

Abstract
The dysregulation of apoptosis is a key step in developing tumours, and mediates resistance to cancer therapy. Many different signals for cell death converge on permeabilization of the outer mitochondrial membrane, which is controlled by the Bcl-2 family of proteins. The importance of this step is becoming increasingly relevant as the first generation of small molecules that inhibit the interaction of Bcl-2 family proteins enters clinical trials as anticancer agents. The Bcl-2 family can be divided into three classes: BH3-only proteins that are activated by various forms of cellular stress, Bax and Bak proteins that mediate mitochondrial membrane permeabilization, and inhibitory proteins such as Bcl-2 and Bcl-XL. The recently proposed embedded together model emphasizes the fact that many of the regulatory interactions between different classes of Bcl-2 family members occur at intracellular membranes, and binding to membranes causes conformational changes in the proteins that dictate functions in a dynamic manner. Within this context, recent results indicate that Bcl-XL functions as a dominant-negative Bax, a concept that resolves the paradox of similar structures but opposite functions of Bcl-XL and Bax. We have also shown that the conformational change that allows Bax to insert into the outer mitochondrial membrane is the rate-limiting step in the multistep process of Bax activation. Nevertheless, investigating the structure of activated Bax or Bak as monomers and as components of the oligomeric structures that mediate membrane permeabilization is the focus of ongoing research (and controversy) at many laboratories worldwide.
Keywords: apoptosis, model, membranes, Bcl-2, Bcl-XL, Bax
The Bcl-2 family regulates apoptosis—but how?
Irrespective of whether the tumour-initiating cell is a stem cell or has adopted such characteristics (induced multipotency), cancer results when DNA in a single cell acquires mutations that trigger uncontrolled growth. The normal defence to loss of growth control is the induction of apoptosis. The commitment step in apoptosis is regulated by the Bcl-2 family of proteins. In a seminal observation made 20 years ago (McDonnell et al., 1989), overexpression of Bcl-2 resulted in polyclonal expansion of cells that preceded carcinogenesis. This initial insight led to the recognition that abrogation of apoptosis by Bcl-2 family proteins (or other means) is absolutely required for cancer development (Hanahan and Weinberg, 2000). Furthermore, compared with normal cells, it has become apparent that many cancers depend on the antiapoptotic activity of Bcl-2 family proteins, not only for tumour initiation but also for tumour maintenance, such that they are ‘addicted’ to these proteins (Certo et al., 2006; Chonghaile and Letai, 2009). Therefore, understanding the function of Bcl-2 proteins is a critical step in manipulating apoptosis to exploit this differential ‘addiction’ therapeutically.
The three-level hierarchy of the (now very large) Bcl-2 family (Youle and Strasser, 2008) involves (1) BH3 proteins that monitor cellular fitness and signal execution, (2) membrane-permeabilizing proteins Bax and Bak and (3) antiapoptotic proteins Bcl-2, Bcl-XL, Mcl-1, Bcl-w, and Bcl2-A1. There are many different types of BH3 proteins, most of which have unrelated functions in normal cell physiology but monitor different abnormal cellular functions (reviewed in Lomosova and Chinnadurai, 2009).
When the cell is stressed or damaged, these proteins are altered: for example, proapoptotic signalling causes an increase in protein levels because of increased transcription (PUMA; Yu and Zhang, 2009), decreased turnover (Noxa; Ploner et al., 2009), post-translational modification (Bad; Danial, 2009), a caspase (protease)-dependent change in structure (Bid; Billen et al., 2009) or a change in binding partner and subcellular localization (Bim; Piñon et al., 2009). The result of all these changes is that the activated BH3 proteins bind to one or more of the multidomain Bcl-2 family proteins, either pro- or antiapoptotic. As only Bax and Bak permeabilize the outer mitochondrial membrane, signalling from BH3 proteins and from the antiapoptotic Bcl-2 family members is integrated at the level of activation of these two proteins that kill cells by oligomerizing in the membrane, thereby causing mitochondrial outer membrane permeabilization (MOMP). A consequence of MOMP is the release of intermembrane space proteins such as cytochrome c into the cytoplasm, where they allosterically activate the adaptor protein Apaf-1 to initiate the cascade of caspases that cleave substrates leading to cell death.
To understand the principles governing the complex interactions between different classes of Bcl-2 family members, the three current models for the organization of the Bcl-2 family of proteins are outlined in simplified form in Figure 1. Since the past 5 years, this field has been dominated by two major contrasting and competing models: the derepression (Chen et al., 2005; Willis et al., 2005, 2007) and direct activation models (Kuwana et al., 2002; Letai et al., 2002; Kim et al., 2006) (Figures 1a and b). The most contentious point is the activation state of Bax/Bak and therefore the mechanism by which Bcl-XL inhibits MOMP: the derepression model postulates that Bcl-XL must continuously bind to Bax/Bak (which are proposed to always be active) to prevent MOMP, whereas the direct activation model states that Bax/Bak are activated by BH-3 proteins, and that Bcl-XL functions by sequestering these activators from Bax. In the latter model, BH-3 proteins comprise two classes: those that directly activate Bax/Bak (activators such as tBid and Bim) and those that displace activator BH3 proteins from Bcl-XL and activate Bax/Bak indirectly (sensitizers such as Bad).
Figure 1.
