

ABSTRACT
Mycobacterium tuberculosis has evolved to become the single greatest cause of death from an infectious agent. The pathogen spends most of its infection cycle in its human host within a phagocyte. The bacterium has evolved to block the normal maturation and acidification of its phagosome and resides in a vacuole contiguous with the early endosomal network. Cytokine-mediated activation of the host cell can overcome this blockage, and an array of antimicrobial responses can limit its survival. The survival of M. tuberculosis in its host cell is fueled predominantly by fatty acids and cholesterol. The ability of M. tuberculosis to degrade sterols is an unusual metabolic characteristic that was likely retained from a saprophytic ancestor. Recent results with fluorescent M. tuberculosis reporter strains demonstrate that bacterial survival differs with the host macrophage population. Tissue-resident alveolar macrophages, which are biased towards an alternatively activated, M2-like phenotype, are more permissive to bacterial growth than monocyte-derived, inflammatory, M1-like interstitial macrophages. The differential growth of the bacterium in these different phagocyte populations appears to be linked to host cell metabolism.
INTRODUCTION
The foundations of our understanding of intracellular parasitism by a range of eukaryote and prokaryote pathogens has been laid by using tissue culture infection models. These models, using defined cell lines or expanded primary cell cultures, have been invaluable in the generation of the knowledge base on which the field currently relies. However, the models artificially compress the heterogeneity that exists for all these pathogens in their natural in vivo infection cycle. It is the heterogeneity within the pathogen population that enhances a pathogen’s capacity to adapt and survive under the different immune pressures and tissue environments within its host (1–3).
The past few years have seen the development of a new generation of tools that will enable us to better understand the functional consequences of heterogeneity both in the pathogen population and in the subsets of host cells present in vivo (4–6). Mycobacterium tuberculosis is a human pathogen and is the largest single cause of death by a single infectious agent. There are no effective vaccines against infection and no biomarkers for protective immunity (7–10). While there are drugs that are effective against M. tuberculosis, treatment requires a cocktail of three or four drugs taken continuously for 8 to 9 months. Such drug regimens are a serious strain on the resources of the health care systems in many resource-challenged nations, and drug-resistant strains emerge with disturbing frequency in many countries. Understanding the consequences of bacterial heterogeneity in vivo with respect to both drug action and immune containment remains a serious challenge to the field.
THE IMMUNE ENVIRONMENT AT THE SITE OF INFECTION
While not an obligate intracellular pathogen, M. tuberculosis does spend the greatest part of its infection cycle within host phagocytes, and the granuloma, the tissue response to M. tuberculosis infection, is an extremely macrophage-rich structure (11, 12). Recent data indicate that, following inhalation of infectious M. tuberculosis, the bacterium is phagocytosed by alveolar macrophages (AMs) patrolling the airway surface (13). Uptake of M. tuberculosis activates an inflammatory response through the stimulatory capacity of the multiple Toll-like receptor ligands on the bacterial cell wall. The infected AM invades the subtending tissue of the lung, and the proinflammatory response amplifies. This response leads to the generation of chemokines, such as CCL2, that are the primary drivers of the recruitment of interstitial macrophages (IMs) derived from peripheral blood monocytes in the circulatory system (14–17). This proinflammatory response persists until the development of an acquired immune response, which in the murine model system is delayed until 3 to 4 weeks postinfection because it is dependent on dendritic cells carrying M. tuberculosis antigen to draining lymph nodes to prime the initial T-cell response to infection (18, 19).
Upon initiation of a specific immune response against M. tuberculosis, the replication of the bacterium is restricted and the infection transitions into a containment state with a relatively static bacterial burden (Fig. 1A). In non-human primates and, by inference, in humans, the infection is paucibacillary, whereas in mice there is a much greater bacterial burden. This is one of the features of the murine infection that raises concerns regarding its usefulness as a model for human tuberculosis (TB). During this phase of containment and cellular consolidation, new macrophage phenotypes, such as epithelioid macrophages, multinucleated giant cells, and foamy macrophages, appear within the granuloma (20). In non-human primates and humans, the granuloma is a highly stratified structure with distinct transcriptional signatures associated with the different regions (Fig. 1B). The central, caseous region of the granuloma has a proinflammatory signature, while the region surrounding the caseum shows marked enrichment for transcripts associated with anti-inflammatory programs (21, 22). Intriguingly, each granuloma functions like an independent entity, and while the systemic immune response appears to be unchanged, some granulomas may progress to active disease while others continue to control the infection, or even progress to a sterile state (23). The factors that determine the localized progression to active disease have remained elusive (24).
FIGURE 1.
