

Abstract
A hallmark of fusion-positive alveolar rhabdomyosarcoma (aRMS) is the presence of a chromosomal translocation encoding the PAX3-FOXO1 fusion oncogene. Primary cell-based modeling experiments have shown that PAX3-FOXO1 is necessary, but not sufficient for aRMS tumorigenesis, indicating additional molecular alterations are required to initiate and sustain tumor growth. Previously we showed that PAX3-FOXO1-positive aRMS is promoted by dysregulated Hippo pathway signaling, as demonstrated by increased YAP1 expression and decreased MST activity. We hypothesized that ablating MST/Hippo signaling in a genetically engineered mouse model (GEMM) of aRMS would accelerate tumorigenesis. To this end, MST1/2-floxed (Stk3F/F;Stk4F/F) mice were crossed with a previously established aRMS GEMM driven by conditional expression of Pax3:Foxo1 from the endogenous Pax3 locus and conditional loss of Cdkn2a in Myf6 (myogenic factor 6)-expressing cells. Compared to Pax3PF/PF;Cdkn2aF/F;Myf6ICN/+ controls, Stk3F/F;Stk4F/F;Pax3PF/PF;Cdkn2aF/F;Myf6ICN/+ animals displayed accelerated tumorigenesis (p<0.0001) and increased tumor penetrance (88% vs. 27%). GEMM tumors were histologically consistent with aRMS. GEMM tumor-derived cell lines showed increased proliferation and invasion and decreased senescence and myogenic differentiation. These data suggest that loss of MST/Hippo signaling acts with Pax3:Foxo1 expression and Cdkn2a loss to promote tumorigenesis. The rapid onset and increased penetrance of tumorigenesis in this model provide a powerful tool for interrogating aRMS biology and screening novel therapeutics.
Keywords: Hippo, MST1, MST2, PAX3-FOXO1, Rhabdomyosarcoma
Introduction
Alveolar rhabdomyosarcoma (aRMS) is a mesenchymal cancer of skeletal muscle histogenesis that is found most commonly in children and young adults (1). Although initially characterized in the 1950s by appearance under light microscopy, aRMS is now known to be associated with stable reciprocal translocations of chromosomes (1;13), or (2;13), yielding the PAX7-FOXO1 or PAX3-FOXO1 fusion genes. In retrospective analyses, expression of PAX3-FOXO1 portends a poor outcome [Ref (2) and references therein]; this is being studied prospectively in current Children’s Oncology Group clinical trials (ClinicalTrials.gov NCT02567435).
Fusion-positive aRMS tumors have remarkably few mutations other than PAX-FOXO1, suggesting it is the dominant tumorigenic driver [Ref (3) and references therein]. Many groups have attempted to directly target and inhibit the PAX3-FOXO1 protein. However, as a transcription factor it has no catalytic domain, and few pockets to bind small molecules. Therefore, we and others have sought to understand the genes and proteins downstream of PAX3-FOXO1 that execute its oncogenic programs. In previously published work (4), we found that PAX3-FOXO1 controls an upstream regulatory region of the RASSF4 gene, encoding the RASSF4 protein that is one of many accessory proteins modulating Hippo pathway activity. RASSF4 protein, in turn, binds and inhibits the Hippo/MST1 tumor suppressor kinase, restraining its tumor suppressor function. This “PAX3-FOXO1-RASSF4-MST1” axis predicts that downregulation of MST activity is a critical event in aRMS pathogenesis, and should be investigated further. Promoter methylation analysis showing silencing of MST loci in soft tissue sarcomas including RMS (5) further underscores the importance of this signaling node in RMS biology.
To evaluate the role of MST loss-of-function in PAX3-FOXO1 fusion-positive aRMS in vivo, we created a genetically engineered mouse model of fusion positive-aRMS lacking MST1/2 expression. Based on our prior work suggesting MST signaling plays an important tumor suppressor role, we hypothesized this would accelerate tumorigenesis and provide insight into the role of MST in aRMS.
Materials and Methods
Mouse Colony
Animals were maintained on a mixed C57BL/6(J) and 129 background. Stk3F/FStk4F/F mice (JAX stock #017635) were purchased from The Jackson Laboratory (6). Pax3PF/PF mice (B6;129-Pax3tm1Mrc/Nci) and Myf6ICN/ICN mice (B6;129-Myf6tm2(cre)Mrc/Nci) were obtained from the NCI mouse repository (7). For clarification, Myf6 is the designated HUGO gene nomenclature, but it is also sometimes referred to as Mrf4. Cdkn2aF/F (Ink4a/Arflox/lox) mice were provided by Ron DePinho, and have been described (8,9). Animals were housed at 72+/−2°F,30–70% humidity, and freely fed reproduction-supportive chow (Purina LabDiet 5053). This study was approved by the Institutional Animal Care and Use Committee (IACUC) of Duke University.
