

Summary
In response to infection, naive CD4+ T‐cells proliferate and differentiate into several possible effector subsets, including conventional T helper effector cells (TH1, TH2, TH17), T regulatory cells (Treg) and T follicular helper cells (TFH). Once infection is cleared, a small population of long‐lived memory cells remains that mediate immune defenses against reinfection. Memory T lymphocytes have classically been categorized into central memory cell (TCM) and effector memory cell (TEM) subsets, both of which circulate between blood, secondary lymphoid organs and in some cases non‐lymphoid tissues. A third subset of memory cells, referred to as tissue‐resident memory cells (TRM), resides in tissues without recirculation, serving as ‘first line’ of defense at barrier sites, such as skin, lung and intestinal mucosa, and augmenting innate immunity in the earliest phases of reinfection and recruiting circulating CD4+ and CD8+ T‐cells. The presence of multiple CD4+ T helper subsets has complicated studies of CD4+ memory T‐cell differentiation, and the mediators required to support their function. In this review, we summarize recent investigations into the origins of CD4+ memory T‐cell populations and discuss studies addressing CD4+ TRM differentiation in barrier tissues.
Keywords: protective immunity, T helper
Introduction
CD4+ T lymphocytes are a key element of adaptive immunity, acting to direct and enhance functions of innate cells, B‐cells and CD8+ T‐cells in response to diverse pathogens. Following infection, naive CD4+ T‐cells proliferate and have the potential to differentiate into at least seven functionally distinct T helper (TH) subsets with unique effector functions within the circulation, secondary lymphoid organs (SLO) and infected tissues.1 Depending on the type of immunological threat, early host–pathogen interactions result in an infection milieu that directs differentiation of naive CD4+ T‐cells to acquire the helper functions to ensure that each class of pathogen is countered with the appropriate immune response.2 Once infection has cleared, the majority of the effector cells die via apoptosis during a contraction phase, while a small proportion persists and differentiates into long‐lived memory cells. This memory population enables a rapid and robust secondary response against recurring pathogens and, thus, is pivotal in conferring lasting cellular immunity particularly against pathogens where neutralizing antibodies alone are insufficient at providing long‐term protection.
While significant advances have been made in understanding the generation and maintenance of memory CD8+ T‐cells and B‐cells, the molecular mechanisms underlying the generation of memory CD4+ T‐cells remain relatively elusive. Two major obstacles have contributed to this knowledge deficit. First, CD4+ T‐cells are inherently less proliferative and the CD4+ memory T‐cell population appears to decline following antigen clearance, while the CD8+ memory T‐cell population, if established, is typically stable3, 4, 5 and thus, fewer cells are available for study. Second, the existence of functionally distinct effector TH cell subsets hinders the ability to characterize a common CD4+ memory T‐cell precursor. Further, TH effector and memory T‐cells also exhibit significant plasticity and can interconvert between lineages, both in vivo and in vitro, adding an additional layer of complexity to identifying memory precursor cells in CD4+ T memory studies.6, 7, 8, 9, 10, 11
Memory T‐cells have been conventionally divided into central memory (TCM) cells, which circulate between the blood and SLO, and effector memory (TEM) cells, which can migrate from the blood into non‐lymphoid tissues 1, 11. Over the past decade, evidence of a novel subset of memory cells called tissue‐resident memory T‐cells (TRM) has emerged. TRM are seeded in non‐lymphoid tissues, particularly at barrier sites like the skin and intestinal mucosa.12, 13, 14, 15 TRM have limited recirculation out of tissues and serve as sentinels at sites of potential reinfection, where they coordinate the initial response to pathogens and provide a substantial boost to tissue immunity via direct antigen recognition and recruitment of circulating immune cells.13, 16 The majority of studies on TRM have focused on CD8+ tissue‐resident lymphocyte differentiation, survival and function, while less is known about their CD4+ counterparts, which are also known to mediate antiviral responses.17, 18 The relationship between circulating and tissue‐resident CD4+ memory T‐cell origins and TH effectors subsets is currently unknown. A better understanding of the precursors of CD4+ TRM and the molecular mechanisms mediating their differentiation will allow us to harness the protective capacity of this memory population and modulate their activity in the context of infection or inflammatory diseases.
Despite the challenges in studying memory CD4+ T‐cells, efforts in recent years focusing on different aspects of memory development have begun to elucidate a more comprehensive picture for the generation and maintenance of memory CD4+ T‐cells. In this review, we summarize recent studies addressing the identity of memory CD4+ T‐cell populations and their precursors in both the periphery and non‐lymphoid tissues.
CD4+ T‐cell memory in secondary lymphoid organs
Despite clear differences between memory CD4+ and CD8+ T‐cell populations, including the range of effector cell heterogeneity,3 the models for memory CD8+ T‐cell formation have served as a useful framework for investigation of memory CD4+ T‐cells. During the primary response of antigen‐specific cytotoxic CD8+ T‐cells (CTL), two effector CD8+ T‐cell populations can be identified based on surface expression of killer cell lectin‐like receptor subfamily G member 1 (KLRG1) and interleukin‐7 receptor‐α (CD127).19 The KLRG1hiCD127lo population, termed terminal effector cells (TE), is predominantly lost during the contraction phase, while the KLRG1loCD127hi subset contains memory‐precursor cells (MP), which can differentiate into long‐lived memory CD8+ T‐cells.19 CD4+ T‐cells also express KLRG120 and CD127.21 However, the roles of these molecules in memory CD4+ populations are not well established nor are clear strategies for distinguishing shorter‐lived effector and precursors of memory TH populations.
Evidence for long‐lived CD4+ memory T‐cells capable of responding to pathogen re‐challenge has been documented in studies of adoptive transfer of T‐cell receptor (TCR) transgenic T‐cells22, 23, 24, 25 and endogenous immune responses.23 However, the diversity of functional TH phenotypes has made identification of distinct CD4+ TE and MP effector populations challenging. Additionally, it is unclear whether all CD4+ TH effector T‐cells possess the same potential to differentiate into long‐lived memory cells. A separate MP may exist for each subset or there may be a unique effector subset with an inherent memory programme that can give rise to memory populations with the potential to generate TH subsets with all or some effector functions [TH1, TH2, TH17, T follicular helper cells (TFH), T regulatory cells (Treg)] in a secondary infection. An elegant study by Tubo et al.26 addressed this issue by following the differentiation of individual CD4+ T‐cells responding to infection. Utilizing over 80 distinct TCR clones that can specifically respond to Listeria monocytogenes (LM) infection, they demonstrated that all microbe‐specific naive CD4+ T‐cells have the potential to give rise to memory cells following acute infection.26 Different individual naive CD4+ T‐cells generated antigen‐specific effector populations with varying frequencies of TH1 and TFH effector cells. Notably, the relative frequencies of these subsets were preserved into the memory phase, suggesting that both TH1 and TFH effector populations contain precursors of memory cells that retain their effector TH characteristics (Fig. 1a). These data favour the idea that some CD4+ memory cells are relatively lineage‐committed; however, a range of expansion potential and plasticity among progeny was also observed, suggesting that not all CD4+ memory precursor cells may be equivalent.
