

Abstract

Mycobacterium tuberculosis protein-tyrosine-phosphatase B (MptpB) is a secreted virulence factor that subverts antimicrobial activity in the host. We report here the structure-based design of selective MptpB inhibitors that reduce survival of multidrug-resistant tuberculosis strains in macrophages and enhance killing efficacy by first-line antibiotics. Monotherapy with an orally bioavailable MptpB inhibitor reduces infection burden in acute and chronic guinea pig models and improves the overall pathology. Our findings provide a new paradigm for tuberculosis treatment.
Introduction
Tuberculosis (TB) remains a major health problem and leading cause of death worldwide. Antibiotic resistance is a main obstacle in the cure and eradication of TB, with over half a million new cases of drug-resistant TB per year. More alarming is the increasing number of extensive or untreatable drug-resistant TB cases (WHO TB Report 2017). Critical to the pathogenesis of Mycobacterium tuberculosis, the causative agent of TB, is the secretion of virulence factors that subvert the innate immune response and prevent bacterial control by host macrophages.1,2 In this context, antivirulence drugs targeting the pathogen survival mechanisms may represent an efficient complementary strategy to antibiotics to increase efficacy and assist in clearing the infection. Antivirulence strategies are now emerging as promising therapies to counterbalance antibiotic resistance in a number of microbial infections including TB,3−7 and yet this is a largely unexploited area in the clinic.
One such virulence factor is the MptpB phosphatase that is secreted into the cytoplasm of host macrophages.8 MptpB is critical for M. tuberculosis intramacrophage survival and for persistence of the infection in animal models9,10 and acts by both attenuating the bactericidal immune responses and promoting host cell survival. We report here that selective inhibition of MptpB impairs survival of multidrug-resistant (MDR) TB in human macrophages and reduces infection burden in acute and chronic guinea pig models. Furthermore, inhibition of MptpB enhances mycobacterial killing by the first-line antibiotics rifampicin (RIF) and isoniazid (INH). Previously, we reported that MptpB dephosphorylates in vitro the key signaling lipids phosphatidylinositol 3-phosphate (PI3P) and phosphatidylinositol 3,5-biphosphate (PI(3,5)P2).11 These lipids control critical steps of phagolysosomal biogenesis and bacterial clearance.12 We hypothesized that inhibition of MptpB activity may thus restore the intrinsic host response compromised by this virulence factor, offering a novel therapeutic mechanism against M. tuberculosis infections. Since MptpB is not essential for extracellular growth,10 its inhibition could prove a distinct therapeutic advantage over antibiotics as it would potentially inflict less selective pressure and reduce acquired drug resistance. An additional advantage is that to block the secreted MptpB there is no need for drug delivery across the complex and poorly permeable mycobacterial cell wall. The lack of a human orthologue also makes MptpB an attractive drug target for specific and selective therapy.
Crucially, we and others have demonstrated that MptpB inhibitors impair mycobacterial survival in macrophages,13−15 supporting our hypothesis. Our initial isoxazole-based MptpB inhibitors displayed modest potency and selectivity.13 Other reported potent MptpB inhibitors showed little efficacy in animal models,15 indicating that optimization of both target affinity and pharmacokinetics are needed to develop compounds with in vivo efficacy.
We report here the rational structure-based development of our initial C1 isoxazole inhibitor13 to generate a new series of MptpB inhibitors with improved potency, selectivity, and cell activity. Furthermore, an analogue from this series showed an excellent pharmacokinetics profile, oral bioavailability, and in vivo efficacy in guinea pig models of tuberculosis infection.
Results and Discussion
The structure of MptpB16 has an unusually large active site, with a primary phosphate-binding pocket (P1) and two unique secondary pockets (P2 and P3) not present in human phosphatases. In the crystallographic structure of MptpB with an oxalylamino-methylene-thiophene sulphonamide inhibitor,17 the oxalylamino group binds to P1 whereas the sulphonamide partially occupies P2. Molecular docking of our C1 inhibitor13 indicated that the isoxazole group binded to P1 and P3, but P2 remained unoccupied (Figure 1). Our strategy to develop this initial hit used a structure-based rational approach aimed to retain binding at P1 and P3 (isoxazole warhead) while exploiting binding at P2 to increase potency and selectivity, see, for example, structures 2 and 5 (Scheme 1, Figure 1).
Figure 1.

Rational structure-based design of MptpB inhibitors. (a) Active site of MptpB with compounds C1 (cyan)13 and 5 (orange) docked. Isoxazole head in C1 and 5 occupies the P1 pocket (P-loop) and neighboring P3 pocket, whereas the additional dichlorophenol group in 5 occupies P2. (b) Poor activity of the isoxazole head alone and that of the intermediates of the series confirms that binding at P2 is essential to achieve higher potency.
Scheme 1. Development of the New Series of Isoxazole-Based Compounds.

P1/P3 binding isoxazole warhead was used as the starting core. Subsequent addition of a 4-phenyl linker and the dichlorophenol fragment generated the parent compound 5 providing higher potency and selectivity over human phosphatase PTP1B.
Computational screening of commercial fragment libraries using a genetic-based algorithm18,19 and the structure of MptpB17 identified >300 motifs interacting at P2, providing suitable starting points for the design of a new series of 4,5-diarylisoxazole-3-carboxylic acids (Scheme 2).
Scheme 2. Isoxazoles Identified for Synthesis.

Synthesis of 4,5-Diarylisoxazole-3-carboxylic Acids
Isoxazoles 1–13 were identified for synthesis (Scheme 2). The synthesis of 4,5-diarylisoxazole-3-carboxylic acids 1–4 is outlined in Scheme 3. Methyl 5-phenylisoxazole-3-carboxylate 14 is well known20,21 and was synthesized from acetophenone by a slight modification of literature procedures (see Experimental Methods). Following bromination of the methyl carboxylate 14, Suzuki couplings of the bromoisoxazole 15(22) with phenyl-, 3-methoxyphenyl-, and 3-hydroxyphenyl-boronic acids gave the 4,5-diarylisoxazole-3-carboxylates 16,2317, and 18. Saponification of these and the isoxazolecarboxylate 14 gave the corresponding carboxylic acids 1–4.
Scheme 3. Synthesis of the 4,5-Diarylisoxazole-3-carboxylic Acids 1–4.

Reagents and conditions: (i,) NaOH, MeOH, THF, rt, 1 h (1, 65%; 2, 60%; 3, 50%; 4, 55%); (ii) NBS, TFA, heat under reflux 48 h (64%); (iii) ArB(OH)2, Na2CO3, DMF, bis(triphenylphosphine)palladium(II) dichloride or Pd(Ph3P)4, 90 °C, 1–3 h (16, 80%; 17, 42%; 18, 49%).
The triflate 19 prepared from the phenol 18 was coupled with the boronate 21 that was available from 4-bromo-2,6-dichlorophenol 20 to give the methyl 4,5-diarylisoxazole-3-carboxylate 22. This was hydrolyzed to give the isoxazole-3-carboxylic acid 5 ready for screening, see Scheme 4.
Scheme 4. Preparation of the Isoxazole-3-carboxylic Acid 5.

Reagents and conditions: (i) (CF3SO2)2O, py., DCM, 0 °C to rt, 90 min (94%); (ii) bis(pinacolato)diboron, [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride, dioxane, 90 °C, 18 h (59%); (iii) 19, 21, DMF, Na2CO3, Pd(PPh3)4, 90 °C, 3 h (45%); (iv) NaOH, MeOH, rt, 1 h (81%).
The analogous isoxazole-3-carboxylic acids 6–12 were similarly obtained from the triflate 19 and the requisite aryl boronic acid or boronate. These were commercially available except that they required the preparation of the isoxazole 12. This was prepared from the corresponding bromide (see Experimental Methods). Analogous chemistry was used to prepare the 5-methylisoxazole-3-carboxylic acid 13 from the known methyl 4-bromo-5-methylisoxazole 24, see Scheme 5.
Scheme 5. Preparation of the 5-Methylisoxazole-3-carboxylic Acid 13.

Reagents and conditions: (i) NBS, TFA, 0 °C, heat under reflux, 16 h; (ii) ArB(OH)2, aq. NaHCO3, DMF, Pd(Ph3P)2Cl2, 90 °C, 5 h (28%); (iii) (CF3SO2)2O, py., DCM, 0 °C to rt, 6 h (66%); (iv) 21, Pd(Ph3P)4, DMF, 90 °C, 3 h; (v) NaOH, MeOH, 0 °C, 1 h (65%).
Activity of the 4,5-Diarylisoxazole-3-carboxylic Acids and Biochemical Validation
The initial isoxazole fragment alone 1 showed very poor inhibition, but the introduction of a phenyl ring at the 4-position reduced the IC50 from >500 μM 1 to 17 μM 2. Extending the aromatic ring with a dichlorophenol fragment, identified in the computational screen, afforded a further reduction to 0.9 μM 5. Improved binding for the new compounds was accompanied with increased selectivity over the human phosphatase hPTP1B from 1.6-fold for compound C1 to 5.9-fold for compound 2 and 141-fold for compound 5 (Figure 1b), supporting the notion that binding at P2 is critical for selectivity.
Computational docking analysis of the new series of compounds showed a good correlation between the estimated free energy of binding and the experimental activity (Supplementary Table 1). Molecular docking confirmed that additional binding at P2 was responsible for the significant increase in potency of 5 compared to compounds 2–4. Subsequently, we explored variations of the dichlorophenol fragment substituents in the derivatives 6–12 (Table 1).
Table 1. Inhibitory Activity of the 4,5-Diarylisoxazole-3-carboxylic Acids and Selectivity over M. tuberculosis and Human Phosphatasesa.
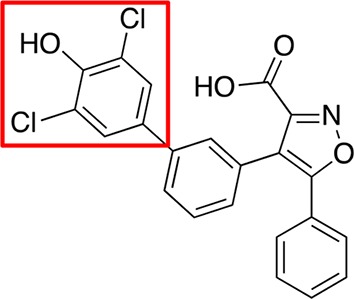

Scaffold of the new series of isoxazole-based compounds is shown at the top. Substitutions of the dichlorophenol head (red square) were explored during development of compound 5. Activities (IC50 values in μM) toward M. tuberculosis MptpB, MptpA, and human PTP1B and Vaccinia H1-related (VHR) phosphatases are shown.
The double- and triple-substituted aromatic groups showed similar IC50 to 5. The introduction of the NO2 group at the meta position of the phenyl ring 12 increased potency by 50% with respect to 5, resulting in an IC50 of 0.4 μM, and afforded an excellent selectivity over human phosphatases (900-fold for PTP1B) and the M. tuberculosis phosphatase MptpA (Table 1).
Our models suggested that the para OH group of the phenyl ring (P2 head) could form hydrogen bonds with the Oε1 of E129 (Figure 2a) and that the meta NO2 group in 12 could form additional interactions with R136 or H94, thus explaining its increased potency. Mutational analysis of the P2 pocket residues validated this mode of binding. Mutations of E129 to alanine showed a 2–5-fold decrease in inhibition (Figure 2b), while mutation of H94 or R136 had a milder effect (∼2-fold decrease).
Figure 2.

Binding at the P2 pocket is important for the efficacy of the new series of MptpB inhibitors. (a) Mode of binding of the representative compound 5 into the active site of MptpB, as suggested from the molecular docking. (b) Mutation of key interacting residues at the P2 pocket resulted in a loss of 2–5 fold the IC50 values (μM). (c) Cell activity of the new series results in substantial (81–87%) reduction in the mycobacterial (BCG) burden of infected mouse macrophages (J774) 72 h post infection, compared to DMSO-treated macrophages. Reduction in bacterial burden is already observed at 24 h postinfection (Supporting Information Figure 2). Plots represent the average CFUs (+SEM) per well (from a 96-well plate, see Experimental Methods for details) of at least three independent experiments, with statistical significance relative to the control (DMSO treated) established using one-way ANOVA, Dunnett’s test (****p value < 0.0001, **p = 0.012). Inset shows viability of treated macrophages.
