

Summary
Plasmodium sporozoites inoculated by mosquitoes migrate to the liver and infect hepatocytes prior to release of merozoites that initiate symptomatic blood-stage malaria. Plasmodium parasites are thought to be restricted to hepatocytes throughout this obligate liver-stage of development, and how liver-stage expressed antigens prime productive CD8 T cells responses remain unknown. We found that a subset of liver-infiltrating monocyte-derived CD11c+ cells co-expressing F4/80, CD103, CD207 and CSF1R, acquired parasites during the liver-stage of malaria, but only after initial hepatocyte infection. These CD11c+ cells found in the infected liver and liver-draining lymph nodes exhibited transcriptionally and phenotypically enhanced antigen-presentation functions; and primed protective CD8 T cell responses against Plasmodium liver-stage restricted antigens. Our findings highlight a previously unrecognized aspect of Plasmodium biology and uncover the fundamental mechanism by which CD8 T cell responses are primed against liver-stage malaria antigens.
Graphical Abstract
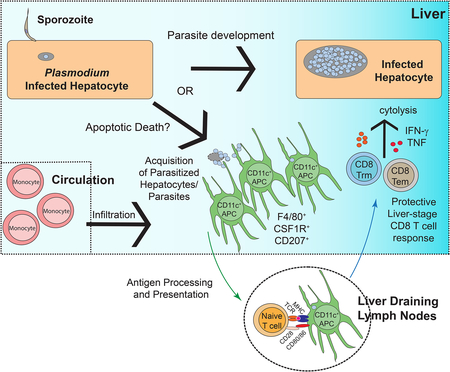
There is an urgent need for effective vaccine strategies to protect against malaria. Kurup et al. describe the fundamental mechanisms by which protective CD8 T cell responses are elicited against the liver-stage of malaria, possibly helping to refine the rational design of anti-malarial vaccines.
Introduction
Malaria is a global health burden, with an estimated 217,000,000 infections and 435,000 fatalities in 2017 (WHO, 2018). Infected mosquitoes deliver Plasmodium sporozoites into the skin, and these sporozoites travel through the blood-stream to the liver where they infect individual hepatocytes. Upon hepatocyte entry, the liver-stage of malaria ensues with the replication and differentiation of Plasmodium sporozoites into merozoites, which are later released from hepatocytes to initiate the symptomatic blood-stages of malaria (Crompton et al., 2014).
Plasmodium sporozoites delivered by mosquito bites interact with three distinct host cell populations: CD11c+ antigen-presenting cells (APC) in the skin draining lymph nodes (dLN), hepatocytes and Kupffer cells. A fraction of sporozoites inoculated into the skin using a needle, or during mosquito bite do not enter the circulation and are acquired and processed by resident CD11c+ APCs within the skin dLN (Chakravarty et al., 2007; Radtke et al., 2015). Acquisition of parasites by dLN CD11c+ APC does not result in productive infection; however, it helps prime protective CD8 T cells against sporozoite expressed antigens such as the circumsporozoite protein (CSP). Indeed, CD11c+ APC are also required to prime protective CD8 T cell responses against radiation attenuated sporozoites (RAS) delivered into the blood-stream (Jung et al., 2002).
A fraction of skin-deposited sporozoites reach the liver where they infect hepatocytes, directly or after traversal through Kupffer cells. Hepatocyte infection is obligatory for replication and differentiation of parasites into merozoites capable of infecting red blood cells and initiating the symptomatic blood-stages of infection (Cowman et al., 2016). Parasites undergo developmental changes in infected hepatocytes resulting in substantial alterations in protein expression during Plasmodium liver-stage infection, generating liver-stage-specific antigens that could potentially serve as targets of protective immune responses (Holz et al., 2016). RAS, which infect hepatocytes but cannot complete differentiation to merozoites, are capable of eliciting responses in both humans and mice that prevent the parasite from completing liver-stage differentiation and CD8 T cells are major mediators of RAS-induced immunity in mouse models (Holz et al., 2016; Seder et al., 2013). Genetically attenuated Plasmodium parasites (GAP), tailored to arrest late during hepatocyte infection, induce better protective immunity and evoke larger Plasmodium-specific CD8 T cell responses than RAS or early arresting GAP without modulating CD8 T cell responses directed at the sporozoite stage (Butler et al., 2011; Vaughan and Kappe, 2017). These data strongly suggest that protective CD8 T cells can be primed not only against sporozoite antigens, but also against antigens expressed only after hepatocyte infection. Consistent with this, Plasmodium parasites that complete full liver-stage replication and development but are eliminated after a single round of blood-stage infection by chloroquine pretreatment induce sterilizing protection to rechallenge (Bijker et al., 2013). Together, these data suggest that the capacity of whole parasite vaccines to carry out most or all of liver-stage differentiation will be an important characteristic to induce sterilizing immunity to malaria mediated by the broadest array of protective CD8 T cells.
The host-pathogen interactions that help prime CD8 T cell responses against Plasmodium antigens expressed after hepatocyte infection remain unknown; and present a conundrum, since expression of liver-stage antigens is expected to be largely constrained to developing Plasmodium in the hepatocytes that are not professional antigen presenting cells. It remains unknown how antigens expressed by Plasmodium exclusively during the infection of hepatocytes prime productive CD8 T cells responses. The anatomical site for priming these CD8 T cell responses also remains unknown.
Here, we show that a subset of liver-infiltrating monocyte-derived CD11c+ cells with a distinct phenotypic and transcriptional signature acquired Plasmodium parasites after hepatocyte infection but prior to merozoite release. These cells co-express F4/80, CSF1R, CD207 and CD103 and primed protective CD8 T cell responses in liver-draining lymph nodes. These data reveal aspects of host-parasite interactions in the liver that help resolve how and where liver-stage specific malaria antigens are accessed by APC to prime CD8 T cell responses. Collectively, our results describe the cellular mechanism by which CD8 T cells are primed against the developmentally essential liver-stage of malaria.
Results
CD11c+ cells invade the liver, harbor Plasmodium during liver-stage malaria.
Immunization using late-liver-stage arresting GAP induced superior protective immunity over early arresting GAP (Butler et al., 2011), suggesting that antigens expressed during the liver-stage of malaria prime CD8 T cell responses and serve as target antigens for protective immunity. The immunological pathways leading to CD8 T cell responses against Plasmodium liver-stage antigens remain undefined. To gain insights into the cellular dynamics in the liver after Plasmodium infection, we infected C57BL/6 (B6) mice with sporozoites of P. yoelii (Py) via intravenous (i.v.) injection and analyzed the myeloid compartments at 36–40 hours post infection (p.i.), a time point where blood-stage merozoites have not yet been released from infected hepatocytes (Prado et al., 2015). Based on traditionally used phenotypic markers, the total numbers of Kupffer cells (CD45+ CD11c−MHC-II+F4/80+CD11bintCX3CR1lo-int), macrophages (CD45+CD11c−F4/80+ CD11bhiCX3CR1hi) and monocytes (CD45+MHC-IIintCD11c−F4/80−Ly6C+) extracted from Py infected livers remained unchanged. However, as previously shown (Parmar et al., 2018), neutrophils (CD45+Ly6G+MHC-IIlo-int) and CD11c+ dendritic cells (CD45+CD11c+CD11bint-hiMHC-II+) were increased in Py infected livers by 36–40 hours p.i. (Figure 1A). Consistent with this, the density of YFP+ cells in B6.CD11c-YFP reporter mice (Lindquist et al., 2004) was increased in Py infected livers as determined by in situ 2-photon microscopy of the parenchyma, below the liver-capsule (Figure 1B). These data suggested a potentially relevant influx of CD11c+ cells into the liver after Plasmodium infection.
Figure 1: CD11c+ cells are recruited to liver after Plasmodium infection and harbor parasites.

(A) Total number of indicated myeloid cell populations in naïve or 36h Py infected livers of B6 mice.
(B) Left panels are representative, pseudo-colored multiphoton live-microscopy images showing CD11c+ DCs in naïve or 36h Py infected B6.CD11c-YFP livers. Panel on the right summarizes the density of CD11c+ cells. Each dot represents a microscopic field.
(C) Relative parasite loads in the hepatocyte and non-hepatocyte fractions from 36–44h Py infected B6 mice livers. Each dot represents one mouse.
(D) t-SNE plot displaying the clustering and distribution of indicated myeloid cell populations in Py-GFP infected B6 mice livers. Data analyzed by flow-cytometry and concatenated from >2x105 non-hepatocytes, gated on CD45+ cells. Colors correspond to automated clustering of distinct cell populations based on the indicated markers, in the left and middle panels. Portion of the maps identified with arrowhead indicates Py-GFP distribution. Panel on the right indicates relative expression of the designated markers in Py-GFP+ CD45+ cells in the liver.
(E) Representative, pseudo-colored multiphoton live-microscopy images showing hepatic CD11c+ cells (green) containing Py-GFP (red) in B6.CD11c-YFP mice; indicated by arrows. Qdot 705 staining (white) indicates hepatic sinusoids. All data depict one of 2–5 independent experiments, with at least 3 mice or samples/group, and are presented as mean± s.e.m. Statistical analyses with t-tests with Welch corrections. (*) p ≤ 0.05, (**) p ≤ 0.01, (n.s) p > 0.05.