Figure 1.
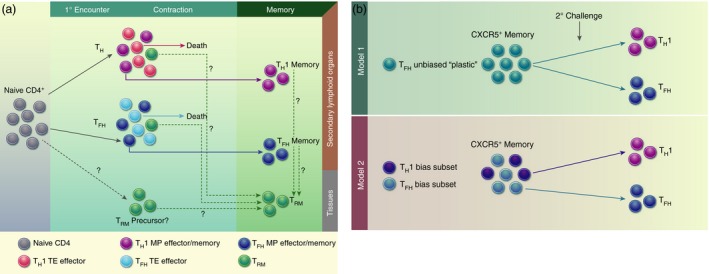
Models of CD4+ memory T‐cell formation. (a) Upon antigen encounter, naive CD4+ T‐cells differentiate into effector subsets based on the type of infection. Within each effector CD4+ subset, there potentially exist terminal effectors (TE) and memory precursor (MP) effectors. The majority of TEs die during the contraction, while MPs can survive and transition into resting memory cells. CD4+ tissue‐resident memory cells (TRM) may differentiate from: (1) the naive subset; (2) MP cells within the effector population; or (3) committed memory cells. (b) Two models for T follicular helper cell (TFH) multi‐potency: (1) TFH memory cells retain cellular plasticity and can differentiate into TH1 or TFH secondary effectors based on signals present during secondary challenge; (2) TFH memory cells are actually a heterogeneous population with subsets that are biased or primed towards a particular secondary effector lineage (TH1 or TFH).
TH1 and TFH CD4+ memory T‐cells
In efforts to address these questions, several groups have used lymphocytic choriomeningitis virus (LCMV) to characterize the response of adoptively transferred SMARTA (SM) cells, which have transgenic expression of an MHC Class II‐restricted T‐cell antigen receptor (TCR) specific for LCMV glycoprotein amino acids 66–77.24, 25, 27, 28 Meanwhile, other investigators have studied the endogenous polyclonal response by utilizing the peptide‐loaded major histocompatibility complex class II (pMHCII) tetramer‐based approach to identify antigen‐specific CD4+ T‐cells.22, 23, 25 During acute infection with LCMV‐Armstrong, antigen‐specific CD4+ T‐cells differentiate into two main helper subtypes in the spleen and lymph nodes: TH1 and TFH. TH1 cells express the transcriptional regulator T‐bet and are known for secreting their signature effector molecule, interferon gamma (IFNɣ), while TFH cells express Bcl6 and their hallmark surface molecule C‐X‐C chemokine receptor type 5 (CXCR5), which allows for homing to germinal centres to support B‐cell responses. To explore the origins of TH1 and TFH memory cells, investigators utilized fluorescence‐activated cell sorting to isolate TH1 and TFH effector and memory cells based on known markers, and studied their characteristics in the context of reinfection.21, 24, 25, 26
Marshall et al.24 found that within the primary effector populations from the spleen at day 8, two CD4+ T‐cell subsets that resembled the CD8+ TE and MP T‐cells were observed. The TE‐like population was marked by high expression of both P‐selectin glycoprotein ligand‐1 (PSGL‐1) and lymphocyte antigen 6 complex (Ly6C), while the MP‐like effector cells were PSGL‐1hiLy6Clo. In contrast to the PSGL‐1hiLy6Chi cells, the PSGL‐1hiLy6Clo MP‐like population exhibited greater longevity in uninfected hosts, increased proliferation following antigen re‐challenge, and similar gene‐expression profiles with day 60 PSGL‐1hi memory CD4+ T‐cells.24 These results led the authors to propose that differential expression of Ly6C can distinguish TE from MP cells within the TH1 subset. At day 8, PSGL‐1loLy6Clo effector cells showed high expression of known TFH markers (ICOS, CXCR5, PD‐1). This PSGL‐1loLy6Clo subset was found along with PSGL‐1hiLy6Chi and PSGL‐1hiLy6Clo TH1 cells within the memory cells at day 150 after infection, suggesting that MP of both TH1 and TFH phenotypes may persist long term.24 Interestingly, while the PSGL‐1hiLy6Clo MP population was thought to be primarily TH1 cells, it was later shown by Choi et al.25 that the PSGL‐1hiLy6Clo MP population actually contains both CXCR5− TH1 and CXCR5+ TFH cells at comparable frequencies. These results highlight the complexity and heterogeneity within CD4+ memory T‐cells and the need for further studies to fully understand the nature of the CD4+ memory T‐cell pool.
To investigate the potential of TFH memory cells for re‐differentiation upon reinfection, Hale et al.27 utilized expression of CXCR5 and Ly6C to distinguish between TH1 (CXCR5−Ly6Chi) and TFH (CXCR5+Ly6Clo & CXCR5+Ly6Cint) memory populations following acute infection with LCMV‐Armstrong, then transferred each of the three subsets into naive hosts for reinfection. TH1 memory cells mostly maintained high Ly6C expression with few effector cells gaining CXCR5 expression, while TFH memory cells were able to give rise to both CXCR5−Ly6Chi TH1 cells and CXCR5+Ly6Clo/int TFH cells. This multi‐potency of TFH memory cells during re‐challenge has also been observed in acute bacterial infection with LM29 as well as in viral influenza infection.28
In a concurrent study, Pepper et al. addressed CD4+ memory T‐cell differentiation using LM infection and the expression of CXCR5 and CC chemokine receptor 7 (CCR7), a marker used in previous studies to identify TCM. During acute infection, antigen‐specific effector cells segregated into a CXCR5− population favouring the TH1 phenotype and a CXCR5+ population.30 A fraction of the CXCR5− TH1 population, which the authors termed TH1 effector memory cells, survived to a memory time point and, upon re‐challenge, produced TH1 effector cells. The CXCR5+ effector population included cells with high expression of the lineage‐defining factor Bcl6, were localized to follicles and were termed TFH, while cells with lower Bcl6 levels showed co‐expression of CCR7 and were termed TCM. It is worth noting that the TFH subset resembled what some studies term germinal centre TFH cells (GC TFH); GC TFH can lose expression of Bcl6 after infection, suggesting that, depending on the time point, the CXCR5+ population can include cells that did not enter the GC as well as those that were transiently in the GC. While both TFH and TCM in this study expressed CXCR5, TCM were not seen in the follicles and, upon re‐challenge, produced both TH1 effector cells and CXCR5+ cells that likely include TFH and GC TFH. 30 Notably, Choi et al.25 found that precursors of TFH or the CXCR5+ populations show greater potential to develop into memory cells compared with TH1 precursors and share gene‐expression signatures with memory CD8+ T‐cells. These results suggest that both TH1 and TFH effector T‐cells can give rise to memory cells, and CXCR5+ TFH‐derived memory cells have greater plasticity in generating secondary effector phenotypes.