Generation of Tumor-Derived Cell Lines
C2C12 cells were obtained from the Duke University Cell Culture Facility (ATCC CRL-1772), validated by short tandem repeat (STR) DNA profiling, and mycoplasma tested. Cell lines except C2C12 were derived from resected tumors from humanely euthanized animals. Tumor tissue was enzymatically digested in a suspension of 5mg/ml collagenase type IV (Gibco), 1.3mg/ml Dispase II (Gibco), and 0.05% Trypsin in sterile PBS with magnesium and calcium in a shaker at 37°C for 1h. Tissue was mechanically digested by serial pipetting, washed two times at 1200rpm for 10min and resuspended in sterile PBS. The cell suspension was filtered through a 70micron cell strainer, pelleted, and resuspended in RPMI medium containing 10%FBS and 1x Anti-Anti antibiotic-antimycotic (Gibco). Cells were plated at high density and assessed for cell death the following morning. Viable cells were passaged in RPMI/FBS plus Anti-Anti. Cell lines generated are listed in Supp.Table 1.
Differentiation, Invasion, and Senescence Assays
Differentiation assays and MF20 immunocytochemistry were performed as described (10). The MF20 antibody recognizes all isoforms of myosin heavy chain in differentiated skeletal muscle and was deposited in the Developmental Studies Hybridoma Bank by Fischman, DA. In vitro invasion assays were performed using Matrigel-coated Transwell inserts and modified from (11). Modifications are described in Supp. Methods. For senescence assays, cells were seeded at 105 cells/well in a 12-well plate and senescence-associated β-galactosidase activity was assessed with a commercially available colorimetric senescence detection kit (Calbiochem) after 24 hours. For all studies cells were imaged on a Leica DMi1 microscope and processed on Leica Application Suite software. Cells were counted manually with the aid of FIJI software package.
BrdU Incorporation
BrdU assays to measure cell proliferation were performed as described (4).
Quantitative Real-Time PCR and Semi-Quantitative PCR
Mouse genotypes were determined by quantitative PCR (qPCR) performed on tail snips from neonates by Transnetyx (Cordova, TN, USA), with the exception of the Ink4a/Arf allele, which was genotyped by semi-quantitative PCR. Details described in Supp. Methods. The primers for PCR detection of Ink4a/Arf and Pax3:Foxo1 allelic recombination have been reported (7,9). RNA isolation from tumor-derived cell lines, conversion to cDNA, and qPCR quantitation were performed as described (4).
Immunoblotting
Cell lysis and immunoblotting were performed as described in Supp. Methods. Antibodies used for immunoblotting experiments are listed in Supp. Table 2.
Immunohistochemistry
Tissue samples were fixed in 10% formalin/70% ethanol and embedded in standard paraffin blocks. 5µm sections were mounted and stained with H&E or antibodies. Antibodies and dilutions are listed in Supp. Table 2. MYOD1, MYOG, MYF5 and GFP immunohistochemistry were performed using the Vectastain Elite ABC-HRP Kit and DAB Peroxidase Substrate Kit (Vector Labs). Antigen unmasking was performed with a citrate buffer pH6.0 by a modified microwave retrieval method, except MYF5, which was unmasked using a Tris/EDTA buffer pH9.0. Images were captured on a Leica DMLB microscope with DFC425 camera and processed on Leica Application Suite software.
Immunocytochemistry (Immunofluorescence)
Glass chamber slides (4-well, Falcon) were coated with poly-d-lysine and laminin and cells were seeded at 20,000 or 40,000 cells/well and allowed to attach for 24 hours prior to fixing with 4% formaldehyde. Cells were permeabilized with 0.1% TritonX-100/PBS, blocked with 5% goat serum/0.1% TritonX-100/PBS, and immunostained. Primary antibodies are included in Supp. Table 2. Alexa 488-conjugated Goat Anti-Rabbit secondary antibody (ThermoFisher), applied at 4µg/ml and Hoechst 33342 (Invitrogen) were also used. Cells were imaged on a Zeiss 710 inverted confocal microscope and counted manually with the aid of FIJI software.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism (GraphPad). Unless otherwise noted, data is presented as the mean and SE. One-way ANOVA, Pearson’s correlation coefficient, and unpaired T-test were used as appropriate. P values were considered significant at *, P< 0.05; **, P<0.01; ***, P<0.001; and ****, P<0.0001.
