

Abstract
Most of the dual nano drug delivery systems fail to enter malignant brain tumors due to the lack of proper targeting systems and the size increase of the nanoparticle after the drug conjugation. Therefore, a triple conjugated system was developed with carbon dots (C-dots) which have an average particle size of 1.5–1.7 nm. C-dots were conjugated with transferrin (the targeted ligand) and two anti-cancer drugs, epirubicin and temozolomide, to build up the triple conjugated system in which the average particle size increased only up to 3.5 nm. In vitro studies were done with glioblastoma brain tumor cell lines of SJGBM2, CHLA266, CHLA200 (pediatric) and U87 (adult). The efficacy of the triple conjugated system (dual drug conjugation along with transferrin) was compared to dual conjugated systems (single drug conjugation along with transferrin), non-transferrin C-dots-drugs, and free drug combinations. Transferrin conjugated samples displayed the lowest cell viability even at a lower concentration. Among the transferrin conjugated samples; the triple conjugated system (C-dots-trans-temo-epi (C-DT)) was more highly cytotoxic to brain tumor cell lines than dual conjugated systems (C-dots-trans-temo (C-TT) and C-dots-trans-epi (C-ET)). C-DT increased the cytotoxicity to 86% in SJGBM2 at 0.01 μM while the C-ET and C-TT reduced only to 33 and 8%, respectively. Not only did triple conjugated C-DT increased the cytotoxicity, but also the two-drug combination in C-DT displayed the synergistic effect.
Keywords: C-dots, Glioblastoma, Cell viability, Cytotoxicity, Drug delivery, Epirubicin, Temozolomide, Transferrin
Graphical Abstract
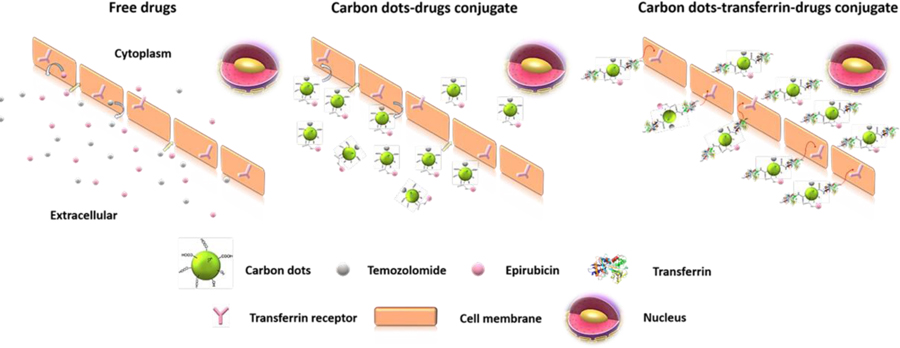
1.0. Introduction
Cancer is one of the leading causes of death by disease worldwide. Although less common than some cancers, malignant brain tumors remain a significant cause of morbidity and mortality in adults and children. In fact, brain tumors are the number one cause of pediatric cancer deaths (1). One of the most common types of brain tumors are gliomas and the prognosis for high-grade gliomas such as glioblastoma (GBM) remain dismal (2–3). Obstacles of successful treatment include: the inability to surgically remove the entire tumor, the lack of effective drugs able to cross the blood-brain barrier (BBB) at therapeutic levels, and the development of multidrug resistance (MDR), which contributes to tumor recurrence and patient relapse (4–5). The development of MDR is multifactorial and includes the increased drug efflux by ATP-dependent pumps, intracellular detoxification, and increased DNA repair anti-apoptosis mechanisms.
Nanoparticles, including carbon dots (C-dots) have been used in drug delivery systems to improve drug solubility, increase drug half-life and improve drug accumulation at the cancerous site (6–8). The Enhanced Permeability and Retention (EPR) effect promotes the accumulation of nanoparticles in the tumor tissues by the help of leaky blood vessels and abnormal lymphatic drainage (9). Therefore, localization of nanoparticle drug delivery systems on tumor reduces drug side effects, enhance the drug bio-availability and improve the drug tolerance (10–12).
However, EPR only enables the accumulation of the nanoparticles in the tumor tissues. The poor cellular uptake of the drug delivery system still limits the anticancer drug dosage which ultimately limits the therapeutic efficacy (13). Furthermore, the physiological structures comprising the BBB, which is composed of endothelial cell monolayer surrounded by pericytes and astrocytes (14–16), significantly decrease the amount of the nanoparticles able to cross the BBB and enter the tumor. To overcome this problem, nanoparticles should conjugate with BBB targeting and tumor-targeting ligands to improve the efficacy of the brain tumor drug delivery systems. Therefore, this nanotechnology is a promising way of cellular imaging and drug delivery (17–18).
The single nano drug delivery system is the most popular drug delivery system. The problem with a single anticancer drug is that, with the long-term delivery, the likelihood of drug resistance increases, which ultimately lowers the therapeutic efficacy. Therefore, to improve treatment response, dual drug delivery systems are currently being investigated. The major advantages of the combinational drug therapy are: the synergistic effect, overcoming the MDR, and reduction of the toxicity. In single drug delivery systems, the drug exerts the anticancer activity only through a specific pathway, but dual drug systems can act in multiple pathways to increase the anticancer activity. Therefore, the overall therapeutic efficacy was found to be greater in combinational drug therapy than the sum of the effects of each individual drug. The combinational therapy therefore provides a synergistic effect on anticancer activity at a lower dose of drugs (19). Conversely, combinational therapy even can be inhibitory, if one drug suppresses the anti-cancer activity of the other drug. Therefore, combinational therapies will not be synergistic for all the drug combinations.
The second advantage of dual drug system is related to the development of multi drug resistance (MDR). Brain tumors demonstrate intra-tumor heterogeneity, which refers to the phenomenon that the cells within the tumor are not identical and can differ regarding gene expression and response to therapy. Therefore, while a single drug may effectively kill a subpopulation of tumor cells, the resistant population will continue to grow. Combinatorial therapy reduces the chances the development of drug resistance by targeting more than one cell population and increasing the potential for tumor cell death and tumor demise.
Also finally, most of the chemotherapeutic agents are pumped out of the cell by drug efflux pumps. In dual drug combination, one of the drugs can be used to block these efflux pumps as an advantage. In contrast, if none of the drugs used in the combinational therapy inhibits the efflux pumps, it is still challenging to deliver the drugs inside the cell. Therefore, a proper targeting system is still required to deliver the chemotherapeutics in to the cell.
