several pharmaceutical molecules.
Introduction
Photoelectrochemical (PEC) cells are widely studied for the conversion of solar energy into chemical fuels.1–3 Typically a photocathode is used to reduce water to hydrogen, and a photoanode is used to oxidize water to oxygen (Figure 1, a). Because high reducing or oxidizing power can be generated in PEC cells under mild conditions upon light illumination, they are well suited to catalyze redox transformation of organic molecules to yield high added-value chemicals (Figure 1, b). However, PEC cells have rarely been used in organic synthesis. Recently the oxidizing power of photoanodes such as BiVO4 and WO3 were exploited for the oxidation of 5-hydroxymethylfurfural (HMF),4 benzylic alcohols,5,6 furan7 and tetralin,5 as well as cyclohexane.8 Despite the conceptual advance, only a handful of simple substrates were tested in these studies. The utilization of PEC cells for broad-scope synthetic methodologies of functional organic molecules remains unexplored.
Figure 1. Photoelectrochemical cells.

a. Cell for water oxidation; b. Cell for oxidative transformations of organic substrates.
Organic compounds containing an aryl C-N moiety are ubiquitous in pharmaceuticals, agrochemicals and natural products. C-H amination has emerged as an attractive method to synthesize these compounds.9–11 Nevertheless, a directing group in the substrates is typically required and the reactions normally occur at an elevated temperature. PhI(OAc)2 was employed as an oxidant for non-directed C-H imidation of arenes.12,13 However, an excess of oxidant and a high temperature (>120 °C) were required. Nicewicz and co-workers14,15 reported an elegant approach harvesting the oxidation power of the photo-excited state of an organic photoredox catalyst, acridinium, for arene C-H amination (Figure 2, a). Oxidation of electron-rich arenes to radical cations followed by nucleophilic addition of nitrogen nucleophiles was the key step in the catalytic cycle. The reactions occurred at room temperature and a high para-selectivity was achieved. This method was further improved by the group of Lei16,17 who developed oxidant-free conditions and the group of König18 who broadened the scope of arenes using 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) as the photoredox catalyst. A challenge of this approach is the limited stability of organic photoredox catalysts. An alternative approach to similar arene C-H amination19–21 was electrochemical oxidation, developed by the group of Yoshida22–25 (Figure 2, b). However, a high oxidation potential is required to oxidize the arenes to the radical cations.
Figure 2. Arene C-H amination.

a. Via organic photoredox catalysis; b. Via direct electrochemical oxidation; c. Via photoelectrocatalysis.
Here we present a photoelectrocatalytic approach to non-directed arene C-H amination. Our strategy involves the use of an Earth-abundant and robust photoanode, hematite (α-Fe2O3), to conduct C-H amination in a PEC setting (Figure 2, c). Hematite has been extensively studied for solar-driven water oxidation thanks to its low cost, high stability, and suitable bandgap of 2.1 eV for strong visible light absorption.26,27 However, it has not been explored for organic synthesis. The valance band position of hematite (2.3 V vs SHE) is comparable to the oxidation potentials of organic photoredox catalysts such as acridinium, suggesting similar reactivity. On the other hand, the stability of hematite and the heterogeneous nature of photoelectrocatalysis in a PEC cell offer potential advantages in catalyst lifetime and product separation. Compared to direct electrocatalysis, photoelectrocatalysis consumes less energy due to light harvesting. Our method is operationally simple and can be used to synthesize a broad range of nitrogen-containing heterocycles relevant to drug discovery. Interestingly, reverse selectivity is achieved for many substrates compared to previously reported photoredox and electrochemical methods.14,15,23 The utility of this method is demonstrated by several examples of late-stage functionalization of pharmaceutical molecules.