New MptpB Inhibitors Reduce Mycobacterial Burden in Macrophage Infections
Next we tested the efficacy of the new compounds in reducing mycobacterial survival in macrophage infections. The new series of compounds showed dose-dependent efficacy in reducing intracellular mycobacterial (BCG) burden in mouse macrophages (J774) up to 87% after 72 h of infection compared to control (DMSO treated) (Figure 2c). These findings are consistent with MtpB inhibition assisting bacterial clearance in host macrophages, even in the absence of INF-γ activation (see methods). The new compounds show increased efficacy at 80 μM (except for 5) compared to our initial C1 isoxazole inhibitor, thus correlating with the higher potency of the new series. Toxicity of the new compounds is low as shown in the cell survival assays (Figure 2c inset), with only compound 10 showing a substantial effect (>23% reduction) on cell viability at >100 μM doses.
The new series of compounds displayed moderate permeability and lipophilicity (logD) and good solubility, they were stable in plasma with moderate clearance in human liver microsomes, and had high plasma protein binding (Suppl. Table 2). They also showed poor in vivo pharmacological properties in guinea pigs, with a bioavailability below 13% (Table 2). We then replaced the phenyl ring at position 5 of the isoxazole by a methyl group to reduce the bulk and number of rings in the compound, generating compound 13. The potency of compound 13, at 3 μM, was lower than the best of the parent series of inhibitors 5 and 12 (by 3–7 fold), possibly due to reduced hydrophobic interactions at the P3 pocket. However, compound 13 showed very good kinetic solubility (200 μM) and a good PAMPA value (78.1 nM/s), suggesting a potential good cell penetration, despite showing a higher lipophilicity than the parent series (logD 4) (see Supporting Information for details). Importantly, compound 13 displayed improved pharmacological properties; it is orally bioavailable and has an excellent phamacokinetic profile (Table 2); therefore, it was selected for further evaluation of its cell activity and efficacy in animal models of infection.
Table 2. Pharmacokinetic Parameters for MptpB Inhibitorsa.
|
13 (lead) |
5 |
8 |
9 |
|||||
|---|---|---|---|---|---|---|---|---|
| guinea
pig |
guinea
pig |
guinea
pig |
guinea
pig |
|||||
| route | IP | oral | IP | oral | IP | oral | IP | oral |
| dose (mg/kg) | 4 | 8 | 2.5 | 5 | 1.25 | 3.5 | 2.5 | 5 |
| Cmax (ng/mL) | 31 519 | 111 099 | 3714 | BLQ | 746 | BLQ | 197 | BLQ |
| t1/2 (h) | 1.4 | 5.1 | 0.7 | BLQ | 1.3 | BLQ | 1.4 | BLQ |
| AUCinf (ng·h/mL) | 71 854 | 230 407 | 4968 | BLQ | 1742 | BLQ | 399 | BLQ |
| bioavailability | 156%b | <4% | <13% | <2.5% | ||||
Compounds 3, 8, 9, and 13 were tested in guinea igs to determine their PK profile. Cmax was observed at 0.5 h after IP (intraperitoneal) dosing and at 0.25 h after PO (oral) dosing.
High bioavailability may be due to prolonged absorption after the oral dose limiting the rate of elimination; BLQ, below limit of quantification.
MptpB Inhibitors Reduce Survival of Multidrug-Resistant Strains in Macrophages and Enhance Killing by First-Line Antibiotics
Investigation of compound 13 desmonstrated that it also exhibits dose-dependent efficacy in reducing intracellular mycobacterial (BCG) burden in mouse macrophages (J774) up to 84% (Figure 3b), yet it does not affect extracellular bacterial growth (Figure 3c), thus confirming that inhibition exclusively targets intracellular mycobacteria, as expected. Critically, treatment with compound 13 also reduces the intracellular mycobacterial burden in human macrophages (THP1) up to 63% when using a drug-susceptible M. tuberculosis strain (H37Rv) or up to 74% when using a MDR strain (Beijing-“W”) (Figure 3d). A similar effect was observed for the initial C1 compound (Supplementary Figure 1), demonstrating that efficacy in reducing MDR-TB survival is a general quality of MptpB inhibitors.
Figure 3.

Compound 13 reduces intracellular bacterial burden of H37Rv and MDR-TB in macrophage infections. (a) Structure of compound 13. (b) Effect of dose-dependent treatment with compound 13 on mycobacterial burden (BCG) in infected mouse macrophages (J774) at 72 h post infection (****p < 0.0001, ***p < 0.0004, *p < 0.04 by one-way ANOVA, Dunnett’s test). Inset shows macrophage viability upon treatment with 13 (**p = 0.01). (c) Extracellular growth of BCG is not affected by treatment with compound 13 (80 μM) respect to DMSO control. (d) Treatment with compound 13 (20 or 100 μM) reduces intracellular survival of an MDR strain (Beijing-W) and of the drug-susceptible M. tuberculosis H37Rv strain in human THP1 macrophages (gray). Controls on the effect on extracellular bacterial growth are shown (black), **p = 0.0021; *** p = 0.0002, ****p < 0.0001 by unpaired t test). (e) Treatment with compound 13 increases the antibacterial activities of RIF and INH antibiotics in infected macrophages (BCG in J774 mouse macrophages, ****p < 0.0001, **p = 0.0017, by one-way ANOVA, Dunnett’s test). (d and e) Data are the mean CFUs at 72 h post infection of at least three biological replicates (+SEM). Limit of detection is <10 cfu.
To test whether compound 13 is compatible with first-line TB drugs, isoniazid (INH) and rifampicin (RIF), we determined doses of these antibiotics that caused <25% reduction in the bacterial burden of macrophages (0.1 μg/mL for INH and 0.3 μg/mL for RIF, Figure 3e). We then used these doses of INH and RIF in combination with a low dose, 5 μM, of compound 13. The combination resulted in >93% reduction in bacterial burden (Figure 3e). Thus, treatment in combination with compound 13 enhances killing by current antitubercular drugs. This is an important finding since tuberculosis treatments rely on drug combination therapies to clear the infection.
MptpB Inhibition Alters Phagosomal Phosphoinositde-3-phosphate Dynamics during Infection
MtptB dephosphorylates PI3P and PI(3,5)P2 in vitro,11 two important anchors of Rab proteins that drive phagosomal maturation and clearance of infection. However, we do not know its effect on cellular PI dynamics. We tested if compound 13 affected PI3P dynamics on M. tuberculosis phagosomes. For that we monitored PI3P localization by live-cell imaging in macrophages expressing a fluorescent PI3P-binding module (FYVE2X-EGFP) after infection with fluorescently labeled M. tuberculosis H37Rv.24 PI3P associated with M. tuberculosis phagosomes immediately after infection, but PI3P signal decreased rapidly after 2–4 min as previously reported.24 However, in the 13-treated macrophages, the peak of PI3P was prolonged up to 12 min (Figure 4). The data indicate that MptpB inhibition extends the presence of PI3P and its association with M. tuberculosis phagosomes, suggesting a role for MptpB in host phosphoinositide metabolism as hypothesized from its in vitro activity.11
Figure 4.
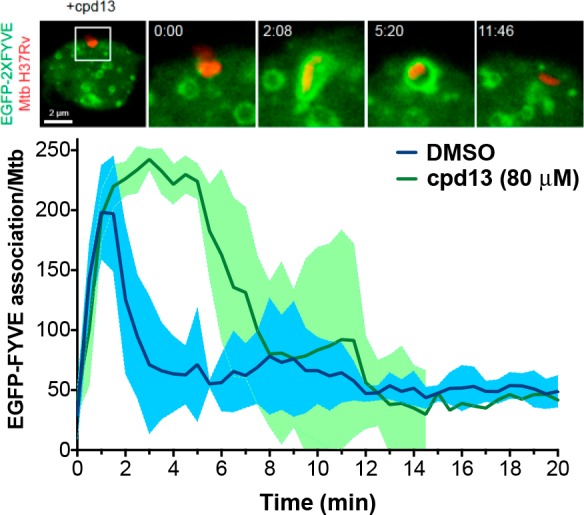
MptpB inhibition alters phagosomal PI3P dynamics. Spatiotemporal dynamics of PI3P (as visualized by expression of EGFP-FYVE) on M. tuberculosis containing phagosomes (top figure). Plot shows the quantitative analysis of the association of EGFP-FYVE to PI3P at the phagosomal membrane during the first 20 min of phagocytosis (data from 3 independent experiments). Treatment with 13 extends the peak of PI3P up to 12 min compared to the rapid decay of the PI3P peak in the untreated phagosomes (DMSO control). DMSO, control for untreated macrophages (RAW264.7); cpd 13, macrophages treated with compound 13.
Monotherapy with an Orally Bioavailable MptpB Inhibitor Reduces Infection Burden in Acute and Chronic Guinea Pig Models
In vivo profiling of compound 13 showed high exposure (Cmax 112 μg/mL, AUC 230 μg·h/mL), long half-life (t1/2 5 h), good oral availability, and relevant tissue distribution in guinea pigs upon parental and oral dosing (Table 2 and Supplementary Figure 3), making it suitable for efficacy studies in animal models of infection. Tolerability studies with 13 at 50 or 100 mg/kg (dosing once daily for 7 days) showed no adverse drug effects, and weight increases of >5% were observed in all animals during the tolerability trial.
Compound 13 was then assessed for efficacy as monotherapy in the acute and chronic guinea pig models of TB infection. For the acute infection, animals were infected with 96 CFU (avg. ± 27 SEM) and after 24 h orally dosed once daily with compound 13 for 4 weeks. Treatment resulted in a 0.9 log reduction of bacterial burden in the lungs relative to vehicle. For the chronic infection, guinea pigs were infected with 63 CFU (avg. ± 18 SEM), and treatments were orally administered daily for 4 weeks starting at 28 days post infection. Treatment with compound 13 resulted in at least 1 log reduction in bacterial burden in lungs and spleens (Figure 5a and Supplementary Table 3).
Figure 5.

MptpB inhibition reduces bacterial burden in animal models and improves pahtology. (a) Efficacy of monotherapy treatment with 13 for 28 days in the acute and chronic guinea pig models of TB (female Hartley Duncan). Treatment was with 13 (100 mg/kg)) or RIF (50 mg/kg) orally administered once daily; VEH, vehicle control. Bars represent the mean value (±SEM) of CFUs from 4 to 5 animals. Statistical significance is indicated (* p < 0.05 by one-way ANOVA, Dunnett’s test). (b and c) Gross pathological scoring of the TB-infected guinea pig lungs (b) and spleens (c) based on Jain et al.27 The lungs and spleen from individual animals were given a score from 1 to 4 based on the number and size of tubercles, level of involvement, inflammation, and necrosis. Bars represent the mean value (±SEM) of CFUs from 4 to 5 animals. Statistical significance is indicated (* p < 0.05 by one-way ANOVA, Dunnett’s test). (d) Representative images of tissues at 56 days post infection (chronic model) showing reduction of the number granuloma in the compound 13-treated lungs and spleen. (e) Representative histopathological images from lungs and spleens at 56 days post infection from the chronic model. Lungs and spleens fixed in neutral buffer formalin were sectioned (5 μm) and stained with hematoxylin and eosin (H&E) and imaged at 10× magnification. Vehicle-treated lungs and spleen show an increased presence of granulomatous infiltration (black arrows) and pathologic damage relative to compound 13-treated lungs and spleen. Bars represent 2 mm.
Pathological differences were observed between 13 and vehicle-treated groups in both lungs and spleens (Figure 5b–e) from both the acute and the chronic infection studies. Although the total number of tubercles was similar in both 13 and vehicle-treated animals, larger tubercles were consistently present in all vehicle group. Overall, less damage to the spleens and lungs were observed in the 13-treated group relative to the vehicle-only group.
Inspection of the lungs and spleens after 56 days of infection revealed visible necrotic lesions ranging from ∼1 to 4 mm, with more consolidation observed in the vehicle group than in the compound 13-treated group, and with organs presenting up to 19% increase in weight respect to the 13-treated group (Supplementary Table 4). The histopathological analysis of the vehicle-treated lungs and spleens displayed large and diffuse confluent granulomas with necrotizing cores and inflammation throughout the parenchyma, pathology typically associated with a chronic infection. The compound 13-treated lungs and spleens showed an improvement in the pathology with fewer smaller lesions and reduced inflammation both in lungs and in spleens (Figure 5e).