Hepatocyte invasion, colonization and replication by Plasmodium are the hallmarks of their liver-stage of development. Most, if not all of the developing Plasmodium during the entirety of liver-stage of malaria are thought to reside in hepatocytes (Cowman et al., 2016). To investigate the mechanism of CD8 T cell priming in liver-stage malaria, we determined whether parasites could be detected in infiltrating myeloid cells. Disrupted livers were separated at ~40 hours p.i., a time point prior to merozoite release (Prado et al., 2015), into hepatocyte and non-hepatocyte fractions (Austin et al., 2014) and Plasmodium 18s rRNA was quantified by qRT-PCR (Arreaza et al., 1991; Doll et al., 2016). In contrast to the prevailing paradigm, the relative parasite burdens were unexpectedly higher within the non-hepatocyte fraction after Py (Figure 1C) or P. berghei (Pb, data not shown) sporozoite infections. To identify and phenotypically define the Plasmodium containing non-hepatocyte cells, B6 mice were infected with Py sporozoites expressing GFP (Py-GFP), delivered by mosquito bites. We observed a distinct population of CD11c+ cells that co-expressed CD11b, F4/80 and Ly6C and harbored Py-GFP parasites (Figure 1D, arrows). To rule out the possibility that the CD11c+ cells acquired parasites released from dying hepatocytes during extraction, we evaluated CD11c-YFP mouse livers by 2-photon intravital microscopy after infection with Py-GFP parasites. CD11c-YFP+ cells containing Py were observed in infected livers after mosquito bite delivery or direct needle-injection of Py-GFP sporozoites (Figure 1E, Figure 2A, Movie S1, S2), indicating that parasite acquisition occurred in situ. We observed 5.79 (±1.66) % of CD11c cells in the livers of B6 mice to harbor Py-GFP when inoculated with virulent sporozoites. Additionally, Plasmodium was found in hepatic CD11c+ cells after infection with RAS that cannot complete liver-stage development, ruling out uptake of released merozoites (Figure 2C). Thus, a substantial fraction of Plasmodium can be found in hepatic CD11c+ cells at 36–40 hours post sporozoite inoculation.
Figure 2: Hepatocyte infection is a prerequisite for CD11c+ cells harboring Plasmodium in liver-stage malaria.

(A) Representative, pseudo-colored multiphoton live-microscopy image showing 42h Py-GFP mosquito bite infected liver of B6.CD11c-YFP mice. Arrowhead marks CD11c+ cells harboring Py-GFP (see movies S1, S2).
(B) Representative, pseudo-colored multiphoton live-microscopy images showing livers of CD11c-YFP=>B6.CD81−/− bone marrow chimeric mice 42h p.i with Py-GFP (left) or Pb-GFP (right) spz. Qdot705 staining indicates hepatic sinusoids. Arrowheads in the right panel indicate CD11c+ cells harboring Pb-GFP. Data represents one of at least 10 separate areas imaged in 3 separate mice.
(C) Representative, pseudo-colored multiphoton live-microscopy image showing 42h Py-GFP RAS infected liver of B6.CD11c-YFP mice. Arrowhead marks CD11c+ cells harboring Py-GFP.
(D) Relative parasite loads in the hepatocyte and non-hepatocyte fractions from livers at 42h p.i. of CD11c-YFP=>B6.CD81−/− bone marrow chimeric mice co-inoculated with Pb and Py, detected with corresponding species-specific primers. Data represent 2 replicate experiments representing the range of data distribution with the error bars indicating the range and midline representing median of distribution based on ANOVA.
(E) Relative parasite loads in the hepatocyte fractions from livers at 42h p.i. of B6 mice co-inoculated with Pb and Py, detected with corresponding species-specific primers. Data represent 3 replicates representing the range of data distribution with the error bars indicating the range and midline representing median of distribution based on 2-tailed t-test.
(F) Relative parasite loads in the hepatocyte and non-hepatocyte fractions from Py infected B6 mice livers, at the indicated times p.i. (x-axis). Data from 3 separate experiments with 5 mice/group, and presented as mean± s.e.m. Groups compared at each time-point by 2-tailed t-tests. (*) p ≤ 0.05, (**) p ≤ 0.01.
Hepatic CD11c+ cells acquire Plasmodium only after hepatocyte infection
Hepatic CD11c+ cells could acquire Plasmodium sporozoites by direct phagocytosis, similar to uptake of the sporozoites by CD11c + cells in skin draining lymph nodes after mosquito bite infection (Chakravarty et al., 2007; Radtke et al., 2015). Alternatively, hepatic CD11c+ cells (co-expressing F4/80) could acquire Plasmodium from infected hepatocytes, some of which may undergo cell-death during the ~48 h (Leiriao et al., 2005) liver-stage differentiation phase in mice. Consistent with the latter, we observed an initially stable small (<10 μ diameter) developing parasite in the liver that appeared to undergo structural instability before a fragment is seemingly acquired by F4/80+ cells in a span of 24–26h post sporozoite inoculation (Movie S3). This process appeared quite distinct from the structurally stable large (>50 μ diameter) schizhont in an infected hepatocyte at ~48 h p.i. that is releasing budding merosomes, some of which are taken up by immune cells (Movie S4). Expression of the tetraspanin CD81 on hepatocytes is vital for invasion by Py but not Pb (Silvie et al., 2003) in mice. To determine if hepatocyte entry is a prerequisite for acquisition of Plasmodium by hepatic CD11c+ cells, we generated bone marrow chimeras with B6.CD11c-YFP bone marrow in irradiated B6.CD81−/− recipients, which were subsequently infected with Py or Pb sporozoites and evaluated by 2-photon microscopy for the presence of Plasmodium in CD11c-YFP+ cells. Hepatic CD11c-YFP+ cells (that express intact CD81) in the chimeric mice acquired Pb, but not Py (Figure 2B) indicating an obligatory role for hepatocyte infection prior to uptake of Plasmodium by CD11c+ cells. This was corroborated by substantial reduction in parasite burden in the non-hepatocyte fraction after Py compared to Pb infection of the CD81-deficient chimeric mice (Figure 2C). Overall, these data showed that Plasmodium infection was associated with an influx of CD11c+ cells into the infected liver, and that these cells acquired and harbored Plasmodium subsequent to hepatocyte infection.
CD11c+ APCs from Plasmodium infected livers prime CD8 T cell responses
CD11c+ cells, which may represent resident dendritic cells, monocyte-derived inflammatory dendritic cells and macrophages can be potent activators of naïve CD8 T cell responses and are clearly required to elicit Plasmodium-specific CD8 T cells after RAS immunization (Radtke et al., 2015). To determine if the Plasmodium-containing CD11c+ cells in the liver have the capacity to present antigens that directly stimulate Plasmodium-specific CD8 T cells, enriched CD11c+ cells from livers of naïve or Py infected B6 mice were co-cultured with TCR-retrogenic (Rg) CD8 T cells specific for an immunodominant Plasmodium epitope from its glideosome-associated protein-50 (GAP5040–48) (Howland et al., 2013; Van Braeckel-Budimir et al., 2017). Enriched fractions of CD11c+ cells from Py infected, but not naïve livers stimulated robust IFNγ and TNF production by GAP50Rg-CD8 T cells (Figure 3A), indicating their capacity to present Plasmodium antigen. To date, all defined native Plasmodium CD8 T cell epitopes known are either expressed by the sporozoite and liver stages or expressed at all stages of the parasite in its mammalian host. The GAP50 protein is expressed in both the liver and blood-stages of Plasmodium infection. To determine the requirement for CD11c+ cells in CD8 T cell priming against liver-stage antigens in vivo and the potential role of these cells in controlling Plasmodium burden, we employed an infection model using RAS that does not generate blood-stage parasites (Nussenzweig et al., 1967). B6 and B6 mice expressing the primate diphtheria toxin (DT) receptor under control of the CD11c promoter (B6.CD11c-DTR mice (Jung et al., 2002)) were infected with Py-RAS, then treated with DT at 24h p.i. A robust circulating GAP50-specific CD8 T cell response was generated in the DT treated B6 mice (and in the untreated B6.CD11c-DTR mice; data not shown), which as predicted from previous work (Jung et al., 2002; Radtke et al., 2015), was eliminated upon depletion of CD11c+ cells (Figure 3B). Of note, depletion of CD11c+ cells at 24 hours p.i. with Py did not enhance the relative whole liver-parasite loads in the mice (Figure S1A), suggesting that CD11c+ cells may not directly contribute to control of liver-stage malaria. In contrast, the relative liver-parasite burden was reduced (quantified by Plasmodium mRNA signal) after depletion of CD11c+ cells, potentially due to the removal of CD11c+ cells harboring Plasmodium. These data provide additional evidence that CD11c+ cells harbor a substantial parasite burden after sporozoite infection.
Figure 3: CD11c+ cells from Plasmodium infected liver and liver draining lymph nodes prime CD8 T cell responses in mice.

(A) Representative flow plots indicating the frequencies of IFNγ (top row) or TNF (bottom row) producing GAP50Rg-CD8 T cells after co-incubation with hepatic CD11c+ DCs from uninfected mice (left), uninfected mice +GAP50 peptide (middle) or Py infected B6 mice (right).
(B) Representative flow plots indicating frequencies of GAP50 tetramer+ CD8 T cells (blood, 5d p.i.) in Py-RAS infected B6 (top) and B6.CD11c-DTR (bottom) mice treated with diphtheria toxin (DT, −1d, +2d p.i.).
(C) Representative flow plots depicting frequencies of GAP50 tetramer+ CD8 T cells (blood, 5d p.i.) in B6 mice that received hepatic CD11c− (2x106, top) or CD11c+ (2x105, bottom) cells from 36h p.i. Py infected B6 donors. (A-C) Bottom row: Bar graphs summarizes data as mean ± s.e.m, analyzed using 1-way ANOVA with Holm-Sidak correction (A) or two-tailed t-tests (B,C). (*) p ≤ 0.05, (**) p ≤ 0.01.