Several studies have been done for the dual drug delivery systems with different types of nanoparticles for cancer treatments. Still these studies have limitations, which hinder apply those in brain tumors treatments. Shen et al. (20) have studied dual drug system of an anthracyclin drug, doxorubicin and an efflux pump inhibitor, verapamil by using magnetic nanoparticles, which have given a high drug efficacy, but the overall particle size has been increased up to 144 nm. Song et al. (21) have reported the delivery of fluorescein and rhodamine B with mesoporous silica nanoparticles electrospun composite mat, but again the particle size after the drug loading is around 163 nm, which are still too large to cross the BBB (20–21). Aryal et al. (22) have reported dual drug pre-covalent conjugation of paclitaxel and gemcitabine through a hydrolysable ester bond which poses a risk of releasing the drugs before they reach the cancerous site. Furthermore, the lipid coated polymeric nanoparticle they used was 80 nm in size (22). So even though these studies are dual moiety drug delivery systems, most of them are without a proper targeting system, as well as bigger in size. For brain tumor treatment, smaller nanoparticle size is preferred since it has been demonstrated that smaller nanoparticles can more easily cross the blood-brain barrier, and more rapidly penetrate the tumor tissue (23–24). Therefore, an optimal brain tumor drug delivery system should be tumor cell targeted, deliver more than one chemotherapeutic drug, and be smaller in size. In this study, we synthesized a triple conjugated targeted drug delivery system with a dual-drug and non-toxic C-dots as the nanocarrier. The carboxylic acid functionalized C-dots have been covalently conjugated via the amide linkage with the targeting ligand transferrin, and the anti-cancer agents; epirubicin and temozolomide (Figure 1).
Figure 1:

The schematic illustration of the triple conjugated system of transferrin, epirubicin and temozolomide on the carboxylic acid functionalized C-dots. The drawings are not according to the exact scale and the ratio. Only 1:1:1:1 ratio of -COOH: transferrin: epirubicin: temozolomide is shown for clarification.
C-dots are photoluminescent nanomaterials, which are 1–10 nm in size (25–28). They are prominent in many applications due to their unique characteristics. C-dots show high photoluminescence, wavelength dependent/independent emission, water dispersity, high cell membrane permeability, excellent biocompatibility and non-toxicity, which make them superior candidates for the drug delivery process and biomedical application (28–33). On the surface of the C-dots mainly exists C, H and O elements (26). In general, for broad application, the surface of C-dots is functionalized with either carboxylic acids or amine groups, which is beneficial for the covalent conjugation with drugs and other targeted molecules through the amide linkage (26, 34).
Folic acid and transferrin are widely used bio-ligands. Transferrin is a blood plasma glycol protein which contains 679 amino acids with a high molecular mass (MW 80 kDa). Li et al. (30) have identified that C-dots-transferrin can cross the blood-brain barrier and enter the central nervous system (CNS) by using zebrafish as the biological model (30). Recently transferrin has been also identified as a good cancer targeting ligand due to the over expression of the transferrin receptors at the surfaces of many types of cancer cells (35). In this current study, transferrin was used only as a targeted ligand which increased the cell penetration.
One of the most widely used anti-cancer drugs for nanodelivery is doxorubicin which is in the anthracycline family. Epirubicin (epi) is a 4’- epimer of doxorubicin and compared to doxorubicin, is less cardio-toxic and better tolerated. Like other anthracyclines, epi intercalates into DNA inducing DNA cleavage assisted by topoisomerase II and inhibits the synthesis of DNA and RNA. In addition to the formation of reactive oxygen species, which increases oxidative damage (14, 36), epirubicin is also approved for the treatment of breast cancer and is being studied as a treatment for solid malignant tumors of stomach, lungs, ovaries and lymphomas.
Temozolomide is a DNA alkylating/methylating agent which causes the DNA strand breakage and apoptosis of cells. This is a second generation orally administered imidazotetrazine derivative, which has shown to increase overall survival of GBM patients from 12.1 to 14.6 months (37). However, during the activation process, temozolomide readily hydrolyses in the physiological pH in to active 5-(3-methyltriazen-1-yl) and imidazole-4-carboxamide (MTIC) and finally to aminoimidazole-4-carboxamide (AIC) (Figure S1). Even though temozolomide can easily cross the BBB by itself, MTIC or AIC cannot cross (38). Therefore, temozolomide conjugation on C-dots (prior to the hydrolyzation) is more important to increase the therapeutic efficacy.
Herein, this triple conjugated C-dot model is to deliver the chemotherapeutic drugs of epirubicin and temozolomide into glioblastoma brain tumor cells with the help of transferrin that enters to the cell via receptor mediated endocytosis.
2.0. Materials
Carbon nano powder, N-hydroxy succucinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), epirubicine hydrochloide and temozolomide were bought from Sigma Aldrich (St Louis, MO). Iron saturated human transferrin (HOLO) was obtained from MP Biomedicals (Solon, OH). Sulfuric acid (98%) and nitric acid (68–70%) were purchased from ARISTAR (Distributed by VWR, Radnor, PA). ThermoFisher Scientific (Waltham, MA, USA) provided the dialysis tubing with molecular weight cutoff (MWCO) 3500 Da and Sephacryl S-300 was obtained from GE Healthcare (Uppsala, Sweden) for size exclusion chromatography (SEC). Deionized (DI) water Modulab 2020 water purification system (San Antonio, TX) has a resistivity of 18 MΩ·cm and surface tension of 72.6 mN m−1 at 20.0 ± 0.5 0C. Pediatric brain tumor cell lines, SJGBM2, CHLA200 (glioblastoma) and CHLA266 (atypical teratoid/rhabdoid tumor) were procured from Children’s Oncology Group (COG, Lubbock, TX) while the adult glioblastoma cell line, U87 was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines were cultured in RPMI-1640 (ThermoFisher Scientific, Waltham, MA, USA) and supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin–streptomycin; both of them were purchased from Gemini Biosciences (West Sacramento, CA). All cell lines were routinely tested for mycoplasma using LookOut mycoplasma PCR detection kit from Sigma Aldrich (St. Louis, MO) according to the manufacturer’s instructions and maintained at 37 °C in a humidified 5% CO2 incubator.
