

Abstract
The leukodystrophies are a group of genetic metabolic diseases characterized by an abnormal development or progressive degeneration of the myelin sheath. The myelin is a complex sheath composed of several macromolecules covering axons as an insulator. Each of the leukodystrophies is caused by mutations in genes encoding enzymes that are involved in myelin production and maintenance. The lysosomal storage diseases (LSDs) are inborn disorders of compartmentalized cellular organelles with broad clinical manifestations secondary to the progressive accumulation of undegraded macromolecules within lysosomes and related organelles. The over 60 different LSDs are rare diseases; however, collectively, the incidence of LSDs ranges just over 1 in 2,500 live-births. The majority of LSDs are associated with neurological manifestations including developmental delay, seizures, acroparesthesia, motor weakness, and extrapyramidal signs. These inborn organelle disorders show wide clinical variability affecting individuals from all age groups. In addition, several of neurological, also known as neuronopathic, LSDs are associated with some level of white matter disease which often triggers the diagnostic investigation. Most LSDs are autosomal recessively inherited and few are X-linked being females at risk of presenting with mild, but clinically relevant neurological manifestations. Biochemical assays are the basis of the diagnosis and, are usually confirmed by a molecular genetic testing. Novel therapies have emerged. However, most affected patients with LSDs yet only have supportive management to rely upon. A better understanding of the mechanisms resulting in the leukodystrophy will certainly result in innovative and efficacious disease-modifying therapies.
Introduction
The lysosomal storage disorders (LSDs) are ‘inborn organelle defects’ with broad clinical manifestations secondary to progressive accumulation of undegraded macromolecules within lysosomes and related organelles. Individually, each of the almost 60 different LSDs is a rare or orphan; however, their incidence combined ranges from 1 in 2,315 to 7,700 live-births.1, 2 Most LSDs are associated with neurological manifestations including developmental delay, seizures, acroparesthesia, motor weakness, and extra-pyramidal signs. The chronic and later onset forms of LSDs are often misdiagnosed as symptoms may include slowly progressive neuropsychiatric problems that, often, precede other neurological and systemic manifestations. In several LSDs, the clinical subtypes are difficult to determine due to substantial variability of clinical manifestations of these diseases. In the majority of LSDs, if not all, a single disease continuum is observed with different age of onset and rates of disease progression. The first symptoms in affected individuals can occur at any age from birth to late adulthood. In general, the earlier onset of the symptoms and signs, the more rapidly progressive and greater disease severity is noted on the course of the disease. From a genetic standpoint, the majority are transmitted as autosomal recessive traits. A few of LSDs are X-linked inherited and some females may present neurological symptoms as observed in Fabry disease.
The diagnosis of most LSDs is traditionally done through biochemical assays followed by confirmatory molecular genetic testing, in the cases caused by an underlying lysosomal enzyme deficiency. Nowadays, with the increasing use of the whole-exome sequencing (WES) in the investigations for neurogenetic disorders, the diagnosis of several LSDs has been done through identification of pathogenic variants in lysosomal enzyme encoding genes. Interestingly, in the scenario of novel variants found in WES, the biochemical enzymatic assays become confirmatory test to assess the pathogenicity of the identified genetic variants. However, in inherited metabolic clinical practices, the lysosomal enzyme assays and metabolic-based assays, i.e. mucopolysaccharide profile either by thin-layer chromatography or liquid chromatography tandem mass-spectrometry (LC-MS/MS)3, are still used as primary diagnostic tests. In the setting of molecular genetics reports of variants of unknown significance (VUS) in genes encoding lysosomal enzymes and proteins, these biochemical tests including enzymatic activity and measurement of specific accumulated metabolites in patient’s samples become extremely helpful in the interpretation of the molecular results and reaching the final diagnosis. Substantial advances have been made in specific therapies for certain LSDs. Nevertheless, the majority of LSDs still have only supportive and symptomatic treatment available, especially regarding those with prevalent neurological manifestations. Current investigations of the pathogenic mechanisms are generating novel insights for innovative therapeutic strategies. A better understanding of the neurological manifestations in LSDs will allow early diagnosis and identification of clinical endpoints that can be further explored when examining the impact of interventional therapies.
Lysosomes are heterogeneous membrane-enclosed cellular organelles containing several hydrolytic enzymes within an acidified milieu, in which several metabolites are degraded. Lysosomes were originally identified and characterized by Christian De Duve in the mid-1950s.4 Due to recent discoveries and advances, lysosomes are currently considered a key organelle in sensing and signaling environmental changes, including nutrient availability, growth signals and stress, coordinating appropriate cellular responses to diverse types of stimuli. As an example, lysosomes function as key components of both anabolic and catabolic signaling. The anabolic signaling is mediated via the mechanistic target of rapamycin complex 1 (mTORC1), which is the master regulator of cellular growth. Whereas the catabolic signaling is mediated via AMP-activated protein kinase (AMPK), which is the main sensor of energy stress.5, 6 Under conditions favoring proliferation and cell growth, the transcription factor EB (TFEB), which activates the transcription of genes in the Coordinated Lysosomal Expression And Regulation (CLEAR) network, is sequestered away from the nucleus due to its phosphorylation by activated mTORC1 on lysosomes. In contrast, in catabolism-inducing conditions, the TFEB undergoes nuclear translocation and gene expression required for lysosome biogenesis and autophagy induction.5 Therefore, along with having degradation and recycling intracellular function, lysosomes are now viewed as key regulators of cellular homeostasis in response to environmental stimuli.
The deficiency of one or more these hydrolases, or a specific lysosomal membrane protein defect results in the lysosomal accumulation of several substrates, initiating a cascade of various pathogenic processes ultimately leading to cell death. The genetic disorders caused by mutations in genes encoding enzymes or proteins that are essential for physiological function of lysosomes are called lysosomal storage diseases (LSDs). A significant number of LSDs are characterized by a progressive neurodegenerative course, resulting in severe disability and premature death due to disease-related complications.7 As the accumulated metabolites, such as glycosphingolipids and sphingolipids, are abundant and, physiologically important in the central nervous system, the wide range of neurological symptoms/signs are prevalent findings in several of LSDs. Myelination, for instance, is crucial in the first years of life requiring intact function of a number of lysosomal hydrolases for the rapid turnover of glycosphingolipids essential to myelin formation and maintenance.
Leukodystrophy, as a term, was first used by Bielschowski and Henneberg to describe the neuropathological findings, indicating abnormalities in central nervous system (CNS) glial cell types as the primary or fundamental pathological abnormality.8 Several LSDs have been associated with leukodystrophies. Given the lysosomal role in the myelin homeostasis, leukodystrophy as a neuroradiological finding becomes a relevant finding with clinical and pathogenic importance in this group of inherited neurological diseases. This review is focused on LSDs that predominantly present with leukodystrophy as part of their constellation of clinical manifestations at onset or during anytime of the disease course. Given the current availability of therapy for certain LSDs, as well as, the potential associated risks for other family members, early diagnosis and timely initiation of treatment may be of utmost importance for an optimal therapeutic outcome.
Globoid-cell leukodystrophy or Krabbe disease
Krabbe disease, or globoid-cell leukodystrophy (GLD), is a classical neurological LSDs caused by the deficiency of β-galactocerebrosidase (GALC) due to autosomal recessively inherited mutations in the GALC gene. The GALC is the only enzyme able to hydrolyze galactosylsphingosine, mostly known as psychosine, a deacylated analogue of galactosylceramide. Whereas galactosylceramide can also be degraded by the β-galactosidase generating as a product lactosylceramide.9 Sulfatide and galactosylceramide are very important constituents of myelin, which is the insulating lipid-complex sheath produced by oligodendrocytes in CNS, and by Schwann cells in the peripheral nervous system (PNS). At non-physiological elevated levels, psychosine is highly toxic to several cell types from CNS and PNS, particularly to the oligodendrocytes and Schwann cells. The GLD phenotype correlates with the primary involvement of these cells and secondary neuroinflammatory responses. From a clinical standpoint, GLD is characterized by a progressive leukodystrophy with a classical infantile-onset associate with severe neurologic impairment and deterioration and, ultimately, death by two years of age.10, 11 Infantile GLD is divided into four chronologically ordered stages based on levels of disease progression.12 Stage 1 is characterized by irritability, vomiting, and gastroesophageal reflux and dysphagia, and it is followed by stage 2 which includes hypertonia of upper and lower extremities, pyramidal signs, and progression of dysphagia. In stage 3, patients present with severe hypertonia of all extremities, become wheelchair bound and can manifest opisthotonic posturing followed by progressive hyporreflexia, which reflects the progression to polyneuropathy. In addition, bilateral optic atrophy is often noticeable at this stage. Stage 4 is characterized by immobility, either hypotonic or hypertonic, tube feeding dependence, seizures, unexplained fevers, and startle reflexes. These are general stages with significant overlap and some patients do not fit into these stages chronologically. Average survival is around 12 months or less. In terms of neuroradiological findings, patients with the infantile form of GLD present with high signal intensity at T2-weighted MR imaging in the centrum semiovale, periventricular white matter, and deep gray matter bilaterally. The subcortical U-fibers are usually spared until late stages of the disease course.13 In addition, abnormal areas of T2-hyperintensity signal is observed in the cerebellum and pyramidal tract early in the disease course. Severe progressive atrophy occurs as the disease advances with optic nerve hypertrophy also noticeable.14 In the late-onset forms of GLD, brain MRI studies show that the involvement of the dentate and cerebellar white matter are rarely present, whereas extensive involvement of the parieto-occipital white matter, periventricular regions, posterior corpus callosum, and posterior internal capsule are often present.15 Diffuse tension (DTI) allows a quantitative evaluation of white matter damage in GLD. Radial diffusity (RD) seems to be the most sensitive DTI parameter in agreement with the histopathological findings in GLD.16
Hematopoietic stem cell transplantation (HSCT) in newborns with infantile GLD prolongs survival and cognitive decline is greatly slowed.17 Whereas, the PNS function still show progressive deterioration. The clinical outcome of the neurological problems in GLD is dependent on the time the HSCT is performed during the disease course.18, 19 The late-onset forms of GLD occur in 10–12% of patients who present with protracted neurological problems with onset between the late first to the fifth decades.10, 11 The onset and progression of neurological symptoms and signs in the late-onset forms of GLD are quite variable. Patients with late-onset GLD can be clinically normal for years until weakness, vision loss, and intellectual regression become evident which may occur during mid-adulthood. The onset of symptoms and clinical course can be variable even among siblings. In terms of management, the HSCT in presymptomatic infants and older individuals with mild symptoms provides a benefit over symptomatic treatment only.20 Some patients with GLD, who underwent HSCT, show improved and preserved cognitive function.17, 21 However, many show progressive deterioration of the PNS, which is manifested by signs and symptoms of peripheral neuropathy.19, 22 The availability of umbilical cord blood banks have expanded the access to allogenic HSCT to presymptomatic and early diagnosed infants.
The initial success of HSCT in infants with GLD prompted the development of newborn screening (NBS) programs to identify affected infants presymptomatic, i.e. before the neurological symptoms are manifested.19, 23 The NBS has shown that it is difficult to elaborate predictions on GLD clinical forms, disease course without treatment, and long-term consequences of the HSCT.21 The incidence of Krabbe disease in NY is different than was anticipated, which may reflect the diversity of State of New York population. Before NBS, the estimated incidence of GLD was 1 in 100,00012, but the actual incidence of the infantile Krabbe disease in NY detected by NBS is only 1 in 394,000.24 The infantile form of GLD is represents 85–90% of diagnosed GLD patients in North American and European countries. Whereas, in Japan and other countries of Asia, the late-onset forms of GLD are more prevalent reaching 59–60% of GLD cases.25 The data from NBS program from NY state showed only 5 infants with infantile form of GLD. However, the NBS also identified 46 children who are in the moderate or high-risk category for later-onset GLD phenotypes. According to NY NBS program, moderate risk is defined as confirmatory GALC enzymatic activity within 0.16–0.29 nmol/hr/mg protein with two known or potentially pathogenic mutations in the GALC gene.21 The high-risk are newborns identified with confirmatory GALC enzymatic activity < 0.15 nmol/hr/mg protein and also with two known or potentially pathogenic mutations in the GALC gene.21 This data from NY NBS program indicates that the late-onset form of GLD may be more prevalent than previously expected and very likely underdiagnosed owing to its protracted and mild clinical presentation.19, 24 Given the significant clinical variability among individuals with late-onset forms of GLD, and even among those patients sharing the same genotype, evaluation of the effectiveness of HSCT and other therapies can be very difficult.