(D) Representative histograms indicating BrdU incorporation at 5d p.i. in adoptively transferred CS252Rg-CD8+ T cells or GAP50Rg-CD8+ T cells, in popliteal or liver-draining lymph nodes of FTY720-treated CB6F1 recipient mice inoculated in foot-pad with Pb-RAS; numbers represent mean ± s.e.m. Grey histograms are isotype controls. Data represent one of at least 3 separate experiments, with at least 3 mice per group.
To determine if hepatic CD11c+ cells harboring Plasmodium were sufficient to prime antigen-specific CD8 T cell responses in vivo, we enriched CD11c+ or CD11c− cells from livers of Py infected B6 mice at 36 hours p.i. and transferred these cells into separate groups of naïve B6 mice. Despite transfer of 10-times more CD11c− cells, only CD11c+ cell transfer primed a detectable GAP50-specific CD8 T cell response in the recipient mice (Figure 3C). It is noteworthy that transfer of hepatic CD11c+ cells from Pb or Py infected mice did not elicit blood-stage malaria in the recipients (data not shown), suggesting that Plasmodium do not undergo productive development in the hepatic CD11c+ cells. Together, these data argued further that the parasite containing CD11c+ cells were bona fide antigen presenting cells (APCs) and indicated that CD11c+ cells from the Plasmodium infected liver were sufficient for priming specific CD8 T cell responses.
Liver draining lymph node CD11c+ cells prime liver-stage specific CD8 T cell responses.
CD11c+ cells acquire the skin inoculated parasites and prime sporozoite-antigen specific CD8 T cell responses in the skin draining lymph nodes (dLN) (Chakravarty et al., 2007). To determine if hepatic CD11c+ cells can prime liver-stage malaria specific CD8 T cell responses in the celiac and portal liver-dLN (LdLN) (Barbier et al., 2012), we generated naive GAP50 (GAP50Rg-CD8 T cells, H-Db restricted) or circumsporozoite (CS) protein derived CS252 (CS252Rg-CD8 T cells, H-2Kd restricted) -specific CD8-Rg cells in BALB/C x B6 F1 (CB6F1) mice and co-transferred these populations into naive CB6F1 mice prior to Pb-RAS infection. To enhance recovery of cells in reactive lymph nodes, the recipient mice were treated with the S1P1R antagonist FTY720 to inhibit cellular egress from lymphoid tissues (Shiow et al., 2006). On challenge with Pb-RAS in the footpad, both Rg populations underwent substantial proliferation in the footpad-draining, popliteal lymph nodes (pLNs) and in the LdLNs, as determined by BrdU incorporation at 5 days p.i. (Figure 3D). These data indicated that priming of Plasmodium-specific CD8 T cells against its pre-erythrocytic developmental stages occurred in LdLN as well as in the skin-dLN. Additionally, transfer of LdLN cells from Plasmodium-RAS infected mice into congenically distinct B6 mice generated substantial primary CD8 T cell responses against GAP50 and another Plasmodium epitope, PbT (Lau et al., 2014) (Figure S1C). These data indicated that APC in LdLN were likely sufficient to prime Plasmodium-specific CD8 T cell responses.
The GAP50 and CS252 antigens used above are expressed in multiple stages of the Plasmodium life-cycle, preventing us from distinguishing between CD8 T cell priming against antigens expressed in the mosquito-inoculated sporozoites versus in the bona fide hepatic developmental stages. To address this, we compared the capacity of RAS generated from transgenic Pb that only express the model antigen chicken ovalbumin after liver-stage infection (Pb-hep17Ova) (Lin et al., 2014) or have constitutive (Pb-sOva) Ova expression, to stimulate a 1:1 mixture of naïve OT-I TCR-transgenic CD8 T cells specific for Ova257–265 or the irrelevant, control P14 TCR-tg cells specific for LCMV GP31–40 in the same host. While footpad inoculation of Pb-sOva-RAS stimulated OT-I responses in both pLNs and LdLNs (Figure 4A, summarized in Figure 4B), Pb-hep17Ova-RAS infection selectively expanded OT-I cell frequencies only in the LdLNs (Figures 4A-B). Additionally, while OT-I cells (but not the control P14 cells) underwent substantial proliferation, as determined by BrdU incorporation in both pLNs, LdLNs and spleen in response to Pb-sOva-RAS (Figures 4C, S1B), Pb-hep17Ova-RAS induced proliferation of OT-I cells exclusively in the LdLNs (Figures 4C, S1B). These data indicated that CD8 T cell responses generated against an antigen expressed exclusively in the liver-stage of Plasmodium development were primed in the LdLNs. Due to anatomical and numerical constraints, we were not able to image CD11c+ cell interactions with the sparse naïve T cells in situ in the LdLN. However, we were able to detect interactions of numerically increased memory OT-I cells with Pb-hep17Ova containing CD11c+ cells in LdLN explants (Figure 4D). These data demonstrated that while Plasmodium-specific CD8 T cell responses may be primed in the spleen, skin and LdLN, liver-stage-restricted antigens may be selectively primed in the LdLN, likely by the hepatic CD11c+ APCs that acquired Plasmodium that had undergone development in the hepatocytes.
Figure 4: CD8 T cell responses to hepatic parasite antigens are primed in liver-draining lymph nodes.

(A) Representative flow plots indicating the frequencies of transferred OT-I or control P14 cells in popliteal (left) or liver-draining (right) lymph nodes at 5d p.i. of FTY720-treated B6 recipient mice inoculated in the foot-pad with Pb-sOva-RAS or Pb-hep17Ova-RAS.
(B) Data in (A) summarized as mean ± s.e.m; analyzed using two-tailed t-tests. (*) p ≤ 0.05, (**) p ≤ 0.01, (n.s) p > 0.05.
(C) Representative histograms of BrdU incorporation in the transferred OT-I cells shown in (A); presented as mean ± s.e.m. Grey histograms represent isotype controls.
(D) Representative, pseudo-colored multiphoton microscopy images showing OT-I cells in direct apposition with Pb-hep17Ova harboring CD11c+ DCs in the liver-draining lymph nodes. All data represent one of at least 3 separate experiments, with at least 3 mice per group.
Hepatic CD11c+ APCs harboring Plasmodium are phenotypically and transcriptionally distinct
Tissue resident and inflammatory CD11c+ cells manifest overlapping, but distinctive phenotypic attributes (Mildner and Jung, 2014). To further characterize the hepatic CD11c+ cells that acquired Plasmodium, mice were infected with Py-GFP (delivered by mosquito bite or needle) and the hepatic CD45+ cells that contained Plasmodium (Figure 5A) were evaluated for expression of multiple markers; and compared to hepatic CD45+ CD11c+ cells from uninfected mice and those that did not harbor parasites in infected mice. These comparisons were chosen because of the snapshot nature of the analyses, where we could not know if “Plasmodium negative” CD11c+ cells from infected mice may have harbored and processed the parasites at some time point prior to our analysis. Additionally, the influx of CD11c+ cells into the infected liver (Figure 1) suggested they might differ from those found in uninfected mice. Py harboring hepatic CD11c+ cells exhibited high expression of CD11b, Ly6C and CD14, which were absent on CD11c+ cells from uninfected livers (Figure 5B). Ly6G, CD69, CCR2 and Tim4 exhibited modest upregulation on CD11c+ cells harboring Py. Expression of the T cell co-stimulatory molecules CD40 and CD86 were elevated on Py harboring CD11c+ cells relative to their naïve counterparts, whereas MHC class II expression was similar in either CD11c+ population. Of note, these parasite containing CD11c+ hepatic cells also expressed CD103, a marker associated with the ability of DCs to cross-present cellular antigens (Nierkens et al., 2013; Sathe et al., 2011), but unusually co-expressed CSF1R, CD207 and F4/80 (which we now call CSF1R+ CD11c+ cells for simplicity), that does not appear to place them precisely into any of the various current definitions of macrophage or DC subsets (Merad et al., 2013; Mildner and Jung, 2014; Randolph et al., 2008) (Figure 5B). Similar differences in phenotype were observed upon comparing CD11c+ cells with detectable Plasmodium parasites to CD11c+ cells from the same livers without detectable parasites (Figure S2), with the latter looking similar phenotypically to CD11c+ cells from a naïve mouse. Consistent with these data, Py (marked with CellTrace Far Red) were distinctly found within hepatic F4/80+ (Figure 5C), CSF1R+ (Figure 5D) and CD207+ (Figure 5E) cells in situ at ~40 h after sporozoite infection. Plasmodium infection is thought to induce caspase-mediated cell-death in hepatocytes (Kaushansky et al., 2013). There is a possibility that hepatic CD11c+ cells recruited into the liver may acquire Plasmodium, subsequent to hepatocyte cell-death, as suggested by Figure 2F. In support of this notion, mice treated with a pan-caspase inhibitor exhibited reduced frequencies of hepatic CSF1R+ CD11c+ cells that harbored Py-GFP at 42h post-infection (Figure S3). To test if hepatic CD11c+ cells harboring Plasmodium originated from circulating monocytes, we transferred congenically distinct splenic CD11c− monocytes into recipient mice and then infected with Py-GFP RAS (Figure S4A). Transferred monocytes acquired CD11c expression and Py in the liver of infected recipients, indicating that recruited monocyte-derived hepatic CD11c+ cells indeed acquired Plasmodium during liver-stage malaria (Figure S4B). To test if CSF1R had a functional role in recruiting circulating monocytes into the liver, we transferred congenically distinct splenic CD11c− monocytes into recipients as above, infected with Py-GFP RAS and concurrently neutralized the CSF1R-specific ligand, M-CSF. M-CSF neutralization did not alter recruitment of the monocyte derived CD11c+ APCs or their acquisition of Plasmodium in the liver (Figure S4C–E). Additionally, M-CSF neutralization did not impact the priming of Plasmodium (GAP50) specific CD8 T cell responses or protection from a subsequent Py spz challenge (Figure S4F–H). Together, these data implied that CSF1R expressed by the monocyte derived CD11c+ APCs might help in phenotypically refining a cell subset that acquires Plasmodium in the liver, but may not play a functional role in that process.