Results
Optimization of reaction conditions
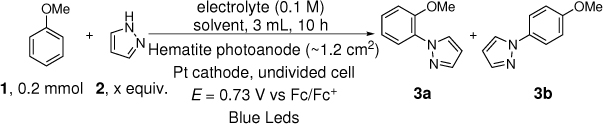
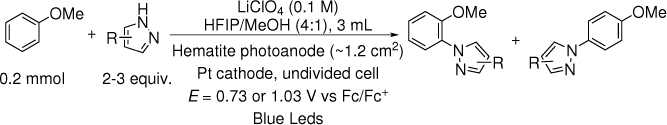
The C-H/N-H coupling of anisole with pyrazole was used as the test reaction for the C-H amination (Table 1). Nanostructured hematite was prepared as described previously.28 The reaction was performed with constant potential under blue LED light. After some initial testing, the potential was set to 0.73 V vs Fc/Fc+ where the photocurrent is at 2~3 mA/cm2 and the reaction time was set to 10 h. The electrolyte was initially tetrabutylammonium hexafluorophosphate (TBAPF6). Inspired by the work of Berlinguette and co-workers which showed the benefit of organic media for PEC oxidation5, various organic solvents were tested. Acetonitrile (CH3CN), the solvent of choice in direct electrochemical oxidation,22–24 was ineffective (Table 1, entry 1). When CH2ClCH2Cl were used as the solvent, 14% of the desired product was obtained (Table 1, entry 2). The group of Waldvogel reported that a mixture of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and methanol as solvent was essential for electrochemical phenol-arene C-C coupling.29–31 Inspired by their finding, we tested this solvent (4:1 HFIP/methanol) and were pleased to find that it led to a high yield of 75% (Table 1, entry 3). Both ortho and para isomers were formed with a ratio of 4:1. Surprisingly, the major product is the ortho isomer 3a, in contrast to the results of photoredox and electrochemical reactions14,15,23 where the major product was the para isomer. HFIP could not be replaced by CF3COOH (Table 1, entry 4). The reaction did not occur with HFIP or methanol as the sole solvent (Table 1, entries 5 and 6). Replacing TBAPF6 by LiClO4 as electrolyte, the yield was slightly higher and the selectivity was improved to 6:1 (Table 1, entry 7). Changing the ratio of HFIP and methanol changed the yields and selectivity but in opposite directions (Table 1, entries 8 and 9). Increasing the loading of the nucleophile to 3 equiv. improved the yield to 86%, but the selectivity decreased to 3:1 (Table 1, entry 10). Control experiments showed that in the absence of light or electricity no amination occurred (Table 1, entries 11 and 12). Without light but under a high applied potential, hematite could serve as a dark electrode. In order to reach the same current density, a potential of E = 1.53 V vs Fc/Fc+ was necessary. The yield was decreased to 58% and the selectivity was decreased to 2:1 (Table 1, entry 13). We also tried a conductive glassy carbon as a dark electrode. A potential of 1.33 V vs Fc/Fc+ was necessary, and the yield was only 38% (Table 1, Entry 14). Moreover, side products such as 1,2,4-trimethoxybenzene (about 5%) and 1-(2,5-dimethoxyphenyl)-1H-pyrazole (about 10%) were detected. These side products were absent when using illuminated hematite as the anode.
Table 1. Optimization of reaction conditions for the photoelectrocatalytic arene C-H amination [a].
 | |||||
|---|---|---|---|---|---|
| Entry | 2 (x equiv.) | electrolyte | solvent | Yield (%)a | 3a:3b |
| 1 | 2.0 | TBAPF6 | CH3CN | 0 | -- |
| 2 | 2.0 | TBAPF6 | CH2ClCH2Cl | 14 | 1:1 |
| 3 | 2.0 | TBAPF6 | HFIP/MeOH (4:1) | 75 | 4:1 |
| 4 | 2.0 | TBAPF6 | CF3COOH/MeOH (4:1) | 0 | -- |
| 5 | 2.0 | TBAPF6 | HFIP | 0 | -- |
| 6 | 2.0 | TBAPF6 | MeOH | 0 | -- |
| 7 | 2.0 | LiClO4 | HFIP/MeOH (4:1) | 77 | 6:1 |
| 8 | 2.0 | LiClO4 | HFIP/MeOH (3:1) | 78 | 4:1 |
| 9 | 2.0 | LiClO4 | HFIP/MeOH (5:1) | 62 | 8:1 |
| 10 | 3.0 | LiClO4 | HFIP/MeOH (4:1) | 86 | 3:1 |
| 11b | 2.0 | LiClO4 | HFIP/MeOH (4:1) | 0 | -- |
| 12c | 2.0 | LiClO4 | HFIP/MeOH (4:1) | 0 | -- |
| 13b,d | 2.0 | LiClO4 | HFIP/MeOH (4:1) | 58 | 2:1 |
| 14b,e | 2.0 | LiClO4 | HFIP/MeOH (4:1) | 38 | 12:1 |
Yiled determined by GC
Without light
Without electricity
Applied potential: E = 1.53 V vs Fc/Fc+
Glassy carbon (~1.2 cm2) was used as the anode. Applied potential: E = 1.33 V vs Fc/Fc+
The time-dependent concentration profile of the reagents and products (under conditions of entry 7) was shown in Supplementary Figure 2. Although analysis of the rate order from this profile was complicated by mass transfer to the photoanode, it was apparent that the reaction started to slow down after about 6 h. About 10% of substrate, anisole, remained after 10 h. Increasing reaction time beyond 10 h would have a limited impact in the yield.
Scope of photoelectrocatalytic arene C-H amination
With the optimized conditions in hand (Table 1, entry 7), the scope and limitation of the photoelectrocatalytic C-H amination method were explored (Table 2). Monosubstituted arenes were first tested. Arenes with one alkoxy substituent were aminated in good yields (3-8). Both para- and ortho- products were formed, and the major products was the ortho isomer. As mentioned above, this selectivity is opposite to analogous photoredox and electrochemical reactions.14,15,23 When diphenyl was used as the substrate, a complete para selectivity was obtained (9). Multisubstituted arenes were also suitable substrates. For 1,4-disubstituted arenes, the reactions afforded single products with a complete site selectivity (10-15). Only isomers with the pyrazole group ortho to the alkoxy group were obtained. For 1,3-disubstituted arenes, two isomers were formed (16-18). Trisubstituted arenes could be aminated as well (19 and 20). In addition, polycyclic aromatic hydrocarbons such as naphthalene and 9,9-dimethyl-9H-fluorene were aminated to generate single isomers (21 and 22). Various functional groups such as ester (6 and 17), halogen (5, 13, 14 and 18), cyano (15) and acyl (16) are tolerated under the reaction conditions.