3.0. Methods
3.1. Synthesis of black C-dots powder
Carboxylic acid functionalized black C-Dots powder was synthesized by following Li et al. (29) via the acidic oxidation method of carbon nano powder. 1 g of carbon nano powder was mixed with 36 mL sulfuric acid and 12 mL nitric acid. The mixture was refluxed for 15 hrs at 110 0C in an oil bath. Then after it cooled down, the unreacted acids were neutralized in an ice bath with saturated sodium hydroxide solution (pH 14). Then the mixture was vacuum filtered to remove the unreacted carbon powder. The supernatant was kept in an ice bath to precipitate the unwanted salts. A piece of sodium sulfate was added to avoid the super-saturation. The above step was repeated one more time to remove the salts further. The excess water was evaporated out from the supernatant and then the mixture was followed by washing with chloroform (60 mL) three times. Then the solution was centrifuged at 3000 rpm for 30 min. Next, the solution was transferred to a dialysis bag (MWCO: 3500 Da) and dialyzed with 4 L of DI water for 5 days. DI water was changed every 4–10 hrs. After that, the solution was again heated around 75–85 0C to concentrate it and then it was placed in the rotovap to evaporate off the water to get the C-dots.
3.2. Synthesis of C-dots-transferrin-epirubicin-temozolomide Complex (Triple system; C-DT)
8 mg C-dots were dissolved in 3 mL 25 mmol L−1 phosphate buffer solution (PBS) (pH 7.4). Then 17.78 mg 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) dissolved in 0.5 mL of PBS was added in to the C-dots solution. The mixture was stirred at room temperature for 20 min. Then 10.7 mg of N-hydroxysuccinimide (NHS) dissolved in 0.5 mL of PBS was added to the mixture. After 20 min of stirring, 1 mL transferrin (3 mg mL−1) solution was added. In another 45 min, 4.0 mg of epirubicin and 13.5 mg of temozolomide were added, which had been dissolved in 0.5 mL of dimethyl sulfoxide (DMSO) each. The entire mixture was stirred at room temperature overnight. Then the mixture was dialyzed with the 3500 MWCO dialysis bag for 4 days. The DI water was replaced every 4–10 hrs. Then the pre-dialyzed sample was further purified by SEC. The resultant eluent was collected to different test tubes and analyzed by UV-vis and fluorescence spectroscopies to identify the samples containing C-dots-epirubicin-temozolomide-transferrin (C-DT). The identified samples were frozen at −80 0C and lyophilized for 4 days to get the powdered product.
3.3. Synthesis of C-dots-epirubicin-transferrin complex (Dual system; C-ET)
The same procedure above was followed by starting the synthesis with 8 mg of C-dots. Then the EDC and NHS addition were done with the same amounts, as described in 3.2 within the same interval of time. Then the same amount of transferrin was added followed by the addition of 4.0 mg of epirubicin after the same interval of time. The purifications were conducted as above, and the powdered product was taken out after lyophilization.
3.4. Synthesis of C-dots-temozolomide-transferrin complex (Dual System; C-TT)
The exact same procedure above was followed by adding 13.5 mg of temozolomide only, instead of 4.0 mg of epirubicin.
3.5. Synthesis of C-dots-epirubicin-temozolomide, C-dots-epirubicin and C dots-temozolomide complexes (Non-transferrin complexes)
8.0 mg of C-dots were activated by EDC and NHS with the same amount as described in 3.2. Then 13.5 mg of temozolomide and 4.0 mg of epirubicin were added in two different reaction vessels separately to synthesize C-dots-temozolomide, C-dots-epirubicin. Finally, 13.5 mg of temozolomide and 4.0 mg of epirubicin both were added together with 8.0 mg of C-dots to synthesize C-dots-epirubicin-temozolomide conjugation.
3.6. Characterization
The synthesized conjugates (20 µg mL−1) were tested with UV-vis spectroscopy in a 1 cm quartz cell by using Shimadzu UV-2600 spectrometer. Then the fluorescent emission spectra of the same samples were recorded by Horiba Jobin Yvon Fluorolog-3 with slit width 5 nm for both excitation and emission. The solid FTIR study was performed on a PerkinElmer FTIR (Frontier) spectrometer using the attenuated total reflection (ATR) technique which uses the ATR prism alone as the background. The matrix-assisted laser desorption ionization time of flight (MALDI-TOF) was measured by a Bruker autoflex speed spectrometer. Transmission electron microscopic (TEM) studies were conducted with the JEOL 1200X TEM and atomic force microscopic (AFM) studies were done by an Agilent 5420 atomic force microscope using the tapping mode. Each characterization study was repeated with different batches of C-dots-conjugates to verify the consistency of the data and the stability of the complexes.
3.7. In vitro applications
The effect of chemotherapy or conjugation to C-dots with and without transferrin was evaluated in the pediatric brain tumor cell lines of SJGBM2, CHLA266, CHLA 200, and adult brain tumor cell line U87. Viability of cells was determined using MTS assay as previously described. Briefly stated, the cells were plated in 96 well plates with 0.5–2 × 104 per well, 24 hrs prior to the drug treatment. Subsequently the cells were treated with 0.01, 0.5, 0.1 μM epirubicin with and without 1 μM temozolomide, or 0.1, 1, 10 μM C-dot conjugates: C-dot-epi, C-dot-temo, C-dot-epi+temo, or 0.01, 0.05, 0.1 μM C-dot-transferrin conjugates: C-dots-trans-epi (C-ET), C-dots-trans-temo (C-TT) and C-dots-trans-dual (both epirubicin and temozolomide) drugs (C-DT). After 72 hrs incubation, viability was examined using CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega) based on manufacturer’s instructions. Absorbance was measured at 490 nm using a BioTek Synergy HT Plate reader. Data is represented as the average of 3 separate experiments in which the viability was calculated as the percent of non-treated cells. + Standard error of the mean (SEM) was calculated. Significance was determined using Students T-test. Consistency of the data were confirmed by different batches of C-dots-conjugates.
