

Abstract
Symbiosis is a major force of evolutionary change, influencing virtually all aspects of biology, from population ecology and evolution to genomics and molecular/biochemical mechanisms of development and reproduction. A remarkable example is Wolbachia endobacteria, present in some parasitic nematodes and many arthropod species. Acquisition of genomic data from diverse Wolbachia clades will aid in the elucidation of the different symbiotic mechanisms(s). However, challenges of de novo assembly of Wolbachia genomes include the presence in the sample of host DNA: nematode/vertebrate or insect. We designed biotinylated probes to capture large fragments of Wolbachia DNA for sequencing using PacBio technology (LEFT-SEQ: Large Enriched Fragment Targeted Sequencing). LEFT-SEQ was used to capture and sequence four Wolbachia genomes: the filarial nematode Brugia malayi, wBm, (21-fold enrichment), Drosophila mauritiana flies (2 isolates), wMau (11-fold enrichment), and Aedes albopictus mosquitoes, wAlbB (200-fold enrichment). LEFT-SEQ resulted in complete genomes for wBm and for wMau. For wBm, 18 single-nucleotide polymorphisms (SNPs), relative to the wBm reference, were identified and confirmed by PCR. A limit of LEFT-SEQ is illustrated by the wAlbB genome, characterized by a very high level of insertion sequences elements (ISs) and DNA repeats, for which only a 20-contig draft assembly was achieved.
Subject terms: Genomic analysis, Molecular biology, Next-generation sequencing
Introduction
A comprehensive understanding of symbiotic evolution remains challenging1. A remarkable example of the biological relevance and universality of symbiotic interactions is that of Wolbachia bacteria. The study of obligate intracellular alpha-proteobacteria Wolbachia symbiont and its host interactions provide a model system for analysis as they are present in a large fraction of invertebrate species on this planet, including nematodes, insects, mites, spiders and crustaceans2–5. While in arthropods they generally act as reproductive parasites6, in their filarial nematodes hosts they have generally taken an alternative evolutionary trajectory as strict mutualists, being obligate for adult and larval worm development and reproduction7,8. Underlying mechanisms of symbiosis remain largely elusive, although for some insects, a pair of genes (cifA and cifB) have been identified as part of the cytoplasmic incompatibility system, one of the arthropod reproductive manipulation phenotypes9,10. Comparative genomic analyses remains a viable strategy to identify candidate genes involved in Wolbachia symbioses11. Obtaining additional genomic data from a variety of Wolbachia clades will help elucidate the nature of the symbiotic mechanisms(s).
Currently, de novo sequence assembly of Wolbachia genomes often confronts several obstacles. First, it remains challenging to produce high quality genome sequences due to the presence of host DNA, which can complicate the assemblies because of low levels of Wolbachia sequence reads relative to host reads. Furthermore, the presence of lateral gene transfers (LGTs) from Wolbachia to the host genome can complicate assembly12,13 as observed for the Wolbachia genome of Drosophila ananassae14. One method to overcome the host DNA problem is to use targeted Wolbachia genome enrichment to capture large DNA fragments, recently developed for short-read paired-end technologies15,16.
A second challenge of de novo genome assembly is the presence of many long repetitive elements17 which inhibit correct assemblies. The first Wolbachia genome studies highlighted the presence of large amounts of repetitive DNA18,19. For example, at least 14% of the genome of Wolbachia from D. melanogaster (wMel) is composed of repetitive DNA and insertion sequences18. Although present at different levels among strains20, Wolbachia genomes often contain numerous transposable elements (Insertion sequences (ISs) and group II introns) and prophage sequences15,18. As opposed to short-read paired-end technologies, long-read sequencing methods, such as PacBio or Nanopore, enable longer sequence reads, often through the repeats and thus can significantly improve de novo assembly17.