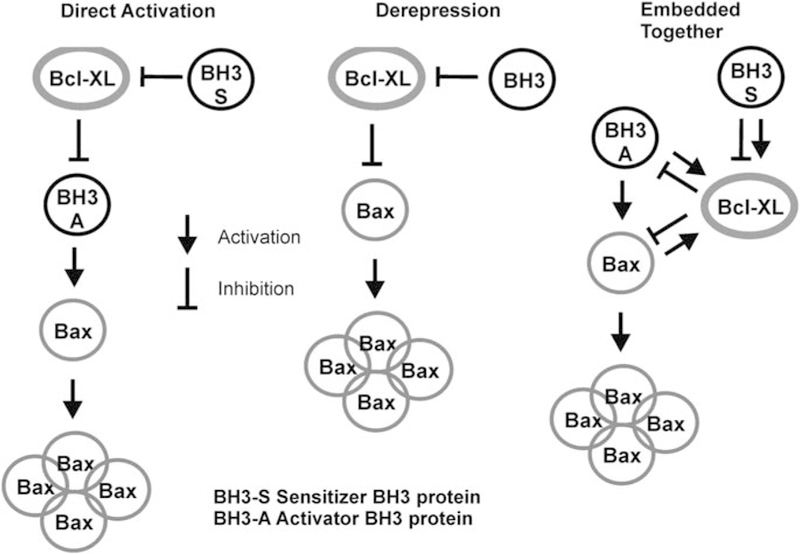
Models of regulation of apoptosis by Bcl-2 family proteins. Simplified versions of the three models of apoptosis regulation to emphasize the different roles postulated for interactions between the three different classes of Bcl-2 proteins (BH3-only proteins, antiapoptotic members such as Bcl-XL and membrane-permeabilizing proteins such as Bax). These diagrams neglect the role of the membrane in permitting a comparison of the direct activation and derepression models (no role for membrane) with the protein interactions of the embedded together model. The direct activation and derepression models postulate different functional states for Bax or Bak; in the former, these proteins are inactive and must be activated by BH3-only proteins, whereas in the latter they are constitutively active and must be repressed by antiapoptotic proteins such as Bcl-XL. Thus, they propose different mechanisms of action for Bcl-XL inhibition of apoptosis. The embedded together model recognizes both of these interactions. In the derepression model, BH3-only proteins are distinguished by their differential affinity to the varying antiapoptotic Bcl-2 family members, but in the direct activation model they are divided into two classes: those that directly activate Bax or Bak (activators) and those that prevent Bcl-XL from binding to activators (sensitizers). The embedded together model also recognizes these two different classes, although it considers the fact that both classes have a role in ‘activating’ Bcl-XL by causing its membrane insertion. The consequence of the subsequent interaction in the membrane differs: BH3-only activator proteins are sequestered by Bcl-XL and therefore cannot activate Bax or Bak, whereas BH3-only sensitizer proteins prevent Bcl-XL from binding to activator proteins, thereby inhibiting Bcl-XL.
By measuring the rate and extent of the critical interactions directly with full-length recombinant proteins, using purified organelles and a liposome system that recapitulates MOMP (schematic and representative results shown in Figure 2), it was possible to evaluate the competing claims of these models. Our data allowed us to propose a model we termed as ‘embedded together’, to emphasize the point that binding to membranes is essential for many of the critical interactions between different classes of Bcl-2 family members (Leber et al., 2007). More recent data (Billen et al., 2008; Lovell et al., 2008) have allowed us to refine the embedded together model (as summarized in Figure 1c); we propose that antiapoptotic Bcl-2 proteins not only have both functions proposed by the other two models (a consensus that is also recognized in recent publications by the proponents of the original models, for example, Brunelle et al., 2009; Mérino et al., 2009), but also with two other unique features not previously included, namely, that BH3-only proteins can both inhibit and activate functions of antiapoptotic Bcl-2 proteins (Figure 1c) and that many of the functional interactions between the proteins occur only in membranes because membrane binding induces the unique conformational changes required for the interactions. Furthermore, recent investigations have uncovered the series of ordered molecular steps by which Bax regulates membrane permeabilization and have identified the rate-limiting step in this process as described below. Here, we will briefly recapitulate the series of experiments that led us to propose the embedded together model and discuss how recent results extend and enrich it.
Figure 2.

Mitochondrial outer membrane permeabilization by tBid-activated Bax is a multistep process. Using fluorescently labelled recombinant proteins and membranes to monitor reactions in real time, our results (Lovell et al., 2008) indicate that Bax activation is a multistep process whereby tBid binds to membranes (step I), then recruits Bax (step II), which causes a conformational change that allows Bax to insert into membranes (step III—the rate-limiting step in the process); membrane associated Bax recruits other cytosolic Bax (step IV), which then oligomerizes in membranes (step V). Membrane permeabilization by oligomerized Bax (step VI–left panel) can be conveniently and reproducibly measured by the release of a fluorescent molecule encapsulated within the liposome that is quenched by a co-encapsulated quencher. Thus, this step can also be measured in real time (step VI—right panel).
Bcl-XL functions as a dominant-negative Bax—a resolution of the paradox of similar structures with opposite functions
In 2004 we published the first evidence that, at the onset of apoptosis, Bcl-2 changes conformation in the membrane of the mitochondria (Kim et al., 2004). This astonishing observation has provided a key to understanding fundamental aspects of the regulation of apoptosis. When we set out to determine the functional significance of this change, we discovered that, in some experimental conditions, insertion of α-helices 5 and 6 into membranes is required for Bcl-2 to inhibit Bax oligomerization and MOMP (Dlugosz et al., 2006; Peng et al., 2006). By introducing two cysteines into Bcl-2, which when oxidized became crosslinked and thereby prevented the insertion of helices 5 and 6, the ability of Bcl-2 to inhibit apoptosis was abolished. However, when the dicysteine bond was reduced, function of Bcl-2 was restored (Dlugosz et al., 2006). The trigger for the conformational change that results in insertion of helix 5 into the bilayer was binding of proapoptotic BH3 proteins or peptides such as tBid or Bim. The consequence of this binding was an antiapoptotic response, because the induced activating conformation change in Bcl-2 preceded the activation of Bax by tBid or Bim. We speculate that this conformational change allows Bcl-2 to stably bind proapoptotic BH3 activator proteins, thereby preventing them from activating Bax. A major goal was then to determine the generality and specificity of this conformational change that is, did this change happen in other antiapoptotic Bcl-2 family members (for example, Bcl-XL), and would the same change also occur after binding with the other class of BH3-only proteins (for example, sensitizers such as Bad).