4.0. Results and discussion
The purpose of this study is to analyze the efficacy of the dual drugs with the targeted ligand on the same nano-carrier (C-dots) (triple system, C-DT) compared to the same drugs conjugated separately on C-dots (dual systems, C-TT and C-ET). Also, drugs have been loaded to C-dots with and without transferrin to prove the efficiency of the samples with transferrin. The samples without transferrin consist of C-dots-epi+temo, C-dots-epi, C-dots-temo, while C-dots-trans-epi+temo (C-DT), C-dots-trans-epi (C-ET), and C-dots-trans-temo (C-TT) are the samples conjugated with transferrin (Figure 2). Prior to the conjugation, carbon dots were synthesized via top-down, acid oxidation method by using carbon nano powder as the precursor which was previously reported by Li et al (29). In the process of conjugation, carboxylic groups on C-dots were first activated by EDC and NHS and then the covalent conjugations were done with transferrin and drugs (30, 33). The conjugated samples were first dialyzed by using 3500 MWCO dialysis bag for 4 days to remove the non-conjugated drugs and other small molecule. DI water was replaced by 4–10 hrs. Finally, the pre-dialyzed C-dots-transferrin conjugated samples were further purified with SEC to remove the unbound transferrin molecules. The conjugated samples were characterized by UV-Vis, fluorescence, FTIR, MALDI-TOF spectroscopies, TEM and AFM images. The in vitro studies were done with pediatric brain tumor cell lines SJGBM2, CHLA200, CHLA266 and adult brain tumor cell line U87.
Figure 2:

The images of transferrin conjugated samples; C-dots-trans-temo (C-TT), C dots-trans-epi (C-ET), and C dots-trans-temo+epi (C-DT) (Left to right respectively) (a) under white light and (b) UV light (365 nm).
4.1. Characterization of C-dots-epirubicin and activation process of temozolomide
From the UV-vis spectra (Figure 3), the presence of epirubicin in triple conjugated C-DT can be confirmed and the activation process of temozolomide can be detected. The epirubicin (red) spectrum displayed a characteristic absorption peak at 487 nm. An absorption peak at the same wavelength (487 nm) can be seen in triple conjugated C-DT (blue) spectrum, indicating the existence of epirubicin in C-DT.
Figure 3:

The UV-vis spectra comparison of free, epirubicin (epi), C-dots, temozolomide (temo), transferrin (trans) and the triple conjugation of C dots-trans-epi-temo (C-DT). The samples (20 μg mL−1) were tested in 1 cm optical quartz cell.
The temozolomide (green) spectrum diplayed a significant absorption peak at 330 nm, which clealy disappeared in the triple conjugated C-DT spectrum (blue). This can be explained by the analysis of electronic features of temozolomide and its metabolites by UV-vis absorption spectroscopy, that has been done by Khalilian et al. (38). During the activation process of temozolomide, the tetrazinone ring distorts with the addition of water to carbonyl moiety and then the tautomerization occurs with the elimination of CO2. The resulting methyltriazenylimidazole-4-carboxamide (MTIC), further converts to aminoimidazole-4-carboxamide (AIC) (Figure S1). Khalilian et al. have further studied the variation of each absorption λmax of temozolomide, and its metabolites of MTIC and AIC in PBS medium. The peak at 330 nm of temozolomide (green), is due to π-π* transition of N=N, which dissappeared in the AIC structure due to the breakage of tetrazinone ring (Figure S1). The absorption peak of AIC appears at 265 nm. However, the new absorption peak of AIC at 265 nm is overwhelmed (in the triple conjugated C-DT spectrum (blue)) by the absorption peak of C-dots (pink). The characteristic absorption peak of C-dots at 265 nm is due to the transition of n-π* of C=O (Figure 3). Therefore the disappearance of the peak at 330 nm in the C-DT (blue) spectrum can be possibly due to the structual change of temozolomide to AIC.
4.2. Characterization of C-dots-transferrin-epirubicin-temozolomide (C-DT)
The best analysis methods to describe the conjugation of transferrin on C-dots are fluorescence and MALDI-TOF spectroscopies.
Transferrin, epirubicin and C-dots have different fluorescence emission spectra (Figure S1-S3 in the supporting information), which are clearly distinguished in the spectrum of the triple conjugated system (C-DT) (Figure 4). The fluorescence emission spectrum of free transferrin has a peak at 346 nm, when excited at 280 nm (Figure S2) while an emission peak observed at 343 nm in triple conjugated C-DT emission spectrum when excited at 280 nm (Figure 4). The minor 3 nm blue shift of transferrin in C-DT spectrum, is only due to the attachment of transferrin on C-dots.
Figure 4:

The fluorescence emission spectra of the triple system, C-dots-trans-epi-temo (C-DT) at different excitation wavelengths. Sample was tested with 5 nm slit width for both the excitation and emission. Inset, normalized fluorescence emission spectra. The C-DT sample was tested at 10 μg mL−1 concentration in 1 cm (optical path length) quartz cell.
The fluorescence spectrum of epirubicin (Figure S3) displayed two emission peaks at 557 and 592 nm when excited at 480 nm. Even in the triple conjugated system (C-DT) (Figure 4), two emission peaks were observed at 551 and 590 nm when excited at 480 nm. The fluorescence spectroscopy further confirms the presence of epirubicin in the conjugated system with a narrow blue shift. Further when the C-DT was excited at different wavelengths (280–460 nm), the wavelength dependency was observed (Figure 4), which is a characteristic feature only of C-dots (Figure S4). Therefore, fluorescence spectroscopy was an analytical method which verified the conjugation of both transferrin and epirubicin on C-dots.
Further characterization was performed by Fourier transform infrared spectroscopy (FTIR-ATR) to observe the structural changes of temozolomide and its conjugation on C-dot.
4.3. Characterization of conjugation of C-dots-temozolomide
The conjugation of temozolomide on C-dots can be characterized by FTIR-ATR spectroscopy. In figure 5(a), the weak N-H stretching peak was observed in C-dots-temo (blue) spectrum at 3380 cm-1. It was sharply overlapped with N-H stretching peak in the spectrum of free temozolomide (black) (as shown as dark green dotted line), which confirmed the presence of temozolomide on C-dots (Figure 5(a)). Also, a new peak was observed at 1338 cm−1 in C-dots-temo (blue) spectrum in figure 5(a), which belongs to C-N stretching peptide bond. This appeared after the covalent conjugation of temozolomide –NH2 with –COOH of C-dots (39–40). Therefore, the formation of the C-N peptide bond further confirmed the conjugation of temozolmide on C-dots.