Metachromatic Leukodystrophy (MLD)
Metachromatic leukodystrophy (MLD) is a LSDs caused by arylsulfatase A (ASA) deficiency characterized by three clinical forms. The late-infantile form of MLD is the most prevalent clinical form (50%−60%) with onset between the age of one and two years. The late-onset forms are the juvenile MLD form (~20%−30%) with disease-onset between age four years and the age of sexual maturity (12–14 years); and adult MLD (~15%−20%), with onset after sexual maturity.26 Affected patients with the late-infantile MLD show motor weakness, hypotonia, incoordination, gait ataxia, ankle spasticity and dysarthria progressing to generalized or partial seizures, hearing and visual loss and peripheral neuropathy. In the final stages of the disease, which is between 4–10 years of age, patients become bedridden and death. In the juvenile form of MLD, patients start to decline in academic performance, behavioral disturbances, and extra-pyramidal symptoms including incoordination, gait ataxia, dysarthria, fecal and urinary incontinence, and recurrent episodes of irritability and aggressiveness during this period. Seizures can be commonly seen. Patients diagnosed with the adult onset of MLD initially present decline in academic or work performance, personality changes and social and emotional labilities, which may occur post-adolescence or even between 3rd and 5th decades. At disease onset, the neurologic symptoms include motor weakness and loss of coordination, progressing to spasticity and incontinence or seizures predominate. The peripheral neuropathy is commonly found in patients with the adult onset form of MLD. The final MLD clinical stages are similar to those from the juvenile and late-infantile onset forms. In terms of diagnosis, the late-infantile form presents almost no ASA activity in peripheral leukocytes. The late-onset forms of MLD are associated with a residual activity of ASA, which can be better determined in cultured fibroblasts from affected patients. In the cultured fibroblasts established from skin biopsy specimens, the mutant ASA activity levels are usually less than 10% of normal controls.27, 28 Because of the high frequency of the so-called pseudodeficiency (Pd) allele, additional studies in all individuals with low ASA activity are required.29 The diagnosis of MLD is confirmed by one or more of the following additional tests: molecular genetic testing of ARSA gene sequencing, and/or urinary excretion of sulfatides. The HSCT is currently the only therapy for primary CNS manifestations, but depending on the neurological manifestations, it still remains controversial because of their substantial risk and uncertain long-term effects.30, 31 In this setting, the brain MRI along with the neurological and neuropsychometric assessments become an important component of the evaluation and decision on therapies. In affected patients, the brain MRI shows diffuse symmetric periventricular myelin with hyperintensities on T2-weighted images. In the initial stages, posterior involvement is observed in most late-infantile cases with subcortical U-fibers and cerebellar white matter spared (Fig.1). At later stages, the brain MRI becomes more pronounced in a rostral-to-caudal progression, and cerebral atrophy develops.32–35 In the juvenile and adult forms, the hyperintensity T2-weighted signals are located in parieto-occipital regions affecting the periventricular and central white matter. In the frontal regions, only central parts of the white matter are slightly involved. Both genu and splenium of the corpus callosum can present T2-signal hyperintensities, with the splenium more severely affected.34 In terms of treatment, the MLD patients undergoing HSCT or BMT before the neurological symptoms arise have a better outcome. More recently, ex vivo lentiviral gene therapy (GT) in hematopoietic stem cells (HSC) has proven to provide clinical benefit in MLD. After promising results obtained in MLD mouse models,36, 37 a non-randomized, open-label, single-arm phase I/II clinical trial of HSC-GT for the treatment of MLD has started and is ongoing.38 In three pre-symptomatic late-infantile children, preliminary results showed stable engraftment of the transduced HSCs in the bone marrow and peripheral blood, with reconstitution of ASA activity in all HSC lineages and in the cerebrospinal fluid (CSF).39 Further results from the first nine treated patients (six late-infantile, two early juvenile and one with an intermediate phenotype) were recently published with a median follow-up of 36 months (range 18–54 months).38 The results showed hematopoietic reconstitution in all analyzed patients, stable engraftment of gene-corrected cells and stable reconstitution of ASA activity that was also restored in the CSF as early as six months after HSC-GT.38 Moreover, this study underlined that the extent of benefit was influenced by the time interval between ex vivo GT and the expected time of disease onset. Other types of therapies are being investigated using different approaches including small molecule therapies.40
Figure 1. Brain magnetic resonance imaging (MRI) of a patient with metachromatic leukodystrophy (MLD).
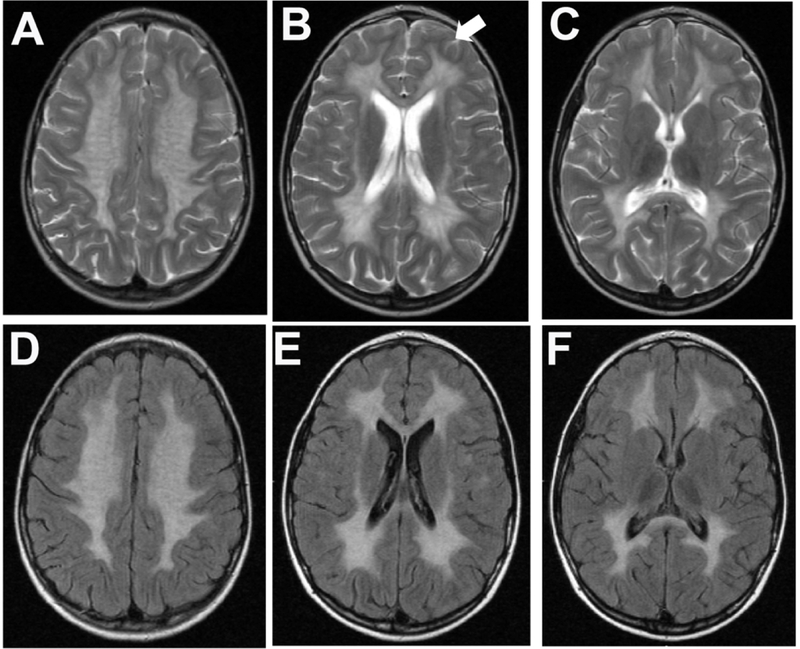
A 7-year old female who had one-year history of progressive ataxia, cognitive impairment and dysarthria which started approximately 18 months before the brain MRI was performed. She was diagnosed with the juvenile form of MLD. Sequences of fluid attenuated inversion recovery (FLAIR; A-C) and T2-weighted turbo spin echo (T2W_TSE; D-F) of transverse sections are shown. (A) The brain MRI shows typical widespread hyperintensity signals affecting the entire centrum semiovale of both hemispheres, frontal, parietal and occipital regions extending bilaterally to the temporal white matter. In panel B, the white-arrow shows subcortical U-fibers in the L frontal lobe, that are initially spared in MLD. (C and F) Brainstem shows small scattered areas of punctate high-signal representing also white matter demyelination, mostly in the thalami and subthalamic regions in the T2W and FLAIR sequences.
Multiple sulfatase deficiency
Multiple sulfates deficiency (MDS) is a LSD caused by the deficiency in processing of an active-site cysteine to formylglycine (alanine-semialdehyde), a proenzyme activation step common to most sulfatases.41, 42 Findings suggesting the diagnosis of multiple sulfatase deficiency include the following: reduced activity of other sulfatases including arylsulfatase B, arylsulfatase C, iduronate sulfatase (enzyme deficient in MPS-2), and heparan-N-sulfamidase in leukocytes or cultured cells. Elevated mucopolysaccharides and sulfatides in the urine are observed. Although a broad clinical variability of MSD is noted, features of both MLD and a mucopolysaccharidosis can be noticeable in affected patients.43 More severe forms of MSD resemble late-infantile MLD. In other cases, MPS-like features such as coarse facial features, visceromegalies, and skeletal abnormalities may be evident in infancy and early childhood, with MLD-like symptoms becoming evident in later childhood. Eventually, the disease course resembles MLD with demyelination dominating the clinical picture. Therefore, the radiological signs consist of diffuse white matter involvement.44 Ichthyosis, common to arylsulfatase C deficiency, is also often present.44 The deficiency in the formylglycine-generating enzyme (FGE) is causative biochemical defect in MSD. The formylglycine-generating enzyme is responsible for the activation of most sulfatases, and a variable degree of arylsulfatase A deficiency occurs in many tissues in its absence.45
Glycosphingolipidosis
Deficiencies of lysosomal enzymes involved in the degradation of glycosphingolipids are the LSDs mostly known as glycosphingolipidoses. The glycosphingolipids (GSLs) are synthesized in the Golgi apparatus in a stepwise manner and play an essential role in the CNS, promoting cell signaling and contributing to the myelin synthesis and maintenance. The major glycosphingolipidosis associated with leukodystrophies are described in table 1.46–54 Gaucher and Fabry diseases are also considered glycosphingolipidoses since the accumulated primary substrate are neutral glycosphingolipids (GSLs).
Table 1.