Therefore, we switched our focus from Bcl-2 to Bcl-XL—a move also motivated by both theoretical and practical considerations. Bcl-XL is 10 times more potent in inhibiting apoptosis in cells exposed to the same cytotoxic stimulus (Fiebig et al., 2006), and therefore the interactions of Bcl-XL with other proteins would be stronger and therefore easier to analyse; moreover, Bcl-XL posed additional mechanisms of action not shared by Bcl-2. Furthermore, Bcl-XL is similar to Bax, that is, it is cytoplasmic or loosely membrane bound in many but not all cell types (Hsu and Youle, 1997; Kaufmann et al., 2003) and therefore may be similarly subjected to a regulatory step that triggers membrane binding in cells undergoing apoptosis. As a likely consequence of its cytoplasmic location, recombinant full-length Bcl-XL can also be purified without using nonionic detergents in a quantity sufficient for biophysical studies, something that has yet to be achieved with Bcl-2. By using recombinant full-length Bcl-XL, Bax, and tBid in an in vitro system with liposomes containing a fluorescent marker and its quencher, those protein(s) that Bcl-XL bound to inhibit membrane permeabilization could be evaluated directly. Using this in vitro system, these interactions can be examined in a manner that is independent of other binding partners and post-translational modifications that may occur in cells. These results then provide a basis for the interpretation of the effect of these factors in future study involving more complex systems such as native membranes.
We showed unequivocally that Bcl-XL bound to both Bax and tBid, but only on the membrane, as the binding to either protein was extremely weak or undetectable when membranes were absent. That Bcl-XL only binds other proteins on the membrane is a likely explanation of the fact that Bcl-XL lacking its C-terminal membrane insertion sequence is less active and much less specifically targeted to membranes in cells than is full-length Bcl-XL (Fiebig et al., 2006). However, this form of Bcl-XL shows antiapoptotic activity in vitro (Billen et al., 2008) and when expressed in cells (Fiebig et al., 2006), suggesting that there may be as yet undiscovered mechanism(s) by which Bcl-XL inhibits apoptosis. However, for Bcl-XL to be fully effective it must be triggered to bind to the membrane, and this occurs after binding to either tBid or Bax, which are themselves already membrane bound.
By using a variety of mutants that disrupt specific interactions between tBid or Bax and Bcl-XL, it was shown that both interactions occur independently. Thus, the original versions of the direct activation and derepression models are both right—and both incomplete—to the extent that they fail to account for the other interaction. Moreover, for Bcl-XL, it was not possible to clearly show that binding to tBid was ‘more important’ than binding to activated Bax to prevent apoptosis, or vice versa. It is highly likely that in cells one or the other mechanism may predominate, on the basis of the relative expression levels of the activated binding partners of Bcl-XL, as a consequence of cell type, stage of development or differentiation, specific apoptotic stimulus, and so on. Furthermore it seems that all these interactions are in reversible equilibria that can be accommodated by standard biochemical techniques and principles.
By using recombinant full-length proteins labelled with small fluorescent dyes, it was possible to measure in real-time the interaction between Bcl-2 family members (Lovell et al., 2008). Aside from the insights that this has yielded with regard to the process of Bax activation discussed below, it also allowed a direct test of the proposed division of BH3 proteins into activator and sensitizer classes proposed by the direct activation model (Letai et al., 2002; Kim et al., 2006). For example, it was clearly shown that Bad binds to Bcl-XL and can displace a previously bound tBid molecule such that this tBid is free to bind to and activate Bax, thereby causing membrane permeabilization (Lovell et al., 2008). Along with others, we (Kuwana et al., 2002) have also shown that Bad does not activate Bax in vitro: taken together, these results confirm that Bad functions as a sensitizer BH3 protein. It is therefore important to systematically analyse the function of other BH3 molecules in the presence of membranes to determine which class they belong to (or if other functional subclasses exist, as discussed below). In an elegant series of experiments using liposomes and mitochondria from mice with various Bcl-2 family members knocked out, Douglas Green’s laboratory has recently shown that recombinant PUMA is not an activator of Bax/Bak, but functions exclusively as a sensitizer BH3 protein (Chipuk et al., 2008). However, this conclusion remains controversial, as in vitro-translated PUMA binds to Bax in mitochondria and causes conformational changes in Bax associated with activation and pore formation (Kim et al., 2009).
Hence, the embedded together model agrees with the direct activation model in stating that the functional division of BH3 proteins is real and critical to understanding the mechanism of apoptosis. Furthermore, we and others (Jeong et al., 2004) have also shown that recombinant Bad spontaneously inserts into membranes, and it can cause Bcl-XL to insert into membranes as well. The embedded together model predicts that both tBid and Bad recruit supra-stoichiometric quantities of Bcl-XL to membranes, thereby activating it. However, the consequences of the direct interactions in the membrane differ: tBid is prevented from activating Bax or Bak by binding to Bcl-XL, whereas direct binding of Bad is inhibitory to the function of Bcl-XL. Therefore, a prediction of the embedded together model is that the conformation of Bcl-XL bound to tBid in the membrane would be different from that of Bcl-XL bound to Bad.
In the case of both BH3 proteins identified as triggers for insertion of Bcl-XL into membranes, the regulation of their own membrane localization is the focus of ongoing investigations. After Bid is cleaved by activated caspase-8 to generate a cleaved Bid (cBid), no further modifications (such as N-myrisoylation, Zha et al., 2000) or proteins are required to separate the amino-terminal p7 fragment from the p15 functional truncated Bid (tBid) fragment, as long as a membrane is present (Lovell et al., 2008). This suggests that cBid undergoes a substantial conformational change when it binds to membranes. Traditionally, tBid has been generated in vitro from caspase-8 cleaved recombinant Bid by exposure to detergent, which presumably mimics that which happens with physiological membranes. By using recombinant cBid, Bax, and liposomes or reconstituted mitochondria of a defined composition, Kuwana’s laboratory reinvestigated the controversial role of cardiolipin in membrane permeabilization in an elegant series of experiments (Schafer et al., 2009). Cardiolipin was not required to permeabilize liposomes when Bax was activated by Bid (or Bim) BH3 peptides, but was required for activation by cBid, which contains both the N- and C-terminal portions of the molecule. As activation of Bax by cBid is a multistep process (Lovell et al., 2008), it will be interesting to determine whether cardiolipin is involved in the initial membrane binding of cBid, its separation into tBid, or binding to Bax. When mitochondrial membranes are reconstituted in such a way that minimal cardiolipin is present, a protein factor can substitute for cardiolipin in mediating cBid/Bax permeabilization (Schafer et al., 2009). This factor does not seem to be the previously reported tBid-interacting protein, Mtch2 (Grinberg et al., 2005), atleast in the Xenopus-derived mitochondria used for these experiments.