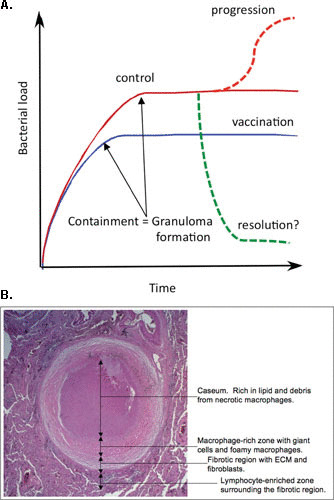
(A) Schematic illustration of the potential outcomes of infection with M. tuberculosis. In most hosts, M. tuberculosis exhibits rapid expansion of the bacterial burden during the first 3 to 4 weeks of infection. At this point, the acquired immune response has developed and controls the bacterial burden at a subclinical level but is unable to clear the infection. In vaccinated hosts, this transition to control of the bacterial burden is achieved at around 1 log fewer bacilli. While resolution of infection is theoretically possible, it is virtually impossible to demonstrate. Progression from latent disease to active disease appears to occur in the face of a robust systemic immune response that is Th1 dominant. While there are candidate indicators of early disease progression, the field lacks immunological markers to detect vaccine-induced protection. Published previously in reference 10. (B) The main features of the human TB granuloma. A fully formed human TB granuloma is an extremely stratified structure. The center of the granuloma is caseous and rich in lipids, thought to be derived from the lipids present in foamy macrophages. The caseum is surrounded by a macrophage-rich layer that contains foamy macrophages, multinucleated giant cells, and epithelioid macrophages. M. tuberculosis bacilli are observed in many of these cells. This structure is frequently encased in a fibrous capsule of collagen and other extracellular matrix proteins. Lymphocytes tend to be restricted to the periphery of the granuloma outside the fibrous outer layer. Published previously in reference 77.
This phenomenon reflects one of the greatest obstacles to combating this disease. There are no reliable biomarkers for protective immunity and therefore no surrogates to inform vaccine development programs (7–9). Increasingly sensitive indicators of early disease progression have been reported (25), but these indicators require initiation of the tissue damage that accompanies actual disease, so they are not useful indicators of protective immune status. Mycobacterial growth inhibition assays are the most utilized peripheral indicator of protective immunity (26). The data look compelling because they show a functional readout linked to bacterial survival. However, recent comprehensive evaluation of extensive data sets for the application of mycobacterial growth inhibition assays to different human populations indicates that, while the data are indicative of trained innate immunity, they do not correlate with the protection status of the individual (27).
LIFE OR DEATH IN THE PHAGOCYTE
M. tuberculosis is internalized by classic phagocytosis. Inert particles phagocytosed by macrophages are delivered to the acidic, hydrolytic environment of the phagolysosome, but M. tuberculosis has evolved strategies to subvert the process of phagosome maturation (28). The compartment in which M. tuberculosis resides is slightly acidified (pH 6.4), remains interactive with the endosomal network, and shows limited acquisition of lysosomal hydrolases. Classic activation of the macrophage with interferon gamma (IFN-γ) prior to infection enables the macrophage to overcome this process and deliver the bacterium to an acidic lysosome (29, 30). The killing of M. tuberculosis by activated macrophages is dependent on multiple factors, most significantly, the production of nitric oxide (NO), the low pH of the lysosome, and the delivery of antimicrobial peptides through the process of autophagy (31–33).
Several publications document the ability of M. tuberculosis to escape the phagosome and access the cytosol of its host cell (34–37). Escape from the phagosome appears to precipitate the necrotic death of the infected macrophage and a marked growth spurt in the intracellular bacterial population (38, 39). This transient event may have significance with respect to the pathology observed in late-stage disease but may be of less significance to long-term survival of the pathogen in its host. Data indicate that, temporally and spatially, the intravacuolar population of M. tuberculosis likely represents the more significant target for therapeutics (40).
UNDERLYING MECHANISMS OF IMMUNE CONTROL AND DISEASE PROGRESSION
Our understanding of immune control of TB is shaped heavily by failed immunity in the form of knockout mouse studies or catastrophic human genetic lesions (41, 42). IFN-γ is known to be important because mice deficient in IFN-γ fail to control M. tuberculosis infection and humans with genetic defects in the IFN-γ receptor are exquisitely susceptible to TB and to infection with M. bovis BCG. IFN-γ release assays have also been used, unsuccessfully, as indicators of a protective immune response or treatment efficacy (43). However, our current knowledge indicates that while a Th1-biased immune response and the production of IFN-γ are required for an effective immune response to M. tuberculosis, they are not sufficient to protect against either infection or disease progression. Moreover, the assumption that disease progression is the consequence of failure of Th1-dependent immune control, while widely held, is actually unsubstantiated.
A recent study used fluorescent M. tuberculosis fitness reporter strains to identify host phagocytes that best controlled M. tuberculosis growth and those that were permissive (44). The strains all expressed mCherry constitutively and expressed green fluorescent protein (GFP) either as a fusion protein with the single-strand binding protein (SSB) as a readout for replication or conditionally under regulation of the NO-responsive promoter for hspX (4–6) (Fig. 2). Studies in vaccinated and naïve mice demonstrated that expression of hspX′::GFP correlated with the development of a Th1 immune response and the expression of iNOS in the host tissue and that fluorescent SSB-GFP foci were less numerous in the face of a Th1 immune response (5). These data were generated with tissue sections from the murine granulomas. The phenotype of the bacterium at individual cell level was determined on cell suspensions from infected tissue (44). At 2 weeks postinfection, the bacteria were present predominantly in neutrophils, AMs, and IMs. Upon characterization of the reporter M. tuberculosis strains, it was found that the levels of stress induction (hspX′:: GFP) were higher in M. tuberculosis in IMs and neutrophils than in bacilli in AMs. Conversely, the SSB-GFP puncta were more frequent in M. tuberculosis in AMs and neutrophils than they were in M. tuberculosis in IMs. These data suggested that M. tuberculosis in AMs experienced less stress and replicated more actively than those in IMs. This result was corroborated with M. tuberculosis expressing a clock plasmid, pBP10, which is lost from the bacteria at a fixed rate linked to replication (45, 46).