Corroborative reports affirming the increased plasticity of TFH memory relative to TH1 memory upon re‐challenge suggest that TFH memory populations may retain a greater cellular ‘stem‐ness’ and are capable of providing a more comprehensive and robust secondary response during re‐infection. Two possible models can explain the multi‐potency demonstrated by CXCR5+ memory cells (Fig. 1b). One possible explanation is that TFH memory cells are inherently more plastic compared with other TH memory cells and, therefore, retain the ability to differentiate into alternative helper lineages upon reinfection. A second possibility is that the CXCR5+ memory population actually contains distinct subsets that are programmed or biased towards a specific TH lineage upon secondary challenge. In this case, CXCR5+CCR7+ could distinguish memory cells with the greater potential for re‐expansion, while CXCR5+CCR7− cells may be long‐lived TFH/GC TFH cells that have downregulated Bcl6 and PD‐1, and are more similar to long‐lived effector subsets. Based on the data currently available, neither hypothesis can be eliminated and further characterization of TFH memory cells, perhaps using single‐cell approaches, is needed to determine whether the multi‐potency of TFH memory is the result of cellular plasticity or population heterogeneity, or both.
In line with this idea, a recent study by Ciucci et al.31 utilized single‐cell RNA sequencing to investigate the heterogeneity of antigen‐specific CD4+ effector T‐cells in response to acute LCMV infection. Visualization with t‐distributed stochastic neighbour embedding (t‐SNE) of day 7 effector T‐cells yielded multiple transcriptionally distinct clusters showcasing the heterogeneity exhibited by TH1 and TFH effector cells. At 30 days post‐infection, single cell analysis also showed multiple distinct transcriptional clusters with shared TFH features, supporting the idea that memory CXCR5+ TFH multi‐potency may be the result of population heterogeneity.
TH2 CD4+ memory T‐cells
TH2 memory cells have been best characterized in the context of allergic inflammatory disorders,32 though some studies have highlighted this population's role in defense against parasitic worm infection. As mentioned previously, antigen‐experienced CD4+ TH cells contract more rapidly after pathogen clearance compared with CD8+ T‐cells,4 which is why early investigations into TH2 memory relied on adoptive cell transfers of in vitro polarized TH2 effectors.33 This system involved activating CD4+ T‐cells in vitro with antigen and antigen‐presenting cells (APCs) followed by culturing in TH polarizing conditions33 and subsequent adoptive transfer. Interestingly, in vitro generated TH1 and TH2 cells retained their expression of lineage‐defining transcription factors (TFs), T‐bet and GATA3, respectively, for months after transfer into naive hosts.34 However, upon viral infection with LCMV, in vitro‐derived TH2 memory cells were able to adapt a TH1 phenotype and persist as a ‘hybrid’ memory cell with combined TH1 and TH2 characteristics.34 Utilizing a similar in vitro polarization system, Endo et al.35 identified an interleukin‐5 (IL‐5)‐producing subset of TH2 memory cells in the spleen that is primarily responsible for asthmatic symptoms, such as eosinophilic infiltration into the airway, airway hyper‐responsiveness and mucus hyper‐production in a murine model of TH2‐driven allergic airway inflammation. These studies provided early evidence of the potential existence of TH2 memory populations, but data demonstrating direct in vivo generation were lacking until recently.
A study by Hondowicz et al.36 provided key insights into TH2 memory studying the endogenous allergen‐specific CD4+ T‐cells induced in response to house dust mite (HDM) inoculation. Using pMHCII tetramers to follow antigen‐specific CD4+ T‐cells, the authors showed an expansion of allergen‐specific CD4+ TH2 cells in SLOs and the lung following intranasal HDM administration. Notably, this allergen system induces both antigen‐specific TH2 and TFH cells, analogous to the TH1 and TFH response against LCMV‐Armstrong. The allergen‐specific memory pool in the SLOs consisted of CXCR5+ and CXCR5− cells that also expressed CCR7+, consistent with the earlier observations that memory T‐cells retain characteristics of TH effector phenotypes. The authors further explored characteristics of allergen‐specific TH2 resident in the lung, which will be discussed in the lung CD4+ TRM section below.
TH17 CD4+ memory T‐cells
Though not as extensively characterized as other helper subsets, memory TH17 cells have been documented in both humans and mice, primarily in the context of autoimmunity.37 Early memory experiments using LM infection showed that TH17 cells existed only transiently following intranasal infection.23 However, it is worth noting that LM may not be an optimal infection for TH17 studies as it is an intracellular pathogen38 and most efficiently induces TH1 cells. Muranski et al.39 reported on long‐lived memory TH17 cells but, similar to early TH2 studies, these cells required in vitro polarization prior to transfer into host mice. In a recent study of dry eye disease, Chen et al.40 utilized a pre‐clinical murine model of autoimmune ocular disease, where mice were subjected to 14 days of environmental desiccating stress followed by rest in normal conditions for 14 days, and found disease‐specific pathogenic memory TH17 cells in both the inflamed site and draining lymph nodes. Two cytokines associated with CD4+ memory T lymphocytes, IL‐7 and IL‐1541 were shown to be crucial in the maintenance of these pathogenic TH17 cells. Neutralization of these cytokines with topical application of anti‐IL‐7 or anti‐IL‐15 antibody decreased the number of TH17 cells in both the conjunctivae and lymph nodes, offering a potential therapy for autoimmune disorders. One crucial caveat to note is that these ‘memory’ TH17 cells were studied under the chronic inflammatory environment of autoimmunity, perhaps under prolonged or recurrent exposure to antigen; therefore, this population's identity as true resting memory cells remains uncertain.