Table 2. Scope of arenes for the photoelectrocatalytic arene C-H amination.a.
 | |||
|---|---|---|---|
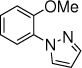 3, 10 h, 77% o:p = 6:1 |
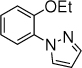 4b, 10 h, 65% o:p = 7:1 |
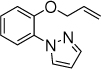 5, 10 h, 56% o:p = 4:1 |
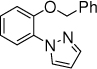 6c, 15 h, 66 % o:p = 5:1 |
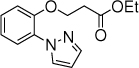 7c, 15 h, 86 % o:p = 4:1 |
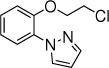 8b, 10 h, 71% o:p = 3:1 |
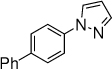 9b,c, 15 h, 50% |
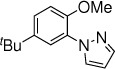 10c, 8 h, 77% |
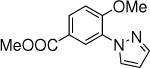 11c,d, 10% (10 h) 37% (24 h), 45% (36 h) |
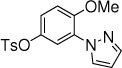 12b,c, 24 h, 54% |
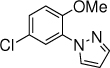 13, 10 h, 56% |
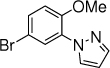 14b, 10 h, 51% |
 15c, 15 h, 52% |
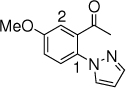 16c, 24 h, 48% P1:P2 =2:1 |
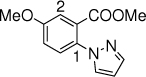 17c, 24 h, 89% P1:P2 =3:1 |
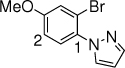 18c, 10 h, 82% P1:P2 =2:1 |
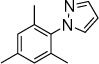 19b,c, 24 h, 50% |
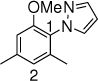 20c,10 h, 78% P1:P2 = 3:1 |
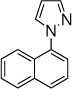 21, 10 h, 62% |
 22, 10 h, 60% |
Reaction conditions: Substrate (0.2 mmol) in HFIP/MeOH (4:1, 3 mL) at ambient temperature. Isolated yields.
Applied potential: E = 0.83 V vs Fc/Fc+
Azoles (0.6 mmol, 3 equiv.) was added.
Applied potential: E = 1.03 V vs Fc/Fc+. HFIP/H2O (9:1, 3 mL) was used.
Substrates with high oxidative potentials gave low to mid yields, likely because the oxidation was inefficient. For example, methyl 4-methoxybenzoate reacted with pyrazole to give 11 in only 37% yield after 24 h, and 57% of methyl 4-methoxybenzoate remained unreacted. Likewise, 1-(3-methoxyphenyl)ethan-1-one reacted with pyrazole to give 16 in 48% yield after 24 h, and 49% of 1-(3-methoxyphenyl)ethan-1-one remained. We investigated the reaction of methyl 4-methoxybenzoate at different reaction times. After 10 h, the yield was only 10%, but after 36 h, the yield was 45%. Apparently the reaction rate became relatively slow after 24 h, making it difficult to achieve a high yield by extending the reaction time further. Improving the photoelectrochemical cell to promote mass transport is a potential future solution to increase the reaction rate for difficult substrates.
The scope of nitrogen nucleophiles was then explored. A diverse set of azoles were suitable reaction partners (Table 3). Pyrazoles with a methyl, chloride, and bromide group at the 4-position gave high yields of amination products (23-25), with high ortho selectivity. When 3-methyl-1H-pyrazole was used as the nucleophile, amination by the sterically less encumbered N was slightly favored (26). Triazoles including 1,2,3-triazole, 1,2,4-triazole and benzotriazole were also successful nucleophiles, giving products with ortho selectivity (27-29). Imidazole was not a suitable nucleophile (30).
Table 3. Scope of nitrogen nucleophiles for the photoelectrocatalytic arene C-H amination.[a].
 | ||||
|---|---|---|---|---|
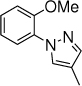 23b, 10 h, 41% o:p = 7:1 |
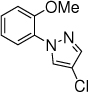 24, 15 h, 58% o:p = 14:1 |
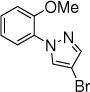 25, 10 h, 44% |
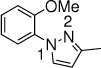 26, 15 h 38% o:p = 7:1 |
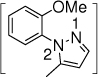 26', 27% o:p = 1:1 |
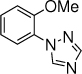 27c, 24 h, 43% o:p = 2:1 |
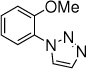 28, 24 h, 74% o:p = 4:1 |
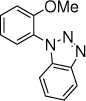 29c, 15 h, 59% o:p = 4:1 |
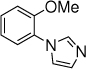 30, 10 h, 0% |
|
Reaction conditions: Substrate (0.2 mmol) and azoles (0.6 mmol, 3 equiv.) in HFIP/MeOH (4:1, 3 mL) at ambient temperature. Isolated yields.
Azoles (0.4 mmol, 2 equiv.) was added.
Applied potential: E = 1.03 V vs Fc/Fc+.