Figure 5: Hepatic CD11c+ cells that harbor Plasmodium in liver-stage malaria are phenotypically discrete.

(A) Representative flow plots showing liver (CD45+) cells in the non-hepatocyte fraction from uninfected (left) or 36h Py-GFP infected (middle) mice. Numbers inset indicate the mean frequency of the gated population ± s.e.m.
(B) First panel, CD11c expression on the indicated CD45+ hepatic cells. Remaining panels represent histograms depicting the expression of the indicated markers on the Py-GFP harboring hepatic CD11c+ DCs versus CD11c+ DC from uninfected livers; 36–42h p.i.
(C-E) Representative, pseudo-colored multiphoton live-microscopy images showing hepatic F4/80+ (C) CSF1R+ (D) or CD207+ (E) cells harboring Py in the liver of 36–42h infected mice. All data represent at least 3 separate biological replicates.
Hepatic CD11c+ cells that acquire Plasmodium may have unique characteristics that promote the process of parasite uptake. To transcriptionally define the CSF1R+ CD11c+ subset observed in Plasmodium infected liver, B6 mice were infected for 36–40 h with Py sporozoites and the differentially expressed genes of CSF1R+ or CSF1R− CD11c+ subsets were analyzed by RNA-seq. The transcriptional differences observed between CSF1R+ and CSF1R− CD11c+ hepatic cells promptly separated them into distinct hierarchical clusters (Figure S5A). Relative expression of various regulatory molecules and associated pathways were altered between subsets, with predicted enhanced phagocytosis and phagocyte recruitment functions being associated with the hepatic CSF1R+ CD11c+ cells (Figure S5B). Further analysis of differentially expressed genes resulted in identification of immunological functions in CSF1R+ CD11c+ cells, as demonstrated by the enrichment of genes associated with phagocytosis, antigen presenting cellular responses and phagocyte recruitment (Figure 6B). Together, these data demonstrated that liver-stage parasites were present in a subset of hepatic CSF1R+ CD11c+ cells, that exhibited a unique phenotypic and transcriptional profile characterized by enhanced cellular recruitment, phagocytosis and antigen presentation functions.
Figure 6: Hepatic CSF1R+ CD11c+ cells in liver-stage malaria have a distinct transcriptional identity.

(A) Heat-map depicting the functionally distinct identity of CSF1R+ CD11c+ DCs based on differential expression of the indicated genes derived from the top predicted pathways of immunologically relevant functional molecular interactions (1.5x change, activation Z score ≥2, p ≤ 1.1e−10) in the whole transcriptome.
(B) Significantly enriched network of functional interactions evaluated by Ingenuity pathway analysis comparing CSF1R+ and CSF1R− (CD11c+) DCs from the liver of 36–42h Py infected B6 mice. The nodes in (B) represent the key factional outcomes predicted based on the transcriptional identity of the CSF1R+ DCs.
Hepatic CSF1R+ CD11c+ cells prime protective CD8 T cell responses
In light of the distinct phenotypic and transcriptional profile of CSF1R+ CD11c+ cells that corroborated their capacity for efficient antigen presentation, we examined the possible role of this subset in priming CD8 T cell responses to liver-stage malaria. The relative parasite burden was substantially higher in the CSF1R+, compared to the CSF1R− subset of hepatic CD11c+ cells (Figure 7A). 2-photon microscopy supported this finding, with Py being distinctly associated with the hepatic CSF1R+ CD11c-YFP+ cells (Figure 7B) ~ 40 hours post infection. The CSF1R+ CD11c+ cells co-expressing F4/80 were distinct from the traditionally described F4/80+ Kupffer cells in the liver; treatment with an anti-CSF1R depleting antibody (Sierro et al., 2017) that depleted the CSF1R+ CD11c+ cells, left a discernable population of (CSF1R−) F4/80+ cells that are possibly the Kupffer cells in the liver (Figure 7C). We next examined if CD8 T cell priming in liver-stage malaria requires CSF1R+ CD11c+ cells. CSF1R expressing cells were selectively depleted in Py-RAS inoculated B6.Csf1r− mice, which express human FK506 binding protein 1A under control of the CSF1R promoter, by treating with a dimerizing agent (Burnett et al., 2004), and CD8 T cell responses were compared to the control treated B6 mice at 7 days p.i. The frequency of antigen experienced CD8+ T cells (CD8αlo CD11ahi) (Rai et al., 2009) induced by infection was reduced nearly 10-fold in the absence of CSF1R+ cells (Figure 7D) and CD8 T cell responses to the Py specific GAP50 and PbT epitopes were greatly diminished (Figures 7E–F). As noted, Plasmodium containing CD11c+ cells also expressed CD207 (langerin) (Figure 5B and5E). Consistent with this, selective depletion of CD207 expressing cells (using DT treatment in Lang-DTREGFP mice (Kissenpfennig et al., 2005)) after Py-RAS infection also resulted in a substantial reduction in the frequency of antigen experienced and GAP50 specific CD8 T cells (Figure S6A). Further, depletion of CSF1R+ or CD207+ cells prior to RAS immunization resulted in significant defect in the establishment of liver resident memory CD8 T cells (Fernandez-Ruiz et al., 2016) that are critical to protection from subsequent sporozoite challenges (Figure S6B). Although these data indicated that the subset of cells expressing CSF1R or CD207 may be vital in priming CD8 T cell responses to Plasmodium infection, our depletion of these cellular subsets were not specific to the liver. To determine if liver derived CSF1R+ CD11c+ cells were capable to induce Plasmodium-specific CD8 T cell responses, CSF1R+ or CSF1R− CD11c+ cell fractions from the livers of 36 hour Py or Pb infected mice were enriched, transferred to naïve hosts, and Plasmodium-specific CD8 T cell responses were examined. Substantially higher GAP50 specific responses were observed in recipients of CSF1R+ CD11c+ cells (Figure 7G–H). Similarly, transfer of CSF1R+ CD11c+ cells from LdLNs of 36 hours p.i. Py infected mice into naïve recipients primed Py specific CD8 T cell responses (Figure S6B). Despite the CSF1R+ CD11c+ APCs harboring parasites, blood-stage infection was not detected in any recipient mice (not shown), indicating that this model evaluates priming only against liver-stage antigens and not antigens from blood-stage parasites.
Figure 7: Hepatic CSF1R+ CD11c+ cells prime protective CD8 T cell responses.

(A) Relative parasite loads in the CSF1R+ and CSF1R− CD11c+ fractions from 36–44h Py sporozoite infected B6 mice livers. Each data point represents an individual mouse
(B) Representative, pseudo-colored multiphoton live-microscopy images showing F4/80+ CSF1R+ hepatic cells harboring Cell Trace Far Red-labeled Py.
(C) Representative, pseudo-colored multiphoton live-microscopy image showing depletion of hepatic APC with anti-CSF1R antibody.
(D-F) Representative flow plots showing frequencies of activated (CD8αloCD11ahi) (D), GAP50 tetramer+ (E) and PbT tetramer+ (F) CD8 T cells at d7 after Py-RAS immunization of dimerizing agent treated B6 (left) or B6.Csf1r- (middle) mice. Data summarized as scatter plots in the panel on right. Each data point represents an individual mouse
(G-H) Representative flow plots depicting frequencies of splenic GAP50 tetramer+ CD8 T cells (5d p.i.) in B6 mice that received hepatic CSF1R− (top) or CSF1R+ (middle) APC from 36h Py (left) or Pb (right) infected B6 donors (G). Data summarized in scatter plots (H). Each data point represents an individual mouse.
(I) Schematic representation of Py-RAS immunization and challenge in dimerizing agent treated C56BL/6 or B6.Csf1r- mice. Scatter plot in the right panel indicates relative parasite loads in hepatocytes after Py spz infection. Each data point represents an individual mouse. All data summarized here as mean ± s.e.m and represents one of at least 3 separate experiments, with a minimum of 3 mice/group. Statistical analyses with two-tailed student t-tests (A, D, G) or ANOVA with Tukey’s correction (I). (*) p ≤ 0.05, (**) p ≤ 0.01, (n.s) p > 0.05.
CD11c+ APCs (Figure 3C) or the more specific CSF1R+ CD11c+ APC subset (Figures 7G–H, S6B) from Plasmodium infected liver or LdLNs may have directly primed CD8 T cell responses in the recipient mice or served as antigen-delivery vehicles for subsequent cross-priming by recipient DCs. To address this, hepatic CSF1R+ CD11c+ APCs from Py infected mice were transferred into B6.CD11c-DTR recipients where the endogenous CD11c+ cells had been depleted by DT treatment. Depletion of endogenous CD11c+ cells in B6.CD11c-DTR recipients did not alter the Plasmodium specific CD8 T cell responses generated after the transfer of CSF1R+ APCs (Figure S6C). These data demonstrated that hepatic CSF1R+ CD11c+ APCs from Plasmodium infected mice can directly prime CD8 T cells in vivo.