Lysosomal storage diseases associated with white matter abnormalities
| Disease | Deficient Enzyme | Primary storage metabolite | Gene, locus (inheritance) | Neurological Symptomatology | Non-neurological Symptoms | |||
|---|---|---|---|---|---|---|---|---|
| Common Neurological LSDs | ||||||||
| Gaucher disease (Types I, II, III) | Glucosylceramidase | glucosylceramide | GBA 1q21 (AR) | Type I – (adult) parkinsonism, peripheral neuropathy Type II – (fetus – 12-months) stridor, oculomotor apraxia, dysphagia, dystonia, pyramidal tract signs, and sometimes opisthotonus Type III – (>1-year old) myoclonic and tonic-clonic seizures, horizontal supranuclear gaze palsy, ataxia | Hematological – anemia, leucopenia, thrombocytopenia Visceromegaly – hepatosplenomegaly General -low energy pulmonary Hypertension Dermatological – neonatal ichthyosis | |||
| Fabry disease | α-Galactosidase A | globotriasylceramide | GLA Xq22 (X-linked) | Acroparesthesia, dysesthesia, recurrent acute and chronic pain, hearing impairment, tinnitus, recurrent cerebrovascular disease | Renal -chronic renal disease Cardiac -hypertrophic cardiomyopathy arrhythmias, Ocular – corneal verticillata Dermatological – angiokeratomas, distal edema (hands and feet) | |||
| Mucopolysaccharidoses (MPSs) | ||||||||
| MPS I (Hurler, Scheie, Hurler/Scheie) | α-Iduronidase | Dermatan sulfate, heparan sulfate | IDUA 4p16.3 (AR) | Hurler-Global developmental delay, carpal tunnel syndrome, myelopathies, spinal cord compression Scheie-Normal neurodevelopment, carpal tunnel syndrome, myelopathies, spinal cord compression | Skeletal – dysostosis multiplex and multiple joint contractures Organomegaly Obstructive Sleep Apnea Corneal clouding Cardiac valvulopathies and hypertrophic cardiomyopathy | |||
| MPS II (Hunter) | Iduronate sulfatase | Dermatan sulfate, heparan sulfate | IDS Xq28 (X-linked) | Severe form-Global developmental delay, carpal tunnel syndrome, myelopathies, spinal cord compression Attenuated form-Normal neurodevelopment, carpal tunnel syndrome, myelopathies, spinal cord compression | Skeletal – dysostosis multiplex and multiple joint contractures Organomegaly Obstructive Sleep Apnea Corneal clouding Cardiac valvulopathies and hypertrophic cardiomyopathy | |||
| MPS III Sanfilippo syndrome | MPS-IIIA -heparan sulfamidase | Heparan sulfate | SGSH 17q25.3 (AR) | Global neurodevelopmental delay (1st stage), behavior problems characterized by temper tantrum, aggressive behavior, and extreme restlessness (2nd stage). Severe dementia, decline motor functions (3rd stage). | Coarse facies, obstructive air way, dysostosis multiplex (thoracolumbar kyphosis, abnormally shaped vertebrae and ribs, spatulate ribs, hypoplastic epiphyses, thickened diaphyses, and bullet-shaped metacarpals). Brain MRI with ventricular dilatation and enlargement of subarachnoid spaces, thin corpus callosum, enlarged perivascular spaces. | |||
| MPS-IIIB -acetyl α-glucosaminidase | Heparan sulfate | NAGLU 17q21.2 (AR) | ||||||
| MPS-IIIC -acetyl CoA: α-glucosaminide N-acetyltransferase | Heparan sulfate | HGSNAT 8p11.21 (AR) | ||||||
| MPS-IIID N-acetyl glucosamine-6-sulphatase | Heparan sulfate | GNS 12q14.3 (AR) | ||||||
| MPS IVA (Morquio A) | Acetyl galactosamine-6-sulphatase | Keratan sulfate, chondroiotin 6-sulphate | GALNS 16q24.3 (AR) | Normal neurodevelopmental, carpal tunnel syndrome, myelopathies, spinal cord compression | Skeletal – dysostosis multiplex and multiple joint contractures Organomegaly Obstructive Sleep Apnea Corneal clouding Cardiac valvulopathies and hypertrophic cardiomyopathy | |||
| MPS IVB (Morquio B) | β-Galactosidase | Keratan sulfate | GLB1 3p22.3 (AR) | Normal neurodevelopment, carpal tunnel syndrome, myelopathies, spinal cord compression | Skeletal – dysostosis multiplex and multiple joint contractures Organomegaly Obstructive Sleep Apnea Corneal clouding Cardiac valvulopathies and hypertrophic cardiomyopathy | |||
| MPS VI (Maroteaux-Lamy) | Acetyl galactosamine 4-sufatase (arylsulfatase B) | Dermatan sulfate | ARSB 5q14.1 (AR) | Normal neurodevelopmental, carpal tunnel syndrome, myelopathies, spinal cord compression | Skeletal – dysostosis multiplex and multiple joint contractures Organomegaly Obstructive Sleep Apnea Corneal clouding Cardiac valvulopathies and hypertrophic cardiomyopathy | |||
| MPS VII (Sly) | β-Glucuronidase | Dermatan sulfate, heparan sufate,
chondroiotin 6-sulphate |
GUSB 7q11.21 (AR) | Global developmental delay, carpal tunnel syndrome, myelopathies, spinal cord compression | Skeletal – dysostosis multiplex and multiple joint contractures Organomegaly Obstructive Sleep Apnea Corneal clouding Cardiac valvulopathies and hypertrophic cardiomyopathy | |||
| Glycosphingolipidosis | ||||||||
| Niemann-Pick (type A, type B) | Acid sphingomyelinase | Sphingomyelin | SMPD1 11p15.4 (AR) | Psychomotor development progresses no further than the 12-month level, after which neurologic deterioration is relentless. NP-B: Normal neurodevelopment | NP-A: hepatosplenomegaly with progressive hypersplenism and stable liver dysfunction, interstitial pulmonary disease, osteopenia, atherogenic lipid profile. | |||
| Niemann-Pick type C Disease | NPC1 and NPC2 | Unesterified cholesterol and several glycosphingolipids | NPC1 and NPC2 (AR) | Progressive impairment of psychomotor development, Vertical supranuclear gaze palsy (VSGP), followed by progressive ataxia, dysarthria, dystonia, and, in some cases, seizures and gelastic cataplexy | hepatosplenomegaly with progressive hypersplenism, and neonatal jaundice, interstitial pulmonary disease, osteopenia, atherogenic lipid profile. | |||
| Farber Disease | Acid ceramidase | Ceramide | ASAH1 8p22 (AR) | severe progressive impairment of psychomotor development and neurologic deterioration with epilepsy | progressively deformed joints, subcutaneous nodules, and progressive hoarseness (laryngeal involvement) Upper airway obstruction | |||
| Gangliosidosis GM1 (Types I, II, III) | GM1-β-galactosidase | GM1 ganglioside, Keratan sulphate, oligos, glycolipids | GLB1 3p22.3 (AR) | Type I: rapidly progressive with hypotonia, severe Type II: neurodegeneration extrapyramidal signs, gait disturbance | short stature, kyphosis, and scoliosis of varying severity Cardiomyopathy | |||
| Gangliosidosis GM2, | β-Hexosaminidase A (Tay-Sachs) β-Hexosaminidase A + B (Sandhoff) | GM2 ganglioside, oligos, glycolipids | HEXA 15q23 (AR) HEXB 5q13 (AR) | Progressive weakness, loss of motor skills, decreased attentiveness, and increased startle response at 3–6 months with seizures, blindness, spasticity. In late-onset forms: progressive dystonia, spinocerebellar degeneration, motor neuron disease, and, in some individuals with adult-onset disease, a bipolar form of psychosis. | hepatosplenomegaly, coarse facial features, cardiac involvement, cherry red spot and dysostosis multiplex | |||
| Lysosomal leukodystrophies* | ||||||||
| Krabbe | β-Galactosylceramidase (GALC) | Galactosylceramide, galactosylsphingosine (psychosine) | GALC 14q31.3 (AR) | Infantile onset: progressive leukodystrophy with a classical infantile-onset associate with severe neurologic impairment and deterioration and ultimately death by two years of age Late onset GLD: neuropsychiatric disturbances, motor weakness, vision loss, and intellectual regression | Most symptoms are related to the neurological complications including dysphagia, recurrent pneumonias and multiple joint contractures | |||
| Metachromatic Leukodystrophy | Arylsufatase A (ASA) | Sulfatides | ARSA 22q13.33 (AR) | Late-infantile: motor weakness, hypotonia, clumsiness, frequent falls, toe walking and slurred speech generalized or partial seizures, hearing and visual loss and peripheral neuropathy. Late onset: neuropsychiatric and behavioral disturbances, motor weakness, spasticity and incontinence and peripheral neuropathy is common. | Gallbladder abnormalities (polyposis, wall thickening, cholelithiasis, sludge) | |||
| Multiple Sufatase Deficiency | Multiple sulfatase | Sulfatides, glycolipids, GAGs | SUFM1 3p26.1 (AR) | hypotonia, developmental regression and progressive neurodegeneration, nystagmus, dysmyelinating motor sensory neuropathy | coarse facial features, 3athepsin3aly, corneal
clouding, upper airway obstruction, dysostosis multiplex |
|||
| Olygosaccharidoses (glycoproteinoses) | ||||||||
| Galactosialidosis | Protective protein 3athepsin A (PPCA) | Sialyloligosaccharides | CTSA 20q13.12 (AR) | Hypotonia, spasticity and seizures, cognitive decline | hydrops fetalis, angiokeratomas, hepatosplenomegaly, coarse facial features, cardiomyopathy, ‘cherry red spot’ and dysostosis multiplex | |||
| Fucosidosis | α-Fucosidase | Glycoproteins, glycolipids, Fucoside-rich oligosaccharides | FUCA1 1p36.11 (AR) | seizures, cognitive impairment, seizures, spasticity and motor weakness | coarse facial features, short stature, dysostosis multiplex, angiokeratoma corporis diffusum, hepatosplenomegaly, upper airway obstruction, recurrent pneumonias | |||
| α-Mannosidosis | α-Mannosidase | Mannose-rich oligosaccharides | MANSA 19p13.2 (AR) | incoordination, ataxic gait, metabolic myopathy, and incoordination. Spastic paraplegia spasticity, rigidity, and dyskinesia slight strabismus, hydrocephalus sensorineural deafness | Facial coarseness, lumbar gibbous, hepatomegaly, and dysostosis multiplex | |||
| Sialidosis | Neuraminidase | Oligos, glycopeptides | NEU1 6p2s1.33 (AR) | developmental delay, myoclonic epilepsy, visual impairment and ataxia, generalized tonic-clonic or myoclonic seizures, progressive visual impairment along with night blindness, nystagmus | hydrops fetalis, retinal cherry-red spot, coarse facial features, hepatomegaly, dysostosis multiplex, corneal opacities | |||
Lysosomal storage diseases (LSDs) primarily associated with leukodystrophies. These LSDs are caused by lysosomal enzyme deficiencies or defected proteins whose primary substrates have a key role on myelin metabolism.
AR, autosomal recessive
Fabry disease
Fabry disease (FD), also known as Anderson-Fabry disease or angiokeratoma corporis diffusum, currently recognized as the most prevalent LSD, is caused by the deficiency of α-galactosidase A (α-Gal A), mapped to chromosome Xq22 and transmitted an X-linked trait. Affected males and female patients carry monoallelic deleterious variants in GLA gene affecting the α-Gal A enzymatic activity. The α-Gal A deficiency results in accumulation of globotriaosylceramide also known as ceramide:trihexoside (GL-3), which occurs mostly in endothelial and smooth muscle cells of the medium-and micro-vasculature, along with renal podocytes and neuronal cells of autonomic nervous systems.55, 56 The incidence of the disease is estimated to range from 1:3,800 to 1:40,000 live births.57
The clinical course is heterogeneous and may differ also within affected members of the same family who share the same genotype. So far over 800 GLA mutations have been identified.56 The disease is caused by mutations, mainly missense and nonsense, but also small and large deletions. Disease severity is related to residual α-Gal A enzymatic activity, whereas a clear genotype-phenotype correlation has not been demonstrated, given the high disease inter-and intra-familial variability. Females may develop symptoms that can be more variable in severity when compared with affected males. The pathogenesis of symptomatology in FD is still not fully understood. In histological studies, the gradual GL-3 accumulation in dorsal root ganglion neurons is postulated to result in neuronal apoptosis and ‘Wallerian’ degeneration. Another hypothesis is the continuous accumulation of GL-3 in the endothelial cells of the vasa vasorum leads to chronic nerve hypoxia and ischemia. Gb3 deposits may also interfere with cellular membrane proteins altering neuronal excitability, resulting in nerve fiber damage and death. The paresthesia is characterized by the burning or prickling pain that, usually begins in childhood and, tends to spread out from proximal to distal areas of extremities. The incidence of neuropathy is as high as 80 % in FD. Extreme ambient temperatures, including fever or freezing temperatures may induce the pain crisis. The episodic pain in FD is a severe and disabling paroxysmal pain starting in the hands and feet with centripetal radiation associated with dysautonomic symptoms lasting from hours to several days.58 Chronic pain may induce psychiatric symptoms such as mood disorders, behavioral changes, and severe depression with a substantial impact on quality-of-life. Hypohydrosis is also a common symptom.