Here, we demonstrate a large fragment targeted enrichment capture method using SeqCap® EZ probes (Roche) and PacBio sequencing for Wolbachia de novo assembly (LEFT-SEQ - Large Enriched Fragment Targeted Sequencing). We tested this method on three different Wolbachia strains: wBm, from the nematode Brugia malayi, for which a previous complete genome sequence was available; wAlbB, from the mosquito Aedes albopictus, for which different draft genomes were available; and two isolates of wMau, from Drosophila mauritiana, for which no genome draft was available.
Results
De novo assembly and coverage
The LEFT-SEQ method was implemented to capture relatively large DNA fragments for long-read NextGen sequencing to more efficiently enable genome assemblies. The library preparation workflow was optimized, in particular with an additional exonuclease treatment step, modified PCR conditions and lower ratio of AMPure® PB bead/DNA clean-up (Supplementary Methods 1 & Supplementary Fig. 1), and applied to insect or nematode samples harboring Wolbachia symbionts.
We used LEFT-SEQ (Fig. 1) and bioinformatic analysis (Fig. 2) to produce de novo drafts of three Wolbachia symbiont genomes. The method created complete circular sequences for two of the genomes (wBm, wMau) and a set of 20 contigs for the third (wAlbB). For wMau, the Wolbachia from D. mauritiana, the analysis of 28,840 PacBio CCS (Circular consensus sequence) reads of fly population 177 and 45,984 CCS reads of the fly population 181 (3 SMRT cells each barcoded samples) produced, respectively, a circularized genome of 1,273,527 bp and 1,273,530 bp (Table 1). For wBm, the Wolbachia from B. malayi, the analysis of 40,241 PacBio CCS reads (2 SMRT cells) produced a 3 contig draft while 76,216 reads (3 SMRT cells) produced a circularized genome of 1,080,939 bp (Table 1). For wAlbB, the Wolbachia from Aedes albopictus, the analysis of 81,233 reads (2 SMRT cells) produced a 42 contig draft and 290,028 PacBio CCS reads (12 SMRT cells) produced a 20 contig draft (Table 1). For wBm and wMau, the single circular contigs were validated by PCR amplification (Tables S1, S2 and S3). Regarding genome coverage, enrichment provided a reduction in host sequences and only a few areas not covered at a depth of 20X (Table 1). The entire wBm genome was captured at an average depth of 78X. Likewise, the entire wMau genome was captured for both populations with average coverage of 71X for population 181 and 44X for population 177 (Table 1). The 20 contigs draft of wAlbB was obtained at an average depth of 266X (only 1.6% of the draft had coverage <20X) (Table 1).
Figure 1.

Workflow overview of LEFT-SEQ (Large Enriched Fragment Targeted Sequencing) library preparation.
Figure 2.

Overview of the bioinformatics pipeline.
Table 1.
Information of produced genomes.
| wBm | wMau (pop 181) | wMau (pop 177) | wAlbB | |||
|---|---|---|---|---|---|---|
| Number of SMRT cell | 2 | 3 | 3 (barcoded) | 3 (barcoded) | 2 | 12 |
| Number of reads | 40,241 | 76,216 | 45,984 | 28,840 | 81,233 | 290,028 |
| Number of contigs | 3 | 1 | 1 | 1 | 42 | 20 |
| Size of the largest contig | 416,288 | 1,080,939 | 1,273,588 | 1,273,527 | 186,312 | 249,386 |
| Total length (bp) | 1,082,170 | 1,080,939 | 1,273,588 | 1,273,527 | 1,466,139 | 1,492,731 |
| N50 | 415,815 | 1,080,939 | 1,273,588 | 1,273,527 | 54,550 | 145,461 |
| L50 | 2 | 1 | 1 | 1 | 7 | 4 |
| Number of reads mapped to wb reference | 23,742 | 45,107 | NA | NA | 61,930 | 246,276 |
| % reads mapped to wb reference | 59 | 59 | NA | NA | 76 | 84.9 |
| Number of reads mapped to produced wb | 23,733 | 45,103 | 44,734 | 27,842 | 71,203 | 249,032 |
| Number of reads mapped to produced wb | 59 | 59 | 97.28 | 96.5 | 87 | 85.8 |
| Average depth | 52X | 78X | 71X | 44X | 75X | 266X |
| bases with coverage <20X | 35,393 | 1,586 | 28,128 | 63,096 | 129,224 | 24,200 |
| % bases with coverage <20X | 3.2 | 0.14 | 2.2 | 4.9 | 8.5 | 1.6 |
Summary of de novo assembly using canu processed in the current study (statistics using QUAST36). Lines 1–7, summary statistics; lines 8 to 11, summary of mapping using ngmlr; lines 12 to 14, coverage statistics across the produced genomes using the SAMtools depth38. Abbreviations: bp: bases pair; wb: Wolbachia; pop: population.