For all reactions in Tables 2 and 3, only unreacted substrates, but no side products, were detected after the reactions (by GC). Overoxidation of electron-rich arenes to give trimerization of oligomerizaion was common in electrosynthesis.30,32,33 From the mass balance (typically > 85% for substrate and product), the overoxidation was insignificant, if it ever occurred in our system. The hematite photoanode was very stable during photoelectrocatalysis. The photoelectrode was always re-used. After being used for more than 10 times, the photoanode still gave similar results of amination reactions compared to a freshly made photoanode.
Late-stage functionalization of pharmaceuticals
To further demonstrate the potential utility of this photoelectrocatalytic arene C-H amination method, we tested it for late-stage functionalization of pharmaceuticals. Clofibrate is a lipid-lowering agent used for controlling the high cholesterol level in the blood.34 Using the present method, Clofibrate reacted with pyrazole to give the amination product 32 in 62% yield (Figure 3, a). Likewise, Metaxalone, a muscle relaxant,35 reacted with pyrazole to give the corresponding product 34a (49%) and 34b (24%) (Figure 3, b). Moreover, antimicrobial agent benzethonium chloride36 reacted with pyrazole to give 36 in 87% yield (Figure 3, c).
Figure 3. Late-stage Functionalization of Pharmaceuticals.

a. C-H Amination of clofibrate; b. C-H Amination of metaxalone; c. C-H Amination of benzethonium chloride.
(Photo)electrochemical measurements
Some (photo)electrochemical measurements were performed to probe the properties and role of hematite in this photoelectrocatalysis. Linear sweep voltammetry (LSV) curves of the hematite photoanode were recorded in the reaction medium in the dark and under LED illumination (Figure 4, a). In the absence of light, oxidation was observed only at potentials higher than 0.9 V vs. Fc/Fc+. Upon illumination, oxidation already started at Eonset = 0 V vs Fc/Fc+. Thus, the photo voltage supplied by hematite for this reaction was about 0.9 V. An applied voltage (e.g., 0.73 V vs Fc/Fc+) was necessary to enhance the separation of electrons and holes in hematite and give a reaction rate relevant to synthesis. Electrochemical impedance spectroscopy (EIS) was conducted to estimate the flat-band potential of hematite in the reaction medium. According to the Mott-Schottky curve derived from EIS data (Figure 4, b) and Supplementary Figure 4), the flat-band potential was Efb = -0.12 V vs Fc/Fc+. The difference between the Eonset and Efb (Eonset - Efb = 0.12 V) might be considered as the overpotential of the oxidation. Apparently the reaction has a small overpotential. The incident photon-to-electron conversion efficiency (IPCE) of hematite was also determined in the reaction medium (Figure 4, c). The IPCE was similar to that measured in aqueous solutions,28 indicating similar photophysical properties of hematite in aqueous and organic solutions. The Faradaic yield of C-H amination of anisole was determined by photoelectrolysis. A yield of about 35% was consistently obtained. Judging from the LSV curve of hematite in the presence of MeOH (Supplementary Figure 5), some of the photo generated holes from hematite were used to oxidize MeOH. The LSV curves of hematite in the presence of a single reaction partner, i.e., either anisole or pyrazole, indicated that the photo generated holes oxidized anisole but not pyrazole (Supplementary Figure 6).
Figure 4. (Photo)electrochemical measurements.

a. LSV curves of a hematite photoelectrode under LED illumination (red line) and in the dark (black line) in HFIP/MeOH (4:1, 3 mL) containing LiClO4 (0.1 M), anisole (0.2 mmol) and pyrazole (0.4 mmol). Scan rate: 30 mV/s; b. Mott-Schottky plot measured in HFIP/MeOH (4:1); c. IPCE of hematite measured under catalytic conditions at E = 0.73 V vs Fc/Fc+.
Discussion
According to the above preliminary investigations and previous work on arene C-H amination, we proposed the following mechanism for the photoelectrocatalysis (Figure 5, a). Upon illumination of hematite, holes are generated at the valence band. The holes oxidize an electron-rich arene to a radical cation, which is electrophilic and reacts with an azole to give intermediate A. Loss of a proton from A yields intermediate B, which upon oxidation and removal of one proton gives the amination product. The oxidation of B might occur at the photo anode by the photo generated holes, or through a methoxy radical which might be generated at the photoanode as well.29 The photoelectrocatalysis is completed when the photoexcited electrons migrate from the conduction band of hematite to the counter electrode to reduce protons into dihydrogen gas.
Figure 5. Mechanistic hypothesis.

a. Proposed Mechanism of C-N bond formation; b. Proposed hydrogen bonding among anisole, HFIP and pyrazole.