FIGURE 2.
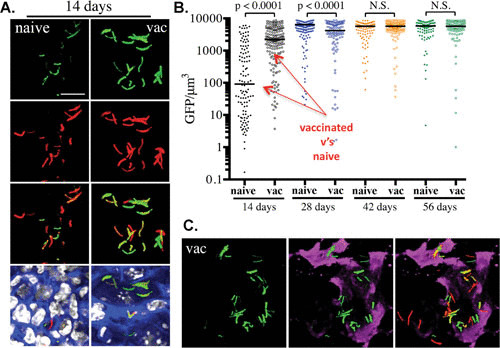
Usefulness of the hspX′::GFP reporter strain in assessing and reporting on the localized induction of inducible nitric oxide synthase at the site of infection. Phosphate-buffered saline-immunized (naïve) mice and mice vaccinated with heat-killed M. tuberculosis (vac) were infected with an hspX′::GFP smyc′:: mCherry Erdman M. tuberculosis reporter strain. Fluorescence induction of the hspX promoter-dependent GFP is higher at 14 days in the vaccinated animals, as assessed by confocal microscopy of thick tissue sections (A), which were scored subsequently by Volocity (B). (C) The thick tissue sections were probed with antibodies against murine NOS2 (magenta), demonstrating the colocalization between GFP induction and NOS2 expression at the site(s) of infection. N.S., not significant. Data are from reference 5).
Clodronate liposome-mediated depletion of the macrophage subsets was conducted to demonstrate the functional significance of the IM and AM host cell populations. Delivery of clodronate liposomes to the lung airways depleted the AM population, and intravenous inoculation of clodronate liposomes depleted the blood monocytes and therefore the IM population. In the mice with depleted AMs, the bacterial burden was reduced by approximately 1 log, while in the mice with depleted IMs, the bacterial burden was increased by 1 log (Fig. 3). These data demonstrate that by altering the relative proportion of IMs and AMs available to act as host phagocytes, one can impact the bacterial load in the mice either positively or negatively, an observation consistent with previous macrophage depletion studies (14, 16, 17).
FIGURE 3.
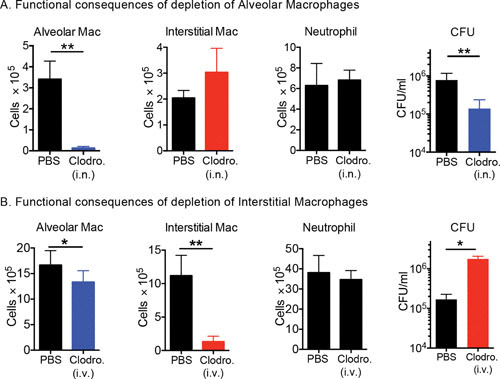
Selective depletion of AMs and IMs results in a decrease and an increase in bacterial burden, respectively. Mice were treated with clodronate (Clodro.) liposomes delivered either intranasally (i.n.) (A) or intravenously (i.v.) (B) to deplete the AMs or the circulating monocytes, which depleted the recruited IMs. Neither treatment impacted the neutrophil population within the infected lung tissue. Interestingly, depletion of AMs led to a reduction in bacterial burden, while depletion of IMs led to an increase in bacterial burden. The data demonstrate how modulation of the relative dimensions of the permissive (AM) and controller (IM) macrophage populations directly impacts bacterial burden. Data are from reference 44.
IMs AND AMs ADOPT MARKEDLY DIFFERENT METABOLIC STATES IN RESPONSE TO M. TUBERCULOSIS INFECTION
Analysis of the transcriptional profiles of both M. tuberculosis-infected and uninfected IMs and AMs showed that all four phagocyte populations had their own discrete signatures (44). Pathway analysis of the M. tuberculosis-infected AMs and IMs indicated that infected AMs were enriched in transcripts associated with fatty acid metabolism and cholesterol homeostasis. In contrast, infected IMs were up-regulated in transcripts linked to inflammatory responses, glycolysis, IFN-γ signaling, and hypoxia. Treatment of infected mice with the nonhydrolyzable glucose analog 2-deoxyglucose led to a decrease in IM number without impacting the AM population. The reduction in IM number was accompanied by an increase in bacterial burden, providing an independent demonstration that the reduction in the relative proportion of IMs and AMs drives an expansion in the bacterial burden.
A functional link between host cell metabolism and bacterial growth was demonstrated through the manipulation of M. tuberculosis-infected bone marrow-derived macrophages with the metabolic inhibitors 2-deoxyglucose and the fatty acid oxidation inhibitor etomoxir in vitro. Inhibition of glycolysis in the infected bone marrow-derived macrophages enhanced bacterial growth, while inhibition of fatty acid oxidation with etomoxir led to a reduction in bacterial growth. Neither compound had any impact on bacterial growth in rich Middlebrook 7H9 bacterial broth.
BASIS OF THE DIFFERENCE BETWEEN AMs AND IMs
Until very recently, it was thought that all macrophages in the body were differentiated from peripheral blood monocytes derived from hematopoietic precursors in the bone marrow. This is now known not to be the case; most tissue-resident macrophages, including AMs, derive from fetal yolk sac and fetal liver stem cells during embryogenesis (47, 48). These tissue-resident cells are self-maintaining and capable of replication, albeit at a low rate during homeostasis. Interestingly, recent reports suggest that M. tuberculosis infection arrests cell cycle in infected cells while increasing bystander macrophage replication within the infected tissue (44, 49). A similar state of monocytosis has been observed in human TB, indicating that this response is not restricted to the murine infection model (50, 51). The induction of replication within the macrophage populations in the infected lung provides another route for the selective expansion of permissive AM populations.