Results
Mice with dual loss of MST1 and MST2 demonstrate accelerated aRMS tumorigenesis
To investigate the role of MST/Hippo loss-of-function in aRMS, we crossed MST1/MST2-floxed (Stk4F/F,Stk3F/F) mice with an existing aRMS GEMM (7) that for the purpose of this work we termed “MSTWT” aRMS, driven by conditional expression of Pax3:Foxo1 from the endogenous Pax3 locus and conditional loss of Cdkn2a in Myf6-expressing cells (see Supp.Fig.1 for schema). Heterozygous mating pairs were crossed to produce offspring of both the MSTWT and MSTNull genotypes (Pax3PF/PF; Cdkn2aF/F; Myf6ICN/+ or Stk3F/F; Stk4F/F; Pax3PF/PF; Cdkn2aF/F; Myf6ICN/+, respectively), yielding two experimental mouse lines and nine control mouse lines (Supp. Table 3). Mice of experimental cohorts were not used as breeders, and mice that were bred were not included in experimental cohorts. Experimental progeny were born in Mendelian ratios without evidence of compromised fertility or development, and monitored for 365 days for tumors. Compared to MSTWT aRMS controls, MSTNull aRMS mice exhibited accelerated tumorigenesis (median time to tumor 112 vs. 224 days, p<0.0001) and increased tumor penetrance (88% vs. 27%) (Fig.1A, Supp. Table 4). No control mice (mice not homozygous for both Pax3:Foxo1 expression and Cdkn2a loss) developed aRMS tumors, similar to the prior aRMS GEMM modeling studies (7,12). However, beginning at approximately eight months of age, two out of 165 control mice developed non-RMS tumors histologically resembling lymphomas (Supp. Table 3).
Figure 1. Kinetics, histology and anatomic location of MSTNull compared to MSTWT tumors.
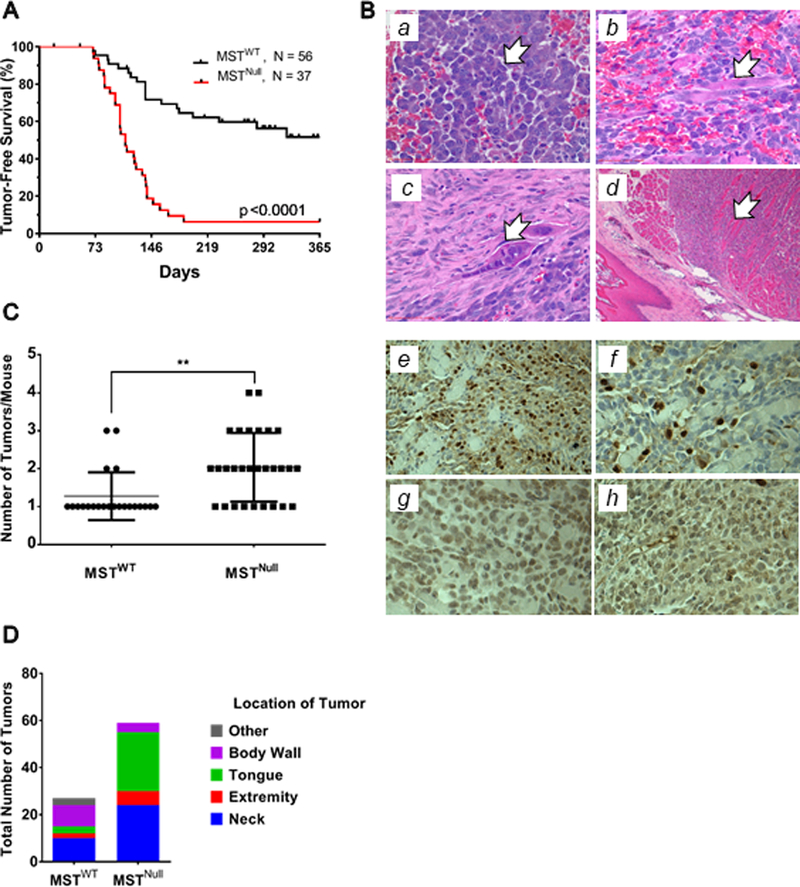
(A) Kaplan-Meier analysis demonstrates a significantly shorter latency of tumor onset in the MSTNull aRMS model compared to the MSTWT aRMS model. Penetrance of the MSTNull model is increased to 88% compared to 27% in the MSTWT model. (B) FFPE tumor samples were examined by light microscopy. H&E reveals classical RMS cell morphologies including (a) small round blue cells (400x), (b) strap cells (400x), (c) multinucleate cells (400x), and (d) entrapped skeletal muscle (100x). Tumor samples were immunostained for (e) MYOD1 (400x), (f) MYOG (400x), (g) GFP (400x), and (h) MYF5 (400x) to confirm aRMS histology. Note that GFP serves as a surrogate for PAX3-FOXO1 protein expression, since an internal ribosomal entry site (IRES) downstream from Foxo1 exon 3 allows for bicistronic expression of YFP with the fusion protein. Representative images shown. (C) Mice of the MSTNull aRMS genotype develop more tumors per animal than MSTWT counterparts. (D) 80% of the MSTNull mice with multiple tumors develop 2–4 masses in the tongue and neck, at the expense of the body wall and other locations.