Figure 5:

FTIR-ATR spectra comparison of (a) C-dots, temozolomide and C-dots-temozolomide (C-dots-temo) conjugation. (b) C-dots, epirubicin and C-dots-epirubicin (C-dots-epi) conjugation.
The structural change of temozolomide can be further detected by FTIR-ATR spectroscopy. In the spectrum of C-dots-temo (blue) in figure 5(a), the N=N vibration peak at 1595 cm−1 barely appeared, when compared with the black spectrum of temozolomide (the dark red dotted line) (40). This is possibly due to the structural change of temozolomide to AIC (Figure S1), because the N=N bond disappeared in AIC due to the breakage of the tetrazinone ring of temozolomide. Therefore, the FTIR-ATR spectroscopy also barely confirms the presence of AIC instead of temozolomide in the conjugation.
Figure 5(b) further confirmed the conjugation of C-dots and epirubicin. In the spectrum of C-dots-epi (blue) (Figure 5(b)), the N-H stretching peak at 2920 cm−1 was overlapped with the N-H stretching peak of free epirubicin (black) and the new stretching peak for C-N bond, appeared at 1330 cm-1. This further confirmed the conjugation of epirubicin on C-dots.
The presence of each compound in C-DT was further confirmed by the FTIR-ATR spectroscopy as shown in figure S7. The peaks at 2920 and 2840 cm−1, confirmed the existence of epirubicin and transferrin in C-DT, C=O stretching peak at 1567 cm−1 verified the presence of temozolomide in C-DT and finally the formation of new C-N stretching peak at 1330 cm−1 further confirmed the formation of peptide bond.
4.4. Characterization of the conjugations by particle size
As shown in Figure 6, the AFM and TEM images further revealed the conjugation of temozolomide, epirubicin, and transferrin on C-dots by the particle size increment. The average particle sizes of AFM and TEM for C-dots (before the conjugation) were 1.5 and 1.7 nm, respectively, while the average particle sizes of TEM and AFM for triple conjugated C-dots (after the conjugation) were 2.6 and 3.5 nm, respectively. The size increment of the conjugated sample further supported the successful conjugation of transferrin, epirubicin and temozolomide on C-dots.
Figure 6:

(a) AFM, (b) TEM topology images of free C-dots, (C) AFM and (d) TEM topology images of C-dots-transferrin-epirubicin-temozolomide (tripple system) (C-DT). Scale bars in both the TEM images represent10 nm. The carbon dot solutions were sonicated for 15 mins before the analysis.
4.5. Characterization of the conjugations by the mass
In addition, conjugated samples were characterized by MALDI-TOF mass spectroscopy. According to Figure 7, all the conjugated samples were over 80,000 g/mol (higher than the free transferrin m/z ratio (Figure S5)) which confirmed the successful conjugation of transferrin on C-dot. The molecular weight of C-dot is only around 850 g/mol (Figure S6). Further, compared to mass of C-dots-transferin (C-dots-trans), the mass of drug conjugated samples on C-dots-trans (C-DT, C-TT and C-ET) are higher. This revealed the successful conjugation of drugs on C-dots-trans.
Figure 7:

MALDI-TOF spectra of transferrin conjugated samples of C-dots-transferrin (C-dots-trans), C-dots-transferrin-eprubicin-temozolomide (C-DT) (triple system), C dots-transferrin-temozlomide (C-TT), and C-dots-transferrin-epirubicin (C-ET). Consistancy of the data has been confirmed by the batch to bach analysis.
Moreover, the triple system (C-DT) displayed a lower mass than the conjugates of each individual drug and transferin on C-dots (dual systems) (C-TT and C-ET). This could possibly be due to the higher sterical hindrance in the reaction mixture of the triple sysytem than in the dual systems. In any case, MALDI-mass spectroscopy suggested, the triple system (C-DT) possibly has a lower number of drug molecules than each dual system (C-TT and C-ET). Further, epirubicin calibration curve analysis indicated C-DT has lower concentration of loaded epirubicin than every other sample (Figure S8 and Table S1). Therefore, in vitro cell studies were performed to analyze the efficacy of the triple system (C-DT).
5.0. In vitro applications with glioblastoma brain tumor cell lines
The in vitro efficacy of triple conjugated C-DT was tested with pediatric and adult brain tumor cells: SJGBM2, CHLA200 (both pediatric glioblastoma), U87 (adult glioblastoma) and CHLA 266 (a typical teratoid/rhabdoid tumor). The efficacy of the triple system (C-DT) was compared to a combination of free drugs (temozolomide and epirubicin), and C-dots-drugs conjugations with and without transferrin.
5.1. Efficacy of the mixture of free drugs of temozolomide and epirubicin
The combination of an anthracycline drug with a DNA alkylating agent drug have been shown to be effective for cancer patients in both in vitro and in vivo models. (41–42). In order to determine the drug efficacy of the combination of temozolomide and epirubicin in brain tumor cell lines, cells were treated with increasing concentrations of epirubicin with and without 1μM temozolomide.
Temozolomide induced only a small decrease in the cell viability of pediatric cell lines: SJGBM2, CHLA200 and CHLA266 (Figure 8A–D), while epirubicin induced a dose-dependent decrease in the viability in all cell lines. Mixtures of epirubicin and temozolomide further reduced viability in all cell lines and at some concentrations, the effect was more than additive, suggesting a possible synergistic effect. The lowest cell viabilities were shown in the mixture of epirubicin and temozolomide at the highest concentration of epirubicin. Combined treatment of 0.1 μM of epirubicin and 1 μM of temozolomide reduced viability approximately to 15–20% of non-treated controls except for the U87 cell line, which appeared to be more resistant and reduced the viability only to 45%. However, the combined treatment of epirubicin and temozolomide was more effective than epirubicin alone in each cell line confirming the epirubicin and temozolomide combination is a better choice for inhibiting these brain tumor cell lines. But the current concern is that we still need high concentrations of drugs to reduce the cancer cell viability potentially harming non-cancerous cells. Next we examined whether C-dots would be a suitable nanocarrier and if the chemotherapies conjugated to C-dots would still be effective.