The larger question emerging from these studies is how the IM and AM lineages, which experience the same immune milieu generated by M. tuberculosis infection, adopt such divergent metabolic states. IMs and AMs are ontologically distinct macrophage lineages, suggesting that ontogeny is the dominant determinant controlling their response to infection. This interpretation is supported by recent data from an acute lung injury model where IMs and AMs exposed to lipopolysaccharide in the lung responded divergently despite experiencing the same insult (52). The accepted tissue culture model for macrophage polarization invokes the adoption of an M1 (inflammatory and antimicrobial) state in response to IFN-γ and progression to an M2 (anti-inflammatory and tissue repair) state following exposure to interleukins 4 and 13 (Fig. 4) (53–55).
FIGURE 4.
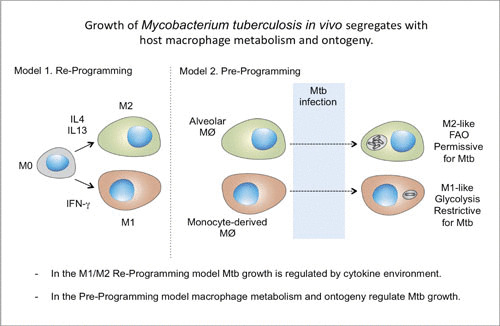
Models of macrophage reprogramming and preprogramming. How macrophages function in the reprogramming model (Model 1) is determined by immune signaling within the tissue niche. In the proposed preprogramming model (Model 2), the function of coexisting macrophage lineages in the lung in M. tuberculosis infection is determined, in large part, by the origin of the macrophage. Mtb, M. tuberculosis; IL, interleukin; FAO, fatty acid oxidation. Published previously in reference 44.
While these definitions provide a useful sense of context, multiple labs report that in vivo, in both humans and mice, different macrophage populations coexpress numerous proteins or transcripts that in vitro are thought to associate exclusively with either M1 or M2 activation states (56, 57). Extensive analysis and modeling of macrophage subsets in TB infection of non-human primates have detailed different populations of macrophages that express M1 (endothelial and inducible nitric oxide synthase)- or M2 (Arg1 and Arg2)-associated markers (58). A subsequent model suggests that the ratio of M1 to M2 macrophage subsets is an accurate predictor of whether any individual granuloma is likely to progress to active disease (59). The model is consistent with data indicating that ontogenically distinct macrophage populations, the AMs and IMs, are actually preprogrammed to respond divergently when experiencing the same immunological milieu during M. tuberculosis infection. Analysis of peripheral blood mononucleocyte-derived macrophages and tissue-resident macrophages under homeostatic conditions suggests that the bias towards M1-like and M2-like phenotypes in these different macrophage lineages exists prior to any insult or infection (60, 61).
BACTERIAL METABOLISM IN THE HOST ENVIRONMENT
The advances in our understanding of host cell metabolism and bacterial control now connect with our appreciation of bacterial metabolism within the host cell environment. M. tuberculosis’s preference for lipids and fatty acids as carbon sources has been discussed since the 1950s, but the central significance of this metabolic dependence for the virulence and pathogenesis of M. tuberculosis has been demonstrated experimentally only recently. In 2000, McKinney and colleagues reported that mutants of M. tuberculosis deficient in expression of isocitrate lyase (icl1) could not sustain an infection in the face of immune pressure (62). Isocitrate lyase of M. tuberculosis is a bifunctional enzyme whose more significant activity is that of a methyl isocitrate lyase that is required for the methylcitrate cycle, which is the primary route for detoxification of propionyl coenzyme A, which accumulates upon degradation of cholesterol (63–66).
M. tuberculosis has specific transport systems dedicated to the acquisition of fatty acids and cholesterol. The Mce family of lipid transporters is conserved across the bacterial kingdom (67) and is present in M. tuberculosis as four distinct multigenic transporter complexes (68). Mce1 and Mce4 are the preferred uptake transporters for fatty acids and for cholesterol, respectively (69). The two transporters share some of their subunit proteins, which stabilize the transporter complexes, and most notably, all Mce transporters use a common motor, the ATPase MceG (69). The linking of fatty acid and cholesterol acquisition is not surprising given the requirement for the balanced production of downstream intermediates to feed the tricarboxylic acid cycle and provide building blocks for the synthesis of complex cell wall lipids (63, 66).
The significance of cholesterol for M. tuberculosis growth was also demonstrated by a large empirical screen to identify compounds active against intracellular M. tuberculosis (70). The screen identified several inhibitors that blocked specific steps in bacterial cholesterol degradation or its regulation (Fig. 5). In addition, transcriptional profiling from a panel of 15 clinical strains of M. tuberculosis that represented the global genetic diversity of the M. tuberculosis complex confirmed that genes involved in the processing of cholesterol and fatty acids were up-regulated during intracellular growth as part of a common core transcriptome shared across all isolates (71).
FIGURE 5.