Mice homozygous for both MST1 and MST2 loss in an aRMS GEMM background developed tumors with high penetrance (Supp. Table 4). Further studies are needed to determine the specific roles of MST1 or MST2 loss in promoting aRMS in this model, since MST1 or MST2 null animals were not evaluated in this study. Previous studies show that MST kinases can homo and heterodimerize with differential impacts on kinase activity (13); the nature of these interactions and their significance to RMS tumorigenesis have yet to be explored. In congruence with previous work describing the MSTWT aRMS model, we found that loss of an additional tumor suppressor (p16/p19) is necessary to facilitate aRMS tumor development (7). While the original studies used Cdkn2a or Tp53 deletion (both are effective, and at least one required), we chose Cdkn2a loss, since there are few TP53 mutations in human PAX3-FOXO1-positive aRMS [Ref (3) and references therein] but evidence of p16INK4A loss (10).
Tumors arising in MSTNull mice histologically resemble aRMS, are transplantable, and show an anatomic distribution distinct from the MSTWT mice
To characterize the tumors arising from the MSTNull cohort, we first analyzed their morphology by H&E. Similar to tumors arising in MSTWT mice, tumors from MSTNull mice contained small round blue cells, strap cells, multinucleate giant cells, and entrapped muscle (Fig.1B,a-d), which are all consistent with histologic characteristics of human aRMS. When transplanted as allografts into SCID/beige recipients (2×105 cells/allograft), five of five samples engrafted and developed tumors. We next examined tumor burden and anatomic location as variables, and found that in the MSTWT model, there were usually one-two tumors per mouse, while in the MSTNull model there were often two or more tumors (Fig.1C). MSTNull mice more often had tumors in the head and neck, especially the tongue, which represented over 40% of the tumors (Fig.1D).
MSTNull tumors resemble MSTWT tumors
To gain insight into the mechanisms underlying the increased penetrance and appearance of tumors in the aRMS mice, we analyzed tumors by IHC. As is standard in the clinical evaluation of RMS, we stained tumors for nuclear MYOD1 and MYOG (Fig.1B,e-f), which are characteristic of aRMS histology. Interestingly, some tumors were negative for both MYOD1 and MYOG. Because loss of MST activity promotes proliferation of stem/progenitor cells (14,15), we hypothesized that in our system this might have shifted the aRMS cells to a more primitive phenotype. Therefore we broadened our diagnostic algorithm (Supp.Fig.1) and sought a marker that appears early in the myogenic commitment lineage. We chose MYF5 since others have shown that RMS tumors not expressing MYOD1 instead express MYF5 (16). Two of the 29 tumors (6.9%) evaluated by IHC were MYOD1-negative and MYOG-negative, but MYF5-positive (Fig.1B,h and Supp.Table 5). If the tumor was MYF5 negative, it was excluded from analysis. Three of the 29 tumors (10.3%) were morphologically similar to lymphomas, and likely reflect the propensity for the Ink/Arf deficient founder mice to develop lymphomas (17).
Tumor-derived cell lines provide insight into role of MST loss-of-function in aRMS biology
From the GEMM tumors, we generated eight cell lines (Supp.Table 1), including two from the Pax3PF/PF;Cdkn2aF/F;Myf6ICN/+ model (MSTWT) and six from the new Stk3F/F;Stk4F/F;Pax3PF/PF; Cdkn2aF/F;Myf6ICN/+ model (MSTNull). Using four of these cell lines (indicated by an asterisk), we verified expression of PAX3-FOXO1 protein and interrogated the impact of MST1/2 loss on the signaling status of the core members of the Hippo pathway. As expected, PAX3-FOXO1 was expressed in all tumor-derived cell lines (Fig.2A, Supp.Fig.2A), consistent with the positive GFP staining in the tumor sections (an IRES site downstream from Foxo1 exon 3 permits bicistronic expression of YFP with PAX3-FOXO1) (Fig.1B,g). Also as expected, cell lines derived from MSTNull animals (911F/F, 914F/F) did not express MST1 or MST2 protein compared to their wild-type counterparts.
Figure 2. Tumor-derived cell lines provide insight into consequences of MST loss-of-function.