Uncovering the origin and identity of resting memory or MP cells within a particular helper T‐cell lineage will lay the foundation for future molecular studies into how each memory TH lineage is uniquely regulated. However, in the next section, we will review two biological requirements crucial for memory formation that appear to be conserved across all TH subsets.
Memory differentiation cues: TCR signalling and IL‐2
A recent comprehensive review of studies aimed at resolving the signals required for CD4+ memory T‐cell formation42 discussed the instructive signals both during the ‘early priming’ phase of initial antigen recognition and activation as well as at ‘late‐acting checkpoints’ prior to contraction that play a role in the effector‐to‐memory transition. Much like the signals important for CD8+ memory T‐cell generation, strengths of TCR and co‐stimulatory signalling also have profound effects on memory TH development.42, 43 Recent results from Snook et al.44 demonstrated that TCR signalling has a direct impact on TH memory formation. Utilizing a panel of TCRs specific for the same viral antigen, the authors showed substantial variability in TCR signal strength, expression of IL‐2‐receptor alpha (CD25) and activation of downstream TFs across the CD4+ memory T‐cell population.44 TCR clones with stronger TCR signalling appear to differentiate towards a more TE state and become largely depleted by memory time points, while clones with comparatively lower signalling were memory‐like and able to persist after antigen clearance. Interestingly, it seems that stronger TCR signalling was associated with higher expression of TH1 markers, while weaker TCR signals correlated with higher expression of TFH markers,44 suggesting that there may be a connection between lineage differentiation and memory potential for CD4+ helper T‐cells.
Utilizing influenza A virus (IAV) as an infection model, McKinstry et al.45 showed that IL‐2 is crucial at a late checkpoint for effector helper T‐cells to survive the contraction phase, allowing for the transition into resting memory cells. To circumvent defects in initial T‐cell priming caused by IL‐2 deficiency, the authors first activated CD4+ T‐cells in vitro with exogenous IL‐2 and then transferred these cells into naive mice for infection. Following IAV challenge, both in vitro primed wild‐type and IL‐2‐deficient donors showed similar cell numbers at the peak of infection and production of IFNɣ; however, the IL‐2‐deficient population quickly declined and was undetectable by day 28 of infection. Exogenous administration of IL‐2 during days 5–7 of infection successfully restored memory cell numbers for IL‐2‐deficient CD4+ T‐cells, demonstrating the importance of IL‐2 for CD4+ memory T‐cell generation in this context. Furthermore, a recent study by DiToro et al.46 with LM infection showed that as early as 20 hr after antigen exposure in vivo, IL‐2 production in CD4+ TH effector cells strongly correlated with TH fate differentiation during infection, again supporting a link between lineage specification and memory formation. To further highlight the importance of IL‐2 in TH memory, Shakya et al.47 identified a role for TF Oct1 and its coactivator OCA‐B in poising the Il2 locus for robust expression in memory CD4+ T‐cells, unveiling an important mechanism by which memory CD4+ T‐cells control IL‐2 production. However, these studies regarding TCR signalling and IL‐2 in CD4+ memory T‐cells were done without investigation of specific TH lineages. Therefore, further investigations into the required transcriptional and epigenetic regulation for generation and maintenance of memory TH subsets are needed. Additionally, while targeting peripheral memory cells in vaccination strategies can provide systemic protection, in some cases a localized strategy in which tissue‐resident cells at barrier surfaces are activated as front‐line defense against recurrent infections may be more effective, thus how these signals pertain to TRM will be informative.48
Tissue‐resident CD4+ memory T‐cells
Much like circulating CD4+ memory T‐cells, studies of tissue‐resident lymphocytes have predominantly focused on CD8+ TRM due to the heterogeneity of CD4+ memory T‐cells and the existing gaps in knowledge regarding mechanisms governing memory CD4+ T‐cell formation. Nevertheless, recent studies have highlighted a prominent population of long‐lived CD4+ T‐cells within many non‐lymphoid tissues, including the lungs,36, 49, 50, 51, 52, 53, 54, 55, 56, 57 small intestine (SI),12, 18, 58, 59, 60 skin,15, 61, 62, 63, 64 and female reproductive tract (FRT)18, 65, 66 (Fig. 2). Similar to their CD8+ T‐cell counterparts, CD4+ TRM have been shown to facilitate rapid immune defense upon re‐exposure to antigen and can supplant innate immunity in recognizing and responding to recurrent infections.67 However, much remains to be explored about the phenotype, function and maintenance of CD4+ TRM during infections. Additionally, differences exist between CD4+ and CD8+ TRM in tissue localization, surface marker expression and cytokine cues driving TRM formation; these outstanding questions in the field must be addressed to better define the identity and differentiation of CD4+ TRM.
Figure 2.
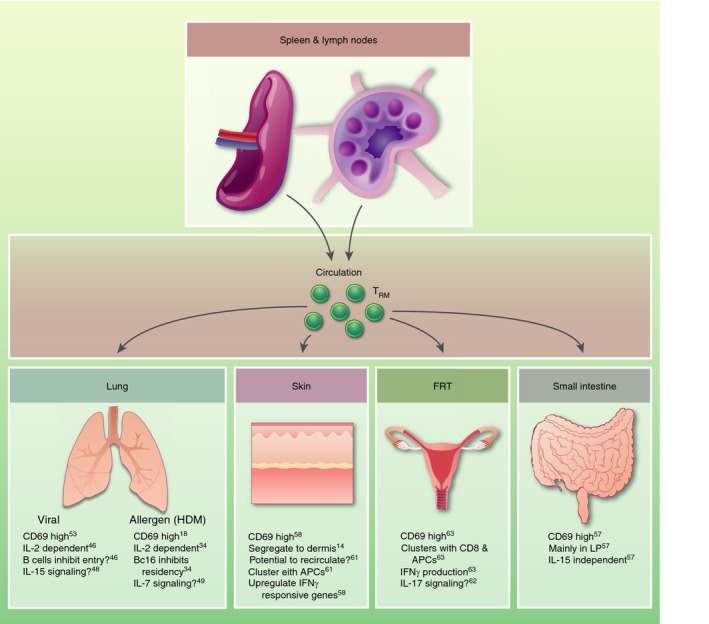
Following infection, CD4+ tissue‐resident memory cells (TRM) are recruited to tissues from circulation and secondary lymphoid organs (SLOs). The majority of CD4+ TRM express high levels of CD69 and can form clusters with other resident immune subsets, including CD8+ TRM, macrophages and antiegn‐presenting cells (APCs). Cytokine signalling may also play a role in recruitment and retention, although specific cytokines are preferred by each tissue.