In the context of immunity to malaria and the ongoing efforts to immunize against this disease, CD8 T cell responses generated against liver-stage of malaria are critical (Coelho et al., 2017; Schwartz et al., 2012). To determine if CD8 T cell responses primed by the CSF1R+ CD11c+ APC subset were pivotal to immunity from malaria infections, mice depleted of CSF1R+ cells or B6 controls were immunized with Py-RAS and then challenged with infectious Py sporozoites at 7 days post immunization. While the relative liver-parasite loads were notably reduced 36 hours post challenge in RAS-immunized B6 mice compared to the unvaccinated controls, CSF1R+ cell depletion markedly compromised RAS-induced control of liver parasite burden (Figure 7I). Together, these data demonstrated that the CSF1R expressing subset of hepatic CD11c+ APCs that harbor Plasmodium parasites were capable of priming CD8 T cell responses against liver-stage malaria; and these cells likely play a crucial role in conferring protection from future challenges.
Discussion
Although malaria remains a global threat, many aspects of its basic biology and host-interactions remain unknown. In contrast to the prevailing notion that most Plasmodium are within hepatocytes during the liver-stage (Cowman et al., 2016), we found a substantial amount of Plasmodium transcript in the non-hepatocyte fraction of infected livers, specifically within hepatic CD11c+ cells, that increase in number after infection. Prior studies reported the infiltration of myeloid cells including inflammatory monocytes and DCs into the liver, during or immediately following Plasmodium infection (Akbari et al., 2018; Parmar et al., 2018). Our data corroborate these findings, while illuminating a fraction of hepatic CD11c+ cells that acquire Plasmodium parasites after initial infection of hepatocytes, revealing a previously unappreciated aspect of Plasmodium biology in its mammalian host. Wild-type or genetically attenuated Plasmodium infections are thought to induce apoptotic death of hepatocytes (Kaushansky et al., 2013; van Dijk et al., 2005). We speculate that the CD11c+ APCs in the liver may be capturing Plasmodium from dying hepatocytes. This notion is supported by the finding that blockade of Caspase-mediated cell-death in Py infected mice reduced the frequency of hepatic CD11c+ APCs containing the parasite. The precise pathways of cell-death evoked by Plasmodium infection remain to be determined.
The majority of Plasmodium containing CD11c+ cells expressed an unusual collection of cell surface markers including CSF1R, CD207, F4/80 and CD103. In addition to Kupffer cells that are predominantly inducers of T cell tolerance in the liver, B220+ plasmacytoid DCs and liver capsular macrophages (LCMs) (Sierro et al., 2017) form key myeloid APC populations in the liver (Ebrahimkhani et al., 2011). Although the Py harboring myeloid cells we identify in this study localize in the liver, are monocyte-derived, and co-express CD11c, F4/80, MHC-II, CD207, CSF1R, CD14 and CD11b, similar to LCMs, their distinct expression of Ly6C, CD103 and Tim4, lack of anatomical restriction to the hepatic sub-capsular space, and detection in the liver-draining lymph nodes clearly separates them from the LCMs. While LCMs directly capture pathogens from peritoneal infections (Sierro et al., 2017), it is clear that Plasmodium sporozoites must initiate an infection of hepatocytes before acquisition by CSF1R+ CD11c+ APCs, suggesting a divergence in pathogen acquisition between these two hepatic APC populations. Despite the unique transcriptional identity of CSF1R+ CD11c+ cells, the ‘snapshot’ nature of our analysis and the inherent plasticity in the DC/macrophage populations prevents us from asserting that CSF1R+ CD11c+ APCs are an independent class of myeloid cells unique to the liver, or to malaria. We also note that neutralizing M-CSF did not prevent monocytes from migrating to the liver and becoming CD11c+ APC that take up Plasmodium, suggesting that CSF1R did not have a functional role in this process.
Although substantial evidence suggests that CD8 T cell responses against liver-stage Plasmodium antigens occur and contribute to protective immunity (Butler et al., 2011; Vaughan and Kappe, 2017), the cellular and anatomical regulation of this response has remained unknown. In general, antigens presented by MHC-I molecules are derived from intracellular proteins, making them available to be identified by CD8 T cells. However, various APC subsets including CD11c+ DC are specialized in the phagocytic uptake and presentation of exogenous antigens on their MHC-I molecules, through the process of cross-presentation (Heath and Carbone, 2001; Joffre et al., 2012). Consistent with their acquisition of Plasmodium after hepatocyte infection and their possible role as APCs, we observed an enrichment of pathways involved in phagocytic, migratory and antigen presentation functions within the hepatic CSF1R+ CD11c+ APCs in liver-stage malaria. Following infection, the Plasmodium harboring CSF1R+ CD11c+ cells were also found in the LdLNs, where they were capable of priming CD8 T cell responses against exclusively liver-stage malaria specific antigens. Additionally, Plasmodium-harboring CSF1R+ CD11c+ APCs were required for optimal RAS-induced CD8 T cell priming and protection against liver-stage malaria.
The priming of CD4 and CD8 T cells in the LdLNs in response to delivery of soluble antigen directly into the liver has been described in the past (Barbier et al., 2012). Additionally, priming of a Plasmodium PbT epitope-specific CD8 T cell response has been observed in the LdLNs, along with the spleen in response to intravenous inoculation of Plasmodium sporozoites (Lau et al., 2014). However, the systemic availability of Plasmodium antigens after intravenous delivery, and expression of PbT in all life-stages of the Plasmodium in mice, prevented identification of the site for priming CD8 T cell responses specific to liver-stage malaria. Our use of Pb-Hep17Ova-RAS, that produces Ova antigen only after hepatocyte infection and development identify LdLNs as the primary lymphoid site for priming CD8 T cell responses to antigens expressed in the liver-stage of malaria. While Plasmodium sporozoites delivered into the skin by mosquito bite or needles can prime CD8 T cell responses directed at sporozoite-specific antigens in the spleen or skin draining LNs (Chakravarty et al., 2007; Radtke et al., 2015), a productive liver-stage infection is required for priming CD8 T cell responses against bona fide liver-stage antigens of Plasmodium. Since the liver-stage of malaria is a critical step in the progression of Plasmodium to the blood-stage, we believe that our finding is a conserved mechanism of priming T cell responses to liver-stage specific antigens, applicable to both natural infections and current sporozoite-based vaccination approaches.
In summary, our results reveal the host cell-parasite dynamics during liver-stage malaria that result in acquisition of Plasmodium by CD11c+ APC after initial colonization of hepatocytes. Elucidating and characterizing the population of APC that prime liver-stage malaria specific T cell responses fills a critical knowledge gap in our understanding of Plasmodium-host interactions; and may aid in tailoring immunization strategies to target these APCs and improve anti-malarial vaccines in the process.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact John T. Harty (john-harty@uiowa.edu)
Experimental Model and Subject Details
Mice and parasites
B6 (C57BL/6J, H-2b), and CB6F1 (BALB/cJ x C57BL/6, H-2bxd) mice were purchased from the National Cancer Institute. CD11c-YFP (B6.Cg-Tg(Itgax-Venys)1Mnz/J) (Lindquist et al., 2004), CD11c-DTR (B6.FVB-1700016L21Rik Tg(Itgax-DTR/EGFP)57Lan/J) (Jung et al., 2002), OT-I (C57BL/6-Tg(TcraTcrb)1100Mjb/J) (Hogquist et al., 1994) and Lang-DTREGFP (B6.129S2-cd207tm3(DTR/GFP)Mal/J) (Sierro et al., 2017) mice were purchased from the Jackson Laboratory. MaFIA (C57BL/6-Tg(Csf1r-EGFP-NGFR/FKBP1A/TNFRSF6)2Bck/J) (Burnett et al., 2004), B6.CD81−/− (CD81tm1Lvy) (Maecker and Levy, 1997), and P14 (B6.P14 (B6;D2-Tg(TcrLCMV)327Sdz/JDvsJ) (Pircher et al., 1989) mice were kindly provided by Drs. Wendy Maury (University of Iowa), Steven Varga (University of Iowa) and Shoshana Levy (Stanford University) respectively and maintained in our laboratory. Male or female mice aged 6–10 weeks were housed with ad libitum feed and water, with appropriate biosafety containment at the University of Iowa Animal Care Unit were used for the study. The animals were treated and handled in accordance with guidelines established by the Institutional Animal Care and Use Committee. Anopheles stephensi mosquitos parasitized with P. yoelii (17XNL strain), Py-GFP and Pb-GFP (ANKA strain) were obtained from New York University. Pb-hep17Ova (Lin et al., 2014) and Pb–sOva (ANKA strain) were generated in our labs and A. stephensi mosquitos parasitized with these were maintained in-house.
Method Details
Isolation of Cells from Lymph Nodes and Spleen
Cells were isolated and stained as previously described (Doll et al., 2016). In brief, spleen, popliteal, portal and celiac lymph nodes were removed, homogenized with glass coverslips in RPMI + 10% FCS, filtered through a 70μ cell strainer and where applicable, MHC-I tetramers for PbT or GAP50 (prepared in house as described (Doll et al., 2016)) were incubated in the presence of Fc-Block (aCD16/32) at 4°C for 45 minutes prior to cell surface staining. Cells were stained for cell surface markers with appropriate antibodies in FACS buffer for 30 minutes and fixed in fix buffer (BD) prior to acquisition on an LSR Fortessa (BD Biosciences). Flow cytometry data was analyzed with Flowjo (Treestar). Peptide restimulation and Intracellular staining for cytokines and BrdU were performed as described before(Martin et al., 2012).