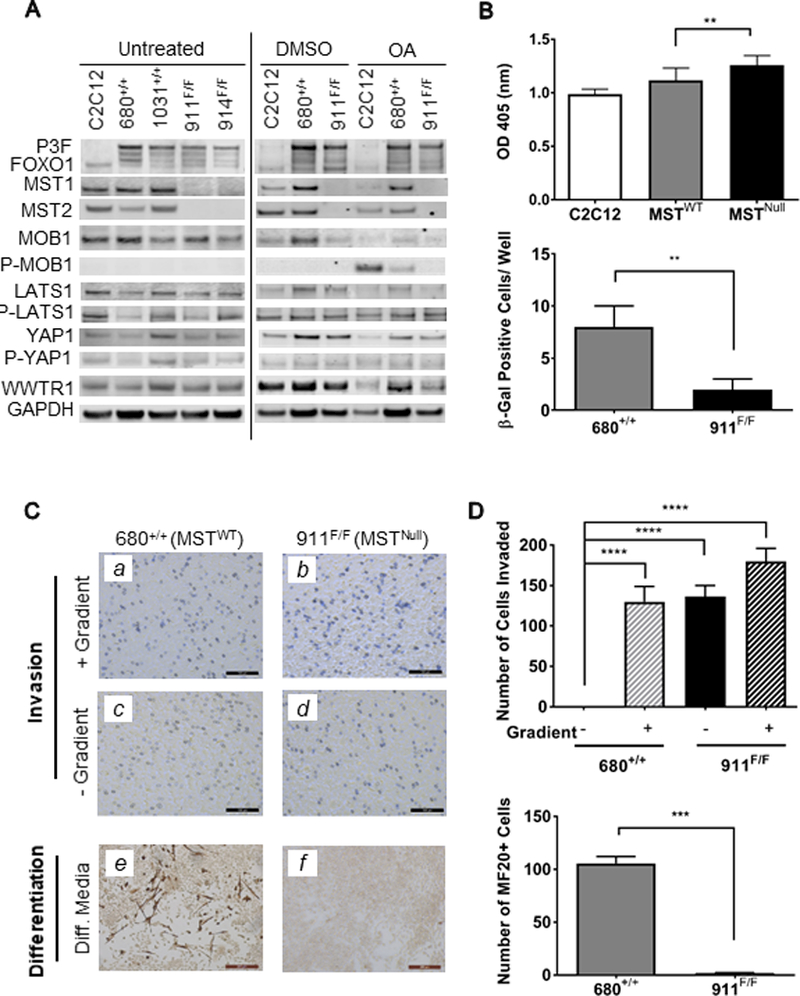
(A) Immunoblot of tumor-derived cell line lysates. Okadaic acid (OA, 150nM) was used as a pharmacologic activator of MST to reveal changes in downstream signaling. (B) Upper: BrdU incorporation is higher in MSTNull versus MSTWT tumor-derived cells. Data shown represents pooled values from three experiments including two cell lines each of MSTWT and MSTNull cell lines. Lower: MSTWT (680+/+) cells show increased β-Gal staining as compared to MSTNull (911F/F), indicating an increased number of senescent cells. Data shown represents four technical replicates. (C) Upper panels: representative images of the invasion potential of (a,c) MSTWT and (b,d) MSTNull cells. Note that MSTNull cells were able to invade even in the absence of a nutritional gradient, as shown by violet staining in (d). Lower panels: as evidenced by MF20 staining, (e) MSTWT cells were able to differentiate down the myogenic lineage, while (f) MSTNull cells were not. Data shown represents four technical replicates. (D) Quantitation of invasion (upper panel) and differentiation (lower panel) assays shown in (C).
MSTNull aRMS cell lines demonstrate increased proliferation, increased invasion, decreased senescence, and decreased differentiation capacity compared to MSTWT aRMS lines
Since the MSTNull animals had an increased number of tumors per animal, which could imply acquisition of aggressive cellular characteristics, we examined the proliferation capacity and relative invasiveness of the tumor-derived cells. Compared to MSTWT cells, MSTNull cells had a statistically significant increase in proliferation rate as measured by BrdU incorporation (Fig.2B, upper panel). Staining of aRMS GEMM tumors (five of each cohort) for the proliferation marker Ki67 also showed a slight increase in Ki67 in MSTNull tumors compared to MSTWT tumors, but this was not statistically significant. A xenograft-based study, in which variables such as tumor cell number, tumor location and duration of tumor growth can be controlled will be better suited to address this question in the future. In Matrigel-coated Transwell invasion assays, MSTWT cells could only invade when a nutritional gradient was present, and remained embedded in the Matrigel matrix on the inner surface when a nutritional gradient was absent. However, MSTNull cells were able to invade and reside on the apical surface independent of the presence of a nutritional gradient between the upper and lower chambers (Fig.2C, a-d, quantitated in Fig.2D, upper panel), consistent with increased invasion capacity.
Prior observations from our laboratory examining RASSF4-MST signaling in aRMS suggested that suppression of MST signaling antagonizes senescence phenotypes and promotes aRMS. To examine their relative propensity for senescence, cells generated from both the MSTWT and MSTNull aRMS models were assessed for β-galactosidase activity. MSTNull cells demonstrated a small but significant decrease in spontaneous senescence by this measure (Fig.2B, lower panel). While these data support the hypothesis that MST signaling plays a role in inhibition of senescence in aRMS, the small magnitude of change observed in these experiments suggests other biological processes are likely involved. Finally, to assess the role of MST/Hippo signaling in the differentiation capacity of aRMS cells, we performed myogenic differentiation experiments in vitro. MSTWT and MSTNull cells were grown in myogenic differentiation-inducing conditions for four days and assessed for the presence of myosin heavy chain. While a portion of MSTWT aRMS cells were myosin heavy chain-positive and demonstrated an elongated, multinucleated phenotype indicative of late-stage myogenesis, MSTNull aRMS cells were unable to differentiate in this setting (Fig.2C, e-f, quantitated in Fig.2D, lower panel). In summary, based on these in vitro studies, it appears that MST loss influences the oncogenic properties of proliferation, invasion, senescence evasion, and loss of differentiation. At this time it is not clear which of these properties (if any) underlie the dramatic difference in tumor penetrance observed in MSTNull mice in vivo, and additional unknown cellular properties may also be involved.