In terms of CNS imaging studies, many patients suffering from FD already have signs of the disease earlier and before the diagnosis. The CNS findings include extensive white matter changes, also known as ‘white matter lesions’, corresponding to leukoariosis (Fig.2), primarily in the occipital areas supplied by the posterior circulation, which is associated with dilatation of the vertebrobasilar vessels up to extensive dolichoectasia.59 In addition, although such abnormalities can have several causes, the observation of high signal intensity of the pulvinar thalami on T1-weighted magnetic resonance imaging (MRI) can be found in up to one quarter of individuals diagnosed with FD (Fig.2).56, 59 In addition, Fabry disease has been identified among patients with ‘cryptogenic stroke’.60 Around 20% of patients with Fabry disease complain of auditory symptoms within the first decade of life. By the 4th to the 5th decade, over to 50–60% report auditory disturbances including tinnitus, hearing impairment and deafness. Histopathologic studies of the VII nerve of FD patients has shown reduced number of spiral ganglion cells supporting the clinical observation of impaired inner ear function.56 Autonomic nerve involvement is assumed for hypohidrosis and gastrointestinal symptoms. Biopsies of the jejunum and rectum revealed storage of glycosphingolipids in the Meissner plexus supporting autonomic nerve involvement.58 Partial corneal clouding can also be present in FD, especially male patients, but this is often not associated with any disturbance in visual acuity. Into the fifth or sixtieth decades, major complications related to cardiac, renal, and/or cerebrovascular involvement become evident.56, 59
Figure 2. Glycosphingolipidosis and white matter alterations.
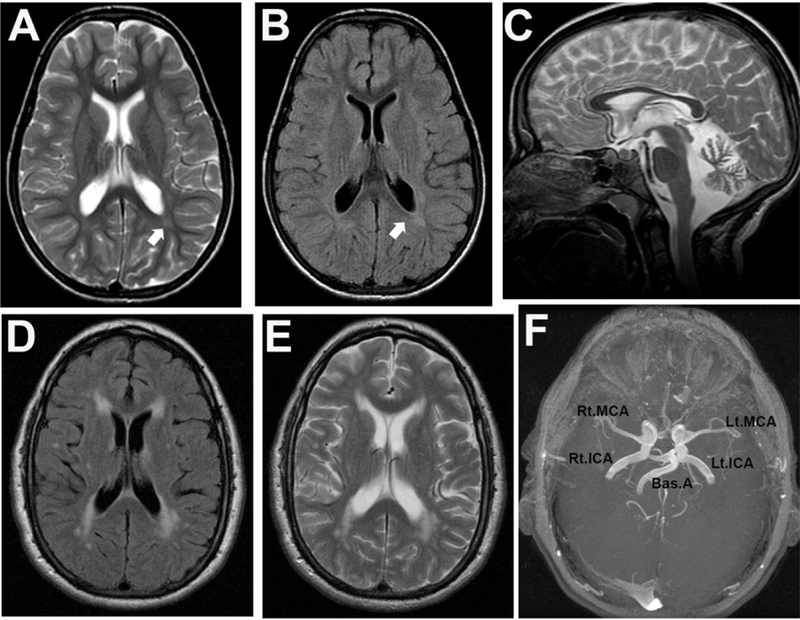
(A-C) This 8.7-year-old girl diagnosed with the juvenile clinical form of GM2 gangliosidosis (Sandhoff variant). She presented with severe cognitive dysfunction, dysarthria, ataxia, muscle wasting, urinary and fecal incontinence at baseline. Psychiatric problems included visual and auditory hallucinations and agitation. She was fully dependent for feeding, bathing, toileting and ambulation when she underwent a brain MRI. (A and B) The brain MRI transverse section showed normal ventricular size with mild FLAIR signal intensity of white matter around the posterior trigones in occipital regions (arrows) as shown in panels A (T2W_TSE) and B (FLAIR). (C) Moderate atrophy of corpus callosum is noticeable as well as moderate diffuse atrophy of cerebellum with a brighter FLAIR signal of globous pallidus. (D-F) A 50-year old man diagnosed with Fabry disease with history of chronic renal disease (stage II), hypertrophic cardiomyopathy, progressive hearing loss and chronic pain and hypohidrosis. Both transverse T2W_TSE (D) and FLAIR (E) sequences show hyperintensity signals in the frontal and occipital periventricular regions. (F) The magnetic resonance arteriography (MRA) revealed dolichoectasia with internal carotids, basilar and middle cerebral arteries as typically observed in Fabry disease. Rt.MCA, right middle cerebral artery; Lt.MCA, left middle cerebral artery; Rt.ICA, right internal carotid artery; Lt.ICA, left internal carotid artery; Bas.A, basilar artery.
Since 2001, ERT for Fabry disease has been available in the form of agalsidase alfa (Replagal®, Shire HGT, Lexington, MA) and agalsidase beta (Fabrazyme®, Genzyme-Sanofi, Cambridge, MA). Both ERT agents are administered intravenously on biweekly infusions. Currently, only agalsidase beta is available in the United States. The ERT has been shown to significantly reduce pain in patients with FD. Several studies have shown that patients with FD on ERT have significant linear decrease in left-ventricular mass index, a reduction in the risk of proteinuria after adjusting for the use of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, and there has been a small increase in eGFR in men and women without pre-treatment proteinuria. In terms of neurological manifestations of FD, long-term studies are currently showing evidence that ERT may have an impact on a variety of neurologic symptoms, including neuropathic pain, impaired sensory function (tactile/temperature), impaired sweating, and hearing impairment.61 In a double-blind, placebo-controlled trial using agalsidase alfa as ERT agent, the severity of neuropathic pain was reduced.62 In the open-labeled extension phase, the Brief Pain Inventory (BPI) significant reduction of the pain scores and improvement of the sweating were observed.63 The positive impact was also noted with another ERT agent, agalsidase beta64, and also confirmed in affected females with FD. The clinical improvement was corroborated by improvement in the function of C-, Adelta-, and Abeta-nerve fibers and intradermal vibration receptors in Fabry neuropathy.65 Longitudinal analyses during ERT in FD affected individuals demonstrate a decline of hearing loss similar to healthy controls. However, hearing loss present before initiation of therapy cannot be reversed. Whether early ERT can prevent hearing loss is unknown.66 However, ERT is unable to alter the risk of stroke and/or improve the ‘white matter lesions’ or hyperintensities observed in brain MRI.67–69 More recently, in a large cohort of patients with FD, no association between ERT and WMH progression was noted, whereas higher total cholesterol at baseline was associated with slower WMH progression, suggesting a complex and still unclear relationship between cholesterol, lowering cholesterol drugs (statins), and cerebral small vessel disease in FD.70 In terms of peripheral neuropathy, ERT may improve proximal skin innervation in patients with good renal function, but does not protect small fiber function in men with impaired renal function.71 Notably, alterations in resting cerebral blood flow was reversible under treatment with agalsidase alfa.72 Further positive effects of both enzyme preparations have been described for cardiac affection, kidney function, and gastrointestinal disturbances. More recently, migalastat (Galafold™, Amicus Therapeutics, Cranbury, NJ), a small molecule drug that assists with the folding of specific GLA mutants has been shown to enhance the residual enzymatic activity of α-galactosidase. Migalastat has been approved for the treatment of Fabry disease in the EU in patients with amenable mutations.73, 74 No significant difference was noted when outcomes were compared between the migalast treated group and those on conventional ERT regimen.74, 75
Gaucher Disease
Gaucher disease (GD) is a LSD caused glucocerebrosidase deficiency resulting in the accumulation of glucosylceramide and glucosylsphingosine. Clinically, GD comprises a continuum of clinical findings from a perinatal lethal disorder to an asymptomatic type.76, 77 Gaucher type 1 is characterized by the symptoms and signs of bone disease including osteopenia, focal lytic or sclerotic lesions, and osteonecrosis, along with hepatosplenomegaly, anemia and thrombocytopenia, lung disease, and no signs of primary CNS disease. Whereas, GD types 2 and 3 are primarily neurological LSD that are distinguished by age of onset and rate of disease progression, however, some of these parameters are not absolute.78 In Tract-Based Spatial Statistics (TBSS) analysis, white matter tracts showed decreased fraction anisotropy (FA) and increased mean diffusity (MD) in GD types 1 and 3, which were primarily in the middle cerebellar peduncles, with some differences also seen in the superior cerebellar peduncles in GD type 3.79 Elevated MD levels and decreased FA levels in white matter are indicative of poorly developed, immature, or structurally compromised white matter. Given the clinical presentation of ataxia seen in GD types 2 and 3 patients, which is not characteristically seen in GD type 1, this finding is clinically relevant and viable to be used when monitoring interventions.80 A further study using DTI supported the microstructural alterations of multiple white matter tracts in type 1 GD patients.81 More recently, cases of brain MRI studies in patients with GD type 1 have shown white matter changes. The brain MRI studies can reveal markedly abnormal patchy T2 hyperintense signal within the supra and infratentorial white and gray matter, including significant swelling and abnormal signal of the bilateral thalami. 82 In terms of therapies, GD was the first LSD to have enzyme replacement therapy (ERT) as the mainstay treatment. The first ERT agent was derived from highly purified GCase out of large pools of human placenta. Currently, there are at least four different ERT agents, administered intravenously (IV) every 2 weeks, which have been approved by regulatory agents in different countries.83, 84 None of the current ERT agents given IV are able to cross the blood-brain barrier (BBB) and, consequently no alteration of neurological symptoms in different GD clinical forms are observed. Another therapeutic modality is substrate reduction therapy (SRT), based on small molecule agents that are able to reduce the biosynthesis of the accumulated substrate of the deficient enzyme.85 The N-butyldeoxynojirimycin, miglustat, (Zavesca®) was approved as an alternative oral treatment for GD with resultant improvement in several clinical endpoints.86 However, the significant gastrointestinal symptoms (diarrhea), tremors, signs of peripheral neuronopathy and weight-loss observed in treated patients has limited its use.87 Another SRT agent, eliglustat (Cerdalga), a d-threo-(1R,2R)-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) analogue with a better safety and tolerability profile has been recently approved for GD.88–90 Unfortunately, neither SRT formulations have been shown to achieve levels in CNS capable of reducing the glycosylceramide levels and reversing the neuropathogenic cascade, accounting for the neurological symptoms in different GD clinical forms.
Other Glycosphingolipidosis
Niemann-Pick disease type C (NPC) is a LSD caused by autosomal recessively inherited mutations in two genes NPC1 and NPC2, which encode NPC1 and NPC2 proteins, respectively. These lysosomal membrane protein NPC1 and NPC2 soluble protein are involved in the intracellular cholesterol trafficking.91 The lipid accumulation is associated with the multisystemic clinical manifestations of NPC. The exact NPC1 and NPC2 protein functions are unknown.92 The mechanisms leading to abnormal storage subsequent cellular malfunction and neurodegeneration have not fully elucidated. A number of hypotheses have been proposed, including the suggestion that sphingosine storage in the endolysosomal compartment leads to impaired calcium homeostasis and plays an integral part in disease pathophysiology.93 In NPC, there is a broad spectrum of clinical presentations. Affected newborns can present with ascites and severe liver disease from infiltration of the liver and/or respiratory failure from infiltration of the lungs. Other infants, without liver or pulmonary disease, have hypotonia and developmental delay. The classic presentation occurs in the mid to late childhood with the insidious onset of ataxia, vertical supranuclear gaze palsy and dementia.92 Dystonia and seizures are common. Dysarthria and dysphagia eventually become debilitating, requiring the initiation of tube feeding. Death usually occurs in the late second or third decade from aspiration pneumonia. Adults are more likely to present with dementia or psychiatric symptoms. The diagnosis of NPC is confirmed by biochemical testing that demonstrates impaired cholesterol esterification and positive Filipin staining in cultured fibroblasts. NPC1 gene mutations are present in 95% of cases and NPC2 mutations are present in approximately 4%; the remainder of patients are biochemically proven cases who do not have identified mutations.52 More than 350 different NPC1 and NPC2 gene mutations have been reported in patients with confirmed diagnose.94 In terms of diagnostic biomarkers, after oxysterol levels noted to be elevated in NPC1 mice models, various studies have shown that plasma oxysterol levels are high in patients with NPC1 and NPC2 mutations.95, 96 The brain imaging studies of affected patients with NPC have shown signs of white matter disease. The fast-spin echo T2-weighted images in brain MRI of patients affected with NPC is characterized by more prominent sulci of both cerebral convexities, especially in the frontal lobes and more increased signal intensity in bilateral parietal–occipital periventricular white matter.97 In addition, diffusion tensor imaging (DTI) of the corpus callosum and brainstem reveal microstructural disorganization, which correlates with NPC1 severity.98 The DTI has now been used to monitor research interventions.99 The demyelination has been reported in patients with infantile-type NPC, which is beyond the extent of neuronal damage. In NPC, the mechanisms of demyelination have not been clearly defined. The GM3 accumulation reportedly disturbs oligodendrocyte development and maintenance and lipid accumulation (particularly of gangliosides) induces demyelination due to the dysfunction of oligodendrocytes.100 Demyelination can also be induced by neuronal cell loss and axonal dystrophy with spheroid formation.101, 102 In terms of therapies, N-butyldeoxynojirimycin (miglustat) (Zavesca®, Actelion Pharmaceuticals Ltd. Basel, Switzerland) is an imino-sugar that partially inhibits glucosylceramide synthase and reduces the biosynthesis of all glucosylceramide-based glycosphingolipids. This therapy modality is called substrate reduction therapy, SRT. The clinical severity composite scale based on ambulation, manipulation, language and swallowing abilities, a marker of disease severity and neurodisability status in many centers, remained stable or even improved in the majority of patients who received continuous miglustat therapy for a period of approximately 2 years.53 Miglustat, however, is not a FDA-approved medication to treat NPC. It is however approved by European Medicine Agency (EMA) and other countries.