Efficiency of the enrichment
The efficiency of the enrichment may impact the assembly quality if low levels of symbiont sequence reads are present, relative to host reads. The enrichment is more efficient for the A. albopictus sample than for the D. mauritiana or B. malayi samples (Fig. 3): 2.52% of the sequenced reads mapped to wBm reference genome without enrichment versus 59% with the enrichment (23X increase); 8.96% of the sequenced reads mapped to produce the wMau genome without enrichment vs. 97.28% with enrichment (11X increase); 0.2% of the sequenced reads mapped to wAlbB drafts or 0.95% to the produced draft without enrichment vs. an average 76.2% or 87.65% with the enrichment (90–340X increase) (Fig. 3). In terms of host-derived sequences, 73.99% of the reads mapped to host B. malayi reference without enrichment vs 41.98% with enrichment (1.76X decrease) with a few reads mapping to the jird (experimental mammalian host of the nematode) draft in both protocols. For A. albopictus, 47.24% of the reads mapped to the draft without enrichment compared to 1.56% with the enrichment (30X decrease). Thus, the variations among the de novo assemblies cannot be explained by different efficiencies of capture.
Figure 3.
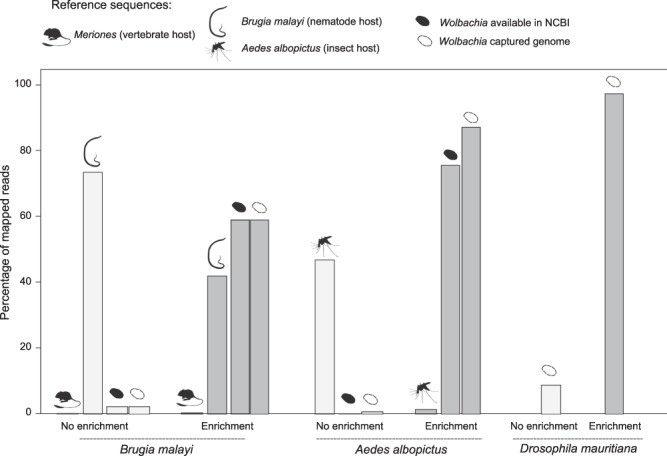
Evaluation of LEFT-SEQ enrichment method. The percentage of the reads mapped across different reference or draft genomes is reported for the three different samples without (pale grey) or with the enrichment method (dark grey). Host and Wolbachia symbionts are indicated with animal symbols.