Figure 8:

Cell viability of brain tumor cell lines; (A) SJGBM2, (B) CHLA200, (C) U87 and (D) CHLA266 exposed to free temozolomide, free epirubicin or the mixture of temozolomide and epirubicin treatment. Data is presented as percent of non-treated control cells +/− SEM. #p<0.05 compared to control, *p<0.05 combined drug treatment compared to free epirubicin.
5.2. Efficacy of C-dots-drugs conjugates (Non-transferrin)
C-dots-drugs conjugated samples of C-dots-epi, C-dots-temo and C-dots-epi+temo were tested at increasing concentrations (0.1–10 μM) on brain tumor cell lines. As shown in Figure 9, C-dots-temo was relatively ineffective in reducing the cell viability in all types of tumor cell lines. On the contrary, all concentrations of C-dots-epi and C-dots-epi+temo significantly reduced viability in all cell lines (Figure 9A–D). Among all three conjugates, C-dots-epi+temo induced substantial loss of cell viability, especially at the concentrations of 1 and 10 μM. The viability of C-dots-epi at 10 μM was 17–30% in all the cell lines (average compared to non-treated controls), while the viability of C-dot-epi+temo was significantly reduced to 7–21%. This reveals the higher efficiency of dual drug conjugation on the same C-dot rather than the single drug conjugation. Anyhow, to get a significant reduction of cell viability, the required concentrations of C-dots-epi+temo are much higher than the free drugs. This may be due to the lack of a cell targeting ligand, and uderscores the necessity for developing cancer cell targeted drug delivery systems (35–37). Using the appropriate targeted system will result in greater cellular uptake and to increase the anticancer activity of the C-dots-drug conjugates. Since most of the tumor cell lines have an over expression of transferrin receptors on the membrane, transferrin will enhance cell penetration since it enters the cell via the receptor mediated endocytosis. Therefore, the next set of samples tested with brain tumor cell lines were transferrin conjugated C-dots-drugs.
Figure 9:

Cell viability of brain tumor cell lines SJGBM2 (A), CHLA266 (B), CHLA200 (C) and U87 (D) exposed to C-dot-temo, C-dot-epi and C-dot-epi+temo. Data is presented as percent of non-treated control cells +/− SEM. #p<0.05 compared to control, *p<0.05 comparing C-dot-epi to C-dot-epi+temo.
5.3. Efficacy of C-dots-drugs-transferrin conjugates (with transferrin)
Three transferrin conjugated samples were analyzed on the same cell lines as above. Transferrin receptor expression in these cell lines was confirmed by immunocytochemistry (Figure S9). The triple conjugated system of C-dots-transferrin-temozolomide-epirubicin (C-DT) and the dual conjugates (but a single drug in each conjugate) of C-dots-transferrin-temozolomide (C-TT) and C-dots-transferrin-epirubicin (C-ET) were tested at the increasing concentration of 0.01–0.1 μM. After the conjugation of transferrin with C-dots-drugs, the cell viability drastically reduced even at a lower concentration than the C-dots-drug conjugations without transferrin (Table 1). Also, among the three transferrin conjugated samples, the triple system (C-DT) displayed lower cell viability than either dual system of C-TT and C-ET.
Table 1:
Average cell viability (All four cell lines) comparison between the non-transferrin and transferrin conjugated samples at their lowest concentrations. All the abbreviations are as below, C-dots-epirubicin (C-dots-epi), C-dots-temozolomide (C-dots-temo), C-dots-epirubicin+temozolomide (C-dots-epi+temo), C-dots-transferrin-temozolomide (C-TT), C-dots-transferrin-epirubicin (C-dots-epi), C-dots-transferrin-epirubicin-temozolomide (C-DT)
| Concentration (μM) | Average cell viability (%) of the cell lines SJGBM2, CHLA266, CHLA200, U87 | |||||
|---|---|---|---|---|---|---|
| Without Transferrin | With Transferrin | |||||
| C-dots-temo | C-dots-epi | C-dots-temo+epi | C-TT | C-ET | C-DT | |
| 10.0 | 98.20 | 23.20 | 13.50 | - | - | - |
| 1.0 | 99.12 | 77.27 | 45.6 | - | - | - |
| 0.1 | 96.12 | 90.65 | 80.5 | 86.60 | 24.30 | 11.60 |
| 0.05 | - | - | - | 91.20 | 36.20 | 27.40 |
| 0.01 | - | - | - | 95.90 | 61.15 | 43.55 |
One of the dual systems, the C-dot-transferrin-temozolomide (C-TT), showed only minimal effect on cell viability. C-TT displayed the lowest cell viability of 75% only at 0.1 μM in U87 (Figure 10D). But the epirubicin conjugated system the C-ET showed cell viability around 20% in the most sensitive cell line SJGBM2 at 0.05 μM and 10% at the concentration of 0.1 μM in CHLA266 (Figure 10A).
Figure 10:

Cell viability of brain tumor cell lines SJGBM2 (A), CHLA266 (B), CHLA200 (C) and U87 (D) exposed to C-dot-temo-trans (C-TT), C-dot-epi-trans (C-ET) and C-dot-epi+temo-trans (C-DT). Data is presented as percent of non-treated control cells +/− SEM. #p<0.05 compared to control, *p<0.05 comparing C-ET to C-ET.
On the other hand, the triple system C-DT showed the lowest cell viability of 14% even at the lowest concentration of 0.01 μM in the most sensitive cell line of SJGBM2 and 18% at 0.05 μM in CHLA266. Even though C-DT and C-ET both displayed the lowest cell viability in CHLA200 and U87 at 0.1 μM, C-ET only reduced the cell viability to 30 and 50%, while C-DT decreased the viability to 15, and 20% in CHLA200 and U87, respectively (Figure 10).
Overall, the addition of transferrin increased the cytotoxicity even at a lower concentration than without transferrin (Table 1). Similarly, we observed nuclear epirubicin fluorescence of transferrin conjugated C-dots at much lower concentrations compared to the non-transferrin conjugated C-dots (Figures S10, S11) indicating that the addition of transferrin increases cellular uptake of the C-dot drug conjugates and therefore cell death. Further, among transferrin conjugated samples, the triple conjugated (C-DT) increased the cytotoxicity at a lower concentration than other two conjugates.