GM1 gangliosidosis (GM1g) is a LSD caused by the deficiency of the β-galactosidase enzyme, which catalyzes the hydrolysis of terminal β-galactosyl moiety from GM1 ganglioside, glycoproteins, and glycosaminoglycans.103 The incidence of GM1 gangliosidosis has been estimated to be 1 in 100,000–200,0000 live births. Although the disorder is panethnic, higher prevalence has been noted in Brazil (1:17,000), Roma population (1:10,000), in the Maltese Islands (1:3700) and in the Cypriot population.103 The clinical manifestations of GM1g result from the progressive storage of GM1ganglioside and related glycoconjugates in different tissues and particularly in the CNS. However, the molecular mechanisms leading to the disease pathogenesis are still incompletely understood. In terms of diagnostic approach, GM1g can be classified into three types based on the clinical phenotype: GM1g type 1 or infantile form with onset between 0 −6 months, rapidly progressive with hypotonia, severe neurodegeneration and death by 1–2 years of age; Gm1 type 2 late infantile or juvenile with onset between 10–12 months and 3 years of age, delay in motor and cognitive development, and slower progression; GM1 type 3 adult or chronic variant with late-onset (3–30 years of age), a extrapyramidal signs, gait disturbance, and cardiomyopathy. The latter clinical form can be misdiagnosed as Parkinson disease. Intellectual impairment is common late in the disease. The skeletal involvement includes short stature, kyphosis, and scoliosis of varying severity. In terms of CNS imaging, brain MRI of affected patients show signs of delayed myelination and DTI abnormalities in the subcortical and periventricular regions, centrum semiovale, and internal and external capsules. Abnormal signal is frequently present in the globus pallidus and thalami bilaterally.104 In terms of therapy, only symptomatic and supportive therapy are available for patients with GM1g. Different strategies have been explored to treat GM1g and they include BMT, gene therapy approaches, and substrate reduction therapy (SRT). Allogenic BMT has been attempted in infantile GM1 gangliosidosis but it has not been successful and beneficial to patients. Imino-sugars inhibiting ganglioside biosynthesis have been investigated in rodents and have resulted in reduction of gangliosides accumulation in the CNS, suggesting that SRT may be a potentially effective early intervention therapy for GM1g. A novel strategy using pharmacological chaperones, which may be rapidly available for clinical translation, has been used such as N-octyl-4-epi-β-valienamine (NOEV).105 This small molecule has been used to stabilize the mutant β-galactosidase protein for restoration of enzyme activity. Oral treatment with NOEV in mice carrying the mutant gene resulted in marked increase of the enzyme activity, reduction of the GM1ganglioside content in CNS, preventing the neurological deterioration. In addition, SRT with miglustat showed improved in movement and speech control, the distance covered during the 6-minute walk test.106
GM2 gangliosidosis (GM2g), also known as Tay-Sachs or Sandhoff diseases, are caused by β-hexosaminidase A (Hex-A) deficiency in lysosomes, resulting in the accumulation of GM2 and other gangliosides from the neuronal cell membrane. The GM2 storage can result in morphological changes in neurons including meganeurites with ectopic dendritogenesis and axonal spheroids. In addition, the GM2, a complex polar glycosphingolipid, may alter neuron-to-neuron microconnectivity initially before neuronal death occurs. The Hex-A deficiency is caused by biallelic mutations in HEXA (Tay-Sachs disease) or HEXB genes (Sandhoff disease). The Hex-A is soluble hydrolase formed by α and β subunits encoded by HEXA and HEXB genes, respectively.107 Clinically, the phenotypes of GM2g can be categorized in 3 major forms: acute infantile with rapid progression and death by 4–5 years of age; juvenile (subacute) with later onset and survival into late childhood or adolescence; chronic and adult-onset with long-term survival. Affected individuals have several different neurological symptoms including: progressive dystonia, spinocerebellar degeneration, motor neuron disease with muscle weakness and fasciculations, and/or psychosis.108 Neuroimaging findings in TSD include cerebellar atrophy in most patients although cerebral atrophy may be present in up to a quarter of patients.109 In GM2g patients, decreased N-Acetyl Aspartate (NAA)/Creatinine (Cr) was noticeable both in the corpus striatum and occipital region including cortex and white matter, while increased myo-Inositol (mI)/Cr was shown in the latter. Serial MR images showed progressive mild brain atrophy and hyperintensity on T2-weighted images in the bilateral white matter including the centrum semiovale and optic radiation (Fig.2). Other case series have shown that cortical neuronal dysfunction, secondary axonal degeneration and myelin destruction had taken place in the occipital region, while neuronal dysfunction without myelin destruction had occurred in the corpus striatum.110 Diagnosis is based on the deficiency of Hex-A in peripheral leukocytes. In GM2g, neuropsychiatric presentations occur in up to half of late-onset TSD patients described in unselected series and the most common neuropsychiatric presentation is psychosis.108, 111–113 The rate of schizophrenia-like psychosis in late-onset patients with GM2g ranges up to 50%.108, 111–113 Catatonia may be the initial presenting feature. Mania or depression has also been described. The treatment of GM2g is mostly supportive and focus in treating the seizures and neuropsychiatric symptoms. The main obstacle for the disease-modifying therapeutic strategies is how to remove the accumulated lysosomal ganglioside once in CNS including neurons and glia. Early experimental intravenous enzyme replacement trials were unsuccessful, as the large molecular weight enzyme did not cross the BBB. Intracranial neuronal-corrective gene therapy has shown promising results although some pitfalls have been identified.114, 115 A clinical trial with SRT agents as N-deoxynojirimycin, showed no improvement pharmacologic chaperone therapy using pyrimethamine, an active-site inhibitor of Hex-A activity showed that this small molecule produces enhancement of leukocyte Hex A activity in patients with late-onset GM2 gangliosidosis at doses lower than those associated with unacceptable side effects.47 The same observation was noted in other early clinical studies.116
There are also much rarer glycosphingolipidoses associated with leukodystrophies, including Farber disease. This glycosphingolipidosis is characterized by a spectrum of clinical signs ranging from the classical triad of painful and progressively deformed joints, subcutaneous nodules, and progressive hoarseness (due to laryngeal involvement) presenting in infancy with a combination of respiratory and neurologic involvement. Farber disease is caused by mutations in the N-acylsphingosine amidohydrolase (ASAH1) gene (8p22), which encodes acid ceramidase, a lysosomal enzyme that hydrolyzes ceramide into sphingosine and free fatty acid, with resultant accumulation of ceramide in most tissues. The brain MRI studies usually show signs of deep white matter loss, diffuse brain atrophy along with demyelinating polyneuropathy in nerve conduction velocity studies.117 The diagnosis is based on clinical and laboratory findings, confirmed by assaying the activity of acid ceramidase in peripheral blood leukocytes, cultured lymphoid cells or cultured skin fibroblasts. Alternatively, diagnosis can be performed by determining ceramide concentration in cultured cells or tissues or by studying lysosomal ceramide catabolism in cultured live cells, which are usually primary patient fibroblasts.
Identification of mutations in the ASAH1 gene by molecular genetic testing usually allows for diagnostic confirmation. No disease-modifying therapy exists for Farber disease. Symptomatic treatment is based on analgesics, corticosteroid therapy, and esthetic surgery for subcutaneous nodules. However, allogeneic HSCT provides a promising approach for patients with limited neurological involvement. The prognosis varies including patients with the severe neonatal form who die within the first few days of life and, others with late onset forms living until adolescence or early adulthood. The enzyme replacement therapy has shown promising results in the mouse model for Farber disease.118
Oligosaccharidosis
Oligosaccharidosis are a group of lysosomal diseases, also called glycoproteinoses, biochemically characterized by storage of protein-bound oligosaccharides within lysosomes and excess urinary excretion of the corresponding sugars. Storage of oligosaccharides results from the absence or, the defective function of a specific lysosomal enzyme. The oligosaccharidosis that can present with involvement of the white matter as part of their symptomatology includes α-and β-mannosidoses, fucosidosis, sialidosis types I and II, and galactosialidosis. In general, the hypomyelination pattern predominates in these group of LSDs. In hypomyelination, T2-weighted signal is hypointense and milder than those observed in demyelinating disorders. In addition, the T1-weighted signal is mildly hyperintense, isointense or mildly hypointense relative to the cortex in hypomyelinating disorders as oligosaccharidosis.119 Galactosialidosis is characterized by the deficiency of β-galactosidase and α-neuraminidase with the presence of oligosaccharides in patient urine. This LSDs has been included among oligosaccharidoses, but it is better classified as a lysosomal enzyme protection defect concerning its primary defect of a non-enzymatic cathepsin A-protective protein.120 The clinical spectrum of the conditions varies widely, as is common in LSDs. Patients frequently have neurological symptoms, but, in rare cases, the clinical presentation in adulthood may be subtle. Psychiatric presentations have been described in affected adult patients. The brain MRI in galactosialidosis follows the hypomyelination pattern as above described. Other findings CNS imaging findings reported in patients include with enlarged ventricles, hyperintense white matter striato thalamic vasculopathy, widened periencephalic spaces.121 Table 1 describes the common features of galactosialidosis and other oligosaccharidoses with their significant symptoms and signs.