These results provide proof-of-concept that MptpB inhibitors, used as monotherapy, can be effective in controlling TB infection in animal models, despite not having direct bactericidal activity. Particularly significant for clinical applications is the efficacy and improved pathology observed in the chronic model because guinea pigs develop a similar immunopathology and inflammatory response to the infection as in humans, forming granulomas that contain persistent bacteria.25,26
Conclusions
In summary, our structure-guided drug development approach, exploiting unique features in the MptpB structure, has delivered an orally bioavailable compound with excellent therapeutic properties. We have demonstrated that MptpB inhibitors are selective and effective against MDR-TB and that they increase the intracellular killing efficacy of first line antibiotics RIF and INH, indicating their suitability for combination therapies. Notably, reduction of intracellular survival occurs in the absence of macrophage preactivation with IFNγ, suggesting that this strategy may be particularly advantageous when impaired macrophage activation fails to control the infection (i.e., immuno-compromised patients).
Importantly, our lead compound showed efficacy in reducing bacterial burden in a clinically relevant guinea pig model of infection as well as improvement in the pathology of lungs and spleen. This is also consistent with the enhanced pathology and reduced inflamation observed when the mptpB gene is mutated.9 This is the first proof of concept that MptpB inhibitors, used as monotherapy, can significantly reduce infection burden. A previous study15 showed a mild effect (<0.5 log reduction) when both MptpA and MptpB inhibitors where added to a cocktail of three first-line antibiotics, isoniazid–rifampicin–pyrazinamide (HRZ), effectively a 5-drug combination. However, there was no effect on bacterial burden when only MptpA or MptpB inhibitors were added to the HRZ cocktail. No data were presented on monotherapy treatments with either MptpA or MptpB inhibitors; thus, no direct comparison can be made with our results.
Renewed interest in developing antivirulence agents for TB treatment (reviewed in refs (3−7)) makes this study timely. Furthermore, MptpB inhibitors could be developed into broad-spectrum antivirulence drugs, as MptpB orthologues are present in more than 50 human pathogens including C. difficile, E. faecalis, K. pneumoniae, Yesinia spp, and L. monocytogenes.28,29
Antivirulence drugs hold great promise for the management of TB infections. The current challenges in eradicating TB include the following: prevention with the existing BCG vaccine has limited efficacy; standard treatments with antibiotics are long and complex, and chronic drug exposure over a prolonged period is linked to development of drug resistance; and host-directed therapies are difficult to control because they are patient dependent. Antivirulence agents offer advantages to overcome these challenges because their action is independent of the host fitness and they have the potential to limit drug resistance. Our findings suggest that MptpB inhibition offers a new paradigm for TB therapy with the potential to treat MDR-TB and improve antibiotic efficacy and overall pathology.
Experimental Methods
Synthetic Chemistry
General Experimental Details
All reactions were carried out under an atmosphere of dry nitrogen unless otherwise stated. Low-resolution mass spectra were recorded on a Micromass Trio 200 spectrometer using electron impact (EI) ionization or electrospray in positive (ES+) or negative modes (ES–). High-resolution mass spectra were recorded on a Kratos Concept IS spectrometer. Infrared spectra were recorded on a Genesis FTIR as evaporated films on sodium chloride plates. Proton NMR spectra (1H NMR) and carbon NMR spectra (13C NMR) were recorded on Bruker (500 MHz), Varian Unity 500 (500 MHz), Varian INOVA 400 (400 MHz), or Varian INOVA Unity 300 (300 MHz) spectrometers. Residual nondeuterated solvent was used as an internal standard. Chemical shifts (δH and δC) are quoted in parts per million (ppm) downfield from tetramethylsilane (TMS).
Flash column chromatography was carried out using silica gel 60H from Merck. Light petroleum refers to the fraction that boils between 40 and 60 °C and was distilled prior to use. Ether refers to diethyl ether that was used without further purification. Tetrahydrofuran was dried over sodium/benzophenone and distilled under a nitrogen atmosphere. DCM was dried over calcium hydride and distilled under an atmosphere of nitrogen. All other reagents and solvents were used as purchased unless otherwise stated. The purity of compounds submitted for screening was determined to be greater than 95% by HPLC (see Supplementary Information for copies of the HPLC traces)
5-Phenylisoxazole-3-carboxylic Acid (1): Standard Procedure30,31
Aqueous sodium hydroxide (2 M, 2.7 mL, 5.4 mmol) was added to methyl 5-phenylisoxazole-3-carboxylate 14 (219 mg, 1.08 mmol) in MeOH:THF (1:1 v/v, 60 mL) at rt, and the mixture was stirred for 1 h. Water was added (60 mL), and the mixture was acidified to pH 1 using aqueous hydrogen chloride (2 M) and then concentrated under reduced pressure to give a slurry that was extracted with EtOAc (3 × 60 mL). The organic extracts were dried (NaSO4) and concentrated under reduced pressure to give an off-white solid that was recrystallized (light petroleum:EtOAc, 100:1 v/v) to give the title compound 1 as a white solid (132 mg, 0.7 mmol, 65%); mp 162.8–163.8 °C (lit.31 160–161 °C); TLC (light petroleum:EtOAc, 50:50 v/v, with 0.1% TFA) Rf = 0.3; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.27 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 14.13 (br. s, 1H), 7.96 (d, J 8.0 Hz, 2H), 7.59–7.54 (m, 3 H), 7.44 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 170.8, 160.9, 157.8, 130.9, 129.3, 126.2, 125.8, 100.9; IR (film) 3126, 3055, 2611, 2512, 1703, 1474, 1445, 1261, 995, 914, 820, 760, 686 cm–1.
4,5-Diphenylisoxazole-3-carboxylic Acid (2)23
Following the procedure outlined for the preparation of carboxylic acid 1, methyl 4,5-diphenylisoxazole-3-carboxylate 16 (90 mg, 0.32 mmol) in MeOH:THF (1:1 v/v, 6 mL) and aqueous sodium hydroxide (2 M, 0.8 mL, 1.6 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 2 as a white solid (51 mg, 0.2 mmol, 60%); mp 134.5–135.3 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.36; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.18 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 13.65 (br. s, 1H), 7.51–7.32 (m, 10H); 13C NMR (125 MHz, DMSO-d6) δ 177.9, 161.0 151.8, 130.0, 129.1, 128.6, 128.4, 126.6, 116.2; IR (film) 3058, 1711, 1429, 1225, 968, 770, 693 cm–1; LRMS (m/z, ES–) 220 ([M – 0.45]−, 100%); HRMS (m/z) [M + H]+ calcd for C16H12NO3, 266.0812; found 266.0817.
4-(3-Methoxyphenyl)-5-phenylisoxazole-3-carboxylic Acid (3)
Following the procedure outlined for the preparation of carboxylic acid 1, 4-(3-methoxyphenyl)-5-phenylisoxazole-3-carboxylate 17 (100 mg, 0.32 mmol) in MeOH:THF (1:1 v/v, 6 mL) and aqueous sodium hydroxide (2 M, 0.8 mL, 1.6 mmol), after recrystallization (MeOH:H2O, 50:1 v/v), gave the title compound 3 as a white solid (47 mg, 0.16 mmol, 50%); mp 162.5–163.5 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.25; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.20 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 13.94 (br. s, 1H), 7.49–7.42 (m, 5H), 7.35 (t, J 8.0 Hz, 1H), 7.01 (dd, J 2.0, 8.0 Hz, 1H), 6.95 (m, 1H), 6.91 (d, J 7.5 Hz, 1H), 3.73 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 166.0, 160.9, 159.2, 156.3, 130.6, 130.2, 129.7, 129.0, 126.7, 126.4, 122.2, 115.9, 115.7, 113.8, 55.1; IR (film) 3154, 1727, 1621, 1580, 1439, 1276, 1227, 1189, 1095, 1042, 1008, 966, 878, 826, 805, 768, 689 cm–1; LRMS (m/z, ES–) 250 ([M – 45]−, 100%); HRMS (m/z, ES–) [M – CO2H]− calcd for C16H12NO2, 250.0873; found 250.0861.
4-(3-Hydroxyphenyl)-5-phenylisoxazole-3-carboxylic Acid (4)
Following the procedure outlined for the preparation of carboxylic acid 1, methyl 4-(3-hydroxyphenyl)-5-phenylisoxazole-3-carboxylate 18 (150 mg, 0.52 mmol) in MeOH:THF (1:1 v/v, 10 mL) and aqueous sodium hydroxide (2 M, 1.5 mL, 3 mmol), after recrystallization (MeOH:H2O, 50:1 v/v), gave the title compound 4 as a white solid (80 mg, 0.29 mmol, 55%); TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.5; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.22 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 14.04 (br. s, 1H), 9.56 (s, 1H), 7.49–7.41 (m, 5H), 7.22 (t, J 8.0 Hz, 1H), 6.81 (d, J 7.5 Hz, 1H), 6.75–6.72 (m, 2H); IR (film) 3468, 3243, 3064, 1722, 1629, 1583, 1474, 1444, 1331, 1228, 1176, 971, 867, 795, 766, 687 cm–1; LRMS (m/z, ES+) 282 ([M + 1]+, 100%); HRMS (m/z, ES+) [M + H]+ calcd for C16H12NO4, 282.0761; found 282.0753.
4-(3′,5′-Dichloro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (5)
Following the procedure outlined for the preparation of carboxylic acid 1, methyl 4-(3′,5′-dichloro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate 22 (900 mg, 2.04 mmol) in MeOH:THF (25 mL, 4:1 v/v) and aqueous sodium hydroxide (2 M, 5.11 mL, 10.2 mmol), after recrystallization (light petroleum:EtOAc, 10:1 v/v), gave the title compound 5 as a white solid (700 mg, 1.65 mmol, 81%); mp 187.5–188.0 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.5; HPLC (silica, EtOAc with 0.1% TFA) retention time 2.79 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 14.02 (br. s, 1H), 10.35 (s, 1H) and 7.75–7.32 (m, 11H); 13C NMR (125 MHz, DMSO-d6) δ 166.3, 160.9, 156.2, 148.6, 137.5, 132.4, 130.6, 129.6, 129.2, 129.0, 128.1, 126.8, 126.6, 126.5, 126.3, 122.8, 116.0; IR (film) 3056, 1717, 1468, 1430, 1291, 1161, 1024, 868, 794, 768, 691 cm–1; LRMS (m/z, ES–) 426 ([M – 1]−, 6%), 424 ([M – 1]−, 8), 382 ([M – 45]−, 65), 380 ([M – 45]−, 100); HRMS (m/z, ES–) [M – H]− calcd for C22H12NO435Cl2, 424.0148; found 424.0154.
4-(4′-Hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (6)
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (300 mg, 0.7 mmol) and 4-hydroxyphenylboronic acid (107 mg, 0.77 mmol) in DMF (7 mL) together with Pd(PPh3)4 (8 mg, 0.007 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v, then with DCM), gave methyl 4-(4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (159 mg, 0.427 mmol, 61%); mp 62.7–63.5 °C; TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.14; 1H NMR (500 MHz, CDCl3) δ 7.61–7.57 (m, 3H), 7.54 (s, 1H), 7.48 (t, J 7.5 Hz, 1H), 7.43 (d, J 9.0 Hz, 2H), 7.39 (d, J 7.5 Hz, 1H), 7.36–7.33 (m, 2H), 7.29 (d, J 7.5 Hz, 1H) 6.89 (d, J 8.5 Hz, 2H), 5.88 (s, 1H), 3.90 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.4, 160.6, 155.7, 155.0, 141.3, 132.9, 130.5, 129.2, 129.1, 128.9, 128.5, 128.4, 128.3, 127.1, 126.9, 126.8, 116.8, 115.8, 52.9; IR (film) 3374, 1741, 1612, 1519, 1445, 1335, 1221, 1175, 1103, 1018, 838, 770, 694 cm–1; LRMS (m/z, ES–) 370 ([M – 1]−, 100%); HRMS (m/z, ES+) [M + H]+ calcd for C23H18NO4, 372.1231; found 372.1223.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate (160 mg, 0.43 mmol) in MeOH (4 mL) and aqueous sodium hydroxide (2 M, 1.1 mL, 2.15 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 6 as a white solid (90 mg, 0.25 mmol, 59%); mp 186.0–187.3 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.49; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.23 min, 100%; 1H NMR (400 MHz, DMSO-d6) δ 13.99 (br. s, 1H), 9.60 (s, 1H), 7.67–7.41 (m, 10H), 7.25 (d, J 7.5 Hz, 1H), 6.83 (d, J 8.5 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.2, 161.1, 157.4, 156.3, 140.4, 130.7, 130.2, 129.4, 129.2, 129.1, 128.0, 127.8, 127.6, 126.8, 126.5, 126.0, 116.2, 115.8; IR (film) 3058, 1754, 1655, 1612, 1545, 1512, 1441, 1222, 1177, 836, 803, 760 cm–1.