The regulation of Bad targeting to membranes has also been the subject of intensive investigation. Here, the relevant activating event is phosphorylation of serine residues, which prevents binding to the ubiquitous cytoplasmic 14–3-3 protein family, and facilitate binding to particular subcellular membranes (Hekman et al., 2006). There is now a bewildering variety of kinases (including PKA, C-Raf/PAK, Akt and B-Raf) that have been reported to differentially phosphorylate these serine residues (Polzien et al., 2009). This panoply of regulators may be required because of the multiple roles of Bad in cellular metabolism, such as gluconeogenesis (reviewed in Danial, 2009), and in control of mitochondrial calcium flux (Roy et al., 2009), which are independent of inhibiting Bcl-XL.
If the cell takes such trouble to ensure that Bid and Bad are present at membranes to orchestrate Bax and/or Bcl-XL insertion into membranes, what is the evidence that other BH3 proteins also bind to membranes? As activators, both tBid and Bim would be expected to bind to Bax, but it is not certain that the mechanism would be identical, and there is significant evidence that they differ. Unlike the other BH3 proteins that seem to be intrinsically unstructured, the structure of Bid is similar to that of Bax, as well as being the closest of all the BH3 proteins in primary sequence to both Bax and Bcl-XL/Bcl-2. Indeed, it has been suggested that the sequence of conformational events that lead to membrane insertion is shared by Bax and Bid, including relief of N-terminal inhibition by unfolding or cleavage that leads to initial membrane binding and exposure of the BH3 region, followed by insertion of the central pair of hydrophobic helices to form a hairpin in the bilayer (Billen et al., 2009). In contrast, Bim without its carboxyl-terminal hydrophobic sequence is largely unstructured in solution until it binds to prosurvival Bcl-2 family members, also engineered to be soluble by removal of their carboxyl-terminal tails. On binding to Bcl-2, Bim undergoes a limited conformational change, whereby the BH3 region assumes an α-helical configuration (Hinds et al., 2007).
Recombinant Bad and Bmf (without the C-terminal hydrophobic sequences) were also reported to be unstructured in solution (Hinds et al., 2007). An intriguing extension of this concept of an unstructured protein folding after binding to its Bcl-2 family partner is that a necessary (previous?) structural change may be triggered by membrane binding, and it is this intermediate conformation that is further modified by binding to other membrane-associated Bcl-2 proteins. Thus, all classes of the Bcl-2 family, including BH3 proteins, may change (or acquire) structure on membrane binding. This feature may explain the much lower potency of His-tagged recombinant Bim to activate Bax, which lacks its C-terminal sequence but is artificially attached to the membrane through a lipid–NTA–Ni linkage, compared with full-length tBid (Terrones et al., 2008). The latter may undergo a conformational change after membrane binding that is not recapitulated by the artificial membrane linkage of the former. Other BH3 proteins such as PUMA (Yu and Zhang, 2009) and Noxa (Ploner et al., 2009) also have C-terminal hydrophobic sequences that may mediate binding to the membrane as a facilitating step before activating conformational changes.
Whatever the further details that are learnt about the membrane interactions of other BH3 proteins and the consequences that this has for activating the two other classes of Bcl-2 proteins, experiments with tBid have shown significant similarity between the behaviour of Bcl-XL and Bax. Both proteins are cytoplasmic and can be recruited to the membrane by tBid. In doing so, both are recruited in excess over the amount of tBid and both proteins change conformation through interactions with the membrane. Both Bax and Bcl-XL can also be recruited to the membrane by activated Bax, and bind to it. However, the consequences of these ‘behaviours’ is different: by binding to membrane-bound tBid, Bcl-XL sequesters it and prevents further activation of Bax, and Bcl-XL when bound to activated, membrane-bound Bax prevents further recruitment of soluble Bax and thereby halts the homo-oligomerization process necessary for membrane permeabilization. This indicates that Bcl-XL functions in a manner conceptually similar to a dominant-negative Bax: Bcl-XL and Bax share binding partners, but Bcl-XL does not initiate or propagate oligomerization (Billen et al., 2008). Therefore, to fully explain the molecular mechanism by which Bcl-XL inhibits Bax, it is necessary to understand the structure that Bax assumes in the membrane when it forms (or organizes) the pore.
Activating Bax at the membrane is the rate-limiting step in apoptosis
When the integration of Bax into membranes was examined, it was discovered that Bax inserts as multi-spanning monomers that then oligomerize to permeabilize membranes (Annis et al., 2005). This mechanism differs from that used by many other pore-forming proteins that oligomerize before inserting into and thereby permeabilizing membranes. Once inserted into the membrane, Bax monomers recruit and activate cytoplasmic Bax by a novel Bcl-2 inhibitable autoactivation mechanism (Tan et al., 2006). However, the sequence of events that starts with Bax in the cytoplasm as a monomer and ends with Bax oligomers in the membrane has been difficult to study. To solve this problem, a combination of fluorescent dyes was used to label tBid, Bax, the membrane, and to monitor membrane permeabilization. These dyes were chosen such that three of them could be used together, and by using fluorescence resonance energy transfer and other fluorescence techniques it was possible to examine in real-time the various discrete steps that lead to MOMP. In these experiments, tBid bound rapidly to the membrane first, followed by tBid binding to soluble Bax (Lovell et al., 2008). On the basis of a previous study (Yethon et al., 2003), it is likely that Bax interacts transiently with the membrane to expose the 6A7 epitope as part of a conformational change that allows soluble Bax to bind to membrane-bound tBid. After binding to tBid, Bax inserts into the membrane and then oligomerizes to form pores. By measuring the progress of these reactions simultaneously, it was shown that they occur as an ordered series of events that ultimately culminate in membrane permeabilization (Lovell et al., 2008). Significantly, insertion of Bax into the membrane was the rate-limiting step in this process.