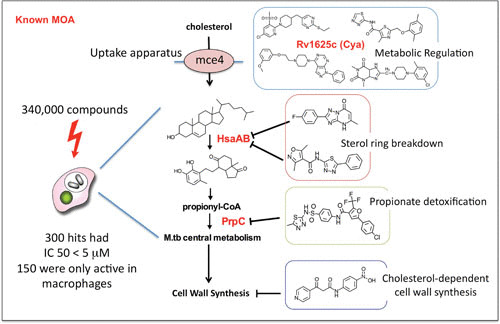
Major classes of cholesterol-dependent anti-M. tuberculosis compounds identified in a screen against intracellular M. tuberculosis (M.tb). The primary screen of 340,000 compounds identified 300 hits with 50% inhibitory concentrations (IC 50) less than 5 μM, 50% of which showed activity only against intracellular bacteria and had no activity against M. tuberculosis in rich broth. However, the majority of these compounds recovered their activity when M. tuberculosis was grown in medium with cholesterol or fatty acids as the limiting carbon source. Major targets or functions inhibited by the compounds are shown. Activators of an adenylate cyclase (rv1625c [Cya]) were shown to be involved in regulation of cholesterol utilization, as well as specific inhibitors of the enzymes HsaAB and PrpC, which are involved in cholesterol breakdown or propionyl coenzyme A (propionyl-CoA) detoxification. Data are from reference 70.
COUPLED METABOLISM OF HOST AND PATHOGEN
While it is clear that the metabolism of M. tuberculosis is shifted towards heavy dependence on fatty acids and cholesterol and that the predisposition of the AM population towards fatty acid oxidation appears to provide M. tuberculosis with a permissive host cell population, the modulation of host metabolism extends beyond the host cell to the surrounding tissue.
Figure 1B illustrates the caseous center of the human TB granuloma. Thin-layer chromatography and mass spectrometry analysis of the lipid species in the human granuloma demonstrated that the major lipid species were triacylglycerols, cholesterol, and cholesterol ester (21). The presence of abundant cholesterol ester in the caseum is strong evidence that these lipids came from lipid droplets present in the foamy macrophages that typically surround the caseous center of the granuloma (72, 73). When cells accumulate cholesterol, they usually esterify the sterol prior to transport from the endoplasmic reticulum and incorporation into the lipid droplet. This esterification is proposed to reduce the toxicity of the cholesterol. M. tuberculosis infection in culture induces a foamy macrophage phenotype in the infected cell and in uninfected bystander macrophages in the same culture. The mycobacterial cell wall lipid trehalose dimycolate has been shown to induce this behavior (72). Trehalose dimycolate is recognized by the scavenger receptor MARCO and signals through both TLR2 and Mincle (74, 75). It is thought that the prolonged, chronic activation of the proinflammatory pathways in macrophages drives this transformation into foamy cells (Fig. 6), similar to the cascade invoked in atherosclerotic plaques.
FIGURE 6.
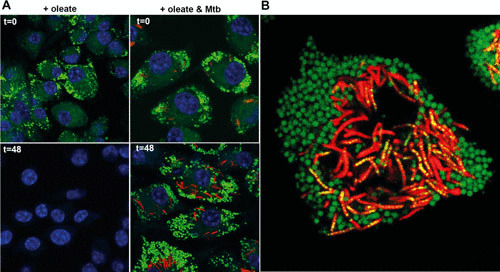
M. tuberculosis infection leads to retention of the foamy macrophage phenotype and facilitates bacterial access to host-derived lipids. (A) Murine bone marrow-derived macrophages were induced to form foamy cells through incubation with 400 μM oleate for 24 h. The cells were subsequently infected with M. tuberculosis or left uninfected. At 0 h and 48 h after infection (t=0 and t=48), cells were fixed and stained with BODIPY 493/503. M. tuberculosis organisms are displayed in red, BODIPY 493/503 is displayed in green, and DAPI (4′,6-diamidino-2-phenylindole)-stained nuclei are shown in blue. The absence of green stain in uninfected cells at 48 h indicates loss of oleate-induced lipid droplets. (B) Visualization of trafficking of host-acquired lipids into intracellular M. tuberculosis. Murine bone marrow-derived macrophages were infected with M. tuberculosis for 5 days and treated with 400 μM oleate for 24 h. The cells were incubated with the fluorescent fatty acid BODIPY FL-C16 for 60 min prior to analysis by confocal microscopy. M. tuberculosis organisms are displayed in red, BODIPY FL-C16 is displayed in green, and colocalization of M. tuberculosis with the fluorescent lipid appears in yellow. Data from reference 78.
CONCLUDING REMARKS
It is interesting to see how evolution appears to have driven M. tuberculosis to exploit the nutrient sources that it has the capacity to enhance at the site of infection, thus maximizing its chance of success. But the odds are not entirely in favor of M. tuberculosis. Of immune-competent individuals that acquire an M. tuberculosis infection, during the course of their lifetime, only 5 to 10 % will progress to develop active disease. These constitute good odds for the human species. However, a problem arises with the bacterium’s extraordinary efficiency of transmission, which enables a single individual with active TB to infect a large number of people. Recent estimates indicated that approximately 25% of the world’s population is subclinically infected with M. tuberculosis (76). In areas of high HIV endemicity, such as sub-Saharan Africa, this constitutes a major challenge to human health. Not only is M. tuberculosis the largest single cause of death by an infectious agent, it is also the single greatest cause of death in individuals living with HIV. The challenges remain, but our increased knowledge of the physiology and metabolism of intracellular M. tuberculosis, and its interplay with different host macrophage populations, will likely provide new avenues to combat this pathogen.