In terms of radiological aspects, in fucosidosis, the brain MRI findings show hypointense signal on T2-weighted images in the globus pallidi and substantia nigra. Diffuse, confluent, symmetrical hyperintensity on T2-weighted images are present in the periventricular and subcortical white matter and, also in the globo pallidi, which are consistent with hypomyelination.122–124 In α-mannosidosis, the CNS imaging studies show also evidence of reduced myelination including elevated mannose complexes, gliosis, elevated CSF-biomarkers, elevation of Cho/Cr and mI/Cr in the white matter.125 In Salla disease, typical MRI findings are hypoplasia of the corpus callosum and homogeneously high signal from cerebral white matter, due to hypomyelination, on T2-weighted sequence images of the brain. The signal from periventricular white matter may be even higher than that from peripheral white matter, as a sign of demyelination.126
Mucopolysaccharidosis
Mucopolysaccharidosis (MPSs) is a group of specific lysosomal hydrolases deficiencies, resulting in progressive accumulation of partially degraded mucopolysaccharides (glycosaminoglycans, GAG) including heparan sulfate, dermatan sulfate or keratan sulfate. Each lysosomal hydrolyase cause one specific type of MPS (Table 1). For instance, deficiency of α-L-iduronidase deficiency (MPS-I; 14p16.3) causes MPS-I also known as Hurler disease (severe subtype) and Scheie disease (attenuated subtype). Mucopolysaccharidosis account for approximately one-third of known LSDs. A significant number of these disorders have onset during infancy or early childhood. Interestingly, in early-onset forms of MPS, increased species of certain glycosphingolipids are consistently elevated despite the underlying biochemical defects being not directly related to the sphingolipid metabolism. In terms of the symptomatology, these are disabling, multisystem disorders with the severe forms resulting in shortened life expectancy. The MPS combined prevalence is approximately 1:22,000.1, 57
Patients with MPS present broad clinical manifestations, reflecting the progressive and multi-level accumulation of GAGs in several tissues and organs. Each MPS have specific and manifestations which are defined in table 1. The radiographic skeletal abnormalities in MPS are collectively labeled ‘dysostosis multiplex’, which is a term encompassing many bone abnormalities such as large skull with, thickened calvarium and prematurely fused sutures, short stature with short trunk, oar-shaped ribs, bullet-shaped phalanges and a hook-like appearance to thoracolumbar vertebrae. It is now clear that MPSs manifest a broad continuum from fatal prenatal hydrops fetalis to individuals with normal intelligence, physical appearance, and, near-normal lifespan.127 With regards to the neurological problems, MPS patients present with a wide constellation of central and peripheral nervous system manifestations. Storage of glycosaminoglycans found in the spiral ganglion and vestibulocochlear nerves may be responsible for sensorineural hearing impairment.128 Carpal tunnel syndrome is a typical manifestation of MPS patients, and its early recognition can substantially impact the outcome of the carpal tunnel surgical release.129 Intellectual disabilities vary in different types of MPS and within certain sub-types. The MPS-II, known as Hunter syndrome, is the only X-linked MPS and caused by iduronate-2-sulfatase (IDS) deficiency with similar symptomatology found in the MPSI form. The neuronopathic form is the most severe and occurs in approximately 75% of MPS-II patients. These patients present substantial neurologic involvement manifested by global delay, cognitive impairment, and even neuroregression, which occurs in the second decade of life. Patients with an attenuated phenotype may have normal intelligence, and typically survive into adulthood. In MPS-III, mostly known as Sanfilippo syndrome, a distinct neurological clinical manifestation is observed. There are four MPS-III subtypes, which are caused by different lysosomal enzyme deficiencies involved in the degradation of heparan sulfate (Table 1). In MPS-III, early development is typically normal. The developmental delay, behavioral problems, or a combination of these concerns are usually the first symptoms at variable age of onset.130 In terms of other MPS types, sensory impairment may result from a range of factors such as corneal clouding, retinopathy and mixed hearing loss and can compound developmental delay or cause regression. Cognitive impairment is not usually observed in MPS-IVA (Morquio A syndrome), and MPS-VI (Marotaux-Lamy syndrome).
The brain MRI in patients with MPS usually show signal abnormalities predominantly in the periventricular white matter.131 In addition, enlargement of typical perivascular (Virchow-Robin) spaces, caused by thickening of the leptomeninges are usually found in routine CNS imaging assessments. The GAGs accumulation in the meninges may interfere with CSF absorption via Pacchionian granulations leading to communicating hydrocephalus.132 Attempts to correlate cerebral MRI findings and cognitive impairment in patients with different MPS have failed. These lesions are denominated cribriform changes or spotty lesions considered to be the result of the abnormal enlargement of perivascular spaces. They were encountered in the corpus callosum, basal ganglia and white matter and, most frequently in severely affected patients with MPS II. They were also found in the patients with MPS I and VI. The degree of the changes are almost parallel to the severity of the disease, although diffuse perivascular lesions and white matter abnormalities can be present in patients with intact intellectual abilities and cognition. Some lesions improve after the HSCT. The NAA/Cr value by MRS of the area with spotty lesions in are normal. However, in some case series, the patchy white matter changes have been associated with MPS-I, MPS-II and MPS-VI as patchy lesions.133 These spotty white matter lesions are suggestive of gliosis or demyelination. Interestingly, in some patients who underwent HSCT, the patchy lesions seem to subside or decrease in number.133 One patient with MPS-IIIB was reported to have a diffuse hyper-intensity T2 signal in which significantly decreased NAA/Cr were noted, indicating neuronal loss.133
In terms of specific therapies, ERT is available for some affected patients with MPSs. MPS-I was the first MPS to have an ERT agent approved, laronidase (Aldurazyme, Genzyme-Sanofi Inc., Cambridge, MA). Although HSCT is still recommended for the Hurler phenotype – early and rapidly progressive form of MPS-I. The ERT agent for MPS-I consists of human α-L-iduronidase, which is administered at 0.58 mg/kg body-weight intravenously once weekly. The major outcome is the control of progressive visceromegaly, increased growth velocity, the stability of cardiac manifestations, and improved joint motion of the upper extremities.134, 135 Obstructive sleep apnea or recurrent hyponoeic episodes improved in 60% of treated patients with MPS-I. There was evidence that the ERT effects on cardiac, ophthalmological or otological involvement.134, 135 Similar outcomes were observed for other ERT agents for other MPSs, including MPS-II with idursulfase (Elaprase, Shire Lexington, MA), MPS-IV with elosulfase alfa (VIMIZIM, Biomarin Pharm. Inc., Novato, CA)136 and MPS-VI with galsulfase (Naglazyme, Biomarin Pharm. Inc., Novato, CA).137, 138 Unfortunately, ERT has not resulted in alterations of the CNS involvement, or associated with amelioration of white matter lesions, due to its large molecular-weight and inability to penetrate through the blood-brain barrier (BBB). For this reason, the outcome of ERT has been restricted to the non-neurological symptoms. Intrathecal administration of α-L-iduronidase (1.74 mg monthly) was studied in patients with MPS-I who were symptomatic for cervical spinal stenosis. No significant changes were noted in brain or cervical spine MRI. However, the study lasted only 12 months.139, 140 In terms of early onset MPS-I, patients diagnosed early when presenting with initial signs of somatic and neurological problems have significant stabilization of the developmental/intelligence quotient (DQ/IQ) if the HSCT is performed before 24-months of age.141 Among all the LSDs, MPS-I is the only LSDs in which the HSCT can alter the neurological outcome of the disease.
Conclusion and future directions
The LSDs are a group of inborn organelle disorders that affect lysosomal function and lead to multisystemic manifestations. Among the over 60 different LSDs, the neurological symptoms are predominant in at least 80%. Some of these neurological LSDs are associated with white matter alterations (Table 1). Symptoms are usually insidious and progressive, with age of onset spanning from prenatal to the elderly. Wide variability and heterogeneity of clinical presentation are observed not only among the different LSDs, but also within affected individuals who share the same pathogenic genetic variants. From a clinical standpoint, diagnosis is based on clinical findings and family history, supported by radiological and laboratory assessments. The investigations for LSDs are based on four major areas including histological and ultra-structural studies, specific biochemical enzyme assays, detection of accumulated metabolites and molecular genetic studies. The current understanding of the pathogenesis of several LSDs has allowed the development of many therapies. Currently, one of the major challenges is the access of the specific therapeutic agents to the CNS and PNS systems, which has limited the efficacy on neurological course of several LSDs. Unfortunately, some patients are late diagnosed due to the atypical neurological presentation at disease onset and/or also slow progression of neurological manifestations. Therefore, when investigating the etiology of inherited neurological disorders, white matter involvement in neuroradiological studies along with the presence of neurological and non-neurological symptoms may indicate a potential underlying inborn lysosomal disorder.
Acknowledgements
We are thankful and privileged to have follow and maintain contact with many patients and their families, who have participated over the years in our natural history studies. Their gracious participation has been allowing a better understanding of several lysosomal storage diseases.
References
- 1.Meikle PJ, Hopwood JJ, Clague AE and Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281(3): 249–54. [DOI] [PubMed] [Google Scholar]
- 2.Metz TF, Mechtler TP, Merk M, et al. Evaluation of a novel, commercially available mass spectrometry kit for newborn screening including succinylacetone without hydrazine. Clin Chim Acta 2012;413(15–16): 1259–64. [DOI] [PubMed] [Google Scholar]
- 3.Langereis EJ, Wagemans T, Kulik W, et al. A Multiplex Assay for the Diagnosis of Mucopolysaccharidoses and Mucolipidoses. PLoS One 2015;10(9): e0138622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Duve C From cytases to lysosomes. Fed Proc 1964;23: 1045–9. [PubMed] [Google Scholar]
- 5.Napolitano G and Ballabio A. TFEB at a glance. J Cell Sci 2016;129(13): 2475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang CS, Jiang B, Li M, et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab 2014;20(3): 526–40. [DOI] [PubMed] [Google Scholar]
- 7.Lloyd-Evans E and Haslett LJ. The lysosomal storage disease continuum with ageing-related neurodegenerative disease. Ageing Res Rev 2016;32: 104–21. [DOI] [PubMed] [Google Scholar]
- 8.Bielschowsky MH and Henneberg R. Uber familiare diffuse sklerose (leukodystrophia cerebri progressiva hereditaria). J Psychol Neurol (Lpz) 1928;36: 131–81. [Google Scholar]
- 9.Suzuki K Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res 1998;23(3): 251–9. [DOI] [PubMed] [Google Scholar]
- 10.Wenger DA, Rafi MA and Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat 1997;10(4): 268–79. [DOI] [PubMed] [Google Scholar]