The unusual selectivity of the present arene C-H amination method warrants some explanation. Generally, nucleophilic functionalization of mono-substituted arene radical cations would occur at both ortho and para positions of the substituent. The para isomer is expected to dominate due to the steric hindrance at the ortho position. This selectivity was indeed observed in previous photoredox and electrochemical reactions.14,15,23 However, in this work high ortho selectivity was achieved. We speculate that the solvent 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) plays an important role in the observed selectivity. HFIP has some unique solvent properties37 such as high polarity and weak nucleophilicity. HFIP can stabilize radical cations,38,39 which might be responsible for the high efficiency of the present photoelectrocatalysis. Moreover, HFIP is a strong hydrogen bonding donor and acceptor.40–44 It was reported that HFIP formed H-bonding with oxygenated compounds such as ethers40 and phenyliodine diacetate41. Furthermore, H-bonding from HFIP in the transition states was proposed to promote epoxidation of olefins using H2O243,44 and electrophilic aromatic chlorination42. Based on these literature precedents, we hypothesize that a hydrogen bonding network is formed among anisole, HFIP and pyrazole (Figure 5, b). The hydrogen bonding would favor the amination at the ortho position. When the hydrogen bonding was not available, e.g., in the case of diphenyl (Table 2, 9) and 9,9-dimethyl-9H-fluorene (Table 2, 22) as substrates, only para-product was generated. In previous work invoking hydrogen bonding from HFIP40,44, CF3CH2OH was shown to have a similar role as HFIP. Thus, we conducted the reaction in Table 1 using CF3CH2OH/MeOH (4:1, 3 mL) as the solvent. The yield was 61% and the selectivity was 2:1. While this selectivity was inferior to that obtained using HFIP/MeOH, the ortho product was still favored. If a non-fluorinated solvent such as CH2ClCH2Cl2 was used, no selectivity was found (o:p = 1:1). This result is consistent with the hypothesis that hydrogen bonding from a fluorinated alcohol leads to higher ortho selectivity.
While both processes utilizes light generated oxidative power to generate the arene radical cations, the photoelectrocatalysis described here is fundamentally different from photoredox catalysis. The light absorption is achieved by a solid-state semiconductor and the photogenerated hole is located at the delocalized valence band. The catalyst is regenerated by migration of photoexcited electrons to a counter electrode to form H2. The catalytic process is heterogeneous. On the contrary, in photoredox catalysis the light absorption and charge separation are done by a single molecule and the catalyst is regenerated by quenching the oxidized photocatalyst with a reductant. The catalytic process is homogeneous. The unique features of photoelectrocatalysis can lead to some potential advantages such as high stability and easy separation of catalysts as well as de-coupling of oxidative and reductive reactions. Moreover, as shown in photoelectrocatalytic water splitting,45additional catalytic layers might be deposited onto the surface of photoelectrodes to catalyze reactions that are otherwise sluggish or impossible by direct electron transfer.
The photoelectrocatalytic approach described here is also fundamentally different from electrochemical synthesis. In this approach, the hole is transferred from the edge of the valence band to the substrate for oxidation. The energy level of the edge is independent of the external bias, which is used to improve charge separation, or to drive the reaction at the metal counter electrode, or both. The charge transfer occurs through the surface states of the semiconductor. While the nature of these surface states remains challenging to characterize, the kinetics of direct charge transfer can be very fast. In electrochemical synthesis, the electron is transferred from the substrate to the Fermi level of the conductive (metallic) electrode. The potential is used to shift the Fermi level in order to match the redox potential of the substrate. The charge transfer is typically an outer sphere event, which can be limited by kinetics. Because of the different nature of the charge transfer processes, different reactivity patterns may be observed using photoelectrocatalytic or electrochemical synthesis. Thus, energy saving due to light harvesting is not the only advantage of photoelectrocatalysis over electrochemical synthesis. Finally, the external bias required in this work can become unnecessary under one or more of the following conditions: (i) A tandem device composed of two light-harvesting components provides enough voltage for the reactions; (ii) The redox couple at the counter electrode matches the flat-band potential of the semiconductor. Clearly further development is required to realize the potential benefits of photoelectrocatalysis.
Conclusion
A photoelectrocatalytic method for non-directed arene C-H amination has been developed, employing abundant and robust hematite as the catalyst. A wide range of heterocycles containing an aryl C-N moiety can be prepared from simple arenes and azoles without pre-functionalization of substrates. Unusual ortho-selectivity is achieved from many substrates that typically exhibit para-selectivity under previously reported methods. The method has been successfully applied for late-stage functionalization of several pharmaceutical molecules. The works opens a new avenue in using PEC cells for organic synthesis.
Methods
General
Supplementary Methods for experimental details and characterization data as well as Supplementary Figures for additional results and NMR spectra can be found in the Supplementary Information.
Materials
All manipulations were carried out under nitrogen. The following chemicals were purchased and used as received: Iron pentacarbonyl (Fe(CO)5, Aldrich), tetraethoxysilane (TEOS, Aldrich), LiClO4 (Aldrich), tetrabutylammonium hexafluorophosphate (TCI), aromatic compounds (Aldrich or TCI), azoles (TCI). Hematite46 and substrates (ethyl 3-phenoxypropanoate47 and 4-methoxyphenyl 4-methylbenzenesulfonate48) were prepared according to previously reported procedures. All other reagents and solvents were purchased from commercial sources and used without purification.