ACKNOWLEDGMENTS
D.G.R., E.V.N., and L.H. are supported by awards from the National Institutes of Health (AI 118582 and AI134183) and by funds from the Bill and Melinda Gates Foundation (OPP1108452 and OPP1156451).
REFERENCES
- 1.Aldridge BB, Fernandez-Suarez M, Heller D, Ambravaneswaran V, Irimia D, Toner M, Fortune SM. 2012. Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335:100–104 10.1126/science.1216166. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manina G, Dhar N, McKinney JD. 2015. Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17:32–46 10.1016/j.chom.2014.11.016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 3.Rego EH, Audette RE, Rubin EJ. 2017. Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546:153–157 10.1038/nature22361. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abramovitch RB, Rohde KH, Hsu FF, Russell DG. 2011. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol Microbiol 80:678–694 10.1111/j.1365-2958.2011.07601.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sukumar N, Tan S, Aldridge BB, Russell DG. 2014. Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. PLoS Pathog 10:e1004394 10.1371/journal.ppat.1004394. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tan S, Sukumar N, Abramovitch RB, Parish T, Russell DG. 2013. Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PLoS Pathog 9:e1003282 10.1371/journal.ppat.1003282. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhatt K, Verma S, Ellner JJ, Salgame P. 2015. Quest for correlates of protection against tuberculosis. Clin Vaccine Immunol 22:258–266 10.1128/CVI.00721-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadena AM, Flynn JL, Fortune SM. 2016. The importance of first impressions: early events in Mycobacterium tuberculosis infection influence outcome. mBio 7:e00342-16 10.1128/mBio.00342-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goletti D, Petruccioli E, Joosten SA, Ottenhoff TH. 2016. Tuberculosis biomarkers: from diagnosis to protection. Infect Dis Rep 8:6568 10.4081/idr.2016.6568. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang L, Russell DG. 2017. Protective immunity against tuberculosis: what does it look like and how do we find it? Curr Opin Immunol 48:44–50 10.1016/j.coi.2017.08.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flynn JL, Gideon HP, Mattila JT, Lin PL. 2015. Immunology studies in non-human primate models of tuberculosis. Immunol Rev 264:60–73 10.1111/imr.12258. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell DG. 2007. Who puts the tubercle in tuberculosis? Nat Rev Microbiol 5:39–47 10.1038/nrmicro1538. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, Sherman DR, Gerner MY, Urdahl KB. 2018. Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24:439–446.e4 10.1016/j.chom.2018.08.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonelli LR, Gigliotti Rothfuchs A, Gonçalves R, Roffê E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A. 2010. Intranasal poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest 120:1674–1682 10.1172/JCI40817. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kipnis A, Basaraba RJ, Orme IM, Cooper AM. 2003. Role of chemokine ligand 2 in the protective response to early murine pulmonary tuberculosis. Immunology 109:547–551 10.1046/j.1365-2567.2003.01680.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, van Deventer SJ, van der Poll T. 2001. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J Immunol 166:4604–4611 10.4049/jimmunol.166.7.4604. [PubMed] [DOI] [PubMed] [Google Scholar]
- 17.Samstein M, Schreiber HA, Leiner IM, Susac B, Glickman MS, Pamer EG. 2013. Essential yet limited role for CCR2+ inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife 2:e01086 10.7554/eLife.01086. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. 2008. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med 205:105–115 10.1084/jem.20071367. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf AJ, Linas B, Trevejo-Nuñez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179:2509–2519 10.4049/jimmunol.179.4.2509. [PubMed] [DOI] [PubMed] [Google Scholar]
- 20.Flynn JL, Chan J, Lin PL. 2011. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol 4:271–278 10.1038/mi.2011.14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim MJ, Wainwright HC, Locketz M, Bekker LG, Walther GB, Dittrich C, Visser A, Wang W, Hsu FF, Wiehart U, Tsenova L, Kaplan G, Russell DG. 2010. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med 2:258–274 10.1002/emmm.201000079. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, Daudelin IB, Chen PY, Booty MG, Kim JH, Eum SY, Via LE, Behar SM, Barry CE III, Mann M, Dartois V, Rubin EJ. 2016. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med 22:531–538 10.1038/nm.4073. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, Flynn JL. 2014. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 20:75–79 10.1038/nm.3412. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cadena AM, Fortune SM, Flynn JL. 2017. Heterogeneity in tuberculosis. Nat Rev Immunol 17:691–702 10.1038/nri.2017.69. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petruccioli E, Scriba TJ, Petrone L, Hatherill M, Cirillo DM, Joosten SA, Ottenhoff TH, Denkinger CM, Goletti D. 2016. Correlates of tuberculosis risk: predictive biomarkers for progression to active tuberculosis. Eur Respir J 48:1751–1763 10.1183/13993003.01012-2016. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanner R, O’Shea MK, Fletcher HA, McShane H. 2016. In vitro mycobacterial growth inhibition assays: a tool for the assessment of protective immunity and evaluation of tuberculosis vaccine efficacy. Vaccine 34:4656–4665 10.1016/j.vaccine.2016.07.058. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Joosten SA, van Meijgaarden KE, Arend SM, Prins C, Oftung F, Korsvold GE, Kik SV, Arts RJ, van Crevel R, Netea MG, Ottenhoff TH. 2018. Mycobacterial growth inhibition is associated with trained innate immunity. J Clin Invest 128:1837–1851 10.1172/JCI97508. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.VanderVen BC, Huang L, Rohde KH, Russell DG. 2016. The minimal unit of infection: Mycobacterium tuberculosis in the macrophage. Microbiol Spectr 4:TBTB2-0025-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaible UE, Sturgill-Koszycki S, Schlesinger PH, Russell DG. 1998. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J Immunol 160:1290–1296. [PubMed] [PubMed] [Google Scholar]