4-(3′-Hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (7)
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (300 mg, 0.7 mmol) and 3-hydroxyphenylboronic acid (107 mg, 0.77 mmol) in DMF (7 mL) together with Pd(PPh3)4 (8 mg, 0.007 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v, then with DCM), gave methyl 4-(3′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (50 mg, 0.135 mmol, 19%); mp 59.0–59.6 °C; TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.14; 1H NMR (300 MHz, CDCl3) δ 7.54 (dt, J 2.0, 8.0 Hz, 1H), 7.48–7.44 (m, 3H), 7.38 (t, J 8.0 Hz, 1H), 7.29–7.14 (m, 5H), 7.05–6.85 (m, 2H), 6.73 (ddd, J 1.0, 3.5, 8.0 Hz, 1H), 5.92 (s, 1H), 3.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.4, 160.4, 156.1, 154.8, 141.9, 141.1, 130.5, 129.9, 129.2, 129.1, 129.0, 128.9, 128.8, 127.3, 127.0, 126.6, 119.3, 116.6, 114.6, 114.1, 52.8; IR (film) 3393, 1741, 1597, 1445, 1332, 1220, 783, 694 cm–1; LRMS (m/z, ES–) 370 ([M – 1]−, 80%); HRMS (m/z, ES+) [M + H]+ calcd for C23H18NO4, 372.1231; found 372.1238.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(3′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate (50 mg, 0.135 mmol) in MeOH (2 mL) and aqueous sodium hydroxide (2 M, 1.1 mL, 2.15 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 7 as a white solid (40 mg, 0.11 mmol, 82%); mp 189.9–190.8 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.49; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.22 min, 100%; 1H NMR (400 MHz, DMSO-d6) δ 14.02 (br. s, 1H), 9.55 (s, 1H), 7.66 (ddd, J 1.5, 2.0, 8.0 Hz, 1H), 7.61 (dd, J 1.5, 2.0 Hz, 1H), 7.52–7.42 (m, 6H), 7.33 (dt, J 1.5, 7.5 Hz, 1H), 7.23 (t, J 8.0 Hz, 1H), 7.03 (d, J 8.0 Hz, 1H), 6.99 (t, J 2.0 Hz, 1H), 6.76 (dd, J 2.0, 8.0 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 171.8, 163.1, 157.8, 157.6, 157.8, 157.6, 140.9, 140.4, 130.7, 130.0, 129.8, 129.2, 129.1, 129.0, 128.2, 126.8, 126.6, 117.4, 114.7, 113.5; IR (film) 3066, 1736, 1598, 1489, 1458, 1429, 1349, 1322, 1240, 1210, 1023, 992, 896, 866, 810, 785, 764, 719 cm–1; LRMS (m/z, ES–) 312 ([M – 45]−, 100%); HRMS (m/z, ES–) [M – CO2H]− calcd for C21H14NO2, 312.1030; found 312.1017.
4-(3′,5′-Dichoro-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (8)
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (290 mg, 0.68 mmol) and 3,5-dichlorophenylboronic acid (143 mg, 0.75 mmol) in DMF (7 mL) together with Pd(PPh3)4 (8 mg, 0.007 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v, then with DCM), gave methyl 4-(3′,5′-dichoro-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (120 mg, 0.28 mmol, 42%); TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.51; 1H NMR (500 MHz, CDCl3) δ 7.52 (d, J 7.5 Hz, 1H), 7.45–7.42 (m, 4H), 7.34–7.30 (m, 4H), 7.27–7.23 (m, 3H), 3.82 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.4, 160.3, 154.7, 143.3, 138.9, 135.2, 130.5, 130.2, 129.7, 129.4, 128.9, 128.8, 127.4, 127.0, 126.6, 125.6, 116.1, 52.8; IR (film) 3074, 1740, 1585, 1560, 1444, 1334, 1220, 1175, 1100, 1028, 973, 857, 794, 770, 693 cm–1.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(3′,5′-chloro-3-biphenyl)-5-phenylisoxazole-3-carboxylate (120 mg, 0.28 mmol) in MeOH (3 mL) and aqueous sodium hydroxide (2 M, 1 mL, 2.15 mmol), after recrystallization (light petroleum:EtOA, 100:1 v/v), gave the title compound 8 as a white solid (55 mg, 0.13 mmol, 48%); mp 169.9–171.0 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.53; HPLC (silica, EtOAc with 0.1% TFA) retention time 4.29 min, 100%; 1H NMR (400 MHz, DMSO-d6) δ 14.00 (br. s, 1H), 7.85–7.81 (m, 2H), 7.74 (d, J 2.0 Hz, 2H), 7.60 (m, 1H), 7.55–7.39 (m, 7H); 13C NMR (100 MHz, DMSO-d6) δ 160.9, 156.1, 143.0, 139.3, 137.2, 134.7, 130.7, 129.8, 129.3, 129.1, 128.9, 127.1, 127.0, 126.9, 126.5, 125.5, 125.4, 115.9; IR (film) 3072, 1716, 1587, 1559, 1484, 1442, 1374, 1274, 1229, 1192, 1026, 987, 918, 856, 770, 720, 692 cm–1.
4-(3′,5′-Dimethyl-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (9)
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (290 mg, 0.68 mmol) and 3,5-dimethyl-4-hydroxyphenylboronic acid (185 mg, 0.75 mmol) in DMF (7 mL) together with Pd(PPh3)4 (8 mg, 0.007 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v, then with DCM), gave methyl 4-(3′,5′-dimethyl-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (154 mg, 0.39 mmol, 57%); TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.20; 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J 7.5 Hz, 1H), 7.66 (d, J 8.0 Hz, 2H), 7.60 (s, 1H), 7.55 (t, J 8.0 Hz, 1H), 7.48 (t, J 7.0 Hz, 1H), 7.43 (t, J 8.0 Hz, 2H), 7.36 (d, J 8.0 Hz, 1H), 7.27 (s, 2H), 4.87 (s, 1H), 3.98 (s, 3H), 2.37 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 167.3, 160.5, 155.0, 152.1, 141.5, 132.5, 130.4, 129.2, 129.0, 128.8, 128.5, 128.3, 127.4, 127.1, 126.9, 123.4, 116.8, 52.8, 16.1; IR (film) 3513, 2954, 1741, 1603, 1444, 1325, 1221, 1102, 1034, 974, 874, 816, 794, 769, 703 cm–1.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(3′,5′-dimethyl-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate (150 mg, 0.38 mmol) in MeOH (4 mL) and aqueous sodium hydroxide (2 M, 1 mL, 2 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 9 as a white solid (79 mg, 0.20 mmol, 54%); mp 182.3–182.9 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.55; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.80 min, 100%; 1H NMR (400 MHz, DMSO-d6) δ 14.01 (br. s, 1H), 8.40 (s, 1H), 7.63 (d, J 8.0 Hz, 1H), 7.57 (s, 1H), 7.52–7.41 (m, 6H), 7.24 (d, J 7.5 Hz, 1H), 7.21 (s, 2H), 2.19 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 163.6, 161.1, 153.3, 140.6, 130.6, 130.3, 129.3, 129.1, 129.0, 127.8, 127.6, 126.8, 126.6, 126.0, 124.7, 124.5, 116.3, 113.2, 16.8; IR (film) 3474, 1713, 1601, 1490, 1433, 1315, 1227, 1204, 1178, 1031, 973, 928, 902, 868, 799, 773 cm–1.
4-(3′-Chloro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (10)
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (300 mg, 0.7 mmol) and 3-chloro-4-hydroxyphenylboronic acid (133 mg, 0.77 mmol) in DMF (7 mL) together with Pd(PPh3)4 (8 mg, 0.007 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v then with DCM), gave methyl 4-(3′-chloro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (180 mg, 0.44 mmol, 63%); mp 60.3–61.9 °C; TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.14; 1H NMR (500 MHz, CDCl3) δ 7.60–7.56 (m, 3H), 7.53–7.48 (m, 3H), 7.43–7.32 (m, 5H), 7.08 (d, J 8.5 Hz, 1H), 5.62 (s, 1H), 3.91 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.4, 160.5, 154.9, 151.0, 140.0, 134.2, 130.5, 129.5, 129.2, 129.1, 128.9, 128.6, 127.6, 127.2, 127.1, 126.9, 126.8, 120.3, 116.6, 116.5, 52.8; IR (film) 3419, 3061, 2955, 1740, 1608, 1508, 1485, 1464, 1444, 1387, 1335, 1287, 1221, 1177, 1102, 1054, 1021, 973, 909, 867, 816, 794, 770, 731, 706 cm–1; LRMS (m/z, ES–) 406 ([M – 1]−, 65%), 404 ([M – 1]−, 100); HRMS (m/z, ES+) [M + H]+ calcd for C23H17NO435Cl, 406.0841; found 406.0845.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(3′-chloro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate (157 mg, 0.39 mmol) in MeOH (4 mL) and aqueous sodium hydroxide (2 M, 1 mL, 2.15 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 10 as a white solid (80 mg, 0.2 mmol, 52% yield); mp 171.8–175.0 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.5; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.20 min, 100%; 1H NMR (400 MHz, DMSO-d6) δ 14.00 (br. s, 1H), 10.38 (s, 1H), 7.68–7.62 (m, 3H), 7.51–7.41 (m, 7H), 7.28 (d, J 7.5 Hz, 1H), 7.03 (d, J 8.5 Hz, 1H); 13C NMR (100 MHz, DMSO-d6) δ 166.2, 161.0, 156.3, 152.9, 138.9, 131.5, 130.7, 129.5, 129.2, 129.1, 128.6, 127.9, 127.8, 126.9, 126.5, 126.3, 126.1, 120.3, 117.0, 116.1; IR (film) 3361, 1712, 1607, 1483, 1425, 1343, 1279, 1229, 1142, 1059, 930, 882, 866, 820, 802, 788, 771 cm–1.
4-(3′-Fluoro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (11)
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (630 mg, 1.45 mmol) and 3-fluoro-4-hydroxyphenylboronic acid (250 mg, 1.6 mmol) in DMF (15 mL) with Pd(PPh3)4 (15 mg, 0.014 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v, then with DCM), gave methyl 4-(3′-fluoro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (260 mg, 0.67 mmol, 42%); mp 63.4–65.0 °C; TLC (Et2O:light petroleum, 30:70 v/v) Rf = 0.12; 1H NMR (400 MHz, CDCl3) δ 7.49–7.37 (m, 5H), 7.32–7.11 (m, 6H), 6.92 (t, J 8.5 Hz, 1H), 5.81 (s, 1H), 3.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.5, 160.6, 154.9, 152.5, 150.1, 143.3, 140.2, 133.5, 130.6, 129.4, 129.3, 129.0, 128.5, 127.1, 127.0, 126.8, 123.3, 117.7, 116.7, 114.4, 52.9; IR (film) 3363, 3059, 2955, 1737, 1621, 1522, 1444, 1215, 1104, 1021, 973, 868, 815, 789, 768, 749, 691 cm–1.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(3′-fluoro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate (70 mg, 0.18 mmol) in MeOH (1.5 mL) and aqueous sodium hydroxide (2 M, 0.5 mL, 1.0 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 11 as a white solid (10 mg, 0.03 mmol, 17%); mp 193.5–194.6 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.09; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.17 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 13.99 (br. s, 1H), 10.03 (s, 1H), 7.69–7.66 (m, 2H), 7.52–7.42 (m, 7H), 7.31 (d, J 8.0 Hz, 1H), 7.28 (d, J 7.5 Hz, 1H), 7.00 (t, J 9.0 Hz, 1H); IR (film) 3261, 1719, 1621, 1522, 1443, 1216, 1118, 871, 796, 751, 691 cm–1.