Analytical Methods
NMR spectra were recorded on Bruker Avance 400 MHz spectrometers. 1H NMR chemical shifts were referenced to residual protio solvent peaks or tetramethylsilane signal (0 ppm), and 13C NMR chemical shifts were referenced to the solvent resonance. Data for 1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet or unresolved, coupling constant (s) in Hz, integration). Data for 13C NMR are reported in terms of chemical shift (δ, ppm). GC measurements were conducted on a Perkin-Elmer Clarus 400 GC with a FID detector. GC-MS measurements were conducted on an Agilent Technologies 7890A GC system equipped with a 5975C MS detector. HRMS-ESI measurements were conducted at the EPFL ISIC Mass Spectrometry Service with a Micro Mass QTOF. LSV and CV curves were recorded with a three-electrode configuration using VMP-3 instrument (Biologic Science Instrument). Hematite or glassy carbon electrode was used as a working electrode with a Ag/AgCl reference electrode and a Pt counter electrode. LSV and CV curves were performed at a scan rate of 30 mV/s without stirring. All potentials (vs Ag/AgCl) were calibrated with ferrocene/ferrocenyl couple (Fc/Fc+) by reducing 0.17 V. Electrochemical impedance spectroscopy measurement was performed by a three-electrode configuration using Autolab PGSTAT302N (Metrohm). A sinusoidal voltage perturbation was superimposed on a bias voltage, with 10 mV amplitude and frequencies ranging from 100,000 to 0.1 Hz. The impedance was measured at bias voltages from -0.77 to 0.63 V vs Fc/Fc+. All EIS measurements were performed in dark. The incident photon-to-charge conversion efficiencies (IPCE) were measured under light from a 300 W xenon lamp through a monochromator (TLS-300XU; Newport). The photo response was compared with that of a calibrated silicon photodiode (FDS100-CAL, Thorlabs) to determine the IPCE at each wavelength.
Supplementary Material
Acknowledgements
This work was supported by the EPFL, a consolidator grant from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (no.681292 to X.H), the PECHouse3 project from the Swiss Federal Office of Energy (no. SI/500090-03, M.G. and J.L.), the Strategic Japanese-Swiss Science and Technology Programme from the Swiss National Science Foundation (no. 514259, M.G. and J.L.), and the Chinese Thousand Talents Program for Young Professionals (to J.L.).
Footnotes
Data Availability
The data that support the plots within this paper and other findings of this study are available from the corresponding author upon reasonable request.
Author Contributions
X.H. directed the project. L.Z. conceived and performed most of experiments. L.L. prepared hematites and conducted electrochemical impedance spectroscopy analysis. J.L. and D.R. measured the incident photon-to-electron conversion efficiency (IPCE). X.H. and L.Z. wrote the paper, with input from others. All authors analysed the results and reviewed the paper.
Competing Interests
The authors declare no competing interests.
References
- 1.Grätzel M. Photoelectrochemical cells. Nature. 2001;414:338. doi: 10.1038/35104607. [DOI] [PubMed] [Google Scholar]
- 2.Tachibana Y, Vayssieres L, Durrant JR. Artificial photosynthesis for solar water-splitting. Nat Photonics. 2012;6:511. [Google Scholar]
- 3.Fujishima A, Honda K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature. 1972;238:37. doi: 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- 4.Cha HG, Choi K-S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat Chem. 2015;7:328–333. doi: 10.1038/nchem.2194. [DOI] [PubMed] [Google Scholar]
- 5.Li T, et al. Photoelectrochemical oxidation of organic substrates in organic media. Nat Commun. 2017;8 doi: 10.1038/s41467-017-00420-y. 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tateno H, Miseki Y, Sayama K. Photoelectrochemical Oxidation of Benzylic Alcohol Derivatives on BiVO4/WO3 under Visible Light Irradiation. ChemElectroChem. 2017;4:3283–3287. [Google Scholar]
- 7.Tateno H, Miseki Y, Sayama K. Photoelectrochemical dimethoxylation of furan via a bromide redox mediator using a BiVO4/WO3 photoanode. Chem Commun. 2017;53:4378–4381. doi: 10.1039/c7cc01190c. [DOI] [PubMed] [Google Scholar]
- 8.Tateno H, Iguchi S, Miseki Y, Sayama K. Photo-Electrochemical C−H Bond Activation of Cyclohexane Using a WO3 Photoanode and Visible Light. Angew Chem Int Ed. 2018;57:11238–11241. doi: 10.1002/anie.201805079. [DOI] [PubMed] [Google Scholar]