Antibodies and T cell analyses
Tg and Rg CD8 T cells were stained for cell surface antigens CD8α (clone 53–6.7) CD90.1 (clone OX-7), CD90.2 (30-H12), CD11a (clone M17/4), CD45.1 (clone A20) or CD45.2 (clone 104). For intracellular cytokine staining cells were restimulated with peptide as previously described (Martin et al., 2012). Cells were fixed, permeabilized (eBioscience, Transcription Factor Staining Buffer Set) and stained for IFNγ (clone XMG1.2) or TNF (clone MP6-XT22). To inhibit cellular egress from lymphoid tissues, Rg and Tg recipient mice were treated with FTY720 (1mg/Kg) injected i.p. each day (Shiow et al., 2006) (Sigma-Aldrich). For the detection of cellular division, BrdU (2mg/mouse) was injected i.p. and mice were administered BrdU in their drinking water (0.75 mg/mL) for 5 days ad libitum per protocol (BD Biosciences). Detection of BrdU incorporation was performed per manufacturer protocol (BD Biosciences) and stained with anti-BrdU (clone BU20A). For liver myeloid populations, liver isolates were stained for cell surface antigens CD45.2 (clone 104), CD11c (clone N418), CD11b (clone M1/70), Ly6C (clone AL-21), F4/80 (clone BM8), CX3CR1 (clone SA011F11), MHC-II (I-A/I-E, clone M5/114.15.2), Ly6G (clone 1A8-Ly6G), CD86 (clone GL-1), CD40 (clone 3–23), CD103 (clone 2E7), CCR2 (clone SA203G11), CD69 (clone H1.2F3), Tim4 (clone F31–5G3), CD14 (clone sa14–2), TNF (clone MP6-XT22), CD207 (clone 4C7), CSF1R (604B5 2E11).
Bone marrow chimeras
Radiation bone marrow chimeras were generated as previously described (Pham et al., 2011). Briefly, the tibia, fibula and os coxa were isolated from CD11c-YFP mice, cleaned and smashed with a mortar and pestle. Pulverized bones were washed with complete RPMI media and isolated bone marrow was filtered through a 70μ cell strainer. RBCs were lysed with Vitalyse (CMDG) and 5x106 bone marrow cells were injected i.v. into sublethally irradiated (9.5Gy) recipient CD81−/− mice. Mice were rested for at least 8 weeks following transplantation and reconstitution was accessed by flow cytometry.
Isolation and enumeration of liver-resident memory CD8 T cells
In brief, livers were removed, homogenized and filtered through a 100μ cell strainer. Hepatocytes were pelleted and separated from the single cell suspension above by centrifugation at 60g, 3 mins. The supernatant was pelleted and purified using 35% Percoll (GE Healthcare) in HBSS (Gibco). RBCs were lysed and enriched lymphocytes were stained with MHC-I tetramer for GAP50 and incubated in the presence of Fc-Block (aCD16/32) at 4°C for 45 minutes prior to cell surface staining. Cells were stained for cell surface markers with appropriate antibodies in FACS buffer for 30 minutes and fixed in fix buffer (BD) prior to acquisition on an LSR Fortessa (BD Biosciences). Live, liver Trm populations were enumerated based on the expression pattern of the following cell surface markers as previously described (CD8α+ CD11ahi, CD62L− CD69+ CXCR3+ CXCR6+) (Fernandez-Ruiz et al., 2016). Flow cytometry data was analyzed with Flowjo (Treestar).
Monocyte enrichment and adoptive transfer
Spleens from naïve CD45.2 B6 mice were isolated, filtered through a 70μ cell strainer, RBCs were lysed as previously described and monocytes were negatively enriched by magnetic bead isolation with AutoMacs (Miltenyi Biotec). APC conjugated antibodies against CD3e, CD19, NK1.1, Ter119, Ly6G, MHC-II and CD11c were used to negatively enrich for splenic monocytes. Approximately 2.0x106 enriched monocytes were adoptively transferred i.v. to congenically distinct naïve CD45.1 mice 24hrs prior to i.v. inoculation with Py-GFP RAS.
In vivo depletion of cells
Various cell populations were depleted as intended in CD11c-DTR or Lang-DTREGFP mice using diphtheria toxin (DT, 200ng/mouse, Sigma), MaFIA mice using the dimerizing agent (BB Homodimerizer, 0.3mg/mouse, Takara) or in wild-type mice with anti-CSF1R antibody (0.5mg/mouse, BioXcell, clone: AFS98). Where indicated, M-CSF1 neutralizing antibody (1mg/mouse, BioXcell, clone: 5A1) was injected i.p. for 3 consecutive days.
In vivo Pan-Caspase Inhibition
Where indicated, wild-type mice were administered DMSO alone or pan-caspase inhibitor (Q-VD-OPH, 20mg/Kg) dissolved in DMSO via i.p.(Kaushansky et al., 2013) 4h prior to infection and 20h post infection
Plasmodium and RAS inoculations
For sporozoite injections, salivary glands of parasitized A. stephensi mosquitos were dissected and sporozoites were isolated as previously described (Doll et al., 2016) and injected i.v. (200μL total volume) in the tail vein or s.c. (20μL total volume) in the footpad as indicated. In some experiments, parasitized A. stephensi mosquitos were allowed to bite the abdomen or back of ketamine anesthetized mice for 6 separate 5-minute intervals consecutively. For the production of radiation attenuated sporozoites (RAS), freshly isolated sporozoites were attenuated with 200 Gy (Doll et al., 2016) by cesium irradiation or parasitized A. stephensi mosquitos were attenuated with 120 Gy (Rieckmann et al., 1979) by cesium irradiation.
Assessment of liver parasite burden
Liver parasite burden was assessed by quantitative real-time RT-PCR for parasite 18s rRNA by modifying the previously described method (Bruna-Romero et al., 2001; Doll et al., 2016). To determine parasite burdens, total RNA was extracted from whole liver or isolated liver cells (please see below) at the indicated time points post Plasmodium infection with TRIzol, followed by DNase digestion/cleanup with RNA clean and concentrator kit (Zymo Research). 2μg liver RNA per samples was used for qRT-PCR analysis for Plasmodium 18S rRNA using Fast Virus One-step qPCR (Applied Biosystems). Data were normalized for input RNA with GAPDH control.
CellTrace Far Red (CTFR) cell labeling
Isolated sporozoites were incubated at 37°C for 20 minutes with 10μM CellTrace Far Red (CTFR, Life Technologies). The labeled sporozoites were then incubated for 5 minutes with 1mL FCS at 37°C to remove any free CTFR and washed once with media prior to i.v. inoculation into mice.
Intravital microscopy
All images were acquired on a SP8 NLO Microscope (Leica) using a 25x / 0.95 NA water immersion objective with coverslip correction as previously described (Kurup et al., 2017). High resolution stacks of 20–35 xy sections sampled with 1–3μ z spacing were acquired at an acquisition rate of 30–60 seconds per stack and merged with Leica software. Where indicated, mice were injected with F4/80-APC (10μg, i.v.) 1 hour prior to imaging or with Qtracker 705 (Qdot 705; 20μl i.v.) or Evans Blue (0.1% solution in PBS, 200μl i.v.) for vascular label immediately prior to imaging.
Mice were anesthetized with Ketamine/Xylazine (87.5/12.5 mg/Kg) prior to their surgical preparation for imaging. Mice were placed in dorsal recumbency on a heating pad maintained at 37°C, an abdominal anterioposterior incision was made in the skin posterior to the diaphragm. Lateral incisions were made on the left side through the skin at either ends of the earlier incision and skin reflected to the side. Subsequently, the peritoneal membrane was incised and reflected as above, to expose the liver. Mice were positioned on dorsal recumbency on the microscope base in a continuously heated (37°C) enclosed chamber (Leica). A custom tissue-suction apparatus (VueBio) was placed on the liver with 20–25mm Hg of negative pressure to gently immobilize the liver tissue against a fixed coverslip. The liver draining lymph nodes were explanted onto a glass slide, kept immersed in media and imaged directly. All Images were acquired using the following Ex/Em parameters for the various proteins or fluorophores: PE 561/567–623, YFP 940/525–552, GFP 940/490–510, Qdot705 1080/695–780, APC 1250 or 633/645–700, CTFR 633/640–680, collagen matrix Secondary Harmonic Generation (SHG) (535–545nm). Sequences of acquired image stacks were transformed into volume-rendered, isosurfaced, 3D images and 4D time-lapse movies with Imaris Version 8.1 (Bitplane).
DC-Lm immunizations
To generate memory eGFP+ OT-I T cells, 5000 naïve eGFP+ OT-I T cells were adoptively transferred into B6 mice. Mice were subsequently immunized using a DC- L. monocytogenes prime-boost regimen (Badovinac et al., 2005). Flt-3L-induced splenic DCs were LPS-matured, isolated after collagenase/DNase digestion, and cultured with 2 M of Ova257–264 peptide for 2 hours in a 37°C shaking incubator (Pham et al., 2009). CD11c+ cells were then enriched using anti-CD11c MicroBeads (Miltenyi Biotec). A total of 5x105 purified DCs in saline were used to prime each mouse i.v. After 7 days, mice were boosted i.v. with 107 CFU of recombinant attenuated actA- inlB-deficient LM-OVA257–264. The number of injected bacteria was verified by plating on tryptic soy agar supplemented with 50 μg/ml streptomycin. Mice were rested for more than 30 days before subsequent Plasmodium infection.