Loss of MST/Hippo in the aRMS GEMM disrupts non-canonical Hippo signaling
To gain insight into the signaling changes caused by loss of MST in this aRMS GEMM, we returned to the MSTWT and MSTNull aRMS GEMM tumor-derived cell lysates (Fig.2A, Supp.Fig.2A) and interrogated them for MOB1, LATS1, YAP1 and WWTR1 expression. Surprisingly, in response to MST loss, there was no decreased LATS1 Ser909 or YAP1 Ser127 phosphorylation, suggesting that canonical Hippo signaling was not affected. Rather, there was decreased MOB1 Thr35 phosphorylation, revealed under conditions of pharmacologic Hippo pathway activation (18) (Fig.2A, OA treated), similar to what we had observed in prior human aRMS cell-line based studies (4). Interestingly, examination of the MSTWT and MSTNull tumors by both IHC (Fig.3A) and immunofluorescence (Fig.3B, Supp.Fig.2B) showed that YAP1 was still highly expressed in the nucleus of both cohorts, again suggesting that MST loss in this setting was not impacting canonical Hippo signaling. Further, biochemically fractionated lysates from tumor-derived cell lines grown at high or low density confirmed a strong YAP1 band in the nuclear but not cytoplasmic fraction, again suggesting active YAP1 (Fig.3C, Supp.Fig.2C). Interestingly, the YAP1 paralog WWTR1 was present in both the nuclear and cytoplasmic fractions of both MSTWT and MSTNull cell lines and showed some correlation to cell density, suggesting that it can be regulated in a manner separate from YAP1, as recently observed (19). Taken together, these findings suggest that in PAX3-FOXO1-positive aRMS, MST loss-of-function specifically suppresses non-canonical Hippo signaling (Fig.4, dashed line), potentially by signaling to MOB1 (4).
Figure 3. Loss of MST in PAX3-FOXO1-aRMS inhibits non-canonical Hippo signaling.
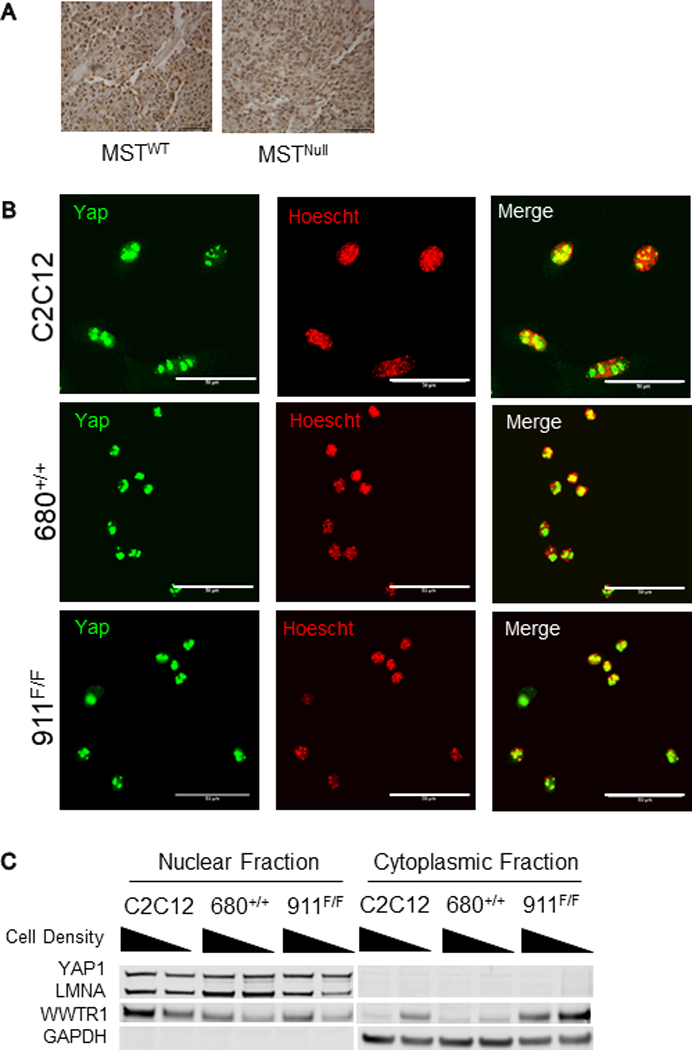
(A) IHC of tumors from MSTWT and MSTNull mice both show strong nuclear YAP1 staining, suggesting that loss of MST1 and MST2 in the MSTNull group is not impacting canonical Hippo signaling, in accord with the immunoblot of Hippo pathway proteins shown in Fig.2A. Representative images shown. (B) Immunofluorescence of tumor-derived cell lines from both MSTWT and MSTNull groups show strong nuclear YAP1 staining. Scale bar: 50µm. (C) Immunoblots of fractionated cell lysates of tumor-derived cell lines show YAP1 in the nuclear but not cytoplasmic fraction, independent of cell density.