- 11.Wenger DA, Rafi MA, Luzi P, Datto J and Costantino-Ceccarini E. Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab 2000;70(1): 1–9. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki K Globoid cell leukodystrophy (Krabbe’s disease): update. J Child Neurol 2003;18(9): 595–603. [DOI] [PubMed] [Google Scholar]
- 13.Provenzale JM, Peddi S, Kurtzberg J, Poe MD, Mukundan S and Escolar M. Correlation of neurodevelopmental features and MRI findings in infantile Krabbe’s disease. AJR Am J Roentgenol 2009;192(1): 59–65. [DOI] [PubMed] [Google Scholar]
- 14.Jones BV, Barron TF and Towfighi J. Optic nerve enlargement in Krabbe’s disease. AJNR Am J Neuroradiol 1999;20(7): 1228–31. [PMC free article] [PubMed] [Google Scholar]
- 15.Abdelhalim AN, Alberico RA, Barczykowski AL and Duffner PK. Patterns of magnetic resonance imaging abnormalities in symptomatic patients with Krabbe disease correspond to phenotype. Pediatr Neurol 2014;50(2): 127–34. [DOI] [PubMed] [Google Scholar]
- 16.Poretti A, Meoded A, Bunge M, et al. Novel diffusion tensor imaging findings in Krabbe disease. Eur J Paediatr Neurol 2014;18(2): 150–6. [DOI] [PubMed] [Google Scholar]
- 17.Wright MD, Poe MD, DeRenzo A, Haldal S and Escolar ML. Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study. Neurology 2017;89(13): 1365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffner PK, Barczykowski A, Jalal K, Yan L, Kay DM and Carter RL. Early infantile Krabbe disease: results of the World-Wide Krabbe Registry. Pediatr Neurol 2011;45(3): 141–8. [DOI] [PubMed] [Google Scholar]
- 19.Wasserstein MP, Andriola M, Arnold G, et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med 2016. [DOI] [PubMed]
- 20.Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med 2005;352(20): 2069–81. [DOI] [PubMed] [Google Scholar]
- 21.Wasserstein MP, Andriola M, Arnold G, et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med 2016;18(12): 1235–43. [DOI] [PubMed] [Google Scholar]
- 22.Wright MD, Poe MD, DeRenzo A, Haldal S and Escolar ML. Developmental outcomes of cord blood transplantation for Krabbe disease: A 15-year study. Neurology 2017. [DOI] [PMC free article] [PubMed]
- 23.Duffner PK, Caggana M, Orsini JJ, et al. Newborn screening for Krabbe disease: the New York State model. Pediatr Neurol 2009;40(4): 245–52; discussion 53–5. [DOI] [PubMed] [Google Scholar]
- 24.Orsini JJ, Kay DM, Saavedra-Matiz CA, et al. Newborn screening for Krabbe disease in New York State: the first eight years’ experience. Genet Med 2016;18(3): 239–48. [DOI] [PubMed] [Google Scholar]
- 25.Hossain MA, Otomo T, Saito S, et al. Late-onset Krabbe disease is predominant in Japan and its mutant precursor protein undergoes more effective processing than the infantile-onset form. Gene 2014;534(2): 144–54. [DOI] [PubMed] [Google Scholar]
- 26.Gieselmann V Metachromatic leukodystrophy: genetics, pathogenesis and therapeutic options. Acta Paediatr Suppl 2008;97(457): 15–21. [DOI] [PubMed] [Google Scholar]
- 27.Conzelmann E and Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci 1983;6(1): 58–71. [DOI] [PubMed] [Google Scholar]
- 28.Leinekugel P, Michel S, Conzelmann E and Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet 1992;88(5): 513–23. [DOI] [PubMed] [Google Scholar]
- 29.Gieselmann V, Fluharty AL, Tonnesen T and Von Figura K. Mutations in the arylsulfatase A pseudodeficiency allele causing metachromatic leukodystrophy. Am J Hum Genet 1991;49(2): 407–13. [PMC free article] [PubMed] [Google Scholar]
- 30.Martin HR, Poe MD, Provenzale JM, Kurtzberg J, Mendizabal A and Escolar ML. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant 2013;19(4): 616–24. [DOI] [PubMed] [Google Scholar]
- 31.Mahmood A, Berry J, Wenger DA, et al. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. J Child Neurol 2010;25(5): 572–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groeschel S, Kuhl JS, Bley AE, et al. Long-term Outcome of Allogeneic Hematopoietic Stem Cell Transplantation in Patients With Juvenile Metachromatic Leukodystrophy Compared With Nontransplanted Control Patients. JAMA Neurol 2016;73(9): 1133–40. [DOI] [PubMed] [Google Scholar]
- 33.Krageloh-Mann I, Groeschel S, Kehrer C, et al. Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. Bone Marrow Transplant 2013;48(3): 369–75. [DOI] [PubMed] [Google Scholar]
- 34.Groeschel S, Kehrer C, Engel C, et al. Metachromatic leukodystrophy: natural course of cerebral MRI changes in relation to clinical course. J Inherit Metab Dis 2011;34(5): 1095–102. [DOI] [PubMed] [Google Scholar]
- 35.Eichler F, Grodd W, Grant E, et al. Metachromatic leukodystrophy: a scoring system for brain MR imaging observations. AJNR Am J Neuroradiol 2009;30(10): 1893–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biffi A, De Palma M, Quattrini A, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest 2004;113(8): 1118–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biffi A, Capotondo A, Fasano S, et al. Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J Clin Invest 2006;116(11): 3070–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016;388(10043): 476–87. [DOI] [PubMed] [Google Scholar]
- 39.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013;341(6148): 1233158. [DOI] [PubMed] [Google Scholar]
- 40.Patil SA and Maegawa GH. Developing therapeutic approaches for metachromatic leukodystrophy. Drug Des Devel Ther 2013;7: 729–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dierks T, Dickmanns A, Preusser-Kunze A, et al. Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 2005;121(4): 541–52. [DOI] [PubMed] [Google Scholar]
- 42.Dierks T, Schmidt B, Borissenko LV, et al. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003;113(4): 435–44. [DOI] [PubMed] [Google Scholar]
- 43.Busche A, Hennermann JB, Burger F, et al. Neonatal manifestation of multiple sulfatase deficiency. Eur J Pediatr 2009;168(8): 969–73. [DOI] [PubMed] [Google Scholar]
- 44.Sabourdy F, Mourey L, Le Trionnaire E, et al. Natural disease history and characterisation of SUMF1 molecular defects in ten unrelated patients with multiple sulfatase deficiency. Orphanet J Rare Dis 2015;10: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dierks T, Schlotawa L, Frese MA, Radhakrishnan K, von Figura K and Schmidt B. Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann-Pick C1 disease -Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochim Biophys Acta 2009;1793(4): 710–25. [DOI] [PubMed] [Google Scholar]
- 46.Barritt AW, Anderson SJ, Leigh PN and Ridha BH. Late-onset Tay-Sachs disease. Pract Neurol 2017;17(5): 396–9. [DOI] [PubMed] [Google Scholar]
- 47.Clarke JT, Mahuran DJ, Sathe S, et al. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol Genet Metab 2011;102(1): 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deodato F, Procopio E, Rampazzo A, et al. The treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression. Metab Brain Dis 2017;32(5): 1529–36. [DOI] [PubMed] [Google Scholar]
- 49.Kannebley JS, Silveira-Moriyama L, Bastos LO and Steiner CE. Clinical Findings and Natural History in Ten Unrelated Families with Juvenile and Adult GM1 Gangliosidosis. JIMD Rep 2015;24: 115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McGovern MM, Dionisi-Vici C, Giugliani R, et al. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet Med 2017;19(9): 967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGovern MM, Wasserstein MP, Kirmse B, et al. Novel first-dose adverse drug reactions during a phase I trial of olipudase alfa (recombinant human acid sphingomyelinase) in adults with Niemann-Pick disease type B (acid sphingomyelinase deficiency). Genet Med 2016;18(1): 34–40. [DOI] [PubMed] [Google Scholar]
- 52.Patterson MC, Hendriksz CJ, Walterfang M, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 2012;106(3): 330–44. [DOI] [PubMed] [Google Scholar]
- 53.Patterson MC, Mengel E, Vanier MT, et al. Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: an observational cohort study. Orphanet J Rare Dis 2015;10: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thurberg BL, Wasserstein MP, Jones SA, Schiano TD, Cox GF and Puga AC. Clearance of Hepatic Sphingomyelin by Olipudase Alfa Is Associated With Improvement in Lipid Profiles in Acid Sphingomyelinase Deficiency. Am J Surg Pathol 2016;40(9): 1232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clarke JT. Narrative review: Fabry disease. Ann Intern Med 2007;146(6): 425–33. [DOI] [PubMed] [Google Scholar]
- 56.Schiffmann R Fabry disease. Handb Clin Neurol 2015;132: 231–48. [DOI] [PubMed] [Google Scholar]
- 57.Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet 2012;379(9813): 335–41. [DOI] [PubMed] [Google Scholar]
- 58.Uceyler N, Ganendiran S, Kramer D and Sommer C. Characterization of pain in fabry disease. Clin J Pain 2014;30(10): 915–20. [DOI] [PubMed] [Google Scholar]
- 59.Kolodny E, Fellgiebel A, Hilz MJ, et al. Cerebrovascular involvement in Fabry disease: current status of knowledge. Stroke 2015;46(1): 302–13. [DOI] [PubMed] [Google Scholar]
- 60.Rolfs A, Fazekas F, Grittner U, et al. Acute cerebrovascular disease in the young: the Stroke in Young Fabry Patients study. Stroke 2013;44(2): 340–9. [DOI] [PubMed] [Google Scholar]
- 61.Beck M, Hughes D, Kampmann C, et al. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis. Mol Genet Metab Rep 2015;3: 21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001;285(21): 2743–9. [DOI] [PubMed] [Google Scholar]
- 63.Schiffmann R, Floeter MK, Dambrosia JM, et al. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve 2003;28(6): 703–10. [DOI] [PubMed] [Google Scholar]
- 64.Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med 2001;345(1): 9–16. [DOI] [PubMed] [Google Scholar]
- 65.Hilz MJ, Brys M, Marthol H, Stemper B and Dutsch M. Enzyme replacement therapy improves function of C-, Adelta-, and Abeta-nerve fibers in Fabry neuropathy. Neurology 2004;62(7): 1066–72. [DOI] [PubMed] [Google Scholar]
- 66.Suntjens EB, Smid BE, Biegstraaten M, Dreschler WA, Hollak CE and Linthorst GE. Hearing loss in adult patients with Fabry disease treated with enzyme replacement therapy. J Inherit Metab Dis 2015;38(2): 351–8. [DOI] [PubMed] [Google Scholar]
- 67.Beck M, Hughes D, Kampmann C, et al. Long-term outcomes with agalsidase alfa enzyme replacement therapy: Analysis using deconstructed composite events. Mol Genet Metab Rep 2018;14: 31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alfadhel M and Sirrs S. Enzyme replacement therapy for Fabry disease: some answers but more questions. Ther Clin Risk Manag 2011;7: 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MG and Hollak CE. Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis 2013;8: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stefaniak JD, Parkes LM, Parry-Jones AR, et al. Enzyme replacement therapy and white matter hyperintensity progression in Fabry disease. Neurology 2018. [DOI] [PMC free article] [PubMed]
- 71.Uceyler N, He L, Schonfeld D, et al. Small fibers in Fabry disease: baseline and follow-up data under enzyme replacement therapy. J Peripher Nerv Syst 2011;16(4): 304–14. [DOI] [PubMed] [Google Scholar]
- 72.Bersano A, Lanfranconi S, Valcarenghi C, Bresolin N, Micieli G and Baron P. Neurological features of Fabry disease: clinical, pathophysiological aspects and therapy. Acta Neurol Scand 2012;126(2): 77–97. [DOI] [PubMed] [Google Scholar]
- 73.Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med 2016. [DOI] [PMC free article] [PubMed]
- 74.Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med 2016;375(6): 545–55. [DOI] [PubMed] [Google Scholar]
- 75.Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet 2017;54(4): 288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grabowski GA, Zimran A and Ida H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol 2015;90 Suppl 1: S12–8. [DOI] [PubMed] [Google Scholar]
- 77.Grabowski GA. Recent clinical progress in Gaucher disease. Curr Opin Pediatr 2005;17(4): 519–24. [DOI] [PubMed] [Google Scholar]
- 78.Weiss K, Gonzalez AN, Lopez G, Pedoeim L, Groden C and Sidransky E. The clinical management of Type 2 Gaucher disease. Mol Genet Metab 2015;114(2): 110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davies EH, Seunarine KK, Banks T, Clark CA and Vellodi A. Brain white matter abnormalities in paediatric Gaucher Type I and Type III using diffusion tensor imaging. J Inherit Metab Dis 2011;34(2): 549–53. [DOI] [PubMed] [Google Scholar]