Generation of TCR retrogenic mice
TCRβ GAP5040–48 and TCRαβ CS252–260 -specific retrogenic (Rg) mice were generated as previously described (Van Braeckel-Budimir et al., 2017) on a B6 or CB6F1 background using retrovirus-mediated stem cell gene transfer (Holst et al., 2006). B6 bone marrow harboring GAP5040–48 -specific constructs was used to reconstitute sublethally irradiated B6 mice. CB6F1 bone marrow harboring Rg CS252–260 or GAP5040–48 constructs were mixed at a 10:1 ratio respectively (optimized to yield a 1:1 precursor frequency of the Rg cells), injected into sublethally irradiated (9.5Gy) CB6F1 recipients and allowed to reconstitute within the same host.
Isolation of liver cells
In anesthetized (Ketamine/Xylazine (87.5/12.5 mg/Kg)) B6 mice, the inferior vena cava was catheterized (BD auto guard, 22G) aseptically to perfuse the liver by draining through the portal vein. As described in detail before (Doll et al., 2016), steady-state perfusion of the liver was performed first with 1x PBS (4mL/min for 5 minutes), then Liver Perfusion Medium (4mL/min for 3 minutes, Gibco), and finally Liver Digest Medium (4mL/minute for 5 minutes, Gibco) and the perfusate was collected. Digested liver was excised and a single cell suspension made and resuspended in a wash solution of 10% FBS (Sigma-Aldrich) in DMEM (Gibco). Hepatocytes were pelleted and separated from the single cell suspension above by centrifugation at 60g, 3 mins. The supernatant and the whole perfusates were combined and purified using 35% Percoll (GE Healthcare) in HBSS (Gibco). Where indicated, these cells were further negatively or positively enriched by magnetic bead isolation with AutoMacs (Miltenyi Biotec). APC conjugated antibodies against CD3e, CD19, NK1.1, and Ter119 were used to negatively enrich DCs, before separating the various indicated subsets within, using positive enrichment.
In vitro restimulation assay
2x105 CD11c+ cells were isolated from the liver of naïve or Py infected (36h) B6 mice as described above and co-incubated in DMEM culture medium with 1x105 GAP50 TCR retrogenic CD8 T cells for 12h. Subsequently, Brefeldin A was added and co-incubated for 4h before surface and intracellular staining to determine cytokine responses in the retrogenic cells.
RNAseq
Freshly enriched CSF1R+ or CSF1R− DCs (as indicated above) were re-suspended in TRIzol and RNA was purified using a RNAeasy kit (Qiagen) as described previously(Vijay et al., 2017). RNA was assessed for purity and quality using an Agilent 2100 Bioanalyzer and RNA seq was performed using the Clontech StrandedRNA Pico Mammalian kit, on the Illumina HiSeq 2500 High-output platform using 2x50 Paired-end Libraries. The sequence reads quality was checked and were aligned to the mouse genome version mm10 using STAR aligner (Dobin et al., 2013). Gene expression profiles were computed using featureCounts (Liao et al., 2014). Next, the raw gene counts were imported to R and analyzed to detect the differentially expressed genes using DESeq2 (Love et al., 2014).Genes with significant changes were filtered with the adjusted p-value of 0.05. Visualization of differentially expressed genes represented as heat maps were generated using Partek GS software and interaction pathways based on analysis were generated using Ingenuity Pathway Analysis software (Qiagen Bioinformatics).
Data and Software Availability
Raw data from RNAseq is available from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the accession number: GSE126110
Quantification and Statistical Analysis
Statistical differences between two study groups were evaluated using unpaired, two-tailed parametric t-tests or the non-parametric Mann-Whitney test. Statistical differences between more than two study groups were evaluated using a one-way ANOVA with the indicated corrections applied. Statistical significance was calculated based on the ‘n’ numbers as indicated in the figure legends and assigned as *p ≤ 0.05, **p < 0.01, n.s: p>0.05. Statistical analyses were performed using Prism 7 software (GraphPad).
Supplementary Material
Movie S1, related to figure 2: Representative, pseudo-colored multiphoton time-lapse live-microscopy images showing hepatic CD11c (YFP)+ cells (green) harboring Py-GFP (red) in B6.CD11c-YFP mice, inoculated by mosquito bites, at 42h p.i. Hepatic sinusoids represented by vascular dye Qdot 705 (white).
Movie S2, related to figure 2: Representative pseudo-colored multiphoton image shown in Movie S1 rotated on its vertical axis to depict hepatic CD11c (YFP)+ cells (yellow) harboring Py-GFP (green) in B6.CD11c-YFP mice, inoculated by mosquito bites, at 42h p.i. Hepatic sinusoids represented by vascular dye Qdot 705 (red).
Movie S3, related to figure 2: Representative, pseudo-colored multiphoton time-lapse live-microscopy images showing hepatic F4/80+ cells (red) capturing Py-GFP (green) in B6 mice in 24–26h p.i. time-window.
Movie S4, related to figure 2: Representative, pseudo-colored multiphoton time-lapse live-microscopy images showing hepatic F4/80+ cells (red) capturing merosomes budding from a Py-GFP (green) infected hepatocyte in B6 mice in at ~48h p.i.
Highlights.
Monocyte-derived CD11c+ cells are recruited during the liver-stage of malaria
Liver-infiltrating CD11c+ cells acquire Plasmodium, which requires hepatocyte infection
Plasmodium harboring CD11c+ cells have distinct phenotypes and transcriptomes
These CD11c+ cells prime protective CD8+ T cells in liver-draining lymph nodes
Acknowledgments
We thank Drs. Vladimir Badovinac and Noah Butler for constructive comments, Dr. Shoshana Levy (Stanford) for CD81-deficient mice, Dr. Wendy Maury (UIowa) for the MaFIA mice, the staff at the New York University Insectary Core, the University of Iowa Flow Cytometry Core and the University of Minnesota Genomics Core for their assistance. This work was supported by grants from the NIH (AI42767, AI85515, AI100527 to J.T.H., T32 5T32HL007 to SMA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests
References
- Akbari M, Kimura K, Bayarsaikhan G, Kimura D, Miyakoda M, Juriasingani S, Yuda M, Amino R, and Yui K (2018). Nonspecific CD8(+) T Cells and Dendritic Cells/Macrophages Participate in Formation of CD8(+) T Cell-Mediated Clusters against Malaria Liver-Stage Infection. Infection and immunity 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arreaza G, Corredor V, and Zavala F (1991). Plasmodium yoelii: quantification of the exoerythrocytic stages based on the use of ribosomal RNA probes. Experimental parasitology 72, 103–105. [DOI] [PubMed] [Google Scholar]
- Austin LS, Kaushansky A, and Kappe SH (2014). Susceptibility to Plasmodium liver stage infection is altered by hepatocyte polyploidy. Cell Microbiol 16, 784–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badovinac VP, Messingham KA, Jabbari A, Haring JS, and Harty JT (2005). Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nature medicine 11, 748–756. [DOI] [PubMed] [Google Scholar]
- Barbier L, Tay SS, McGuffog C, Triccas JA, McCaughan GW, Bowen DG, and Bertolino P (2012). Two lymph nodes draining the mouse liver are the preferential site of DC migration and T cell activation. J Hepatol 57, 352–358. [DOI] [PubMed] [Google Scholar]
- Bijker EM, Bastiaens GJ, Teirlinck AC, van Gemert GJ, Graumans W, van de Vegte-Bolmer M, Siebelink-Stoter R, Arens T, Teelen K, Nahrendorf W, et al. (2013). Protection against malaria after immunization by chloroquine prophylaxis and sporozoites is mediated by preerythrocytic immunity. Proceedings of the National Academy of Sciences of the United States of America 110, 7862–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna-Romero O, Hafalla JC, Gonzalez-Aseguinolaza G, Sano G, Tsuji M, and Zavala F (2001). Detection of malaria liver-stages in mice infected through the bite of a single Anopheles mosquito using a highly sensitive real-time PCR. International journal for parasitology 31, 1499–1502. [DOI] [PubMed] [Google Scholar]
- Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, and Cohen DA (2004). Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. J Leukoc Biol 75, 612–623. [DOI] [PubMed] [Google Scholar]
- Butler NS, Schmidt NW, Vaughan AM, Aly AS, Kappe SH, and Harty JT (2011). Superior antimalarial immunity after vaccination with late liver stage-arresting genetically attenuated parasites. Cell host & microbe 9, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, and Zavala F (2007). CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nature medicine 13, 1035–1041. [DOI] [PubMed] [Google Scholar]
- Coelho CH, Doritchamou JYA, Zaidi I, and Duffy PE (2017). Advances in malaria vaccine development: report from the 2017 malaria vaccine symposium. NPJ Vaccines 2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman AF, Healer J, Marapana D, and Marsh K (2016). Malaria: Biology and Disease. Cell 167, 610–624. [DOI] [PubMed] [Google Scholar]
- Crompton PD, Moebius J, Portugal S, Waisberg M, Hart G, Garver LS, Miller LH, Barillas-Mury C, and Pierce SK (2014). Malaria immunity in man and mosquito: insights into unsolved mysteries of a deadly infectious disease. Annu Rev Immunol 32, 157–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll KL, Pewe LL, Kurup SP, and Harty JT (2016). Discriminating Protective from Nonprotective Plasmodium-Specific CD8+ T Cell Responses. Journal of immunology 196, 4253–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimkhani MR, Mohar I, and Crispe IN (2011). Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 54, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, Lau LS, Mollard V, Cozijnsen A, Collins N, et al. (2016). Liver-Resident Memory CD8(+) T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity 45, 889–902. [DOI] [PubMed] [Google Scholar]