5.4. Synergistic effect of the triple system C-DT
As discussed above in the characterization section, MALDI-Mass spectroscopic data revealed that the triple conjugated system (C-DT) has a lower number of drug molecules (even though both drugs conjugated together on C-dots) than each dual system (with only single drug conjugated on C-dots) (C-TT and C-ET). Moreover, Figure 11 further shows the triple conjugate (C-DT) not only increased the cytotoxicity, even with a lower number of drug molecules, but the two-drug combination displayed a synergistic effect. For instance, 0.01 µM of C-DT in SJGBM2 induced 86% cell death whereas C-TT induced 8% and C-ET induced 33% cell death (Figure 11). The sum of the C-TT and C-ET is only 41%, but C-DT itself increased the cytotoxicity to 86%, which further confirmed the efficiency of the triple conjugated C-DT. The representative morphology images of each cell line of non-treated and C-DT treated are shown in Figure 12. The cell density reduction was clearly seen after treated each cell line with triple conjugated C-DT. The images of each cell line were taken at the lowest effective concentration of C-DT, which showed the highest cell density reduction and the highest synergistic effect.
Figure 11:
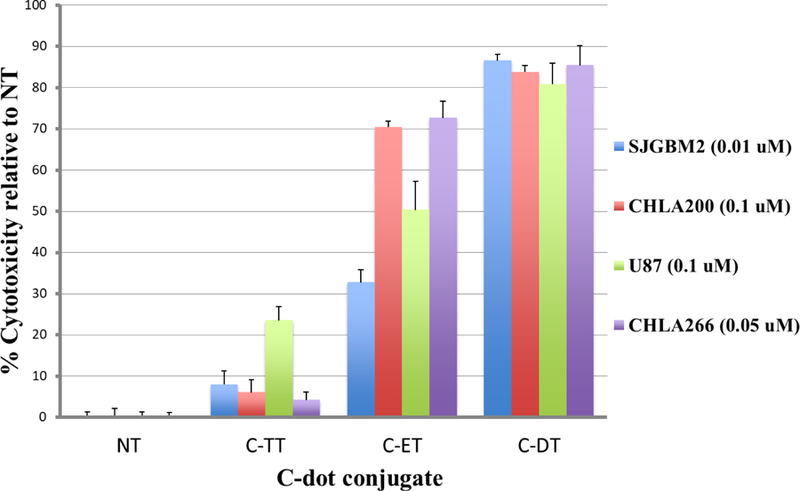
Cytotoxicity profile of the three transferrin conjugated samples; C-TT, C-ET and C-DT in each cell line compare to the non-treated (NT). The concentration in each cell line represents the lowest effective concentration of the triple conjugated C-DT that shows the maximum cytotoxicity and the synergistic effect.
Figure 12:
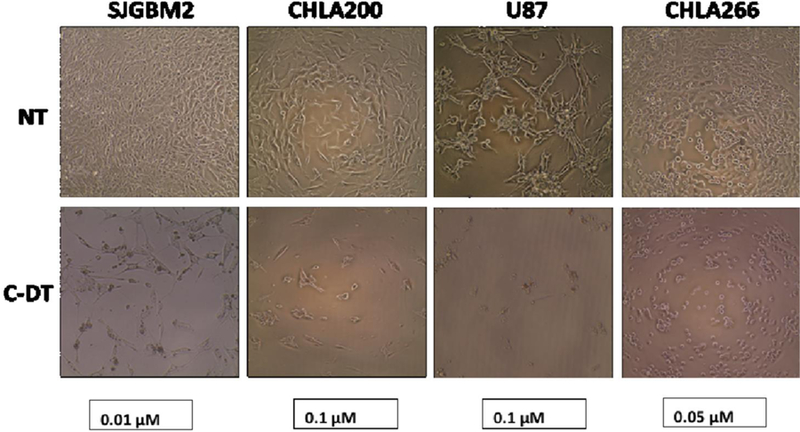
Morphology images of each cell line (SJGBM2, CHLA200, U87, CHLA266) of non-treated (NT) and C-DT treated at the concentrations of 0.01, 0.1, 0.1 and 0.05 μM, respectively.
Conclusion
In this study we have successfully developed a triple conjugated (including two drugs) targeted nano drug delivery system that displays a synergistic effect for the brain tumor cell lines. Transferrin, epirubicin, and temozolomide were conjugated onto the same C-dot. The successful conjugation of each ligand was confirmed by the analysis of UV-vis, fluorescence, FTIR and MALDI-TOF spectroscopies and, TEM and AFM imaging. MALDI analysis further revealed the C-DT might have a lower number of conjugated drugs than C-TT and C-ET. In vitro studies showed that the transferrin conjugated samples drastically reduced the cell viability than the non-transferrin conjugates. Among the three transferrin conjugated samples, the triple system (C-DT) is more cytotoxic to glioblastoma brain tumor cell lines than each dual system of C-TT and C-ET. Only a concentration of 0.01 μM was required from the (triple system) C-DT to reduce the cell viability to 14% in SJGBM2. Finally, we confirmed that the triple conjugated system (epirubicin, temozolomide and transferrin) on C-dots (C-DT) is a better therapeutic agent than its corresponding single drug delivery system.
Supplementary Material
Acknowledgement
We greatly appreciate the generous funding support given by NIH grant GR-009887 and NSF grant GR-011298. Also, we extend our deepest gratitude to the Mystic Force Foundation for providing us with the funding support for cell studies.