- 9.Dieu TL, James R, Olafs D. Directed Amination of Non-Acidic Arene C-H Bonds by a Copper–Silver Catalytic System. Angew Chem Int Ed. 2013;52:6043–6046. doi: 10.1002/anie.201300135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu H, Qiao X, Yang S, Shen Z. Cu-Catalyzed Direct Amidation of Aromatic C–H Bonds: An Access to Arylamines. J Org Chem. 2014;79:4414–4422. doi: 10.1021/jo5003592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsubara T, Asako S, Ilies L, Nakamura E. Synthesis of Anthranilic Acid Derivatives through Iron-Catalyzed Ortho Amination of Aromatic Carboxamides with N-Chloroamines. J Am Chem Soc. 2014;136:646–649. doi: 10.1021/ja412521k. [DOI] [PubMed] [Google Scholar]
- 12.Kim HJ, Kim J, Cho SH, Chang S. Intermolecular Oxidative C–N Bond Formation under Metal-Free Conditions: Control of Chemoselectivity between Aryl sp2 and Benzylic sp3 C–H Bond Imidation. J Am Chem Soc. 2011;133:16382–16385. doi: 10.1021/ja207296y. [DOI] [PubMed] [Google Scholar]
- 13.Kantak AA, Potavathri S, Barham RA, Romano KM, DeBoef B. Metal-Free Intermolecular Oxidative C–N Bond Formation via Tandem C–H and N–H Bond Functionalization. J Am Chem Soc. 2011;133:19960–19965. doi: 10.1021/ja2087085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romero NA, Margrey KA, Tay NE, Nicewicz DA. Site-selective arene C-H amination via photoredox catalysis. Science. 2015;349:1326–1330. doi: 10.1126/science.aac9895. [DOI] [PubMed] [Google Scholar]
- 15.Margrey KA, McManus JB, Bonazzi S, Zecri F, Nicewicz DA. Predictive Model for Site-Selective Aryl and Heteroaryl C–H Functionalization via Organic Photoredox Catalysis. J Am Chem Soc. 2017;139:11288–11299. doi: 10.1021/jacs.7b06715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niu L, et al. Photo-induced oxidant-free oxidative C–H/N–H cross-coupling between arenes and azoles. Nat Commun. 2017;8 doi: 10.1038/ncomms14226. 14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song C, et al. Visible-light-mediated C2-amination of thiophenes by using DDQ as an organophotocatalyst. Chem Commun. 2017;53:3689–3692. doi: 10.1039/c7cc01339f. [DOI] [PubMed] [Google Scholar]
- 18.Somnath D, Palani N, Burkhard K. Teaching Old Compounds New Tricks: DDQ- Photocatalyzed C–H Amination of Arenes with Carbamates, Urea, and N- Heterocycles. Chem Eur J. 2017;23:18161–18165. doi: 10.1002/chem.201705442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauermann N, Mei R, Ackermann L. Electrochemical C−H Amination by Cobalt Catalysis in a Renewable Solvent. Angew Chem Int Ed. 2018;57:5090–5094. doi: 10.1002/anie.201802206. [DOI] [PubMed] [Google Scholar]
- 20.Gao X, Wang P, Zeng L, Tang S, Lei A. Cobalt(II)-Catalyzed Electrooxidative C–H Amination of Arenes with Alkylamines. J Am Chem Soc. 2018;140:4195–4199. doi: 10.1021/jacs.7b13049. [DOI] [PubMed] [Google Scholar]
- 21.Yang Q-L, et al. Copper-Catalyzed Electrochemical C–H Amination of Arenes with Secondary Amines. J Am Chem Soc. 2018;140:11487–11494. doi: 10.1021/jacs.8b07380. [DOI] [PubMed] [Google Scholar]
- 22.Morofuji T, Shimizu A, Yoshida J-i. Electrochemical C–H Amination: Synthesis of Aromatic Primary Amines via N-Arylpyridinium Ions. J Am Chem Soc. 2013;135:5000–5003. doi: 10.1021/ja402083e. [DOI] [PubMed] [Google Scholar]
- 23.Morofuji T, Shimizu A, Yoshida J-i. Direct C–N Coupling of Imidazoles with Aromatic and Benzylic Compounds via Electrooxidative C–H Functionalization. J Am Chem Soc. 2014;136:4496–4499. doi: 10.1021/ja501093m. [DOI] [PubMed] [Google Scholar]
- 24.Morofuji T, Shimizu A, Yoshida J-i. Heterocyclization Approach for Electrooxidative Coupling of Functional Primary Alkylamines with Aromatics. J Am Chem Soc. 2015;137:9816–9819. doi: 10.1021/jacs.5b06526. [DOI] [PubMed] [Google Scholar]
- 25.Tatsuya M, Akihiro S, Jun-ichi Y. Electrochemical Intramolecular C−H Amination: Synthesis of Benzoxazoles and Benzothiazoles. Chem Eur J. 2015;21:3211–3214. doi: 10.1002/chem.201406398. [DOI] [PubMed] [Google Scholar]
- 26.Tamirat AG, Rick J, Dubale AA, Su W-N, Hwang B-J. Using hematite for photoelectrochemical water splitting: a review of current progress and challenges. Nanoscale Horizons. 2016;1:243–267. doi: 10.1039/c5nh00098j. [DOI] [PubMed] [Google Scholar]
- 27.Sivula K, Le Formal F, Grätzel M. Solar Water Splitting: Progress Using Hematite (α-Fe2O3) Photoelectrodes. ChemSusChem. 2011;4:432–449. doi: 10.1002/cssc.201000416. [DOI] [PubMed] [Google Scholar]