Classically, tissue‐resident memory T lymphocytes have been defined using parabiosis experiments in which a naive mouse and an immune mouse, previously exposed to antigen, are surgically joined to create a shared circulatory system.56, 68 Thus, all circulating cells will normalize between both partners while the non‐circulating tissue‐resident cells remain lodged in the tissues of the immune mouse. Alternative methods have been developed and validated to assess whether cells remain in tissues, including intravenous injection of a fluorescently labelled antibody to mark cells in the circulating system.69 Any cells positive for the label are considered ‘circulating’, while unlabelled cells are assumed to have limited access to circulation and are therefore ‘tissue‐resident’.56 To determine the protective functions of tissue‐resident lymphocytes in secondary infection, immune mice are treated with FTY‐720, an agonist of sphingosine‐1‐phosphate receptor 1 (S1PR1), which causes decreased surface expression of S1PR1 and therefore prevents egress of circulating memory cells from lymph nodes.70 When these mice are re‐challenged with the original pathogen, any immune response at the local site of infection will be mediated only by cells resident to that tissue.71
Lung CD4+ TRM cells
The lungs contain a population of CD4+ TRM that have been shown to play a critical role in recruiting CD8+ T‐cells and enhancing secondary immune responses against bacterial, viral and worm infections.51, 55, 56, 72 In an influenza infection model, antigen‐specific memory CD4+ T‐cells migrate to the lungs and are retained in the tissue without recirculation, as demonstrated by parabiosis experiments.56 This subset of CD4+ T‐cells shows a distinct phenotype from circulating populations; specifically, high expression of CD69, a membrane‐bound type II C‐lectin receptor and known marker of tissue retention.73, 74 Functionally, these lung‐resident CD4+ T‐cells provide protection from influenza virus when transferred to naive mice. Similar to its role in directing long‐term memory fate as discussed above, IL‐2 also supports the formation of antigen‐specific lung CD4+ TRM with a transcriptional signature distinct from that of circulating CD4+ T‐cell populations but similar to that of CD8+ TRM. 51, 75 Interestingly, Strutt et al.51 also identified an IL‐2‐independent population of influenza‐specific lung CD4+ TRM following infection, suggesting that IL‐2 may not be the only cytokine regulating lung TRM development and maintenance. In fact, IL‐15 was shown to be essential for these IL‐2‐independent cells, acting as an ‘alarm’ at local sites of infection to promote both CD8+ T‐cell responses and induce long‐lived CD4+ TRM. 42, 51 Using fluorescently labelled antibody injection, Hondowicz et al.49 showed that LCMV‐specific CD4+ T‐cells migrated to the lungs as a Tbethi TH1 subset with CD69 expression similar to influenza experiments. Again, the establishment of LCMV‐specific lung TH1 TRM cells required IL‐2 signalling; however, for long‐term maintenance and survival, additional interactions with B‐cells in tissues were necessary.49
Several groups have shown that allergens in an asthma model also elicit CD4+ TRM responses in the lung. After exposure to HDM, allergen‐specific CD4+ TRM in the lungs were shown to be resident by parabiosis experiments.36 These cells expressed high levels of CD69 and required IL‐2 signalling for their migration to the lungs, similar to the studies above with viral pathogens. Interestingly, the TF Bcl6, the fate‐determining factor for TFH cells, prevented HDM‐specific CD4+ TRM from entering the lungs, and loss of Bcl6 actually increased the TRM population in the tissue. These results suggest a possible antagonistic relationship between TRM and memory TFH cells, and highlight a role for TFs in directing the development of tissue‐resident populations. In another study using the HDM model, lung CD4+ TRM clustered around the airways at rest and rapidly reactivated upon secondary exposure within their clusters to produce IL‐4, IL‐5 and IL‐17 and recruit dendritic cells.50 Additional studies highlight a role for IL‐7, along with IL‐2, in the maintenance of allergen‐specific CD4+ TRM cells in the lung parenchyma and airways. However, these IL‐7‐dependent CD4+ TRM did not express CD69,52 suggesting either that CD69 may not be a conclusive marker for CD4+ TRM or that CD4+ TRM are highly heterogeneous, and further studies must focus on elucidating the different subsets. Additionally, in both viral infection and allergen‐induced asthma models, it remains unclear whether the same cells can become resident in both the parenchyma and airways, and if similar survival signals sustain such subsets.
Skin CD4+ TRM cells
A second well‐studied tissue for CD4+ TRM cells is the skin particularly in a herpes simplex virus (HSV) infection model. Initial studies by Gebhardt et al.15 using intravital microscopy demonstrated that HSV‐specific gDT‐II CD4+ T‐cells showed a migration pattern distinct from that of CD8+ T‐cells in the skin following infection, homing to the dermis as opposed to the preference of CD8+ T‐cells for the epidermis. Additionally, dermal TH cells were significantly more motile and had lower expression of CD103 compared with CD8+ T‐cells. Thus, it appears that CD4+ TRM can migrate between the skin and circulation much more easily than CD8+ TRM and may not be a fully resident population. In another study of skin HSV response, Collins et al. examined skin from naive mice and found resting CD4+ T‐cells preferentially clustered around hair follicles. Upon infection, circulating CD4+ T‐cells were rapidly recruited to the skin, specifically to the perifollicular regions; the majority of recruited cells were lost by day 30, but a small percentage survived in tissue long after initial antigen exposure.64 These skin CD4+ TRM, as demonstrated by parabiosis experiments, also aggregated in APC‐associated clusters, consistent with experiments done in the FRT,66, 76 as discussed below. Additional studies of skin CD4+ TRM following Leishmania major (L. major) infection showed evidence of a L. major‐specific resident population in the dermis with increased expression of IFN genes and chemokine signalling pathways.61 These CD4+ TRM cells were able to control parasite growth following a secondary challenge, presumably through the aforementioned IFN response and recruitment of circulating immune cells. Through FTY‐720 treatment experiments, resident CD4+ T‐cells were found to be the main contributors to protection against L. major, recruiting inflammatory monocytes for production of reactive oxygen species to kill the parasites.62 These results highlight another function of CD4+ TRM in enhancing secondary responses and protection against recurring pathogens.