Size of the sequenced reads
The current enrichment protocol was established after optimization described in the Supplementary files (Supplementary Methods 1) in order to maximize the size of sequenced reads. For all three genome samples, LEFT-SEQ reads are smaller (median between 1–1.3 kb shorter) than those of the control without the capture method (Fig. 4). Tests suggest that the observed shortening occurs during the hybridization bead capture step. Protocols without genomic DNA shearing and without the first PCR step show no clear difference in the median size of reads for all three tested genomes (Fig. S1). The current protocol includes the addition of an exonuclease VII treatment and a DNA damage repair step before the ligation, which reduces the formation of chimeric reads. SeqCap® adaptors (first ligation) are reduced to 3.6% and 6% with the optimized protocol from 14.7% of the reads for B. malayi and 11% for the A. albopictus sample after trimming (Fig. S1). Addition of one or two AMPure® PB bead clean-up steps before the annealing/binding to the SMRT templates increases the median size of the sequenced reads on the tested A. albopictus libraries, but the largest fragments appear to be lost during the library preparation (Fig. S1).
Figure 4.
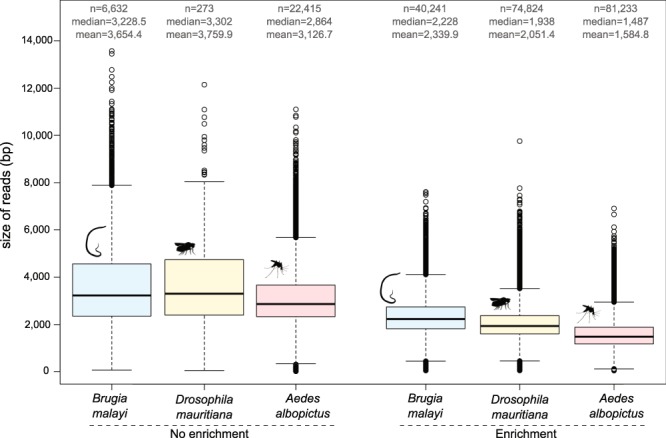
Box-and-whisker plot showing insert size for the three samples with the LEFT-SEQ method and the unenriched control. The different samples are indicated with color (blue for B. malayi, yellow for D. mauritiana and red for A. albopictus) and symbol. Small circles are outlier values. Additional statistics are indicated above the boxplot: number of analyzed reads, the median and the mean.
Presence of mobile genetic elements
The RAST annotation of the wAlbB draft genome highlights the observation that each end of the 20 contigs encoded at least one mobile genetic element: either an insertion sequence element or a group II intron sequence. The number of insertion sequence elements (ISs) or transposases is highly variable among the studied Wolbachia genomes. In all, three partial ISs are detected in wBm genome from two families, IS630 (n = 2) and IS1031 (n = 1); the open reading frames (ORF) lengths are respectively 103 bp and 62 bp (Table S1) and the same ISs are detected in the wBm reference. 46 ISs are detected in wMau from 17 different IS families, with the most represented being IS110 (47.83% of total detected ISs, maximum 1,122 bp), IS5 and IS6 (17.39%, minimum 186 bp) (Table S1). 209 ISs are detected in the wAlbB draft (Table S1), (ORFs from 180 bp to 1,320 bp). The most represented IS families are IS982 (46.86%), IS481 (36.36%), IS66 (8.61%) and IS3 (5.26%). Surprisingly, when the same analysis is performed on three previously published wAlbB drafts, variations are observed: 6 ISs for the 156 contig draft wAlbB (ASM24241v3), 9 ISs for the 131 contig draft wAlbB (ASM237914v1) and 7 ISs for the 177 contig draft wAlbB (ASM237484v1). The difference is more striking for group II intron genes: none are detected in wBm, as previously observed19,20, 7–10 genes are detected in wMau while 72 genes are detected in the wAlbB contigs. Thus, this high level of mobile genetic elements in wAlbB genome compared to the wMau or wBm genomes could explain the inability to generate a single complete consensus sequence.