4-(3′-Chloro-4′-hydroxy-5′-nitro-3-biphenyl)-5-phenylisoxazole-3-carboxylic Acid (12)
A mixture of 4-bromo-2-chloro-6-nitrophenol (1 g, 3.96 mmol), KOAc (1.17 g, 11.88 mmol), bis(pinacolato)diboron (1.21 g, 4.75 mmol), and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride (90 mg, 0.1188 mmol) in dioxane (40 mL) was stirred under nitrogen at 90 °C for 18 h before being poured into EtOAc (100 mL). The mixture was washed with aqueous hydrogen chloride (1 M, 100 mL), dried (MgSO4), and concentrated under reduced pressure. Chromatography (Et2O:light petroleum, 5:95–10:90 v/v) of the residue gave pinacolyl 3-chloro-4-hydroxy-5-nitrophenylboronate as a bright yellow solid (0.46 g, 1.54 mmol, 39%); TLC (Et2O:light petroleum, 20:80, v/v) Rf = 0.69. The crude product was used in the next reaction.
Following the procedure outlined for the preparation of the methyl 4-biphenylisoxazole-3-carboxylate 22, the aryl triflate 19 (295 mg, 0.69 mmol) and pinacolyl 3-chloro-4-hydroxy-5-nitrophenylboronate (310 mg, 1.04 mmol) in DMF (7 mL) with Pd(PPh3)4 (8 mg, 0.007 mmol), after repeated chromatography (Et2O:light petroleum, 20:80–40:60 v/v then with DCM), gave methyl 4-(3′-chloro-4′-hydroxy-5′-nitro-3-biphenyl)-5-phenylisoxazole-3-carboxylate as a white solid (45 mg, 0.103 mmol, 15%); mp 74.5–75.7 °C; TLC (Et2O:light petroleum, 30:70 v/v) Rf = 0.49; 1H NMR (400 MHz, CDCl3) δ 11.00 (s, 1H), 8.23 (d, J 2.5 Hz, 1H), 7.91 (d, J 2.5 Hz, 1H), 7.62 (m, 1H), 7.58–7.54 (m, 4H), 7.43 (d, J 7.5 Hz, 2H), 7.37 (d, J 7.5 Hz, 2H), 3.93 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.6, 160.4, 154.7, 150.7, 137.7, 136.2, 134.6, 132.8, 130.7, 130.4, 130.0, 129.7, 128.9, 128.7, 127.1, 126.9, 126.6, 125.1, 121.6, 116.1, 52.9; IR (film) 3223, 3078, 2948, 1737, 1621, 1539, 1218, 1173, 1110, 1029, 815, 692 cm–1.
Following the procedure outlined for the preparation of the carboxylic acid 1, methyl 4-(3′-chloro-4′-hydroxy-5′-nitrobiphenyl-3-yl)-5-phenylisoxazole-3-carboxylate (45 mg, 0.1 mmol) in MeOH (1 mL) and aqueous sodium hydroxide (2 M, 0.25 mL, 2 mmol), after recrystallization (light petroleum:EtOAc, 100:1 v/v), gave the title compound 12 as a white solid (20 mg, 0.05 mmol, 46%); mp 185.4–185.9 °C; TLC (light petroleum:EtOAc, 50:50 v/v with 0.1% TFA) Rf = 0.15; HPLC (silica, EtOAc with 0.1% TFA) retention time 3.08 min, 100%; 1H NMR (500 MHz, DMSO-d6) δ 13.97 (br. s, 1H), 11.30 (br. s, 1H), 8.16 (m, 1H), 8.14 (m, 1H), 7.84–7.80 (m, 2H), 7.54–7.41 (m, 6H), 7.37 (dd, J 1.0, 8.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 166.3, 160.9, 156.2, 147.5, 138.6, 136.7, 133.1, 131.1, 130.6, 129.8, 129.7, 129.3, 129.1, 128.3, 126.8, 126.4, 124.6, 121.4, 116.0; IR (film) 3256, 3061, 2900, 1716, 1619, 1538, 1429, 1319, 1229, 1110, 906, 795, 763, 691 cm–1.
4-(3′,5′-Dichloro-4′-hydroxy-3-biphenyl)-5-methylisoxazole-3-carboxylic Acid (13)
Trifluoromethanesulfonic anhydride (2.18 g, 7.72 mmol) was added to methyl 4-(3-hydroxyphenyl)-5-methylisoxazole-3-carboxylate (25) (1.2 g, 5.15 mmol) and pyridine (1.01 g, 12.87 mmol) in dichloromethane (30 mL) at 0 °C, and the reaction mixture was stirred at rt for 6 h and then poured into water (30 mL). The mixture was extracted with dichloromethane (2 × 50 mL), and the organic extracts were dried (NaSO4) and concentrated under reduced pressure. Chromatography of the residue (EtOAc:light petroleum, 20:80 v/v) gave the triflate 26 as an off-white solid (1.2 g, 66%); TLC (EtOAc:light petroleum, 10:90 v/v) Rf = 0.8; 1H NMR (400 MHz, CDCl3) δ 7.45 (m, 1H), 7.40–7.10 (m, 3H), 3.90 (s, 3H) 2.45 (s, 3H); LRMS (m/z, ES+) 306 ([M – 59]+, 100%).
Sodium carbonate in water (1.3 g, 12.3 mmol in 1 mL water) was added to a solution of methyl 5-methyl-4-(3-trifluoromethanesulfonyloxyphenyl)-isoxazole-3-carboxylate (26) (1.5 g, 4.10 mmol) and pinacolyl (3,5-dichloro-4-hydroxyphenyl)boronate (21) (1.77 g, 6.16 mmol) in DMF (10 mL) at 0 °C, and the reaction mixture was degassed by bubbling nitrogen through for 15 min. The catalyst Pd(PPh3)4 (470 mg, 0.04 mmol) was added, and the mixture was heated to 90 °C for 3 h, cooled to rt, and poured into ice–water (10 mL). The mixture was extracted with EtOAc (2 × 20 mL), and the organic extracts were dried (NaSO4) and concentrated under reduced pressure to afford the methyl 4-(3′,5′-dichloro-4′-hydroxy-3-biphenyl)-5-methylisoxazole-3-carboxylate (27) as an off-white solid (1.5 g) used without purification; TLC (EtOAc:light petroleum, 5:95 v/v) Rf = 0.5; LRMS (m/z, ES–) 376 ([M(Cl35)2 – 1]−, 100%), 378 ([M(Cl35Cl37) – 1]−, 65%), 380 ([M(Cl37)2 – 1]−, 10%).
Aqueous sodium hydroxide (152 mg in 1 mL water, 3.8 mmol) was added to the methyl ester 27 (500 mg, 1.19 mmol) in MeOH:THF (12 mL, 3:1 v/v) at 0 °C and the mixture stirred for 1 h at 0 °C and then poured into ice–water (5 mL). The mixture was acidified to pH ≈ 1 using aqueous hydrochlogen chloride (2 M) and concentrated under reduced pressure to give an aqueous slurry that was extracted with EtOAc (3 × 10 mL). The organic extracts were dried (NaSO4) and concentrated under reduced pressure to give an off-white solid that was crystallized (light petroleum:EtOAc, 90:10 v/v) to give the title compound 13 (315 mg, 65%) as an off-white solid; TLC (MeOH:dichloromethane, 10:90 v/v) Rf = 0.2; 1H NMR (400 MHz, DMSO-d6) δ 13.87 (br. s, 1H), 10.27 (s, 1H), 7.75–7.60 (m, 4H), 7.50 (t, J 7.6 Hz, 1H), 7.34 (d, J 7.6 Hz, 1H), 2.47 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.6, 162.0, 155.4, 149.1, 137.9, 133.1, 130.0, 129.4, 129.2, 128.1, 127.1, 126.3, 123.3, 116.5 and 11.8; LRMS (m/z, ES+) 364 ([M(Cl35)2 + 1]+, 100%), 366 ([M(Cl35Cl37) + 1]+, 50%), 368 ([M(Cl37)2 + 1]+, 10%).
Methyl 5-Phenylisoxazole-3-carboxylate (14)20,21
Sodium methoxide (18.36 g, 340 mmol) in MeOH (80 mL) was added to acetophenone (20 mL, 161.92 mmol) and dimethyl oxalate (28.66 g, 242.9 mmol) in MeOH (450 mL) at rt, and the solution was stirred at rt for 2 h before being cooled to rt and poured into aqueous hydrogen chloride (2 M, 800 mL). The solid methyl 2,4-dioxo-4-phenylbutanoate was filtered off, washed with water, and used in the next step without further purification.
Hydroxylamine hydrochloride (10.8 g, 154.6 mmol) was added to the methyl 2,4-dioxo-4-phenylbutanoate (21.37 g, 103 mmol) in MeOH (520 mL), and the mixture was stirred under reflux for 24 h. After cooling to rt, the mixture was poured into water (1 L) and cooled to 0 °C. The white precipitate was filtered off, washed with water, and then dissolved in EtOAc. The solution was dried (MgSO4) and concentrated under reduced pressure to give the title compound 14 as a white solid (18.4 g, 90.6 mmol, 88%); mp 81.8–82.5 °C (lit.21 80–82 °C); TLC (Et2O:light petroleum, 10:90 v/v) Rf = 0.2; 1H NMR (300 MHz, DMSO-d6) δ 7.98–7.92 (m, 2H), 7.57–7.52 (m, 3H), 7.51 (s, 1H), 3.92 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ171.1, 159.8, 156.6, 131.0, 129.3, 126.0, 125.8, 100.8, 52.8; IR (film) 3130, 2951, 1724, 1612, 1591, 1571, 1445, 1425, 1246, 1141, 1004, 946, 934, 920, 851, 837, 807, 781, 764 cm–1.
Methyl 4-Bromo-5-phenylisoxazole-3-carboxylate (15)22
N-Bromosuccinimide (6.85 g, 38.47 mmol) was added to methyl 5-phenylisoxazole-3-carboxylate 14 (7.1 g, 34.98 mmol) in trifluoroacetic acid (100 mL), and the solution was stirred under reflux for 48 h before being cooled to rt and poured onto ice. The white precipitate was filtered off and recrystallized (light petroleum:EtOAc, 100:1 v/v) to give the title compound 15 as white crystals (6.35 g, 22.5 mmol, 64%); mp 108–110 °C (lit.22 66–68 °C); TLC (Et2O:light petroleum, 10:90 v/v) Rf = 0.23; 1H NMR (400 MHz, DMSO-d6) δ 8.02–7.98 (m, 2H), 7.65–7.60 (m, 3H), 3.94 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.2, 158.9, 154.6, 131.8, 129.5, 127.1, 125.4, 90.1, 53.3; IR (film) 1732, 1565, 1436, 1414, 1252, 1215, 1183, 1046, 970, 810, 785, 771 cm–1; LRMS (ES+, m/z) 306 ([M + 23]+, 100%), 304 ([M + 23]+, 90%); HRMS (m/z) [M + H]+ calcd for C11H9NO379Br, 281.9761; found 281.9759.
Methyl 4,5-Diphenylisoxazole-3-carboxylate (16)23
A solution of methyl 4-bromo-5-phenylisoxazole-3-carboxylate 15 (100 mg, 0.35 mmol) and phenylboronic acid (52 mg, 0.42 mmol) in DMF (4 mL) was degassed by bubbling nitrogen through it for 30 min. Aqueous sodium carbonate (2 M, 0.5 mL, 1.0 mmol) was added, and nitrogen was bubbled through the mixture for a further 15 min. Bis(triphenylphosphine)palladium(II) dichloride (12 mg, 0.02 mmol) was added, and the mixture was stirred at 90 °C for 3 h. The mixture was cooled to rt and poured into EtOAc (20 mL). The mixture was washed with aqueous hydrogen chloride (2 M, 20 mL), water (20 mL), and brine (20 mL) and then dried (NaSO4) and concentrated under reduced pressure. Chromatography (Et2O:light petroleum, 20:80–40:60 v/v) of the residue gave the title compound 16 as an off-white solid (94 mg, 0.34 mmol, 80%); TLC (Et2O:light petroleum, 20:80 v/v) Rf = 0.40; 1H NMR (400 MHz, DMSO-d6) δ 7.55–7.41 (m, 10H), 3.84 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.6, 159.9, 155.0, 130.8, 130.1, 129.1, 128.7, 128.6, 128.5, 126.8, 126.3, 116.5, 52.8; LRMS (ES+, m/z) 302 ([M + 23]+, 100%), 280 ([M + 1]+, 10%); HRMS (m/z) [M + H]+ calcd for C17H14NO3, 280.0969; found 280.0970.