Figure 4. PAX3-FOXO1 mediates non-canonical Hippo signaling.

Canonical Hippo signaling (gray box) controls the localization of the transcriptional co-activators YAP1 and WWTR1. When the kinase cascade is active, YAP1/WWTR1 are phosphorylated and remain in the cytoplasm; when the kinase cascade is suppressed, YAP1/WWTR1 move to the nucleus where they co-activate the TEAD family of transcription factors to support oncogenic phenotypes. In aRMS, the inhibition of MST by PAX3-FOXO1 (mediated through transcriptional upregulation of the MST inhibitor RASSF4), does not affect canonical Hippo signaling, but rather signals to MOB1, suggesting that it restrains non-canonical Hippo signaling (dashed line).
Discussion
In this work, we investigated and validated a role for MST loss-of-function in vivo in a novel GEMM of fusion-positive aRMS. Loss of MST1 and MST2 accelerated tumorigenesis and increased tumor penetrance. While the microscopic appearance of MSTNull aRMS tumors remained similar to that of MSTWT aRMS tumors, the anatomic location and number of tumors shifted, and there were more tumors per mouse. IHC mostly showed the expected profile of MYOD1 and MYOG expression, however some tumors in both models that stained negative for MYOG and MYOD1 were positive for the early myogenic marker MYF5, suggesting an induction of a more primitive phenotype. Finally, interrogation of tumor-derived cell lines from both models showed that MST1 or MST2 loss does not result in the expected decrease in phosphorylation of LATS1 or YAP1. Rather, cell lines showed decreased phospho-MOB1, as observed in the human aRMS cell line studies examining the impact of PAX3-FOXO1 on Hippo signaling (4), suggesting an effect of PAX3-FOXO1 on non-canonical Hippo signaling.
What does this mean for aRMS biology? This is the first published report of an independent laboratory re-deriving the original aRMS model, demonstrating that with the use of specific founder mice, the combination of Pax3:Foxo1 gain-of-function with loss of Ink4a/Arf loss-of-function under a Myf6-Cre will generate aRMS. Interestingly, our MSTWT model showed ~30% penetrance, which is similar to the first report of the model (7) but different from the second, in which almost 100% penetrance was reported (12). Understanding the variables responsible for this difference may be important to aRMS biology. Additionally, with this MSTNull model, we now have the ability to study how MST loss-of-function drives or supports aRMS tumorigenesis. For example, does the lack of change in LATS-YAP1 signaling in response to MST deletion mean that the requirement for canonical YAP1-TEAD signaling was already met during aRMS tumor initiation? Does the decreased signaling to MOB1 reflect a higher “dosage” of MST loss, and could this non-canonical signaling function as a biomarker of a more aggressive phenotype? It has already been shown that MOB1 is an important tumor suppressor in Mob1a/1b double-mutant mice, which have increased susceptibility to cancers including sarcomas (20), but it will be important to determine whether loss of MOB1 activity correlates with a poorer outcome in aRMS. This model also provides an opportunity to dissect the contributions of MST1 versus MST2 loss in a mouse model, and underscores the need to assess the expression of MST1 and MST2, as well as downstream effectors, in human aRMS tumor tissue. We note that MST loss led to a propensity for tumors in the tongue, suggesting a role for MST activity in the muscle of the tongue. There are many reports of RMS arising in the tongue [Ref (21) and references therein], and an increasing body of literature on the role of Hippo regulation in craniofacial development (22).
What are the implications for aRMS therapy? The Hippo pathway members and associated proteins are being actively investigated as therapeutic targets (23). However, because the kinases of the core Hippo signaling cassette are tumor suppressive, re-activating them is a daunting task. An alternate approach might focus on inhibiting the terminal oncogenic transcriptional co-activators of the pathway, YAP1 and/or WWTR1, and indeed these efforts are ongoing (24). However, our finding that loss of MST1 and MST2 inhibits non-canonical Hippo signaling suggests that additional targets within the Hippo pathway should be studied within the context of aRMS and perhaps other cancers. This new GEMM also sets the stage for the development of additional aRMS models, and provides a powerful tool for interrogating aRMS biology and screening novel therapeutics. A temporally and spatially-inducible model would be ideal for pre-clinical drug studies, but this is not possible with the current aRMS models in which Cre recombinase is expressed from the Myf6 promoter, which is active in satellite cell progenitors during embryogenesis (25).