- 28.David TS, Maurin C, Kevin S, Michael G. Light-Induced Water Splitting with Hematite: Improved Nanostructure and Iridium Oxide Catalysis. Angew Chem Int Ed. 2010;49:6405–6408. doi: 10.1002/anie.201003110. [DOI] [PubMed] [Google Scholar]
- 29.Kirste A, Elsler B, Schnakenburg G, Waldvogel SR. Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: The Benign Role of Water or Methanol. J Am Chem Soc. 2012;134:3571–3576. doi: 10.1021/ja211005g. [DOI] [PubMed] [Google Scholar]
- 30.Anton W, et al. Electrifying Organic Synthesis. Angew Chem Int Ed. 2018;57:5594–5619. doi: 10.1002/anie.201711060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernd E, et al. Source of Selectivity in Oxidative Cross-Coupling of Aryls by Solvent Effect of 1,1,1,3,3,3-Hexafluoropropan-2-ol. Chem Eur J. 2015;21:12321–12325. doi: 10.1002/chem.201501604. [DOI] [PubMed] [Google Scholar]
- 32.Bechgaard K, Parker VD. Mono-, di-, and trications of hexamethoxytriphenylene. Novel anodic trimerization. J Am Chem Soc. 1972;94:4749–4750. [Google Scholar]
- 33.Schubert M, et al. Over-Oxidation as the Key Step in the Mechanism of the MoCl5-Mediated Dehydrogenative Coupling of Arenes. Angew Chem Int Ed. 2016;55:1156–1159. doi: 10.1002/anie.201508035. [DOI] [PubMed] [Google Scholar]
- 34.Grundy SM, Ahrens EH, Salen G, Schreibman PH, Nestel PJ. Mechanisms of action of clofibrate on cholesterol metabolism in patients with hyperlipidemia. J Lipid Res. 1972;13:531–551. [PubMed] [Google Scholar]
- 35.See S, Ginzburg R. Skeletal Muscle Relaxants. Pharmacotherapy. 2008;28:207–213. doi: 10.1592/phco.28.2.207. [DOI] [PubMed] [Google Scholar]
- 36.Sickbert-Bennett EE, et al. Comparative efficacy of hand hygiene agents in the reduction of bacteria and viruses. Am J Infect Control. 2005;33:67–77. doi: 10.1016/j.ajic.2004.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colomer I, Chamberlain AER, Haughey MB, Donohoe TJ. Hexafluoroisopropanol as a highly versatile solvent. Nat Rev Chem. 2017;1 0088. [Google Scholar]
- 38.Lennart E, Ola P, P HM. Detection and Reactions of Radical Cations Generated by Photolysis of Aromatic Compounds with Tetranitromethane in 1,1,1,3,3,3-Hexafluoro-2-propanol at Room Temperature. Angew Chem Int Ed Engl. 1995;34:2268–2269. [Google Scholar]
- 39.Eberson L, Hartshorn MP, Persson O, Radner F. Making radical cations live longer. Chem Commun. 1996:2105–2112. [Google Scholar]
- 40.Berkessel A, Adrio JA, Hüttenhain D, Neudörfl JM. Unveiling the “Booster Effect” of Fluorinated Alcohol Solvents: Aggregation-Induced Conformational Changes and Cooperatively Enhanced H-Bonding. J Am Chem Soc. 2006;128:8421–8426. doi: 10.1021/ja0545463. [DOI] [PubMed] [Google Scholar]
- 41.Colomer I, Batchelor-McAuley C, Odell B, Donohoe TJ, Compton RG. Hydrogen Bonding to Hexafluoroisopropanol Controls the Oxidative Strength of Hypervalent Iodine Reagents. J Am Chem Soc. 2016;138:8855–8861. doi: 10.1021/jacs.6b04057. [DOI] [PubMed] [Google Scholar]
- 42.Ben-Daniel R, de Visser SP, Shaik S, Neumann R. Electrophilic Aromatic Chlorination and Haloperoxidation of Chloride Catalyzed by Polyfluorinated Alcohols: A New Manifestation of Template Catalysis. J Am Chem Soc. 2003;125:12116–12117. doi: 10.1021/ja0364524. [DOI] [PubMed] [Google Scholar]
- 43.Berkessel A, Adrio JA. Kinetic Studies of Olefin Epoxidation with Hydrogen Peroxide in 1,1,1,3,3,3-Hexafluoro-2-propanol Reveal a Crucial Catalytic Role for Solvent Clusters. Adv Synth Catal. 2004;346:275–280. [Google Scholar]
- 44.de Visser SP, Kaneti J, Neumann R, Shaik S. Fluorinated Alcohols Enable Olefin Epoxidation by H2O2: Template Catalysis. J Org Chem. 2003;68:2903–2912. doi: 10.1021/jo034087t. [DOI] [PubMed] [Google Scholar]
- 45.Morales-Guio CG, et al. An Optically Transparent Iron Nickel Oxide Catalyst for Solar Water Splitting. J Am Chem Soc. 2015;137:9927–9936. doi: 10.1021/jacs.5b05544. [DOI] [PubMed] [Google Scholar]
- 46.David TS, Maurin C, Kevin S, Michael G. Light-Induced Water Splitting with Hematite: Improved Nanostructure and Iridium Oxide Catalysis. Angew Chem Int Ed. 2010;49:6405–6408. doi: 10.1002/anie.201003110. [DOI] [PubMed] [Google Scholar]
- 47.Al-Awadi SA, et al. Kinetics and mechanism of thermal gas-phase elimination of β-substituted carboxylic acids. Tetrahedron. 2005;61:5769–5777. [Google Scholar]
- 48.Ankner T, Hilmersson G. Instantaneous Deprotection of Tosylamides and Esters with SmI2/Amine/Water. Org Lett. 2009;11:503–506. doi: 10.1021/ol802243d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.