Methyl 4-(3-Methoxyphenyl)-5-phenylisoxazole-3-carboxylate (17)
A solution of methyl 4-bromo-5-phenylisoxazole-3-carboxylate 15 (6.68 g, 23.7 mmol) and 3-methoxyphenylboronic acid (4.3 g, 28.4 mmol) in DMF (230 mL) was degassed by bubbling nitrogen through it for 30 min. Aqueous sodium carbonate (2 M, 36 mL, 72 mmol) was added, and nitrogen was bubbled through the mixture for a further 15 min. The catalyst Pd(PPh3)4 (300 mg, 0.24 mmol) was added, and the mixture was stirred at 90 °C for 3 h. The mixture was cooled to rt and poured into EtOAc (250 mL). The resulting mixture was washed with aqueous hydrogen chloride (2 M, 250 mL), water (250 mL), and brine (250 mL) and then dried (NaSO4) and concentrated under reduced pressure. Chromatography (Et2O:light petroleum, 20:80–40:60 v/v) of the residue gave the title compound 17 as a pale yellow solid (3.1 g, 9.95 mmol, 42%); mp 110.5–111.9 °C; TLC (Et2O:light petroleum, 40:60, v/v) Rf = 0.37; 1H NMR (500 MHz, CDCl3) δ 7.46 (d, J = 7.0 Hz, 2H), 7.32–7.24 (m, 4H), 6.90 (dd, J 2.5, 8.0 Hz, 1H), 6.85 (d, J 7.5 Hz, 1H), 6.81 (m, 1H), 3.81 (s, 3H), 3.71 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.2, 160.4, 159.7, 155.0, 130.4, 130.2, 129.8, 128.8, 127.0, 126.8, 122.5, 116.6, 115.8, 114.2, 55.3, 52.8; IR (film) 3012, 2956, 2843, 1736, 1587, 1420, 1331, 1243, 1213, 1164, 1094, 1043, 971, 872, 843, 789, 768, 709, 691 cm–1; LRMS (ES+, m/z) 332 ([M + 23]+, 100%); HRMS (m/z) [M + H]+ calcd for C18H16NO4, 310.1074; found 310.1071.
Methyl 4-(3-Hydroxyphenyl)-5-phenylisoxazole-3-carboxylate (18)
A solution of methyl 4-bromo-5-phenylisoxazole-3-carboxylate 15 (9.3 g, 32.95 mmol) and 3-hydroxyphenylboronic acid (5 g, 36.5 mmol) in DMF (330 mL) was degassed by bubbling nitrogen through it for 1 h. Aqueous sodium carbonate (2 M, 49.4 mL, 98.85 mmol) was added, and nitrogen was bubbled through the mixture for a further 15 min. The catalyst Pd(PPh3)4 (380 mg, 0.33 mmol) was added, and the mixture stirred at 90 °C for 90 min. The mixture was cooled to rt and poured into EtOAc (1 L). The mixture was washed with aqueous hydrogen chloride (2 M, 500 mL), water (1 L), and brine (1 L) and then dried (NaSO4) and concentrated under reduced presssure. Repeated chromatography of the residue (Et2O:light petroleum, 20:80–40:60 v/v, then DCM) gave the title compound 18 as an off-white solid (4.76 g, 16.1 mmol, 49%); mp 148.7–149.9 °C; TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.24; 1H NMR (400 MHz, MeOD) δ 7.48–7.45 (m, 2H), 7.38–7.28 (m, 3H), 7.19 (t, J 8.0 Hz, 1H), 6.82 (ddd, J 1.0, 2.5, 8.5 Hz, 1H), 6.72–6.70 (m, 2H), 4.65 (s, 1H), 3.77 (s, 3H); 13C NMR (100 MHz, MeOD) δ 168.4, 161.8, 158.9, 156.6, 131.7, 131.5, 131.2, 131.0, 130.0, 128.2, 128.1, 122.4, 118.1, 116.7, 53.2; IR (film) 3473, 1745, 1590, 1449, 1331, 1216, 1164, 1094, 1035, 985, 865, 811, 788, 774, 706, 691 cm–1; LRMS (m/z, ES+) 318 ([M + 23]+, 100%); HRMS (m/z) [M + H]+, calcd for C17H14NO4, 296.0918; found 296.0913.
Methyl 5-Phenyl-4-(3-trifluoromethylsulfonyloxyphenyl)isoxazole-3-carboxylate (19)
Trifluoromethanesulfonic anhydride (1 M in DCM, 16.27 mL, 16.27 mmol) was added to methyl 4-(3-hydroxyphenyl)-5-phenylisoxazole-3-carboxylate 18 (4 g, 13.56 mmol) and pyridine (2.18 mL, 27.12 mmol) in DCM (135 mL) at 0 °C, and the reaction mixture was stirred at rt for 90 min before being poured into EtOAc (800 mL). The solution was washed with aqueous hydrogen chloride (1 M, 800 mL), dried (MgSO4), and concentrated under reduced pressure. Chromatography (Et2O:light petroleum, 20:80 v/v) of the residue gave the title compound 19 as a white solid (5.47 g, 12.75 mmol, 94%); mp 95.0–96.7 °C; TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.41; 1H NMR (400 MHz, CDCl3) δ 7.55 (t, J 8.0 Hz, 1H), 7.49–7.35 (m, 7H), 7.31 (dd, J 1.5, 2.0 Hz, 1H), 3.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 239.2, 168.0, 160.2, 154.6, 149.5, 131.7, 131.0, 130.6, 129.1, 127.1, 126.2, 123.5, 121.6, 114.6, 53.0; IR (film) 3068, 2968, 1738, 1418, 1248, 1210, 1128, 971, 917, 897, 815, 771, 686 cm–1; LRMS (m/z, ES+) 450 ([M + 23]+, 45%).
Pinacolyl (3,5-Dichloro-4-hydroxyphenyl)boronate (21)
A mixture of 4-bromo-2,6-dichlorophenol 20 (7 g, 28.94 mmol), KOAc (8.52 g, 86.8 mmol), bis(pinacolato)diboron (8.82 g, 34.73 mmol), and [1,1′-bis(diphenylphosphino)ferrocene]palladium(II) dichloride (640 mg, 0.87 mmol) in dioxane (300 mL) was stirred under nitrogen at 90 °C for 18 h before being poured into EtOAc (600 mL). The mixture was washed with aqueous hydrogen chloride (1 M, 600 mL), dried (MgSO4), and concentrated under reduced pressure. Chromatography (Et2O:light petroleum, 10:90 v/v) of the residue gave the title compound 21 as a pale yellow solid (4.9 g, 16.96 mmol, 59%); mp 103.3–104.2 °C; TLC (Et2O:light petroleum, 10:90 v/v) Rf = 0.38; 1H NMR (400 MHz, CDCl3) δ 7.61 (s, 2H), 5.99 (s, 1H), 1.26 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 150.0, 134.5, 128.3, 120.9, 84.3, 24.8; IR (film) 3271, 2980, 1592, 1384, 1349, 1230, 1129, 961, 892, 848, 788, 676 cm–1; LRMS (m/z, ES–) 287 ([M – 1]−, 100%); HRMS (m/z, ES–) [M – H]− calcd for C12H14O335Cl2B 287.0418; found 287.0405.
Methyl 4-(3′,5′-Dichloro-4′-hydroxy-3-biphenyl)-5-phenylisoxazole-3-carboxylate (22): Standard Procedure
A solution of methyl 5-phenyl-4-(3-trifluoromethylsulfonyloxyphenyl)isoxazole-3-carboxylate 19 (1.9 g, 4.47 mmol) and the boronate 21 (1.42 g, 4.91 mmol) in DMF (50 mL) was degassed by bubbling nitrogen through it for 30 min. Aqueous sodium carbonate (2 M, 6.7 mL, 13.41 mmol) was added, and nitrogen was bubbled through the solution for a further 15 min. The catalyst Pd(PPh3)4 (50 mg, 0.045 mmol) was added, and the mixture was stirred at 90 °C for 3 h, cooled to rt, and poured into EtOAc (100 mL). The mixture was washed with aqueous hydrogen chloride (2 M, 100 mL), water (100 mL), and brine (100 mL) and then dried (NaSO4) and concentrated under reduced pressure. Chromatography (Et2O:light petroleum, 20:80–40:60 v/v) of the residue gave an off-white solid that was recrystallized (light petroleum:EtOAc, 70:30 v/v) to give the title compound 22 as a white solid (900 mg, 2.05 mmol, 45%); mp 149.7–150.5 °C; TLC (Et2O:light petroleum, 40:60 v/v) Rf = 0.26; 1H NMR (400 MHz, CDCl3) δ 7.59–7.49 (m, 5H), 7.46 (s, 2H), 7.43–7.34 (m, 4H), 5.91 (s, 1H), 3.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 167.5, 160.4, 154.8, 147.3, 138.8, 134.2, 130.6, 129.7, 129.6, 129.4, 128.9, 128.6, 127.1, 126.9, 126.7, 121.5, 116.3, 104.8, 52.8; IR (film) 3404, 1719, 1500, 1465, 1438, 1340, 1296, 1228, 1156, 1030, 973, 866, 820, 790, 765, 688 cm–1; LRMS (m/z, ES–) 442 ([M – 1]−, 20%), 440 ([M – 1]−, 50), 438 ([M – 1]−, 100); HRMS (m/z, ES–) [M – H]− calcd for C23H14NO435Cl2, 438.0305; found 438.0299.
Methyl 4-(3-Hydroxyphenyl)-5-methylisoxazole-3-carboxylate (25)
Hydroxylamine hydrochloride (1.44 g, 20.8 mmol) was added to methyl 2,4-dioxopentanoate (2 g, 13.89 mmol) in MeOH (50 mL), and the reaction mixture stirred under reflux for 16 h. After cooling to rt the mixture was poured into water (50 mL) and the solution extracted with EtOAc (2 × 50 mL). The extracts were dried (Na2SO4) and concentrated under reduced pressure to afford the methyl 5-methylisoxazole-3-carboxylate (23)32 (1.1 g) which was used without purification; TLC (EtOAc:light petroleum, 20:80 v/v) Rf = 0.50; 1H NMR (400 MHz, CDCl3) δ 6.41 (s, 1H) 3.96 (s, 3H) 2.50 (s, 3H); LRMS (m/z, ES+) 142 ([M + 1] +, 100%).
N-Bromosuccinimide (965 mg, 8.578 mmol) was added to methyl 5-methyl-isoxazole-3-carboxylate (23) (1.1 g, 7.799 mmol) in trifluoroacetic acid (15 mL) at 0 °C, and the solution was heated under reflux for 16 h, allowed to cool to rt, and poured onto ice–water (20 mL). The mixture was extracted with EtOAc (2 × 50 mL), and the organic extracts were dried (Na2SO4) and then concentrated under reduced pressure to afford methyl 4-bromo-5-methylisoxazole-3-carboxylate (24)33 (1.2 g) as a white solid which used without purification; TLC (EtOAc:light petroleum, 20:80 v/v) Rf = 0.40; 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 3H) 2.50 (s, 3H); LRMS (m/z, ES+) 221 ([M + 1]+, 100%).
Aqueous sodium hydrogen carbonate (575 mg, 6.89 mmol in 1 mL water) was added to methyl 4-bromo-5-methylisoxazole-3-carboxylate (24) (500 mg, 2.28 mmol) and 3-hydroxyphenylboronic acid (346 mg, 2.51 mmol) in DMF (5 mL) at 0 °C, and the reaction mixture was degassed for 30 min with argon. The catalyst Pd(PPh3)2Cl2 (160 mg) was added at rt, and the mixture was heated at 90 °C for 5 h, cooled to rt, and poured onto ice–water (20 mL). The mixture was extracted with EtOAc (2 × 50 mL), and the organic extracts were dried (Na2SO4) and concentrated under reduced pressure. Chromatography of the residue (EtOAc:light petroleum, 20:80 v/v) gave the title compound 25 as an off-white solid (150 mg, 28%); TLC (EtOAc:light petroleum, 30:70 v/v) Rf = 0.3; 1H NMR (400 MHz, CDCl3) δ 7.30 (m, 1H) 6.90–6.75 (m, 3H) 4.83 (s, 1H), 3.90 (s, 3H), 2.45 (s, 3H).