References
- 1.Curtin SC, Miniño AM and Anderson RN, Declines in Cancer Death Rates Among Children and Adolescents in the United States, 1999–2014, US Department of Health & Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics, 2016. [Google Scholar]
- 2.Sturm D, Pfister SM and Jones DT, Journal of Clinical Oncology, 2017, 35, 2370–2377. [DOI] [PubMed] [Google Scholar]
- 3.Koshy M, Villano JL, Dolecek TA, Howard A, Mahmood U, Chmura SJ, Weichselbaum RR and McCarthy BJ, Journal of neuro-oncology, 2012, 107, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Q, Yang Z, Nie Y, Shi Y and Fan D, Cancer letters, 2014, 347, 159–166. [DOI] [PubMed] [Google Scholar]
- 5.Yan Y, Bjö M and Caruso F, ACS nano, 2013, 7, 9512–9517. [DOI] [PubMed] [Google Scholar]
- 6.Hanafi-Bojd MY, Jaafari MR, Ramezanian N, Xue M, Amin M, Shahtahmassebi N and Malaekeh-Nikouei B, European Journal of Pharmaceutics and Biopharmaceutics, 2015, 89, 248–258. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Yang L and Shin DM, CA: a cancer journal for clinicians, 2008, 58, 97–110. [DOI] [PubMed] [Google Scholar]
- 8.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R and Langer R, Nature nanotechnology, 2007, 2, 751. [DOI] [PubMed] [Google Scholar]
- 9.Fang J, Nakamura H and Maeda H, Advanced drug delivery reviews, 2011, 63, 136–151. [DOI] [PubMed] [Google Scholar]
- 10.Cheng R, Meng F, Deng C, Klok H-A and Zhong Z, Biomaterials, 2013, 34, 3647–3657. [DOI] [PubMed] [Google Scholar]
- 11.Torchilin V, Advanced drug delivery reviews, 2011, 63, 131–135. [DOI] [PubMed] [Google Scholar]
- 12.Gong J, Chen M, Zheng Y, Wang S and Wang Y, Journal of Controlled Release, 2012, 159, 312–323. [DOI] [PubMed] [Google Scholar]
- 13.Du J-Z, Du X-J, Mao C-Q and Wang J, Journal of the American Chemical Society, 2011, 133, 17560–17563. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Y, Peng Z, Seven ES and Leblanc RM, Journal of Controlled Release, 2017. [DOI] [PubMed]
- 15.Luissint A-C, Artus C, Glacial F, Ganeshamoorthy K and Couraud P-O, Fluids and Barriers of the CNS, 2012, 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Begley DJ, Pharmacology & therapeutics, 2004, 104, 29–45. [DOI] [PubMed] [Google Scholar]
- 17.Matai I, Sachdev A and Gopinath P, ACS applied materials & interfaces, 2015, 7, 11423–11435. [DOI] [PubMed] [Google Scholar]
- 18.Xie J, Lee S and Chen X, Advanced drug delivery reviews, 2010, 62, 1064–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parhi P, Mohanty C and Sahoo SK, Drug discovery today, 2012, 17, 1044–1052. [DOI] [PubMed] [Google Scholar]
- 20.Shen J-M, Gao F-Y, Yin T, Zhang H-X, Ma M, Yang Y-J and Yue F, Pharmacological Research, 2013, 70, 102–115. [DOI] [PubMed] [Google Scholar]
- 21.Song B, Wu C and Chang J, Acta biomaterialia, 2012, 8, 1901–1907. [DOI] [PubMed] [Google Scholar]
- 22.Aryal S, Hu CMJ and Zhang L, small, 2010, 6, 1442–1448. [DOI] [PubMed] [Google Scholar]
- 23.Chauhan VP, Stylianopoulos T, Martin JD, Popović Z, Chen O, Kamoun WS, Bawendi MG, Fukumura D and Jain RK, Nature nanotechnology, 2012, 7, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoshyar N, Gray S, Han H and Bao G, Nanomedicine, 2016, 11, 673–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hornyak GL, Tibbals HF, Dutta J and Moore JJ, Introduction to nanoscience and nanotechnology, CRC press, 2008. [Google Scholar]
- 26.Zhao A, Chen Z, Zhao C, Gao N, Ren J and Qu X, Carbon, 2015, 85, 309–327. [Google Scholar]
- 27.Lai IP-J, Harroun SG, Chen S-Y, Unnikrishnan B, Li Y-J and Huang C-C, Sensors and Actuators B: Chemical, 2016, 228, 465–470. [Google Scholar]
- 28.Li S, Peng Z and Leblanc RM, Analytical chemistry, 2015, 87, 6455–6459. [DOI] [PubMed] [Google Scholar]
- 29.Li S, Wang L, Chusuei CC, Suarez VM, Blackwelder PL, Micic M, Orbulescu J and Leblanc RM, Chemistry of Materials, 2015, 27, 1764–1771. [Google Scholar]
- 30.Li S, Amat D, Peng Z, Vanni S, Raskin S, De Angulo G, Othman AM, Graham RM and Leblanc RM, Nanoscale, 2016, 8, 16662–16669. [DOI] [PubMed] [Google Scholar]
- 31.Wu L, Luderer M, Yang X, Swain C, Zhang H, Nelson K, Stacy AJ, Shen B, Lanza GM and Pan D, Theranostics, 2013, 3, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y-F, Wu H-C, Kuan C-H, Lin C-J, Wang L-W, Chang C-W and Wang T-W, Scientific reports, 2016, 6, 21170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng Z, Miyanji EH, Zhou Y, Pardo J, Hettiarachchi SD, Li S, Blackwelder PL, Skromne I and Leblanc RM, Nanoscale, 2017, 9, 17533–17543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian Z, Ma J, Shan X, Feng H, Shao L and Chen J, Chemistry–A European Journal, 2014, 20, 2254–2263. [DOI] [PubMed] [Google Scholar]
- 35.Pardo J, Peng Z and Leblanc R, Molecules, 2018, 23, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tariq M, Alam MA, Singh AT, Panda AK and Talegaonkar S, Drug delivery, 2016, 23, 2990–2997. [DOI] [PubMed] [Google Scholar]
- 37.Kumari S, Ahsan SM, Kumar JM, Kondapi AK and Rao NM, Scientific reports, 2017, 7, 6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khalilian MH, Mirzaei S and Taherpour AA, Journal of molecular modeling, 2016, 22, 270. [DOI] [PubMed] [Google Scholar]
- 39.Faizan M, Bhat SA, Alam MJ, Afroz Z and Ahmad S, Journal of Molecular Structure, 2017, 1148, 89–100. [Google Scholar]
- 40.Łaszcz M, Kubiszewski M, Jedynak Ł, Kaczmarska M, Kaczmarek Ł, Łuniewski W, Gabarski K, Witkowska A, Kuziak K and Malińska M, Molecules, 2013, 18, 15344–15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villodre ES, Kipper FC, Silva AO, Lenz G and da Costa Lopez PL, Molecular neurobiology, 2018, 55, 4185–4194. [DOI] [PubMed] [Google Scholar]
- 42.Zhang R, Saito R, Shibahara I, Sugiyama S, Kanamori M, Sonoda Y and Tominaga T, Journal of neuro-oncology, 2016, 126, 235–242. [DOI] [PubMed] [Google Scholar]
- 43).Amin ML, Drug Target Insights, 2013, 7, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.