CD4+TRM cells in other mucosal tissues
Through parabiosis experiments, CD4+ TRM have been shown to be active in the FRT in response to HSV‐2 infection.66, 77 Intravaginal HSV‐specific CD69+ CD4+ TRM localized to memory lymphocyte clusters containing CD8+ T‐cells, macrophages and other APCs, and were maintained in these structures by chemokines secreted by macrophages. Upon a secondary re‐challenge with lethal HSV‐2, resident TH cells alone were sufficient for protection, largely mediated by CD4+ TRM production of IFNγ to inhibit viral replication. In this context, FRT‐resident CD4+ T‐cells were more directly involved in clearing the infection as opposed to their classic role in alarming and recruiting circulating populations.
The SI contains a potential subset of tissue‐resident CD4+ T‐cells, though no parabiosis experiment has been performed to definitively characterize this population as resident. In general, intestinal helper T‐cells can be found as both intraepithelial lymphocytes (IELs) and lamina propria lymphocytes (LPLs) with a greater proportion of cells in the LP. The majority of studies of gut‐resident CD4+ T‐cell subsets have focused on endogenous polyclonal IELs that originate from the circulating population.59, 78, 79 Upon entering the SI, these CD4+ IELs downregulate the TF ThPOK responsible for thymic CD4+ T‐cell fate and upregulate Runx3, the fate‐determining factor for differentiating CD8+ thymocytes. This switch in programming towards a more cytolytic function appears to be important in the CD4+ T‐cell response to endogenous gut microbiota.78 Nevertheless, this study characterizes polyclonal CD4+ T‐cells with variable TCR specificity and affinity, and further investigations are necessary to determine whether similar transcriptional changes are seen in antigen‐specific responses. In an LM infection, antigen‐specific CD4+ T‐cells preferentially migrated into the LP (though a small population was seen in the IEL) to form a long‐lived antigen‐specific memory population that was predominantly TH1, expressed high levels of CD69 and, unlike lung‐resident CD4+ T‐cells, was independent of IL‐15 signalling.60 Interestingly, this gut ‘TRM’ population had low Ly6C expression relative to other lymphocytes in circulation, similar to the MP population of circulating TH1 cells discussed in the TH1 and TFH CD4+ memory T‐cells above. This suggests that circulating and resident memory cells may share a common precursor.
As discussed in the studies above, CD4+ TRM differ greatly between infection models and tissues, although some general principles can be drawn (Fig. 2). As is the case with CD8+ TRM, the majority of CD4+ TRM express high levels of CD69, but CD103 expression varies within and between tissues.15, 56, 60, 66 Across multiple tissues – lung, skin, FRT – CD4+ TRM form clusters with other resident immune subsets including CD8+ T‐cells, macrophages and APCs.52, 64, 66 These clusters position CD4+ TRM in close proximity to the cells they need to activate in case of a recurrent infection. This may be an optimized way for helper TRM cells to perform their ‘sense‐and‐alarm’ function,68 initiating the innate response to recruit circulating cells and activating CD8+ T‐cells to fight off pathogen. Additionally, cytokines appear to be critical in the recruitment, formation and maintenance of CD4+ TRM in many tissues.50, 51, 52, 60 However, which cytokines are important for which tissues remain unknown, although IL‐2 may play a key role for all TRM given its known effects in circulating memory populations.80 It is also unclear which CD4+ TH effector subset, if any, gives rise to TRM, or whether the tissue‐resident precursor is distinct from the peripheral population completely, residing in the tissue until the right signals are detected to induce differentiation.
Conclusion
The diversity and plasticity of effector CD4+ T‐cells create a heterogeneous memory pool, making the study of helper T‐cell memory differentiation complex. While there are some promising markers to differentiate between memory TH1 and TFH memory subsets,24, 27 it is still unclear whether both helper memory populations originate from their respective effector cells or whether the ‘stem‐like’ properties of TFH cells make them the primary precursor.25 Likewise, in other infection systems that elicit TH2 or TH17 effector cells, we do not know how these effector populations contribute to the final pool of memory cells. Adding further to the complexity, CD4+ TRM cells are highly variable across tissues, and different cues drive their establishment and retention. It is unclear whether the precursor for CD4+ TRM cells originates from the same memory‐precursor population that yields circulating memory cells or if it is found even earlier, before the effector versus memory decision. TFH cells may also contribute to the CD4+ TRM pool, although currently no evidence exists for a TFH‐like subset in tissues. Lastly, it is clear that surface markers do not accurately identify subpopulations in effector and memory pools, and further work requires examining TFs and regulators that may direct the memory programme. One possible approach to parse the heterogeneity of CD4+ memory T‐cells is through bulk and single‐cell epigenetic and transcriptional profiling of cells in the circulation and tissues over the course of an infection to identify whether an early memory precursor exists or whether memory potential is programmed as effector cells contract and die.75, 81, 82 Paired with adoptive transfers of putative subsets, it will be possible to identify key factors at each step in the effector‐to‐memory transition, and in the formation and survival of the tissue‐resident subset.
Disclosures
None declared.
Acknowledgements
This review was supported by the American Heart Association Predoctoral Fellowship to QPN, and US National Institutes of Health R01 AI072117 and U19 AI109976 to AWG. The authors thank Drs Kyla D. Omilusik and J. Ty Crowl for critical review of the manuscript.