- Heath WR, and Carbone FR (2001). Cross-presentation, dendritic cells, tolerance and immunity. Annu Rev Immunol 19, 47–64. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, and Carbone FR (1994). T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27. [DOI] [PubMed] [Google Scholar]
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, and Vignali DA (2006). Generation of T-cell receptor retrogenic mice. Nature protocols 1, 406–417. [DOI] [PubMed] [Google Scholar]
- Holz LE, Fernandez-Ruiz D, and Heath WR (2016). Protective immunity to liver-stage malaria. Clin Transl Immunology 5, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, and Renia L (2013). Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med 5, 984–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joffre OP, Segura E, Savina A, and Amigorena S (2012). Cross-presentation by dendritic cells. Nature reviews. Immunology 12, 557–569. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. (2002). In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushansky A, Metzger PG, Douglass AN, Mikolajczak SA, Lakshmanan V, Kain HS, and Kappe SH (2013). Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis 4, e762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, et al. (2005). Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22, 643–654. [DOI] [PubMed] [Google Scholar]
- Kurup SP, Obeng-Adjei N, Anthony SM, Traore B, Doumbo OK, Butler NS, Crompton PD, and Harty JT (2017). Regulatory T cells impede acute and long-term immunity to blood-stage malaria through CTLA-4. Nature medicine 23, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LS, Fernandez-Ruiz D, Mollard V, Sturm A, Neller MA, Cozijnsen A, Gregory JL, Davey GM, Jones CM, Lin YH, et al. (2014). CD8+ T cells from a novel T cell receptor transgenic mouse induce liver-stage immunity that can be boosted by blood-stage infection in rodent malaria. PLoS pathogens 10, e1004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiriao P, Mota MM, and Rodriguez A (2005). Apoptotic Plasmodium-infected hepatocytes provide antigens to liver dendritic cells. The Journal of infectious diseases 191, 1576–1581. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin JW, Shaw TN, Annoura T, Fougere A, Bouchier P, Chevalley-Maurel S, Kroeze H, Franke-Fayard B, Janse CJ, Couper KN, and Khan SM (2014). The subcellular location of ovalbumin in Plasmodium berghei blood stages influences the magnitude of T-cell responses. Infection and immunity 82, 4654–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, and Nussenzweig MC (2004). Visualizing dendritic cell networks in vivo. Nature immunology 5, 1243–1250. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecker HT, and Levy S (1997). Normal lymphocyte development but delayed humoral immune response in CD81-null mice. The Journal of experimental medicine 185, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MD, Condotta SA, Harty JT, and Badovinac VP (2012). Population dynamics of naive and memory CD8 T cell responses after antigen stimulations in vivo. Journal of immunology 188, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, and Mortha A (2013). The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31, 563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, and Jung S (2014). Development and function of dendritic cell subsets. Immunity 40, 642–656. [DOI] [PubMed] [Google Scholar]
- Nierkens S, Tel J, Janssen E, and Adema GJ (2013). Antigen cross-presentation by dendritic cell subsets: one general or all sergeants? Trends in immunology 34, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig RS, Vanderberg J, Most H, and Orton C (1967). Protective immunity produced by the injection of x-irradiated sporozoites of plasmodium berghei. Nature 216, 160–162. [DOI] [PubMed] [Google Scholar]
- Parmar R, Patel H, Yadav N, Parikh R, Patel K, Mohankrishnan A, Bhurani V, Joshi U, and Dalai SK (2018). Infectious Sporozoites of Plasmodium berghei Effectively Activate Liver CD8alpha(+) Dendritic Cells. Frontiers in immunology 9, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham NL, Badovinac VP, and Harty JT (2009). A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. Journal of immunology 183, 2337–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham NL, Badovinac VP, and Harty JT (2011). Differential role of “Signal 3” inflammatory cytokines in regulating CD8 T cell expansion and differentiation in vivo. Frontiers in immunology 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, Burki K, Lang R, Hengartner H, and Zinkernagel RM (1989). Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342, 559–561. [DOI] [PubMed] [Google Scholar]
- Prado M, Eickel N, De Niz M, Heitmann A, Agop-Nersesian C, Wacker R, Schmuckli-Maurer J, Caldelari R, Janse CJ, Khan SM, et al. (2015). Long-term live imaging reveals cytosolic immune responses of host hepatocytes against Plasmodium infection and parasite escape mechanisms. Autophagy 11, 1561–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke AJ, Kastenmuller W, Espinosa DA, Gerner MY, Tse SW, Sinnis P, Germain RN, Zavala FP, and Cockburn IA (2015). Lymph-node resident CD8alpha+ dendritic cells capture antigens from migratory malaria sporozoites and induce CD8+ T cell responses. PLoS pathogens 11, e1004637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai D, Pham NL, Harty JT, and Badovinac VP (2009). Tracking the total CD8 T cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. Journal of immunology 183, 7672–7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Ochando J, and Partida-Sanchez S (2008). Migration of dendritic cell subsets and their precursors. Annu Rev Immunol 26, 293–316. [DOI] [PubMed] [Google Scholar]
- Rieckmann KH, Beaudoin RL, Cassells JS, and Sell KW (1979). Use of attenuated sporozoites in the immunization of human volunteers against falciparum malaria. Bull World Health Organ 57 Suppl 1, 261–265. [PMC free article] [PubMed] [Google Scholar]
- Sathe P, Pooley J, Vremec D, Mintern J, Jin JO, Wu L, Kwak JY, Villadangos JA, and Shortman K (2011). The acquisition of antigen cross-presentation function by newly formed dendritic cells. Journal of immunology 186, 5184–5192. [DOI] [PubMed] [Google Scholar]
- Schwartz L, Brown GV, Genton B, and Moorthy VS (2012). A review of malaria vaccine clinical projects based on the WHO rainbow table. Malaria journal 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seder RA, Chang LJ, Enama ME, Zephir KL, Sarwar UN, Gordon IJ, Holman LA, James ER, Billingsley PF, Gunasekera A, et al. (2013). Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365. [DOI] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, and Matloubian M (2006). CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440, 540–544. [DOI] [PubMed] [Google Scholar]
- Sierro F, Evrard M, Rizzetto S, Melino M, Mitchell AJ, Florido M, Beattie L, Walters SB, Tay SS, Lu B, et al. (2017). A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity 47, 374–388 e376. [DOI] [PubMed] [Google Scholar]
- Silvie O, Rubinstein E, Franetich JF, Prenant M, Belnoue E, Renia L, Hannoun L, Eling W, Levy S, Boucheix C, and Mazier D (2003). Hepatocyte CD81 is required for Plasmodium falciparum and Plasmodium yoelii sporozoite infectivity. Nature medicine 9, 93–96. [DOI] [PubMed] [Google Scholar]
- Van Braeckel-Budimir N, Gras S, Ladell K, Josephs TM, Pewe L, Urban SL, Miners KL, Farenc C, Price DA, Rossjohn J, and Harty JT (2017). A T Cell Receptor Locus Harbors a Malaria-Specific Immune Response Gene. Immunity 47, 835–847 e834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk MR, Douradinha B, Franke-Fayard B, Heussler V, van Dooren MW, van Schaijk B, van Gemert GJ, Sauerwein RW, Mota MM, Waters AP, and Janse CJ (2005). Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proceedings of the National Academy of Sciences of the United States of America 102, 12194–12199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan AM, and Kappe SHI (2017). Genetically attenuated malaria parasites as vaccines. Expert Rev Vaccines 16, 765–767. [DOI] [PubMed] [Google Scholar]
- Vijay R, Fehr AR, Janowski AM, Athmer J, Wheeler DL, Grunewald M, Sompallae R, Kurup SP, Meyerholz DK, Sutterwala FS, et al. (2017). Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling. Proceedings of the National Academy of Sciences of the United States of America 114, E5444–E5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2018). World Malaria Report 2018 (World Health Organization; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1, related to figure 2: Representative, pseudo-colored multiphoton time-lapse live-microscopy images showing hepatic CD11c (YFP)+ cells (green) harboring Py-GFP (red) in B6.CD11c-YFP mice, inoculated by mosquito bites, at 42h p.i. Hepatic sinusoids represented by vascular dye Qdot 705 (white).
Movie S2, related to figure 2: Representative pseudo-colored multiphoton image shown in Movie S1 rotated on its vertical axis to depict hepatic CD11c (YFP)+ cells (yellow) harboring Py-GFP (green) in B6.CD11c-YFP mice, inoculated by mosquito bites, at 42h p.i. Hepatic sinusoids represented by vascular dye Qdot 705 (red).
Movie S3, related to figure 2: Representative, pseudo-colored multiphoton time-lapse live-microscopy images showing hepatic F4/80+ cells (red) capturing Py-GFP (green) in B6 mice in 24–26h p.i. time-window.
Movie S4, related to figure 2: Representative, pseudo-colored multiphoton time-lapse live-microscopy images showing hepatic F4/80+ cells (red) capturing merosomes budding from a Py-GFP (green) infected hepatocyte in B6 mice in at ~48h p.i.
Data Availability Statement
Raw data from RNAseq is available from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) with the accession number: GSE126110