Recent results using in vitro-translated tBid on mitochondria from Bax/Bak double knockout mouse embryonic fiboblasts (Bax−/−/Bak−/− MEFs) stably reconstituted with wild-type and mutant forms of Bax have confirmed the multistep activation of Bax in organelles (Kim et al., 2009). A tBid-induced change in the conformation of the N-terminus of Bax that preceded homo-oligomerization was also identified. Taken together, these data underline the importance of Bax insertion into the membrane and oligomerization as the critical decision node in the commitment to apoptosis. Understanding the quaternary structure of activated Bax oligomers in membranes is therefore an important goal.
Since the presentation of the embedded together model in 2007, several publications have examined the nature of the membrane-bound oligomers of Bax or Bak that mediate pore formation. The study that has yielded the highest resolution of this complex to date was reported by Kluck and colleagues (Dewson et al., 2008), in which they screened for loss-of-function mutations of transfected Bak in yeast cells exposed to chemical mutagenesis. The phenotype of the recovered Bak mutations was confirmed in Bax−/−/Bak−/− mouse embryonic fibroblasts. The relevant mutations are concentrated in the α-helix 2–5 region, which, from the crystal structure of other Bcl-2 family members, seems to mediate BH3:BH1–3 groove interactions. Significantly, these mutations neither affected the targeting of Bak to MOM in normal cells nor prevented the conformational change occurring during apoptosis, as detected by the Ab-1 monoclonal antibody. However, these mutations inhibited the homo-oligomerization of Bak that can be detected by disulphide crosslinking. This crosslinking is effective only over an extremely short distance, as the disulphide bond is about 2.05 Å in length, and Bak has two endogenous cysteines at position 166 (near the BH2 region and between helices 6 and 7) and position 14 (at the N-terminal) that are substrates for the crosslinking reagent. The authors postulated that crosslinking normally occurs between residues 166 of adjacent molecules, and that the mutations that prevent this interaction are concentrated in the BH1–3 region because this is the ‘acceptor’ groove for the BH3 motif from neighbouring molecules in other known Bcl-2 family pairs. Confirmatory evidence for this conclusion was obtained from experiments in which ‘compensating’ mutations were made ‘in-trans’ in the BH3 motif that, by charge transfer, could bind to and reverse the loss-of-function mutations in the BH1–3 acceptor groove.
The authors also introduced several single cysteines in either the BH3 region or the BH1–3 groove regions of Bak proteins in which the endogenous cysteines had been removed, and showed that when these mutants were tested together they could generate a disulphide-bonded dimer after oxidation. As the position of these interacting cysteine pairs was chosen on the basis of the crystal structure of the Bcl-XL–Bak BH3 peptide complex and the assumed homology between the structures of Bcl-XL and Bak, the results provide support for a ‘symmetric dimer’ model in which each partner ‘donates’ a BH3 region to bind to the other BH1–3 groove. To form this kind of homodimer, a substantial conformational change would occur in both Bak proteins. The two crystal structures that have so far been solved for the soluble domain of Bak are symmetric dimers. Yet the BH3:BH1–3 groove interface does not exist in either structure. Instead, the dimer interface includes either a zinc ion coordinated by Asp160 and His164 from each monomer or a disulphide bond formed between the Cys166 residues of each monomer (Moldoveanu et al., 2006; Wang et al., 2009). It is important to note that the Cys166 residues in both structures are not located in the BH3:BH1–3 groove interface as predicted by Dewson et al. Although the latter structure was proposed to be a proapoptotic structure, further conformational changes would be required for the Bak dimer to oligomerize and therefore be really proapoptotic. The required conformational change might result in the BH3:BH1–3 groove interface and may only occur when Bak is anchored in the MOM. In fact, Cys166 is not required for Bak oligomerization and function as shown in a follow-up study by the same group (Dewson et al., 2009). This study also revealed a second interface in a Bak oligomer that is formed by helix 6 from two neighbouring molecules. The helix 6:helix 6 interaction was captured by crosslinking of the cysteine engineered at same locations in the two helices, suggesting that the two helices are parallel in the interface. However, this topographical arrangement would be energetically unfavourable, as helix 6 has three charged residues and therefore proximity of the two helices would generate a strong electrostatic repulsion.
The interface of the Bax oligomer at MOM was analysed by a different strategy by Luo’s group (George et al., 2007). Deletion mutants were created to determine the minimal regions of Bax that are required for oligomerization and for the proapoptotic activity of a GFP–Bax fusion protein expressed in Bax−/−/Bak−/− mouse embryonic fibroblasts. Oligomerization was assessed by gel filtration of the cell lysate after solubilization in CHAPS, a detergent that does not cause artifactual Bax activation (but does dissociate Bax from tBid in liposomes (Lovell et al., 2008), even though it does not disrupt this interaction in solution (Kim et al., 2009)). Using this technique, Bax helices 2–5 were shown to be sufficient to cause oligomerization. Furthermore, deletion of helix 3 did not prevent oligomerization; thus, the authors concluded that helices 2, 4, and 5 constitute the minimal regions required for oligomerization. This conclusion was supported by results with an even more drastically engineered GFP fusion protein that contained Bax helices 2, 4, and 5 having six alanines to replace the region between helices 2 and 4, and the membrane anchor sequence of Bcl-XL replacing all the Bax residues C-terminal to helix 5. This fusion protein formed oligomers and caused apoptosis when expressed in Bax−/−/Bak−/− mouse embryonic fibroblasts. However, unlike wild-type Bax, the oligomerization and killing activity of this mutant did not require tBid. Does this mean that the mutant has adopted the appropriate active conformation ‘down-stream’ of tBid binding or that the mutant Bax folds into a completely different structure than the parent protein and hence, causes cell death through a different mechanism? A similar concern is raised by a GFP-Bax mutant with alanine substitutions in three to four residues in helices 2, 4, and 5. The mutations abolished tBid-induced oligomerization and apoptosis, but curiously the protein could still be ‘activated’ by treating the cell with staurosporine.
It is interesting that, despite using a different approach, several of the structural models for Bax and Bak homo-oligomerization are consistent, as helices 2, 4, and 5 overlap with BH1 and BH3 regions. However, additional interactions would be required for the repeating unit to assemble into higher-order oligomers, for example, helix 6, as proposed for symmetric dimers.