References
- 1. McKinstry KK, Strutt TM, Swain SL. The potential of CD4 T‐cell memory. Immunology 2010; 130:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol 2010; 28:445–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses1. J Immunol 2002; 168:1528–32. [DOI] [PubMed] [Google Scholar]
- 4. Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4 + T cell repertoire in the transition from effector to memory. Immunity 2008; 28:533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H et al The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8_ T cell response. J Immunol 2009; 183:7919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou L, Chong MMW, Littman DR. Plasticity of CD4 + T cell lineage differentiation. Immunity 2009; 30:646–55. [DOI] [PubMed] [Google Scholar]
- 7. Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM et al Plasticity of TH17 cells in Peyer's patches is responsible for the induction of T cell‐dependent IgA responses. Nat Immunol 2013; 14:372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakayamada S, Takahashi H, Kanno Y, O’ Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol 2012; 24:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cannons JL, Lu KT, Schwartzberg PL. T follicular helper cell diversity and plasticity. Trends Immunol 2013; 34:200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamane H, Paul WE. Memory CD4 + T cells: fate determination, positive feedback and plasticity. Cell Mol Life Sci 2012; 69:1577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. MacLeod MKL, Kappler JW, Marrack P. Memory CD4 T cells: generation, reactivation and re‐assignment. Immunology 2010; 130:10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 2013; 31:137–61. [DOI] [PubMed] [Google Scholar]
- 13. Carbone FR, Mackay LK, Heath WR, Gebhardt T. Distinct resident and recirculating memory T cell subsets in non‐lymphoid tissues. Curr Opin Immunol 2013; 25:329–33. [DOI] [PubMed] [Google Scholar]
- 14. Carbone FR. Tissue‐resident memory T cells and fixed immune surveillance in nonlymphoid organs. J Immunol 2015; 195:17–22. [DOI] [PubMed] [Google Scholar]
- 15. Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR et al Different patterns of peripheral migration by memory CD4 + and CD8 + T cells. Nature 2011; 477:216–9. [DOI] [PubMed] [Google Scholar]
- 16. Gebhardt T, Mueller SN, Heath WR, Carbone FR. Peripheral tissue surveillance and residency by memory T cells. Trends Immunol 2013; 34:27–32. [DOI] [PubMed] [Google Scholar]
- 17. Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue‐resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev 2018; 283:54–76. [DOI] [PubMed] [Google Scholar]
- 18. Turner DL, Farber DL. Mucosal resident memory CD4 T cells in protection and immunopathology. Front Immunol 2014; 5:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long‐lived memory cells. Nat Immunol 2003; 4:1191–8. [DOI] [PubMed] [Google Scholar]
- 20. Beyersdorf N, Ding X, Tietze JK, Hanke T. Characterization of mouse CD4 T cell subsets defined by expression of KLRG1. Eur J Immunol 2007; 37:3445–54. [DOI] [PubMed] [Google Scholar]
- 21. Kiazyk SAK, Fowke KR. Loss of CD127 expression links immune activation and CD4 + T cell loss in HIV infection. Trends Microbiol 2008; 16:567–73. [DOI] [PubMed] [Google Scholar]
- 22. Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T‐cell progenitors. Nature 2008; 452:356–60. [DOI] [PubMed] [Google Scholar]
- 23. Pepper M, Linehan JL, Pagán AJ, Zell T, Dileepan T, Cleary PP et al Different routes of bacterial infection induce long‐lived TH1 memory cells and short‐lived TH17 cells. Nat Immunol 2010; 11:83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA et al Differential expression of Ly6C and T‐bet distinguish effector and memory Th1 CD4 + cell properties during viral infection. Immunity 2011; 35:633–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B et al Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol 2013; 190:4014–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tubo NJ, Fife BT, Pagan AJ, Kotov DI, Goldberg MF, Jenkins MK. Most microbe‐specific naïve CD4 + T cells produce memory cells during infection. Sci Immunol 2016; 351:511–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS et al Distinct memory CD4 + T cells with commitment to T follicular helper‐ and T helper 1‐cell lineages are generated after acute viral infection. Immunity 2013; 38:805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lüthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM et al The development and fate of follicular helper T cells defined by an IL‐21 reporter mouse. Nat Immunol 2012; 13:491–8. [DOI] [PubMed] [Google Scholar]
- 29. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F et al Transcription factor achaete‐scute homologue 2 initiates follicular T‐helper‐cell development. Nature 2014; 507:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pepper M, Pagán AJ, Igyá BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin‐2 receptor generate T helper 1 central and effector memory cells. Immunity 2011; 35:583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ciucci T, Vacchio MS, Gao Y, Ardori FT, Candia J, Mehta M et al The emergence and functional fitness of memory CD4 + T cells require the transcription factor Thpok. Immunity 2019; 50:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Endo Y, Hirahara K, Yagi R, Tumes DJ, Nakayama T. Pathogenic memory type Th2 cells in allergic inflammation. Trends Immunol 2014; 35:69–78. [DOI] [PubMed] [Google Scholar]
- 33. Nakayama T, Yamashita M. Critical role of the Polycomb and Trithorax complexes in the maintenance of CD4 T cell memory. Semin Immunol 2009; 21:78–83. [DOI] [PubMed] [Google Scholar]
- 34. Hegazy AN, Peine M, Helmstetter C, Panse I, Fröhlich A, Bergthaler A et al Interferons direct Th2 cell reprogramming to generate a stable GATA‐3 + T‐bet+ cell subset with combined Th2 and Th1 cell functions. Immunity 2010; 32:116–28. [DOI] [PubMed] [Google Scholar]
- 35. Endo Y, Iwamura C, Kuwahara M, Suzuki A, Sugaya K, Tumes DJ et al Eomesodermin controls interleukin‐5 production in memory T helper 2 cells through inhibition of activity of the transcription factor GATA3. Immunity 2011; 35:733–45. [DOI] [PubMed] [Google Scholar]
- 36. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT et al Interleukin‐2‐dependent allergen‐specific tissue‐resident memory cells drive asthma. Immunity 2016; 44:155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mcgeachy MJ. Th17 memory cells: live long and proliferate. J Leukoc Biol 2013; 94:921–6. [DOI] [PubMed] [Google Scholar]
- 38. Zúñiga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev 2013; 252:78–88. [DOI] [PubMed] [Google Scholar]