Recombinant Protein Production
The Rv2234 and Rv0153c genes, encoding MptpA and MptpB, were amplified from M. tuberculosis H37Rv and cloned into vector pET28a as previously described.11 Site-directed mutagenesis was performed on wild-type pET28-MptpB, using QuikChange (Stratagene) to generate the mutants H94A, Y125A, E129A, and R136A. His-tagged MptpA, MptpB, and derivatives thereof were expressed in Escherichia coli BL21(DE3), with expression induced at 18 °C with 0.5 mM IPTG for 16 h, purified by sequential nickel affinity (in 50 mM HEPES, 500 mM NaCl, pH 7 buffer) and anion-exchange chromatography using MonoQ column (GE Healthcare) in 20 mM Tris, pH 8, and eluted in a NaCl gradient. Fractions were concentrated and further purified on a Seperdex75 column (Amersham Bioscience) with 20 mM Tris, 300 mM NaCl, pH 8. The VHR construct, in pGEX-4T, was a gift from Prof. Rafael Pulido (BioCruces Health Research Institute, Barakaldo, Spain), and the hPTP1B in pGEX-KG was a gift from Prof. Jeroen der Hertog (Hubrecht Institute, Utrecht, Netherlands). The plasmids were transformed into E. coli and protein expression induced at 18 °C with 0.5 mM IPTG for 16 h. Purification of glutathione S-transferase (GST)-tagged proteins was achieved by GST affinity chromatography in 50 mM HEPES, 500 mM NaCl, pH 7 buffer, and eluted with 20 mM glutathione. GST tag was removed by protease cleavage and subsequently purified using a Superdex 75 column in 50 mM HEPES buffer at pH 7.
Inhibition Assays
Inhibition assays were performed as previously described,13 where each titration experiment was performed in triplicate and in at least three independent assays. Experiments were conducted in 96-well microtiter plates, and each well contained a 100 μL reaction mixture including 0.5 μg of protein, in 50 mM Tris, 50 mM BisTris, 100 mM sodium acetate buffer (pH 7 for MptpB, pH 6.5 for MptpA, pH 6 for hPTP1B, and pH 5 for VHR), and the different compounds were dissolved in DMSO at a concentration range between 0 and 250 μM. Reactions were incubated for 15 min at room temperature before the addition of p-nitrophenyl phosphate (pNPP) to a final concentration of 0.35 mM. After 15 min incubation, the reactions were quenched by the addition of 0.5 M NaOH and the absorbance at 405 nm was measured. Production of p-nitrophenol (pNP) was quantified using a pNP (Sigma) calibration curve (2–2000 μM). Control reactions without enzyme were performed to account for the spontaneous hydrolysis of pNPP. Phosphate release was calculated as a percentage of the specific activity and plotted as a function of inhibitor concentration to determine the IC50.
Computational Virtual Screening and Molecular Docking
The computational fragment screening was done using the massive processing algorithm (MPA), a high-throughput virtual screening genetic-based algorithm18,19 that combines ligand docking to the target using Autodock4,34 with text-based (LINGO) similarity searches of the compound library.35 The MPA algorithm was used to search nine different commercial libraries that contained either building blocks (for rapid synthesis approaches) or fragments from different sources. The crystal structure of MptpB (PDB ID 2OZ5) was used in the screening, and a grid box was defined around the secondary P2 pocket. Compounds were scored according to their calculated binding energy (ΔG), and their mode of binding was analyzed graphically using PyMol (Schrödinger). Synthesized compounds were docked to the crystal structure of MptpB (PDB ID 2OZ5), using AutoDock4.34 All compounds were docked using the default parameters, and a maximum of 10 conformations was generated per ligand. Each docking conformation was scored according to their calculated free energy of binding, ΔG.
Bacterial Strains, Cell Culture, and Infections
M. tuberculosis strain H37Rv (ATCC35837, drug susceptible), the clinical isolate “W_565”36 of multidrug-resistant M. tuberculosis, and Mycobacterium bovis Bacille Calmette-Guerin (BCG, Pasteur) were grown at 37 °C in Middlebrook 7H9 broth containing 0.05% Tween 80 with shaking or on Middlebrook 7H10 agar (both types of medium were supplemented with 10% oleic acid/albumin/dextrose/catalase enrichment and 0.5% glycerol).
For infections using BCG, J774A.1 macrophages (ATCC) were cultured in Dulbecco’s modified Eagle’s medium (Sigma) containing glucose (25 mM) and l-glutamine (4 mM) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen) at 37 °C in a humidified atmosphere with 5% CO2. For infections, J774A.1 cells were seeded in 96-well culture plates (Corning) at a density of 2 × 103 per well (in 200 μL of media) and incubated overnight. Cells were subsequently washed twice in prewarmed Dulbecco’s PBS (Sigma), and 100 μL of fresh culture medium, supplemented with inhibitors (dissolved in dimethyl sulfoxide, DMSO) or DMSO alone, was added before infecting with BCG in 100 μL of medium at a multiplicity of infection (MOI) of 10:1 (bacteria:macrophage). After 4 h of infection, cells were washed four times with Dulbecco’s PBS to remove extracellular bacteria and 200 μL of fresh culture medium (supplemented with inhibitors or DMSO) added; this was defined as time 0 h. At 24 h post infection, the cells were again washed twice with Dulbecco’s PBS and fresh medium (supplemented with inhibitors or DMSO) added. At 72 h post infection, infected cells were lysed in 0.05% (v/v) Tween 80 and the number of viable bacteria in each well determined by plating 10-fold serial dilutions on Middlebrook 7H10 agar plates in triplicates. The plates were incubated for 3 weeks at 37 °C prior to counting the number of viable bacteria. All assays were performed in triplicate in at least three separate experiments.
For infections using M. tuberculosis, bacteria were grown for 1 week at 37 °C with shaking and then washed with PBS and frozen (in 7H9 with 15% of Glycerol) at a concentration of 4 × 107 CFU ml–1. THP1 macrophage cells were subcultured in RPMI 1640 (Gibco), supplemented with 10% FBS and 4 mM l-glutamine, and then 1 × 106 cells were aliquoted per well (6-well plates) and treated with PMA overnight. The media was removed the following day, and the cells were washed with PBS, and 1.5 mL of fresh RPMI (with 10% FBS) and inhibitors (0, 20, or 80 μM in DMSO) was added, before infecting with thawed M.tuberculosis at MOI of 1:1. After 3 h of infection, THP1 cells were washed 4 times with PBS, and fresh RPMI was added. After 24 h the media was removed, and cells were washed with PBS prior to the addition of fresh RPMI and inhibitor (20 or 80 μM, dissolved in DMSO). At 72 h post infection, cells were washed with PBS, lysed with 0.05% SDS, and plated onto 7H10 media to determine bacterial numbers. Plates were counted after 3 weeks of growth at 37 °C (all experimental points were plated as 10-fold serial dilutions in duplicates). Each experiment was done in triplicate, including controls.
Cytotoxicity Assays
Macrophage viability was measured using the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) assay.37 Briefly, J774A.1 cells were seeded in 96-well culture plates (Corning) at a density of 6 × 103 (200 μL/well) and allowed to adhere overnight. Fresh media supplemented with inhibitors (dissolved in dimethyl sulfoxide, DMSO) or DMSO only were subsequently added to each well. Cell viability at 24 h was assessed by adding 50 μL of filter-sterilized MTT (5 mg mL–1 in PBS) to each well followed by a 4 h incubation. Media was removed, and 200 μL of DMSO and 25 μL of Sorensen’s glycine buffer were added. The absorbance at 570 nm was measured in a plate reader.
Live Cell Imaging of M. tuberculosis-Infected Macrophages
RAW264.7 macrophages were grown in complete DMEM + 10% FBS. On the day before infection with RFP, M. tuberculosis (4 × 105 cells) was plated on WillCo glass bottom dishes (22 mm, GWST-3522). Cells were left for approximately 6 h at 37 °C to attach before transfection using JetPEI-macrophage (polyplus) transfection reagents. EGFP-2xFYVE plasmid, 1.5 μg, and 4 μL of JetPEI transfection reagent in 100 μL of 150 mM NaCl were used for transfection of one dish.
The day after transfection a single-cell suspension of RFP-Mtb H37Rv was prepared and added to the dish at MOI 5:1 together with the lead compound 13 (80 μM). Directly after addition of bacteria the cells were imaged. Images were taken every 30 s at 512 × 512 resolution, z = 3, 3× line average.24
Pharmacokinetic Evaluations
Guinea pig males (Durkin Hartley) were used to evaluate the pharmacokinetics and tissue distribution of compounds. The route of administration was either intraperitoneal (ip) or oral (po) with dosage varying from 3.5 to 8 mg kg–1 (op) and 2.5–4 mg kg–1 (ip). In each case 4 samples were collected per time point at 0.1, 0.5, 2, 4, and 8 h post dose for ip and 0.25, 0.5, 2, 4, and 8 h post dose for po. Compounds were prepared in 10% DMSO, 5% Cremaphor, and 85% physiological saline for IP administration or 0.5% (v/v) Tween 80, 99.5% (v/v) 0.5% methylcellulose for oral administration. Tissue samples (n = 4) from lung, liver, and kidney were collected at the end of the study and snap frozen prior to bioanalysis.
Efficacy Studies in Guinea Pigs
All animals were housed in the Public Health Research Institute’s Animal Biosafety Level-2 (tolerability) or Animal Biosafety Level-3 (efficacy) Research Animal Facility (ICPH RAF), a center of the New Jersey Medical School, Rutgers University (NJMS-Rutgers). The animal facility follows the Public Health Service and National Institute of Health Policy of Humane Care and Use of Laboratory Animals. All experimental protocols were approved by the Rutgers Institutional Animal Care and Use Committee (IACUC). Female outbred Hartley Duncan Guinea Pigs (∼400 g) were used in all studies. Tolerability studies were done prior to the infection models. Guinea pigs (n = 5) were orally dosed once daily for 7 days with 13 50 or 100 mg kg–1. No adverse drug effects were observed, and weight increases of >5% were observed in all animals during the tolerability trial. Plasma and lung drug levels were analyzed at 2 and 24 h (peak and trough levels) after the last dose administered. Drug levels for the 100 mg kg–1 orally administered 13 were 1748 ng mL–1 in plasma and 623 ng mL–1 in lungs at 2 h and 35 ng mL–1 in plasma at 24 h.
An acute model of TB infection was used to assess the efficacy of compound 13 as monotherapy in reducing bacterial burden during early acute phase of growth in the lungs. In this infection model guinea pigs were infected with 96 CFU (average, ±27 SEM) of M. tuberculosis H37Rv via aerosol inhalation. The animals were randomized into groups of 4 guinea pigs per treatment or vehicle group. The infection was confirmed by lung bacterial burden enumeration from 3 animals sacrificed at 24 h post exposure. Compound 13 (100 mg kg–1) and the vehicle were orally administered (feeding) daily for 4 weeks starting at 24 h post infection. After 4 weeks of treatment all guinea pigs on the study gained weight and had normal behavior and fecal output.
A chronic model of TB infection was used to assess the efficacy of compound 13 as monotherapy (100 mg kg–1). No morbidity was observed throughout the study. Guinea pigs were infected with low-dose M. tuberculosis with 63 CFU (average, ±18 SEM) by aerosol inhalation. The animals were randomized into groups of 5 guinea pigs per treatment or vehicle group. The infection was confirmed by lung bacterial burden enumeration from 3 animals sacrificed at 24 h post exposure. The remaining animals were left untreated for 4 weeks to develop a steady state of TB burdens in the lungs. After 28 days of infection, 4 animals were sacrificed and bacterial burdens of the lungs and spleens were enumerated to establish a baseline burden level prior to the start of treatment. Compound 13 (100 mg kg –1) and the vehicle were orally administered daily for 4 weeks starting at 28 days post infection. After 4 weeks of treatment, all guinea pigs on the study did have varying increases in weight and exhibited normal behavior and fecal output. For statistical analysis, the CFUs are converted to logarithms and evaluated by a one-way ANOVA followed by a multiple comparison analysis of variance by Tukey and/or Dunnett’s test (Graphpad Prism 6.0 software program). Differences were considered significant at the 95% level of confidence. For both the acute and the chronic infection studies, the lungs and spleens were graded from 1 to 4 based on a modified Mitchison virulence scoring from Jain et al.27 The scoring was based on gross pathological examination of inflammation, extent of involvement and necrosis, number, and relative size of lesions.