Detection of Single-nucleotide polymorphisms
There are several possible sources of error associated with NextGen sequencing protocols, including PCR errors, inherent DNA sequencing errors due to the chemistry21,22 or errors due to the de novo assembly process23. In order to test the accuracy of assembly based on PacBio CCS reads, we compared wBm assemblies processed with different correction methods (described in Methods) with the reference wBm genome available in the database. Variant detection between the wBm assemblies of the current study and the reference wBm genome indicates 8 transitions, 9 transversions and 1 insertion, independent of the application of polishing steps. These differences were confirmed by PCR amplification and sequencing (Fig. S2; Tables S2, S3 and S5). Using the two correction methods, only the deletion detection was variable between them. 21 deletions are in common among the wBm assemblies relative to the wBm reference. It is interesting to note that all the deletions occurred at regions where the identical nucleotide was repeated. Correction using SAMtools/BCFtools provided an assembly most similar to the reference, with fewer deletion events than the correction using Pilon, which had indicated more deletions (54, including 10 representing a total of 533 bp). These polishing methods were developed for use with Illumina data and not PacBio consensus sequences. As the coverage with the Pacbio CCS (circular consensus sequence) is low (78X coverage for wBm), it might explain why better results were obtained with the tuned SAMtools pipeline, known to reduce the effect of reads with excessive mismatches (http://samtools.sourceforge.net/mpileup.shtml). Comparison of the two assemblies of wMau polished with the tuned SAMtools pipeline identified 19 base differences (Table S4), mainly deletion events, which all occurred at repeated bases and only one base mutation event. Four of these differences were tested by PCR amplification and all indicated absence of mutation (Table S5), suggesting these were sequencing read consensus errors.
Discussion
LEFT-SEQ provides an efficient enrichment method for long read sequences derived from the endosymbiont Wolbachia. This enables the production of complete or almost complete Wolbachia genomes amongst a background of host sequences in a stepwise and efficient process. The efficacy of the method is variable according to the Wolbachia strain, as complete genomes of wBm or wMau were produced while only a 20-contig draft wAlbB genome was obtained, due to the presence of a high percentage of repeats in the genome. The analysis of wBm shows that the method can enable SNP detection between samples, as at least 18 SNPs were confirmed by PCR, relative to the original published sequence. However, it is difficult to establish if they are real de novo differences or sequence errors during assembly of the original sequenced genome19. The original wBm genome was completed by Sanger dideoxy sequencing of subclones derived from overlapping bacterial artificial chromosome (BAC) templates19 and it is conceivable this cloning approach introduced a small number of errors. However, while the wBm samples derived from the same source, there is an approximate 20-year gap difference in time between isolation of the DNA samples for sequencing. Only 1 potential missense mutation was observed between the two sequenced wMau genome samples but PCR amplification indicated that this was a sequencing error. These two lineages derived from the same initial mating 8 years before samples were collected for this study, suggesting there was no sequence diversity during this relatively short time frame. It is interesting to point out that PacBio reads are not commonly used for SNP detection or are at least rarely used without polishing with Illumina reads, due to the differences of error rates (often established as <0.8% for Illumina and around 10% for PacBio single-molecule reads)22,24. However as recently reported25,26, the use of PacBio circular consensus reads increases accuracy, as used in the current study.
The limitation of the method to assemble the wAlbB genome is not related to the enrichment efficiency or coverage depth. Indeed, a higher percentage of reads belonging to Wolbachia was observed for the mosquito sample, compared to the nematode sample (Fig. 3). This efficiency difference among the samples may be due to Wolbachia copy number differences in their hosts or a differential number of LGTs, but in each case, LEFT-SEQ provides a highly specific sequence capture. The presence of repetitive elements is very variable among analyzed Wolbachia15,18,19. For cases with a very high percentage of repeats, an increased number of reads even with somewhat longer read lengths improved de novo assembly, but still did not enable the assemblers to produce a complete genome. Even longer fragments will be required to cross the repeats in situations like this. Along these lines, LEFT-SEQ identified a high number of mobile elements (ISs) and group II intron-associated genes in our wAlbB draft, as compared to the previous submissions.