- 30.Via LE, Fratti RA, McFalone M, Pagan-Ramos E, Deretic D, Deretic V. 1998. Effects of cytokines on mycobacterial phagosome maturation. J Cell Sci 111:897–905. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Alonso S, Pethe K, Russell DG, Purdy GE. 2007. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci USA 104:6031–6036 10.1073/pnas.0700036104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766 10.1016/j.cell.2004.11.038. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. 1997. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA 94:5243–5248 10.1073/pnas.94.10.5243. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonough KA, Kress Y, Bloom BR. 1993. Pathogenesis of tuberculosis: interaction of Mycobacterium tuberculosis with macrophages. Infect Immun 61:2763–2773. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myrvik QN, Leake ES, Wright MJ. 1984. Disruption of phagosomal membranes of normal alveolar macrophages by the H37Rv strain of Mycobacterium tuberculosis. A correlate of virulence. Am Rev Respir Dis 129:322–328. [PubMed] [PubMed] [Google Scholar]
- 36.Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J. 2012. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8:e1002507 10.1371/journal.ppat.1002507. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129:1287–1298 10.1016/j.cell.2007.05.059. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Lerner TR, Borel S, Greenwood DJ, Repnik U, Russell MR, Herbst S, Jones ML, Collinson LM, Griffiths G, Gutierrez MG. 2017. Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol 216:583–594 10.1083/jcb.201603040. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahamed D, Boulle M, Ganga Y, Mc Arthur C, Skroch S, Oom L, Catinas O, Pillay K, Naicker M, Rampersad S, Mathonsi C, Hunter J, Wong EB, Suleman M, Sreejit G, Pym AS, Lustig G, Sigal A. 2017. Intracellular growth of Mycobacterium tuberculosis after macrophage cell death leads to serial killing of host cells. eLife 6:e22028. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russell DG. 2016. The ins and outs of the Mycobacterium tuberculosis-containing vacuole. Cell Microbiol 18:1065–1069 10.1111/cmi.12623. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boisson-Dupuis S, Bustamante J, El-Baghdadi J, Camcioglu Y, Parvaneh N, El Azbaoui S, Agader A, Hassani A, El Hafidi N, Mrani NA, Jouhadi Z, Ailal F, Najib J, Reisli I, Zamani A, Yosunkaya S, Gulle-Girit S, Yildiran A, Cipe FE, Torun SH, Metin A, Atikan BY, Hatipoglu N, Aydogmus C, Kilic SS, Dogu F, Karaca N, Aksu G, Kutukculer N, Keser-Emiroglu M, Somer A, Tanir G, Aytekin C, Adimi P, Mahdaviani SA, Mamishi S, Bousfiha A, Sanal O, Mansouri D, Casanova JL, Abel L. 2015. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev 264:103–120 10.1111/imr.12272. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.North RJ, Jung YJ. 2004. Immunity to tuberculosis. Annu Rev Immunol 22:599–623 10.1146/annurev.immunol.22.012703.104635. [PubMed] [DOI] [PubMed] [Google Scholar]
- 43.Clifford V, He Y, Zufferey C, Connell T, Curtis N. 2015. Interferon gamma release assays for monitoring the response to treatment for tuberculosis: a systematic review. Tuberculosis (Edinb) 95:639–650 10.1016/j.tube.2015.07.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 44.Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. 2018. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med 215:1135–1152 10.1084/jem.20172020. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gill WP, Harik NS, Whiddon MR, Liao RP, Mittler JE, Sherman DR. 2009. A replication clock for Mycobacterium tuberculosis. Nat Med 15:211–214 10.1038/nm.1915. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohde KH, Veiga DF, Caldwell S, Balázsi G, Russell DG. 2012. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog 8:e1002769 10.1371/journal.ppat.1002769. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. 2013. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 210:1977–1992 10.1084/jem.20131199. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jakubzick CV, Randolph GJ, Henson PM. 2017. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol 17:349–362 10.1038/nri.2017.28. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Cumming BM, Rahman MA, Lamprecht DA, Rohde KH, Saini V, Adamson JH, Russell DG, Steyn AJC. 2017. Mycobacterium tuberculosis arrests host cycle at the G1/S transition to establish long term infection. PLoS Pathog 13:e1006389. CORRECTION PLoS Pathog 13:e1006490 10.1371/journal.ppat.1006389. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.La Manna MP, Orlando V, Dieli F, Di Carlo P, Cascio A, Cuzzi G, Palmieri F, Goletti D, Caccamo N. 2017. Quantitative and qualitative profiles of circulating monocytes may help identifying tuberculosis infection and disease stages. PLoS One 12:e0171358 10.1371/journal.pone.0171358. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang J, Yin Y, Wang X, Pei H, Kuai S, Gu L, Xing H, Zhang Y, Huang Q, Guan B. 2015. Ratio of monocytes to lymphocytes in peripheral blood in patients diagnosed with active tuberculosis. Braz J Infect Dis 19:125–131 10.1016/j.bjid.2014.10.008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mould KJ, Barthel L, Mohning MP, Thomas SM, McCubbrey AL, Danhorn T, Leach SM, Fingerlin TE, O’Connor BP, Reisz JA, D’Alessandro A, Bratton DL, Jakubzick CV, Janssen WJ. 2017. Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am J Respir Cell Mol Biol 57:294–306 10.1165/rcmb.2017-0061OC. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boscá L, González-Ramos S, Prieto P, Fernández-Velasco M, Mojena M, Martín-Sanz P, Alemany S. 2015. Metabolic signatures linked to macrophage polarization: from glucose metabolism to oxidative phosphorylation. Biochem Soc Trans 43:740–744 10.1042/BST20150107. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Verdeguer F, Aouadi M. 2017. Macrophage heterogeneity and energy metabolism. Exp Cell Res 360:35–40 10.1016/j.yexcr.2017.03.043. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Zhu L, Zhao Q, Yang T, Ding W, Zhao Y. 2015. Cellular metabolism and macrophage functional polarization. Int Rev Immunol 34:82–100 10.3109/08830185.2014.969421. [PubMed] [DOI] [PubMed] [Google Scholar]