A recent publication (Gavathiotis et al., 2008; commentary in Green and Chipuk (2008) and Czabotar et al. (2009)) indicates that symmetric dimer formation may not be the only mode of binding. By using a ‘stapled’ Bim-BH3 peptide and Bax protein, nuclear magnetic resonance analysis of the complex revealed that the binding site for the peptide was located in a previously uncharacterized pocket in Bax formed by helices 1 and 6, and the unstructured loop connecting helices 1 and 2. The interaction of BH3 peptides with this pocket (which we will designate as the rear pocket on the basis of the location relative to the BH3 binding groove) caused several conformational changes in Bax that were distant from the rear pocket, including the flexible regions located N-terminal and C-terminal to the helix 1. The most significant conformational change occurred around the N-terminus of helix 1 that overlaps with the 6A7 epitope, a motif that is exposed during Bax activation (Hsu and Youle, 1997). However, the expected conformational changes at the C-terminal helix 9 that ‘release’ it from the binding groove formed by the BH1–3 regions before membrane insertion were not detected, possibly because changes were not significant under the conditions used for collection of nuclear magnetic resonance spectra. Alternatively, these changes may occur later and are not detectable, as Bax aggregation would hamper the nuclear magnetic resonance measurement. This latter notion is consistent with the embedded together model and our recent data (Lovell et al., 2008), in which multiple sequential conformational changes were shown to be required for Bax activation, and some (such as change in the C-terminal and/or the BH1–3 regions) may occur only when Bax is bound to membranes rather than in solution, as was used for the nuclear magnetic resonance analysis. However, even if the changes described are just the first steps in a sequence, these intriguing results raise several questions for future investigations.
The model predicts that the local conformational change induced by the binding of a BH3-only protein to the rear pocket propagates to the other side of Bax causing helix 9 to pop out of the BH1–3 groove and helix 2 containing the BH3 region to become solvent exposed. Is this new BH3 ‘ligand’ enough to activate a second Bax monomer? One possibility is that this BH3 region can now bind to the rear pocket of the second Bax to activate it. This mechanism is consistent with the observation that the BH3 region of Bax is homologous to that of Bim. This would represent one structural explanation for how Bax binding to Bax leads to autoactivation (Tan et al., 2006). Alternatively, the exposed BH3 helix may bind to the BH1–3 groove of the second Bax (as proposed for Bak by Dewson et al., 2008). In this case, is the affinity of the free BH3 helix for the BH1–3 groove strong enough to displace the α-9 helix that covers the groove, or must the second Bax also be preactivated by another BH3-only protein through the rear pocket? This immediately leads to the question as to how the activated Bax proteins oligomerize. One possibility is that the bound BH3-only protein must leave Bax before Bax can form oligomers, particularly if the BH1–3 groove is used to bind the second Bax as the ‘symmetrical’ model predicts (Dewson et al., 2008). The formation of large oligomers from these symmetric dimers requires another binding surface to link them, as the BH3:binding groove interface would be occluded in the dimer.
In a recent study investigating Bak oligomerization, the endogenous and engineered cysteines in the a-6 helix of Bak could be crosslinked, but only after activation by tBid (Dewson et al., 2009). In contrast, the autoactivation model of interaction would allow the addition of the second Bax to the BH1–3 groove of the first Bax without dissociating the BH3-only protein, as these two interactions occur at opposite ‘sides’ of Bax. The second Bax would then bind to the rear pocket on a third Bax and thus propagate autoactivation to form the oligomer without requiring the initial BH3 protein to dissociate. As each monomer in the Bax oligomer uses two different surfaces to interact with neighbouring Bax proteins, we designate such a Bax oligomer as ‘asymmetric’ to distinguish it from the ‘symmetric’ oligomer, as proposed by Dewson and Kluck on the basis of the homology between Bak and Bax (Dewson and Kluck, 2009; Dewson et al., 2009). For both models, the binding of the BH3 protein is ‘catalytic’ in causing a conformational change in Bax. However, binding of a BH3 protein is required for each step during oligomerization in the symmetric dimer model, but only to initiate oligomerization in the asymmetric autoactivation model.
Because studying the conformation of activated Bax in its ‘normal’ membrane-bound environment is very difficult, having an alternative system to investigate activated Bax would be an enormous advantage in allowing many more techniques to be used. Is detergent exposure (Hsu and Youle, 1997) a reliable way to generate authentic activated Bax? As a membrane surrogate, detergent micelles can mimic the membrane environment for certain proteins. It is encouraging that, for Bax, many of the properties caused by exposure to certain nonionic detergents are also detected in biological membranes. Thus, micelles of Triton X-100 or octylglucoside change Bax conformation, exposing the 6A7 epitope (Yethon et al., 2003); detergent-treated Bax releases cytochrome c from mitochondria in a manner similar to that of tBid-activated Bax (Antonsson et al., 2000); Bcl-2 binds Bax in detergent as occurs in membranes (Zhang et al., 2004; Dlugosz et al., 2006; Peng et al., 2006). Furthermore, Bax oligomerizes in detergent as detected by both gel-filtration chromatography and chemical crosslinking, in a manner similar to that of solubilization of membrane-embedded Bax using CHAPS (Billen et al., 2008). It is therefore surprising that, on the basis of data obtained by two sensitive techniques, that is, fluorescence correlation spectroscopy and fluorescence intensity distribution analysis, a fluorescent dye-labelled Bax mutant lacking the C-terminal helix 9 was found as a monomer in micelles of several detergents including octyl-glucoside and Triton X-100 (Ivashyna et al., 2009). This group reported that the relative molecular mass of detergent-treated, fluorescently labelled Bax, as determined by fluorescence correlation spectroscopy, was close to that of a Bax monomer. Furthermore, the fluorescence intensity per micelle determined using fluorescence intensity distribution analysis was similar to that of monomeric Bax in a detergent-free buffer. Both results suggest that, in detergent micelles, Bax remains as a monomer. However, these experiments were conducted with a truncated Bax in which the carboxyl terminus is missing, and the authors assumed that there is no self-quenching or other change in fluorescence in the labelled Bax in the micelles. When full-length Bax was used in a photo-crosslinking study, Bax dimer-specific adducts were detected in Triton X-100 micelles (Zhang et al., 2010). As adducts were formed through photoreactive probes located at multiple sites on the monomer, these adducts are most likely formed between two Bax molecules in an oligomer. Moreover, this study revealed that the Bax oligomer is formed by two separate, yet interdependent, surfaces. It will be important to determine whether Bax uses a similar mechanism to form oligomers in membranes after activation by BH3-only protein.