The different protocols tested during the study to attempt to obtain larger fragments for PacBio sequencing suggested that the fragment length obtained during the library preparation (average 2.3 kb for B. malayi; 2 kb for D. mauritiana and 1.6 kb for A. albopictus) is not related to the initial fragmentation or PCR amplification steps (Fig. S1). We suspect the limiting step may be in the bead hybridization step where longer DNA fragments are selectively eliminated either due to multiple probes hybridizing on the same large DNA fragment or shearing due to large DNA interacting with the beads. Even when size selection systems are utilized, (e.g Sage ELF, Sage Science, Beverly MA) size selection) which can increase the average read size27, the problem still remains and while more DNA input might be helpful to increase the average DNA size for capture, this may be problematic for many studies, where DNA is limited. A future goal will be the modification of the capture step for longer fragment isolation (enhanced, LEFT-E-SEQ).
Materials and Methods
Source of materials
Three invertebrate species were used to test the method of Wolbachia long DNA fragment capture: the filarial nematode B. malayi grown in Meriones unguiculatus (Mongolian jird is the experimental mammalian host) naturally infected with wBm (TRS Labs, Georgia, USA), the fruit fly Drosophila mauritiana infected with wMau (flies from Frydman Lab, Boston University, lab stocks 177 and 181 generated by single pair crosses from the same wMau infected stock)28 and the mosquito Aedes albopictus with an artificial single-infection by wAlbB (mosquitoes from the Rasgon lab, Pennsylvania State University)29. The DNA of the different samples was extracted using the DNeasy Blood and Tissue kit following the manufacturer’s recommendations (Qiagen, Germany) with overnight incubation at 56 °C with proteinase K. DNA was eluted into 1X TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The number of specimens pooled for the DNA extraction was variable: 12 female nematodes for B. malayi, 10 female flies for D. mauritiana and 5 mosquitoes for A. albopictus. In the case of D. mauritiana, two different lineages were sequenced using the same hybridization reaction (see below). Each of these two lineages came from a single pair mating derived from the same wMau infected stock in 2010.
Design of capture-based target enrichment DNA probes
The targeted DNA probes were designed by Roche NimbleGen (Madison, US) based on 25 complete or draft Wolbachia sequences (total of 121,300 Tiled regions: average size 997 bp): Wolbachia endosymbiont of Pratylenchus penetrans wPpe (ASM175266v1;GCF_001752665.1); Wolbachia endosymbiont of B. malayi (ASM838v1;GCF_000008385.1); Wolbachia endosymbiont of Onchocerca ochengi wOo (ASM30688v1;GCF_000306885.1); Wolbachia endosymbiont of O volvulus (W_O_volvulus_Cameroon_v3;GCF_000530755.1); Wolbachia endosymbiont of Dirofilaria immitis (wDiv2; http://dirofilaria.nematod.es); Wolbachia endosymbiont of Litomosoides sigmodontis (wLs2; http://litomosoides.nematod.es); Wolbachia endosymbiont of Wuchereria bancrofti (ASM33839v1;GCF_000338395.1); Wolbachia endosymbiont of Cimex lectularius wCle (ASM82931v1;GCF_000829315.1); Wolbachia endosymbiont of Culex quinquefasciatus Pel wPip (ASM7300v1;GCF_000073005.1); Wolbachia endosymbiont of D. melanogaster wMel (ASM802v1;GCF_000008025.1); Wolbachia endosymbiont of D. simulans wRi/wNo/wHa/wAu (ASM2228v1;GCF_000022285.1/ ASM37658v1;GCF_000376585.1/ASM37660v1;GCF_000376605.1/Wau001;GCF_000953315.1); Wolbachia endosymbiont of D. simulans (ASM16749v1;GCA_000167495.1); Wolbachia endosymbiont of Diaphorina citri (wACP3;GCF_000331595.1); Wolbachia endosymbiont of D. ananassae (ASM16747v1;GCF_000167475.1); Wolbachia endosymbiont of D. willistoni (ASM15358v1;GCF_000153585.1); Wolbachia endosymbiont of D. suzukii (ASM33379v2;GCF_000333795.1); Wolbachia endosymbiont of Muscidifurax uniraptor wUni (ASM198363v1;GCF_001983635.1); Wolbachia endosymbiont of Hypolimnas bolina wBol1-b (ASM33377v1;GCF_000333775.1); Wolbachia endosymbiont of Glossina morsitans (wGmm_version4;GCF_000689175.1); Wolbachia endosymbiont of Nasonia vitripennis wVitA/wVitB (ASM198361v1;GCF_001983615.1/WVB_1.0;GCF_000204545.1); Wolbachia endosymbiont of A. albopictus wAlbB (ASM24241v3;GCF_000242415.2).