Histopathology
For histology, portions of guinea pig lungs and spleens were fixed in neutral buffered formalin and then embedded in paraffin. Sections (∼5 μm) were cut and stained with hematoxylin and eosin.
Acknowledgments
This work was supported by funding from MRC (G0701233) to L.T., E.J.T., and J.C., a MRC confidence in concept award, a BBSRC spark impact award, and a Manchester Chemical Biology Network award to L.T. This work was partially supported by NIH U19AI109713 to D.S.P., MINECO BIO2016-78006-R to M.P., Catalan Government 2009PIV 00100 grants to L.T. and M.P., and the Francis Crick Institute to M.G.G., which receives its core funding from Cancer Research UK (FC001092), the UK Medical Research Council (MC_UP_1202/11, FC001092), and the Wellcome Trust (FC001092). We thank Peter Warn and Lloyd Pane at Euprotec/Evotec for advice and PK/PD analysis, William Hope (Liverpool University) and Redx Pharma Ltd. for initial evaluation of compounds and DMPK analysis, and LifeArc for evaluation of compound 13 and support to the project, and Cele Abad-Zapatero, Scott Franzblau, and Larry Klein (University of Illinois in Chicago) for advice and useful discussions, Nicola Beresford for her early work in the project, Alexander Bruce and James Adams for support with inhibition experiments, Trevor Perrior (Domainex) and John Dixon for advice on medicinal chemistry, John Horton and Chris Reilly for advice, and David Denning for constant support, advice, and encouragement.
Glossary
Abbreviations Used
- DCM
dichloromethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- NBS
N-bromosuccinimide
- TFA
trifluoroacetic acid
- py
pyridine
- MDR
multidrug resistant
- PI
phosphoatidylinositol
- hPTP1B
human protein tyrosine phosphatase 1B
- MptpB
mycobacterium protein tyrosine phosphatase B
- MptpA
mycobacterium protein tyrosine phosphatase A
- VHR
Vaccinia H1-related phosphatase
- CFU
colony forming unit
- PAMPA
parallel artificial membrane permeability assay
- THP1
human monocytic cell line
- H37Rv
M. tuberculosis laboratory strain
- J774
murine macrophage cell line
- BCG
Bacillus Calmette-Guerin
- INH
isoniazid
- RIF
rifampicin
- ANOVA
analysis of variance
- HRZ
isoniazid–rifampicin–pyrazinamide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00832.
Experimental methods; molecular docking of the new series of compounds into the MptpB structure; DMPK profiles of selected MptpB inhibitors; M. tuberculosis infection burden in the lungs and spleen for the acute and chronic guinea pig models after 28 days of treatment; guinea pig lung and spleen weights after 28 days of treatment; C1 compound reduces bacterial burden of a MDR strain (Beijing-w) and drug-sensitive (DS) H37Rv in infected human THP1 macrophages; cell activity of the new series of isoxazole-based compounds results in reduction in the mycobacterial (BCG) burden of infected mouse macrophages (J774) at 24 h postinfection, compared to DMSO-treated macrophages; tissue distribution of compound 13 in guinea pigs after administration (PDF)
Molecular Formula Strings (CSV)
Author Contributions
§ C.F.V. and A.P.G.S.: These authors have contributed equally to the study.
Author Contributions
C.F.V., A.P.G.S., A.C., P.F., N.K., C.S., and Y.N. generated compounds, tested their activity in vitro and in macrophage infections, and supported computational screening. L.S.S. performed live imaging experiments and S.P the animal studies. M.P., M.G.G, B.N.K., D.S.P., E.J.T. J.S.C., and L.T. supervised the work and analyzed data. J.S.C. and L.T wrote the paper; L.T. conceived and managed the project from the beginning.
The authors declare no competing financial interest.
Supplementary Material
References
- Armstrong J. A.; Hart P. D. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 1971, 134, 713–740. 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde K.; Yates R. M.; Purdy G. E.; Russell D. G. Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 2007, 219, 37–54. 10.1111/j.1600-065X.2007.00547.x. [DOI] [PubMed] [Google Scholar]
- Rasko D. A.; Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discovery 2010, 9, 117–128. 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- Silva A. P.; Tabernero L. New strategies in fighting TB: targeting Mycobacterium tuberculosis-secreted phosphatases MptpA & MptpB. Future Med. Chem. 2010, 2, 1325–1337. 10.4155/fmc.10.214. [DOI] [PubMed] [Google Scholar]
- Cole S. T. Inhibiting Mycobacterium tuberculosis within and without. Philos. Trans. R. Soc., B 2016, 371, 20150506. 10.1098/rstb.2015.0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey S. W.; Cheung G. Y. C.; Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discovery 2017, 16, 457–471. 10.1038/nrd.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. K.; Abramovitch R. B. Small molecules that sabotage bacterial virulence. Trends Pharmacol. Sci. 2017, 38, 339–362. 10.1016/j.tips.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul A.; et al. Cloning and characterization of secretory tyrosine phosphatases of Mycobacterium tuberculosis. J. Bacteriol. 2000, 182, 5425–5432. 10.1128/JB.182.19.5425-5432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan P.; et al. Secretory phosphatases deficient mutant of Mycobacterium tuberculosis imparts protection at the primary site of infection in guinea pigs. PLoS One 2013, 8, e77930. 10.1371/journal.pone.0077930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; et al. Disruption of mptpB impairs the ability of Mycobacterium tuberculosis to survive in guinea pigs. Mol. Microbiol. 2003, 50, 751–762. 10.1046/j.1365-2958.2003.03712.x. [DOI] [PubMed] [Google Scholar]
- Beresford N.; et al. MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem. J. 2007, 406, 13–18. 10.1042/BJ20070670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G.; De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Beresford N. J.; et al. Inhibition of MptpB phosphatase from Mycobacterium tuberculosis impairs mycobacterial survival in macrophages. J. Antimicrob. Chemother. 2009, 63, 928–936. 10.1093/jac/dkp031. [DOI] [PubMed] [Google Scholar]
- Zhou B.; et al. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4573–4578. 10.1073/pnas.0909133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta N. K.; et al. Mycobacterial protein tyrosine phosphatases A and B inhibitors augment the bactericidal activity of the standard anti-tuberculosis regimen. ACS Infect. Dis. 2016, 2, 231–239. 10.1021/acsinfecdis.5b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundner C.; Ng H. L.; Alber T. Mycobacterium tuberculosis protein tyrosine phosphatase PtpB structure reveals a diverged fold and a buried active site. Structure 2005, 13, 1625–1634. 10.1016/j.str.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Grundner C.; et al. Structural basis for selective inhibition of Mycobacterium tuberculosis protein tyrosine phosphatase PtpB. Structure 2007, 15, 499–509. 10.1016/j.str.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal D.; Blobel J.; Perez Y.; Thormann M.; Pons M. Structure-based discovery of new small molecule inhibitors of low molecular weight protein tyrosine phosphatase. Eur. J. Med. Chem. 2007, 42, 1102–1108. 10.1016/j.ejmech.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Vidal D.; Thormann M.; Pons M. A novel search engine for virtual screening of very large databases. J. Chem. Inf. Model. 2006, 46, 836–843. 10.1021/ci050458q. [DOI] [PubMed] [Google Scholar]
- Tanaka A.; et al. Inhibitors of acyl-CoA:cholesterol O-acyltransferase. 2. Identification and structure-activity relationships of a novel series of N-alkyl-N-(heteroaryl-substituted benzyl)-N’-arylureas. J. Med. Chem. 1998, 41, 2390–2410. 10.1021/jm9800853. [DOI] [PubMed] [Google Scholar]
- Potkin V. I.; Petkevich S. K.; Lyakhov A. S.; Ivashkevich L. S. Mononuclear heterocyclic rearrangement of 5-arylisoxazole-3-hydroxamic acids into 3,4-substituted 1,2,5-oxadiazoles. Synthesis 2013, 45, 260–264. 10.1055/s-0032-1317947. [DOI] [Google Scholar]
- Melo T. M. V. D. P. E.; Lopes C. S. J.; Gonsalves A. M. D. R.; Storr R. C. Reactivity of 2-halo-2H-azirines. Part II. Thermal ring expansion reactions: Synthesis of 4-haloisoxazoles. Synthesis 2002, 2002, 605–608. 10.1055/s-2002-23542. [DOI] [Google Scholar]
- Watterson S. H.; et al. Potent and Selective Agonists of Sphingosine 1-Phosphate 1 (S1P(1)): Discovery and SAR of a novel isoxazole based series. J. Med. Chem. 2016, 59, 2820–2840. 10.1021/acs.jmedchem.6b00089. [DOI] [PubMed] [Google Scholar]
- Schnettger L.; et al. A Rab20-dependent membrane trafficking pathway controls M. tuberculosis replication by regulating phagosome spaciousness and integrity. Cell Host Microbe 2017, 21, 619–628.e5. 10.1016/j.chom.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. L. Lessons from experimental Mycobacterium tuberculosis infections. Microbes Infect. 2006, 8, 1179–1188. 10.1016/j.micinf.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Helke K. L.; Mankowski J. L.; Manabe Y. C. Animal models of cavitation in pulmonary tuberculosis. Tuberculosis (Oxford, U. K.) 2006, 86, 337–348. 10.1016/j.tube.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Jain R.; et al. Enhanced and enduring protection against tuberculosis by recombinant BCG-Ag85C and its association with modulation of cytokine profile in lung. PLoS One 2008, 3, e3869. 10.1371/journal.pone.0003869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beresford N. J.; Saville C.; Bennett H. J.; Roberts I. S.; Tabernero L. A new family of phosphoinositide phosphatases in microorganisms: identification and biochemical analysis. BMC Genomics 2010, 11, 457. 10.1186/1471-2164-11-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner R.; et al. LipA, a tyrosine and lipid phosphatase involved in the virulence of Listeria monocytogenes. Infect. Immun. 2011, 79, 2489–2498. 10.1128/IAI.05073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan W. R.; Spencer J. L. 5-Phenyl-2-isoxazoline-3-carboxylic acid. J. Org. Chem. 1960, 25, 1160–1164. 10.1021/jo01077a022. [DOI] [Google Scholar]
- King G. S.; Rzepa H. S.; Magnus P. D. Reductive and oxidative cleavage of 5-phenyl-delta-2-isoxazoline-3-carboxylic acid. J. Chem. Soc., Perkin Trans. 1 1972, 1, 437–443. 10.1039/P19720000437. [DOI] [Google Scholar]
- Dannhardt G.; et al. Regioisomeric 3-aminomethyl, 4-aminomethyl and 5-aminomethyl isoxazoles - synthesis and muscarinic activity. Eur. J. Med. Chem. 1995, 30, 839–850. 10.1016/0223-5234(96)88303-6. [DOI] [Google Scholar]
- Lindsay-Scott P. J.; Clarke A.; Richardson J. Two-step cyanomethylation protocol: convenient access to functionalized aryl- and heteroarylacetonitriles. Org. Lett. 2015, 17, 476–479. 10.1021/ol503479g. [DOI] [PubMed] [Google Scholar]
- Morris G. M.; et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal D.; Thormann M.; Pons M. LINGO, an efficient holographic text based method to calculate biophysical properties and intermolecular similarities. J. Chem. Inf. Model. 2005, 45, 386–393. 10.1021/ci0496797. [DOI] [PubMed] [Google Scholar]
- Bifani P. J.; et al. Origin and interstate spread of a New York City multidrug-resistant Mycobacterium tuberculosis clone family. JAMA 1996, 275, 452–457. 10.1001/jama.1996.03530300036037. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.