In conclusion, we report the generation and characterization of a novel GEMM of fusion-positive aRMS additionally bearing MST1 and MST2 loss-of-function. This model validates the important contribution of MST loss-of-function in an autochthonous mouse model of aRMS, and provides a foundation for the further study of the Hippo pathway in fusion-positive aRMS.
Supplementary Material
Acknowledgements
We thank members of the Armstrong, Becher, Kirsch (especially Dr. David Van Mater and Dr. Rebecca Dodd), Linardic and Wechsler laboratories for helpful conversations and experimental suggestions, Paul Ferrell and Dr. Robin Bachelder (Duke University, Department of Pathology) for immunoblot expertise and resources, Dr. Erin Rudzinski (Seattle Children’s, Department of Pathology) for histopathological consultation and Margaret Demonia and Elaine Justice (Duke University) for technical support.
Financial support
This research was supported by an ALSF Innovation Award (to C. M. Linardic), an ALSF Pediatric Oncology Student Training Program Grant (to N. Kuprasertkul), an ALSF Unrestricted Alex’s Million Mile Grant (to K. M. Oristian), a Duke Undergraduate Research Support Independent Study Grant (to N. Kuprasertkul), 1R35CA197616 (to D. G. Kirsch), and a Duke Cancer Institute Bridge Funding Award (to C. M. Linardic).
Footnotes
Conflicts of interest
The authors declare no potential conflicts of interest
References
- 1.Huh WW, Skapek SX. Childhood rhabdomyosarcoma: new insight on biology and treatment. Current oncology reports 2010;12:402–10 [DOI] [PubMed] [Google Scholar]
- 2.Skapek SX, Anderson J, Barr FG, Bridge JA, Gastier-Foster JM, Parham DM, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatric blood & cancer 2013;60:1411–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nat Commun 2015;6:7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crose LE, Galindo KA, Kephart JG, Chen C, Fitamant J, Bardeesy N, et al. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppression. The Journal of clinical investigation 2014;124:285–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seidel C, Schagdarsurengin U, Blumke K, Wurl P, Pfeifer GP, Hauptmann S, et al. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog 2007;46:865–71 [DOI] [PubMed] [Google Scholar]
- 6.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proceedings of the National Academy of Sciences of the United States of America 2010;107:1437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes & development 2004;18:2614–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes & development 2003;17:3112–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y, et al. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Molecular cancer therapeutics 2013;12:1906–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linardic CM, Naini S, Herndon JE, 2nd, Kesserwan C Qualman SJ Counter CM The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer research 2007;67:6691–9 [DOI] [PubMed] [Google Scholar]
- 11.Becciolini L, Meacci E, Donati C, Cencetti F, Rapizzi E, Bruni P. Sphingosine 1-phosphate inhibits cell migration in C2C12 myoblasts. Biochim Biophys Acta 2006;1761:43–51 [DOI] [PubMed] [Google Scholar]
- 12.Nishijo K, Chen QR, Zhang L, McCleish AT, Rodriguez A, Cho MJ, et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer research 2009;69:2902–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galan JA, Avruch J. MST1/MST2 Protein Kinases: Regulation and Physiologic Roles. Biochemistry 2016;55:5507–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 2014;15:642–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Loforese G, Malinka T, Keogh A, Baier F, Simillion C, Montani M, et al. Impaired liver regeneration in aged mice can be rescued by silencing Hippo core kinases MST1 and MST2. EMBO Mol Med 2017;9:46–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tenente IM, Hayes MN, Ignatius MS, McCarthy K, Yohe M, Sindiri S, et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger JH, Bardeesy N. Modeling INK4/ARF tumor suppression in the mouse. Curr Mol Med 2007;7:63–75 [DOI] [PubMed] [Google Scholar]
- 18.Hata Y, Timalsina S, Maimaiti S. Okadaic Acid: a tool to study the hippo pathway. Mar Drugs 2013;11:896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plouffe SW, Lin KC, Moore JL 3rd, Tan FE Ma S Ye Z et al. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. The Journal of biological chemistry 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishio M, Hamada K, Kawahara K, Sasaki M, Noguchi F, Chiba S, et al. Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. The Journal of clinical investigation 2012;122:4505–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatopadhayay R, Tiwari P, Gangopadhyay AN, Pandey V. Respiratory Distress Secondary to Rhabdomyosarcoma of the Tongue and Co-existent Choanal Atresia. J Maxillofac Oral Surg 2016;15:258–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Xiao Y, Hsu CW, Martinez-Traverso IM, Zhang M, Bai Y, et al. Yap and Taz play a crucial role in neural crest-derived craniofacial development. Development 2016;143:504–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park HW, Guan KL. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci 2013;34:581–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan P, Han X, Zheng B, DeRan M, Yu J, Jarugumilli GK, et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat Chem Biol 2016;12:282–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambasivan R, Comai G, Le Roux I, Gomes D, Konge J, Dumas G, et al. Embryonic founders of adult muscle stem cells are primed by the determination gene Mrf4. Dev Biol 2013;381:241–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.