Library preparation protocol
DNA fragmentation
DNA was fragmented using a Covaris® g-TUBETM (Covaris, US) to produce 8-kb fragments. About 1μg DNA (quantified by Nanodrop) was centrifuged twice at 6,500 rpm (Fig. 1). The sheared DNA was purified using 0.8X AMPure® PB beads (PacBio, US) to remove smaller fragments. Elution was in a 57 μL volume of 1X TE.
DNA repair and large insert library preparation
Large insert libraries were constructed using an adaptation of NEBNext® UltraTM II DNA Library Prep protocol (New England Biolabs, US) with preliminary steps of single strand DNA elimination using Exonuclease VII treatment and DNA damage repair using PreCR® repair mix (New England Biolabs, US) (Fig. 1). A 55 μL reaction volume contained the fragmented DNA (~500 ng), 6 μL of NEBNext® UltraTM II End Prep Reaction buffer, 1 μL of NAD+, and 1 μL of Exonuclease VII and was incubated 15 minutes at 37 °C. 2 μL of PreCR® enzyme mix was added to the reaction and incubated for 30 minutes at 37 °C. The sheared DNA was end repaired and A-tailed by addition of 3 μL of NEBNext® UltraTM II End Prep Enzyme Mix and incubated for 5 minutes at 25 °C and then 30 minutes at 65 °C. Following this, SeqCap® adaptors (Roche, NimbleGen) were ligated to both ends of DNA using the NEBNext® UltraTM II Ligation Module (New England Biolabs) (Fig. 1). A 96 μL reaction volume contained 60 μL of the end repaired reaction mixture (previous step), 30 μL NEBNext® UltraTM II Ligation Master Mix, 1μL NEBNext® Ligation Enhancer and 4 μL SeqCap® Adapter A (10 μM stock). The reaction was incubated 20 °C for 15 minutes followed by a 0.5X AMPure® PB bead clean-up and elution in 27 μL water. 1μL amplified DNA was electrophoresed using a DNA 12,000 chip on the 2100 Bioanalyser system (Agilent, US) to determine the concentration.
Library amplification
The resultant insert library was PCR amplified using One TaqTM Hot Start DNA Polymerase (New England Biolabs) (Fig. 1) in a 25 μL reaction containing: Adaptor Ligated DNA Fragments (between 50 to 150 ng), 0.5 μM of each PCR oligo (PCR oligo 1-5′-AAT GAT ACG GCG ACC ACC GAG A- and PCR oligo 2-5′-CAA GCA GAA GAC GGC ATA CGA G-), 1X OneTaq Buffer, Mg-free, 1.5 mM MgCl2, 0.4 mM each dNTP, 2.5 U OneTaq Hot Start enzyme. PCR was performed using the following conditions: 94 °C for 2 minutes, 7 cycles of 94 °C 20 seconds, 56 °C 20 seconds and 68 °C 8 minutes, followed by 68 °C for 10 minutes. The amplified DNA was purified by a 0.45X AMPurePB bead (PacBio) purification. 1 μL amplified DNA was electrophoresed using a DNA 12,000 chip with the 2100 Bioanalyser system (Agilent, US) to determine the concentration.