Determining the structure of Bax/Bak oligomers in membranes will not only be invaluable in determining the mechanism of membrane permeabilization and its inhibition but will also shed light on the other functions of these proteins in mitochondrial physiology, including fission and fusion. This complex topic has recently been reviewed in depth (Suen et al., 2008); therefore, we will simply note that the seemingly paradoxical findings of Bax and Bak promoting either fission or fusion in different contexts may be a consequence of the different amounts and conformation of Bax and Bak proteins in healthy versus apoptotic cells. As the number and conformation of Bax and Bak proteins are in equilibria, as determined by binding partners, both antiapoptotic multi-BH region proteins and proapoptotic BH3-only proteins of the Bcl-2 family would be expected to indirectly regulate mitochondrial fusion and fission. Therefore, elucidating the structure and stoichiometry of Bax/Bak oligomers binding to other family members will shed light on both apoptosis and mitochondrial dynamics.
Conclusion: past progress and future goals
The importance of apoptosis in cancer and the crucial role of the Bcl-2 family of proteins have been intensively investigated for >15 years (Cotter, 2009). Although many other components of the apoptotic machinery have been identified, results with cells derived from mice lacking important effector caspases have reemphasized specific roles for antiapoptotic (for example, Bcl-2) and proapoptotic (for example, Bax) proteins in determining clonal survival after cytotoxic insult (Marsden et al., 2002), the sentinel lesion determining tumour development and treatment resistance. The field has advanced remarkably since the role of Bcl-2 in apoptosis was first recognized. The first phase of identifying the other Bcl-2 family members that are components of the core machinery of apoptosis is at a mature, but not yet complete, stage (Chipuk et al., 2008; Youle and Strasser, 2008). Identifying other potential BH3 proteins that activate Bax/Bak is an important step in completing this phase. The second phase of categorizing Bcl-2 family members into structural and functional subgroups, and identifying relevant interacting partners, has now reached a broad consensus (Youle and Strasser, 2008). Many of these key interactions are modified in a dynamic manner by membranes, as we have emphasized here. Furthermore, the recognition that Bcl-2 family members interact differently in membranes indicates that novel drug targets exist that would not have been evident in the small-molecule screens using truncated proteins that led to the development of the first generation of Bcl-2-inhibiting drugs. The third phase of understanding the molecular mechanisms that mediate interactions between Bcl-2 family members requires a detailed understanding of the structure of omponents (Annis et al., 2005; Denisov et al., 2007; Dewson and Kluck, 2009; Kim et al., 2009) and how various intracellular organelles such as the mitochondria and endoplasmic reticulum (Zhu et al., 1996) modify the structures. However, a deep understanding of how the entire process is regulated can be achieved only with rigorous quantitative analysis of these interactions, and by investigating the manner in which they interface with the signalling pathways that lead to their initiation. To this end, mathematical models based on experimental data are needed (Albeck et al., 2008) to supplant current schematic models.
Acknowledgements
Work from our laboratories cited in this review was supported by a Canadian Institute of Health Research (CIHR FRN12517) Grant to DWA and BL, by a Tier-I Canada Research Chair in Membrane Biogenesis to DWA and by NIH Grant GM062964 to JL.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Albeck JG, Burke JM, Spencer SL, Lauffenburger DA, Sorger PK. (2008). Modeling a snap-action, variable-delay switch controlling extrinsic cell death. PLoS Biol 6: 2831–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B et al. (2005). Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J 24: 2096–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. (2000). Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 345: 271–278. [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. (2008). Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 10: e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billen LP, Shamas-Din A, Andrews DW. (2009). Bid: a Bax-like BH3 protein. Oncogene 27: S93–S104. [DOI] [PubMed] [Google Scholar]
- Brunelle JK, Ryan J, Yecies D, Opferman JT, Letai A. (2009). MCL-1-dependent leukemia cells are more sensitive to chemotherapy than BCL-2-dependent counterparts. J Cell Biol 187: 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA et al. (2006). Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9: 351–365. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG et al. (2005). Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17: 393–403. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Fisher JC, Dillon CP, Kriwacki RW, Kuwana T, Green DR. (2008). Mechanism of apoptosis induction by inhibition of the anti-apoptotic BCL-2 proteins. Proc Natl Acad Sci USA 105: 20327–20332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonghaile TN, Letai A. (2009). Mimicking the BH3 domain to kill cancer cells. Oncogene 27: S149–S157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter TG. (2009). Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 9: 501–507. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Colman PM, Huang DC. (2009). Bax activation by Bim? Cell Death Differ 16: 1187–1191. [DOI] [PubMed] [Google Scholar]
- Danial NN. (2009). Bad: undertaker by night, candyman by day. Oncogene 27: S53–S70. [DOI] [PubMed] [Google Scholar]
- Denisov AY, Sprules T, Fraser J, Kozlov G, Gehring K. (2007). Heat-induced dimerization of BCL-xL through alpha-helix swapping. Biochemistry 46: 734–740. [DOI] [PubMed] [Google Scholar]
- Dewson G, Kluck RM. (2009). Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci 122: 2801–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Czabotar P, Day CL, Adams JM, Kluck RM. (2009). Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol Cell 36: 696–703. [DOI] [PubMed] [Google Scholar]
- Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM et al. (2008). To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3:groove interactions. Mol Cell 30: 369–380. [DOI] [PubMed] [Google Scholar]
- Dlugosz PJ, Billen LP, Annis MG, Zhu W, Zhang Z, Lin J et al. (2006). Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J 2006: 25:2287–25:2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig AA, Zhu W, Hollerbach C, Leber B, Andrews DW. (2006). Bcl-XL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer 6: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG et al. (2008). BAX activation is initiated at a novel interaction site. Nature 455: 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George NM, Evans JJ, Luo X. (2007). A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev 21: 1937–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Chipuk JE. (2008). Apoptosis: Stabbed in the BAX. Nature 455: 1047–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinberg M, Schwarz M, Zaltsman Y, Eini T, Niv H, Pietrokovski S et al. (2005). Mitochondrial carrier homolog 2 is a target of tBID in cells signaled to die by tumor necrosis factor alpha. Mol Cell Biol 25: 4579–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. (2000). The hallmarks of cancer. Cell 100: 57–70. [DOI] [PubMed] [Google Scholar]