- 80.Davies EH, Surtees R, DeVile C, Schoon I and Vellodi A. A severity scoring tool to assess the neurological features of neuronopathic Gaucher disease. J Inherit Metab Dis 2007;30(5): 768–82. [DOI] [PubMed] [Google Scholar]
- 81.Kang H, Zhang M, Ouyang M, et al. Brain white matter microstructural alterations in children of type I Gaucher disease characterized with diffusion tensor MR imaging. Eur J Radiol 2018;102: 22–9. [DOI] [PubMed] [Google Scholar]
- 82.Long V, Soares B and Shankar S. Baffling brain MRI findings in a patient with Gaucher disease. Molecular Genetics and Metabolism 2016;117 (2): S74–S5. [Google Scholar]
- 83.Hollak CE and Weinreb NJ. The attenuated/late onset lysosomal storage disorders: Therapeutic goals and indications for enzyme replacement treatment in Gaucher and Fabry disease. Best Pract Res Clin Endocrinol Metab 2015;29(2): 205–18. [DOI] [PubMed] [Google Scholar]
- 84.Hollak CE and Wijburg FA. Treatment of lysosomal storage disorders: successes and challenges. J Inherit Metab Dis 2014;37(4): 587–98. [DOI] [PubMed] [Google Scholar]
- 85.Vairo F, Netto C, Dorneles A, et al. Enzyme Replacement Therapy in a Patient with Gaucher Disease Type III: A Paradigmatic Case Showing Severe Adverse Reactions Started a Long Time After the Beginning of Treatment. JIMD Rep 2013;11: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cox TM. Substrate reduction therapy for lysosomal storage diseases. Acta Paediatr Suppl 2005;94(447): 69–75; discussion 57. [DOI] [PubMed] [Google Scholar]
- 87.Machaczka M, Hast R, Dahlman I, et al. Substrate reduction therapy with miglustat for type 1 Gaucher disease: a retrospective analysis from a single institution. Ups J Med Sci 2012;117(1): 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Belmatoug N, Di Rocco M, Fraga C, et al. Management and monitoring recommendations for the use of eliglustat in adults with type 1 Gaucher disease in Europe. Eur J Intern Med 2016. [DOI] [PubMed]
- 89.Cox TM, Drelichman G, Cravo R, et al. Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet 2015;385(9985): 2355–62. [DOI] [PubMed] [Google Scholar]
- 90.Ode T, Podyma-Inoue KA, Terasawa K, et al. PDMP, a ceramide analogue, acts as an inhibitor of mTORC1 by inducing its translocation from lysosome to endoplasmic reticulum. Exp Cell Res 2017;350(1): 103–14. [DOI] [PubMed] [Google Scholar]
- 91.Pentchev PG. Niemann-Pick C research from mouse to gene. Biochim Biophys Acta 2004;1685(1–3): 3–7. [DOI] [PubMed] [Google Scholar]
- 92.Vanier MT. Complex lipid trafficking in Niemann-Pick disease type C. J Inherit Metab Dis 2015;38(1): 187–99. [DOI] [PubMed] [Google Scholar]
- 93.Lloyd-Evans E, Morgan AJ, He X, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 2008;14(11): 1247–55. [DOI] [PubMed] [Google Scholar]
- 94.McKay Bounford K and Gissen P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C. J Neurol 2014;261 Suppl 2: S569–75. [DOI] [PMC free article] [PubMed]
- 95.Reunert J, Fobker M, Kannenberg F, et al. Rapid Diagnosis of 83 Patients with Niemann Pick Type C Disease and Related Cholesterol Transport Disorders by Cholestantriol Screening. EBioMedicine 2016;4: 170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reunert J, Lotz-Havla AS, Polo G, et al. Niemann-Pick Type C-2 Disease: Identification by Analysis of Plasma Cholestane-3beta,5alpha,6beta-Triol and Further Insight into the Clinical Phenotype. JIMD Rep 2015;23: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang JY, Peng SF, Yang CC, Yen KY, Tzen KY and Yen RF. Neuroimaging findings in a brain with Niemann-Pick type C disease. J Formos Med Assoc 2011;110(8): 537–42. [DOI] [PubMed] [Google Scholar]
- 98.Lau MW, Lee RW, Miyamoto R, et al. Role of Diffusion Tensor Imaging in Prognostication and Treatment Monitoring in Niemann-Pick Disease Type C1. Diseases 2016;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ory DS, Ottinger EA, Farhat NY, et al. Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial. Lancet 2017;390(10104): 1758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee H, Lee JK, Bae YC, et al. Inhibition of GM3 synthase attenuates neuropathology of Niemann-Pick disease Type C. by affecting sphingolipid metabolism. Mol Cells 2014;37(2): 161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ohara S, Ukita Y, Ninomiya H and Ohno K. Degeneration of cholecystokinin-immunoreactive afferents to the VPL thalamus in a mouse model of Niemann-Pick disease type C. Brain Res 2004;1022(1–2): 244–6. [DOI] [PubMed] [Google Scholar]
- 102.Ohara S, Ukita Y, Ninomiya H and Ohno K. Axonal dystrophy of dorsal root ganglion sensory neurons in a mouse model of Niemann-Pick disease type C. Exp Neurol 2004;187(2): 289–98. [DOI] [PubMed] [Google Scholar]
- 103.Brunetti-Pierri N and Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab 2008;94(4): 391–6. [DOI] [PubMed] [Google Scholar]
- 104.Tuteja M, Bidchol AM, Girisha KM and Phadke S. White matter changes in GM1 gangliosidosis. Indian Pediatr 2015;52(2): 155–6. [DOI] [PubMed] [Google Scholar]
- 105.Hossain MA, Higaki K, Shinpo M, et al. Chemical chaperone treatment for galactosialidosis: Effect of NOEV on beta-galactosidase activities in fibroblasts. Brain Dev 2016;38(2): 175–80. [DOI] [PubMed] [Google Scholar]
- 106.Deodato F, Procopio E, Rampazzo A, et al. The treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression. Metab Brain Dis 2017. [DOI] [PubMed]
- 107.Tropak MB and Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase A activity in GM2 gangliosidosis cells. FEBS J 2007;274(19): 4951–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maegawa GH, Stockley T, Tropak M, et al. The natural history of juvenile or subacute GM2 gangliosidosis: 21 new cases and literature review of 134 previously reported. Pediatrics 2006;118(5): e1550–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Frey LC, Ringel SP and Filley CM. The natural history of cognitive dysfunction in late-onset GM2 gangliosidosis. Arch Neurol 2005;62(6): 989–94. [DOI] [PubMed] [Google Scholar]
- 110.Imamura A, Miyajima H, Ito R and Orii KO. Serial MR imaging and 1H-MR spectroscopy in monozygotic twins with Tay-Sachs disease. Neuropediatrics 2008;39(5): 259–63. [DOI] [PubMed] [Google Scholar]
- 111.Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004;34(3): 236–42. [DOI] [PubMed] [Google Scholar]
- 112.Navon R Late-onset GM2 gangliosidosis and other hexosaminidase mutations among Jews. Adv Genet 2001;44: 185–97. [DOI] [PubMed] [Google Scholar]
- 113.Neudorfer O and Kolodny EH. Late-onset Tay-Sachs disease. Isr Med Assoc J 2004;6(2): 107–11. [PubMed] [Google Scholar]
- 114.Golebiowski D, van der Bom IMJ, Kwon CS, et al. Direct Intracranial Injection of AAVrh8 Encoding Monkey beta-N-Acetylhexosaminidase Causes Neurotoxicity in the Primate Brain. Hum Gene Ther 2017;28(6): 510–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bradbury AM, Cochran JN, McCurdy VJ, et al. Therapeutic response in feline sandhoff disease despite immunity to intracranial gene therapy. Mol Ther 2013;21(7): 1306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Osher E, Fattal-Valevski A, Sagie L, et al. Pyrimethamine increases beta-hexosaminidase A activity in patients with Late Onset Tay Sachs. Mol Genet Metab 2011;102(3): 356–63. [DOI] [PubMed] [Google Scholar]
- 117.Chedrawi AK, Al-Hassnan ZN, Al-Muhaizea M, et al. Novel V97G ASAH1 mutation found in Farber disease patients: unique appearance of the disease with an intermediate severity, and marked early involvement of central and peripheral nervous system. Brain Dev 2012;34(5): 400–4. [DOI] [PubMed] [Google Scholar]
- 118.He X, Dworski S, Zhu C, et al. Enzyme replacement therapy for Farber disease: Proof-of-concept studies in cells and mice. BBA Clin 2017;7: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schiffmann R and van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72(8): 750–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Takiguchi K, Itoh K, Shimmoto M, Ozand PT, Doi H and Sakuraba H. Structural and functional study of K453E mutant protective protein/cathepsin A causing the late infantile form of galactosialidosis. J Hum Genet 2000;45(4): 200–6. [DOI] [PubMed] [Google Scholar]
- 121.Caciotti A, Catarzi S, Tonin R, et al. Galactosialidosis: review and analysis of CTSA gene mutations. Orphanet J Rare Dis 2013;8: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oner AY, Cansu A, Akpek S and Serdaroglu A. Fucosidosis: MRI and MRS findings. Pediatr Radiol 2007;37(10): 1050–2. [DOI] [PubMed] [Google Scholar]
- 123.Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133(10): 2971–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jain P, Sharma S, Kumar A and Aneja S. Hypomyelination with T2-hypointense globi pallidi in a child with fucosidosis. J Child Neurol 2014;29(7): 988–9. [DOI] [PubMed] [Google Scholar]
- 125.Borgwardt L, Danielsen ER, Thomsen C, et al. Alpha-mannosidosis: characterization of CNS pathology and correlation between CNS pathology and cognitive function. Clin Genet 2015. [DOI] [PubMed]
- 126.Linnankivi T, Lonnqvist T and Autti T. A case of Salla disease with involvement of the cerebellar white matter. Neuroradiology 2003;45(2): 107–9. [DOI] [PubMed] [Google Scholar]
- 127.Wraith JE. Mucopolysaccharidoses and mucolipidoses. Handb Clin Neurol 2013;113: 1723–9. [DOI] [PubMed] [Google Scholar]
- 128.Lin HY, Shih SC, Chuang CK, et al. Assessment of hearing loss by pure-tone audiometry in patients with mucopolysaccharidoses. Mol Genet Metab 2014;111(4): 533–8. [DOI] [PubMed] [Google Scholar]
- 129.Baumer T, Buhring N, Schelle T, Munchau A and Muschol N. Nerve ultrasound in clinical management of carpal tunnel syndrome in mucopolysaccharidosis. Dev Med Child Neurol 2016;58(11): 1172–9. [DOI] [PubMed] [Google Scholar]
- 130.Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ and Wijburg FA. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 2008;31(2): 240–52. [DOI] [PubMed] [Google Scholar]
- 131.Pastores GM and Maegawa GH. Clinical neurogenetics: neuropathic lysosomal storage disorders. Neurol Clin 2013;31(4): 1051–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muenzer J, Wraith JE, Clarke LA, International Consensus Panel on M and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123(1): 19–29. [DOI] [PubMed] [Google Scholar]
- 133.Seto T, Kono K, Morimoto K, et al. Brain magnetic resonance imaging in 23 patients with mucopolysaccharidoses and the effect of bone marrow transplantation. Ann Neurol 2001;50(1): 79–92. [DOI] [PubMed] [Google Scholar]
- 134.Laraway S, Mercer J, Jameson E, Ashworth J, Hensman P and Jones SA. Outcomes of Long-Term Treatment with Laronidase in Patients with Mucopolysaccharidosis Type I. J Pediatr 2016;178: 219–26 e1. [DOI] [PubMed] [Google Scholar]
- 135.Kunin-Batson AS, Shapiro EG, Rudser KD, et al. Long-Term Cognitive and Functional Outcomes in Children with Mucopolysaccharidosis (MPS)-IH (Hurler Syndrome) Treated with Hematopoietic Cell Transplantation. JIMD Rep 2016;29: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hendriksz CJ, Burton B, Fleming TR, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis 2014;37(6): 979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)−−10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A 2014;164A(8): 1953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Harmatz PR, Garcia P, Guffon N, et al. Galsulfase (Naglazyme(R)) therapy in infants with mucopolysaccharidosis VI. J Inherit Metab Dis 2014;37(2): 277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Dickson PI, Kaitila I, Harmatz P, et al. Data from subjects receiving intrathecal laronidase for cervical spinal stenosis due to mucopolysaccharidosis type I. Data Brief 2015;5: 71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dickson PI, Kaitila I, Harmatz P, et al. Safety of laronidase delivered into the spinal canal for treatment of cervical stenosis in mucopolysaccharidosis I. Mol Genet Metab 2015;116(1–2): 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood 2015;125(13): 2164–72. [DOI] [PubMed] [Google Scholar]