- 39. Muranski P, Borman ZA, Kerkar SP, Klebanoff CA, Ji Y, Sanchez‐Perez L et al Th17 cells are long lived and retain a stem cell‐like molecular signature. Immunity 2011; 35:972–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Y, Chauhan SK, Tan X, Dana R. Interleukin‐7 and ‐15 maintain pathogenic memory Th17 cells in autoimmunity. J Autoimmun 2017; 77:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pepper M, Jenkins MK. Origins of CD4 + effector and central memory T cells. Nat Immunol 2011; 12:467–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dhume K, McKinstry KK. Early programming and late‐acting checkpoints governing the development of CD4 T cell memory. Immunology 2018; 155:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gasper DJ, Tejera MM, Suresh M. CD4 T‐cell memory generation and maintenance. Crit Rev Immunol 2014; 34:121–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Snook JP, Kim C, Williams MA. TCR signal strength controls the differentiation of CD4 + effector and memory T cells. Sci Immunol 2018; 3:eaas9103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM et al Effector CD4 T‐cell transition to memory requires late cognate interactions that induce autocrine IL‐2. Nat Commun 2014; 5:5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. DiToro D, Winstead CJ, Pham D, Witte S, Andargachew R, Singer JR et al Differential IL‐2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science 2018; 361:eaao2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shakya A, Goren A, Shalek A, German CN, Snook J, Kuchroo VK et al Oct1 and OCA‐B are selectively required for CD4 memory T cell function. J Exp Med 2015; 212:2115–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilk MM, Mills KHG. CD4 TRM cells following infection and immunization: implications for more effective vaccine design. Front Immunol 2018; 9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hondowicz BD, Kim KS, Ruterbusch MJ, Keitany GJ, Pepper M. IL‐2 is required for the generation of viral‐specific CD4 + Th1 tissue‐resident memory cells and B cells are essential for maintenance in the lung. Eur J Immunol 2018; 48:80–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Turner DL, Goldklang M, Cvetkovski F, Paik D, Trischler J, Barahona J et al Biased generation and in situ activation of lung tissue‐resident memory CD4 T cells in the pathogenesis of allergic asthma. J Immunol 2018; 200:1561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strutt TM, Dhume K, Finn CM, Hwang JH, Castonguay C, Swain SL et al IL‐15 supports the generation of protective lung‐resident memory CD4 T cells. Mucosal Immunol 2017; 11:668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yeon SM, Halim L, Chandele A, Perry CJ, Kim SH, Kim SU et al IL‐7 plays a critical role for the homeostasis of allergen‐specific memory CD4 T cells in the lung and airways. Sci Rep 2017; 7:11 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smith NM, Wasserman GA, Coleman FT, Hilliard KL, Yamamoto K, Lipsitz E et al Regionally compartmentalized resident memory T cells mediate naturally acquired protection against pneumococcal pneumonia. Mucosal Immunol 2017; 11:220–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wilk MM, Misiak A, McManus RM, Allen AC, Lynch MA, Mills KH. Lung CD4 tissue‐resident memory T cells mediate adaptive immunity induced by previous infection of mice with Bordetella pertussis . J Immunol 2017; 199:233–43. [DOI] [PubMed] [Google Scholar]
- 55. Sakai S, Kauffman KD, Schenkel JM, McBerry CC, Mayer‐Barber KD, Masopust D et al Control of Mycobacterium tuberculosis Infection by a subset of lung parenchyma‐homing CD4 T cells. J Immunol 2014; 192:2965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrançois L, Farber DL. Tissue‐retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol 2011; 187:5510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chapman TJ, Topham DJ. Identification of a unique population of tissue‐memory CD4 + T cells in the airways after influenza infection that is dependent on the Integrin VLA‐1. J Immunol 2010; 184:3841–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Steinfelder S, Rausch S, Michael D, Kühl AA, Hartmann S. Intestinal helminth infection induces highly functional resident memory CD4 + T cells in mice. Eur J Immunol 2017; 47:353–63. [DOI] [PubMed] [Google Scholar]
- 59. Faria AMC, Reis BS, Mucida D. Tissue adaptation: implications for gut immunity and tolerance. J Exp Med 2017; 214:1211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Romagnoli PA, Fu HH, Qiu Z, Khairallah C, Pham QM, Puddington L et al Differentiation of distinct long‐lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal Immunol 2017; 10:520–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, Scott P. Skin‐resident memory CD4 + T cells enhance protection against Leishmania major infection. J Exp Med 2015; 212:1405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Glennie ND, Volk SW, Scott P. Skin‐resident CD4 + T cells protect against Leishmania major by recruiting and activating inflammatory monocytes. PLoS Pathog 2017; 13:e1006349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shin H, Iwasaki A. Skin TRM mediates distributed border patrol. Cell Res 2012; 22:1325–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO et al Skin CD4 + memory T cells exhibit combined cluster‐mediated retention and equilibration with the circulation. Nat Commun 2016; 7:11 514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bagri P, Anipindi VC, Nguyen PV, Vitali D, Stämpfli MR, Kaushic C. Novel role for interleukin‐17 in enhancing type 1 helper T cell immunity in the female genital tract following mucosal herpes simplex virus 2 vaccination. J Virol 2017; 91:pii: e01234‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Iijima N, Iwasaki A. A local macrophage chemokine network sustains protective tissue‐resident memory CD4 T cells. Science 2014; 346:93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gray JI, Westerhof LM, MacLeod MKL. The roles of resident, central and effector memory CD4 T cells in protective immunity following infection or vaccination. Immunology 2018; 154:574–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing & alarm function of resident memory CD8 + T cells. Nat Immunol 2013; 14:509–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anderson KG, Mayer‐Barber K, Sung H, Beura L, James BR, Taylor JJ et al Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 2014; 9:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hofmann M, Brinkmann V, Zerwes H‐G. FTY720 preferentially depletes naive T cells from peripheral and lymphoid organs. Int Immunopharmacol 2006; 6:1902–10. [DOI] [PubMed] [Google Scholar]
- 71. Schenkel JM, Masopust D. Tissue‐resident memory T cells. Immunity 2014; 41:886–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thawer S, Horsnell WG, Darby M, Hoving JC, Dewals B, Cutler AJ et al Lung‐resident CD4 + T cells are sufficient for IL‐4Rα‐dependent recall immunity to Nippostrongylus brasiliensis infection. Mucosal Immunol 2014; 7:239–48. [DOI] [PubMed] [Google Scholar]
- 73. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 2009; 10:524–30. [DOI] [PubMed] [Google Scholar]
- 74. Masopust D, Vezys V, John Wherry E, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol 2006; 176:2079–83. [DOI] [PubMed] [Google Scholar]
- 75. Milner JJ, Goldrath AW. Transcriptional programming of tissue‐resident memory CD8 + T cells. Curr Opin Immunol 2018; 51:162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Iijima N, Iwasaki A. Tissue instruction for migration and retention of TRM cells. Trends Immunol 2015; 36:556–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012; 491:463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Reis B, Rogoz A, Costa‐Pinto F, Taniuchi I, Mucida D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4 + T cell immunity. Nat Immunol 2013; 14:271–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Reis BS, Hoytema Van Konijnenburg DP, Grivennikov SI, Mucida D. Transcription factor T‐bet regulates intraepithelial lymphocyte functional maturation. Immunity 2014; 41:244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ross SH, Cantrell DA. Signaling and function of interleukin‐2 in T lymphocytes. Annu Rev Immunol 2018; 36:411–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott‐Browne JP et al Epigenetic landscapes reveal transcription factors that regulate CD8 + T cell differentiation. Nat Immunol 2017; 18:573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ et al Early transcriptional and epigenetic regulation of CD8 + T cell differentiation revealed by single‐cell RNA sequencing. Nat Immunol 2017; 18:422–32. [DOI] [PMC free article] [PubMed] [Google Scholar]