- 56.Rückerl D, Campbell SM, Duncan S, Sutherland TE, Jenkins SJ, Hewitson JP, Barr TA, Jackson-Jones LH, Maizels RM, Allen JE. 2017. Macrophage origin limits functional plasticity in helminth-bacterial co-infection. PLoS Pathog 13:e1006233 10.1371/journal.ppat.1006233. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, Yokoyama WM, Hawkins WG, Fields RC, Randolph GJ, DeNardo DG. 2017. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47:323–338.e6. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, Via LE, Barry CE III, Klein E, Kirschner DE, Morris SM Jr, Lin PL, Flynn JL. 2013. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol 191:773–784 10.4049/jimmunol.1300113. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marino S, Cilfone NA, Mattila JT, Linderman JJ, Flynn JL, Kirschner DE. 2015. Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infect Immun 83:324–338 10.1128/IAI.02494-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gibbings SL, Goyal R, Desch AN, Leach SM, Prabagar M, Atif SM, Bratton DL, Janssen W, Jakubzick CV. 2015. Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 126:1357–1366 10.1182/blood-2015-01-624809. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gibbings SL, Thomas SM, Atif SM, McCubbrey AL, Desch AN, Danhorn T, Leach SM, Bratton DL, Henson PM, Janssen WJ, Jakubzick CV. 2017. Three unique interstitial macrophages in the murine lung at steady state. Am J Respir Cell Mol Biol 57:66–76 10.1165/rcmb.2016-0361OC. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKinney JD, Höner zu Bentrup K, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, Swenson D, Sacchettini JC, Jacobs WR Jr, Russell DG. 2000. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 406:735–738 10.1038/35021074. [PubMed] [DOI] [PubMed] [Google Scholar]
- 63.Lee W, VanderVen BC, Fahey RJ, Russell DG. 2013. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem 288:6788–6800 10.1074/jbc.M112.445056. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muñoz-Elías EJ, McKinney JD. 2005. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med 11:638–644 10.1038/nm1252. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muñoz-Elías EJ, Upton AM, Cherian J, McKinney JD. 2006. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol 60:1109–1122 10.1111/j.1365-2958.2006.05155.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. 2008. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J Bacteriol 190:3886–3895 10.1128/JB.01767-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ekiert DC, Bhabha G, Isom GL, Greenan G, Ovchinnikov S, Henderson IR, Cox JS, Vale RD. 2017. Architectures of lipid transport systems for the bacterial outer membrane. Cell 169:273–285.e17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang F, Xie JP. 2011. Mammalian cell entry gene family of Mycobacterium tuberculosis. Mol Cell Biochem 352:1–10 10.1007/s11010-011-0733-5. [PubMed] [DOI] [PubMed] [Google Scholar]
- 69.Nazarova EV, Montague CR, La T, Wilburn KM, Sukumar N, Lee W, Caldwell S, Russell DG, VanderVen BC. 2017. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. eLife 6:e26969 10.7554/eLife.26969. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, Wang T, Locher CP, Russell DG. 2015. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog 11:e1004679 10.1371/journal.ppat.1004679. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Homolka S, Niemann S, Russell DG, Rohde KH. 2010. Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog 6:e1000988 10.1371/journal.ppat.1000988. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffé M, Emile JF, Marchou B, Cardona PJ, de Chastellier C, Altare F. 2008. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog 4:e1000204 10.1371/journal.ppat.1000204. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. 2009. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 10:943–948 10.1038/ni.1781. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bowdish DM, Sakamoto K, Kim MJ, Kroos M, Mukhopadhyay S, Leifer CA, Tryggvason K, Gordon S, Russell DG. 2009. MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog 5:e1000474 10.1371/journal.ppat.1000474. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, Kinoshita T, Akira S, Yoshikai Y, Yamasaki S. 2009. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J Exp Med 206:2879–2888 10.1084/jem.20091750. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Houben RM, Dodd PJ. 2016. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med 13:e1002152 10.1371/journal.pmed.1002152. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim MJ, Homolka S, Niemann S, Rohde KH. 2010. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 8:68–76 10.1016/j.chom.2010.06.002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Podinovskaia M, Lee W, Caldwell S, Russell DG. 2013. Infection of macrophages with Mycobacterium tuberculosis induces global modifications to phagosomal function. Cell Microbiol 15:843–859 10.1111/cmi.12092. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]