- Hekman M, Albert S, Galmiche A, Rennefahrt UE, Fueller J, Fischer A et al. (2006). Reversible membrane interaction of BAD requires two C-terminal lipid binding domains in conjunction with 14–3-3 protein binding. J Biol Chem 281: 17321–17336. [DOI] [PubMed] [Google Scholar]
- Hinds MG, Smits C, Fredricks-Short R, Risk JM, Bailey M, Huang DCS et al. (2007). Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Diff 14: 128–136. [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. (1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272: 13829–13834. [DOI] [PubMed] [Google Scholar]
- Ivashyna O, Garcia-Saez AJ, Ries J, Christenson ET, Schwille P, Schlesinger PH. (2009). Detergent activated BAX protein is a monomer. J Biol Chem 284: 23935–23946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SY, Gaume B, Lee YJ, Hsu YT, Ryu SW, Yoon SH et al. (2004). Bcl-x(L) sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers. EMBO J 23: 2146–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. (2003). Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 160: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ et al. (2006). Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol 8: 1348–1358. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP et al. (2009). Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36: 487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW. (2004). During apoptosis bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol Cell 14: 523–529. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R et al. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342. [DOI] [PubMed] [Google Scholar]
- Leber B, Lin J, Andrews DW. (2007). Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 12: 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. (2002). BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192. [DOI] [PubMed] [Google Scholar]
- Lomosova E, Chinnadurai G. (2009). BH3-only proteins in apoptosis and beyond: an overview. Oncogene 27: S2–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell JF, Billen LP, Bindner S, Shamas-Din A, Fradin C, Leber B et al. (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135: 1074–1084. [DOI] [PubMed] [Google Scholar]
- Marsden VS, O’Connor L, O’Reilly LA, Silke J, Metcalf D, Ekert PG et al. (2002). Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419: 634–637. [DOI] [PubMed] [Google Scholar]
- McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP et al. (1989). bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57: 79–88. [DOI] [PubMed] [Google Scholar]
- Mérino D, Giam M, Hughes PD, Siggs OM, Heger K, O’Reilly LA et al. (2009). The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol 186: 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K. (2006). The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688. [DOI] [PubMed] [Google Scholar]
- Peng J, Tan C, Roberts GJ, Nikolaeva O, Zhang Z, Lapolla SM et al. (2006). tBid elicits a conformational alteration in membrane-bound Bcl-2 such that it inhibits Bax pore formation. J Biol Chem 281: 35802–35811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñon JD, Labi V, Egle A, Villunger A. (2009). Bim and Bmf in tissue homeostasis and malignant disease. Oncogene 27: S41–S52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploner C, Kofler R, Villunger A. (2009). Noxa: at the tip of the balance between life and death. Oncogene 27: S84–S92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polzien L, Baljuls A, Rennefahrt UEE, Fischer A, Schmitz W, Zahedi RP et al. (2009). Identification of novel in vivo phosphorylation sites of the human pro-apoptotic protein Bad: pore-forming activity of Bad is regulated by phosphorylation. J Biol Chem 284: 28004–28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SS, Madesh M, Davies E, Antonsson B, Danial N, Hajnockzy G. (2009). Bad targets the permeability transition pore independent of Bax or Bak to switch between Ca2+ -dependent cell survival and death. Mol Cell 33: 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer B, Quispe J, Choudhary V, Chipuk JE, Ajero TG, Du H et al. (2009). Mitochondrial outer membrane proteins assist Bid in Bax-mediated lipidic pore formation. Mol Biol Cell 20: 2276–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Norris KL, Youle RJ. (2008). Mitochondrial dynamics and apoptosis. Genes Dev 22: 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C, Dlugosz PJ, Peng J, Zhang Z, Lapolla SM, Plafker SM et al. (2006). Auto-activation of the apoptosis protein Bax increases mitochondrial membrane permeability and is inhibited by Bcl-2. J Biol Chem 281: 14764–14775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrones O, Etxebarria A, Landejuela O, Landeta O, Antonsson B, Basanez G. (2008). BIM and tBid are not mechanistically equivalent when assisting BAX to permeabilize bilayer membranes. J Biol Chem 283: 7790–7803. [DOI] [PubMed] [Google Scholar]
- Wang H, Takemoto C, Akasaka R, Uchikubo-Kamo T, Kishishita S, Murayama K et al. (2009). Novel dimerization mode of the human Bcl-2 family protein Bak, a mitochondrial apoptosis regulator. J Struct Biol 166: 32–37. [DOI] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI et al. (2005). Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19: 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE et al. (2007). Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315: 856–859. [DOI] [PubMed] [Google Scholar]
- Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. (2003). Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278: 48935–48941. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L. (2009). PUMA, a potent killer with or without p53. Oncogene 27: S71–S83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ et al. (2000). Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290: 1761–1765. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lapolla SM, Annis MG, Truscott M, Roberts GJ, Miao Y et al. (2004). Bcl-2 homodimerization involves two distinct binding surfaces, a topographic arrangement that provides an effective mechanism for Bcl-2 to capture activated Bax. J Biol Chem 279: 43920–43928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhu W, Lapolla SM, Miao Y, Shao Y, Falcone M et al. (2010). Bax forms an oligomer via separate, yet interdependent, surfaces. J Biol Chem 285: 17614–17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW. (1996). Bcl-2 mutants with restricted subcellular localization reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 15: 4130–4141. [PMC free article] [PubMed] [Google Scholar]