Target enrichment hybridization
1μg of the library, 10 μL SeqCap EZ Developer Reagent (Roche NimbleGen,), 1 μL SeqCap HE Universal Oligo (1 mM) and 1μL SeqCap HE Index Oligo (1 mM) were combined and vacuum dried at 60 °C. The hybridization of DNA with EZ library probes was performed according to SeqCap EZ HyperCap protocol (NimbleGen, User’s guide v1.0) (Fig. 1). However, DynabeadsTM M-270 Streptavidin beads (Invitrogen, US) were used to capture the DNA. The captured DNA fragments were then amplified with the same PCR conditions as the first PCR with the only difference being that the number of cycles was increased to 15. The amplified DNA was purified with 0.5X AMPurePB beads. 1 μL amplified DNA was electrophoresed using a DNA 12,000 chip with the 2100 Bioanalyser system (Agilent) to analyze capture success.
PacBio library preparation
A preliminary step of single strand removal and DNA damage repair was performed using Exonuclease VII and the NEBNext® FFPE DNA Repair kit (New England Biolabs, MA, US) in a 48 μL reaction volume containing: the captured DNA (~500 ng), 5 μL of NEBNext® FFPE DNA repair buffer and 1 μL of exonuclease VII (NEB) (Fig. 1). The reaction was incubated 15 minutes at 37 °C. 2 μL of NEBNext® FFPE DNA Enzyme mix was added to the reaction and incubated 20 minutes at 37 °C. 5 μL of NEBNext® End Repair enzyme mix was then added to the reaction and incubated at 25 °C for 5 minutes. This was followed by a 0.45X AMPurePB bead clean-up step. Next, PacBio Blunt Adapters were ligated in a 40 μL reaction volume containing the end repaired reaction mixture of the previous step using 0.5 μM Annealed PacBio Blunt Adapters, 1X NEB T4 Ligase buffer and 2,000 units of T4 DNA Ligase. The reaction was incubated at 25 °C for 1 hour and at 65 °C for 10 minutes to inactivate the ligase. 100 units of Exonuclease III (NEB) and 10 units of Exonuclease VII NEB) were added to the reaction and incubated 37 °C for 45 minutes. This was followed by one 0.5X AMPure® PB bead clean-up and a second 0.45X AMPure® PB bead clean-up. The size and the concentration of the library was assayed on an Agilent Bioanalyzer using a DNA 12,000 chip according to manufacturer’s instructions.
Annealing and binding to the produced PacBio SMRTbell Template was performed according to the manufacturer’s recommendations (PacBio, US). For each sample, a control library without capture was also produced: only the shearing and the PacBio library preparation steps were utilized.
Bioinformatics analysis
PacBio circular consensus sequences (CCS) were generated using SMRT® pipe RS_ReadsOfInsert Protocol (PacBio) with a minimum 3 full passes and minimum predicted accuracy superior at 99 (Fig. 2). It was first necessary to remove the SeqCap adapter sequences by trimming off the first and last 65 bp of the reads using seqtk (github.com/lh3/seqtk) (Fig. 2). Reads smaller than 20 bp and reads containing residual adaptor sequences (potential chimeric reads) were detected and removed using seqkt (analyses were performed with an in-house shell script). The size of reads was calculated and their means, medians and boxplots were analyzed using R30.
A first de novo assembly was done using Canu31 with the standard overlap algorithm by varying the minimum reads length (100 to 2,200 bp) and the minimum overlap length (100 to 2,000 bp) (Fig. 2). The contigs belonging to Wolbachia symbionts were identified by nucleotide similarity using BLASTn32. If multiple contigs were obtained, a filtering was performed: the circular consensus sequences (CCS) were mapped against the best produced draft (having the largest contig size and/or the highest total length) using ngmlr33 with the PacBio preset settings (Fig. 2). A second de novo assembly was performed with the new selection of CCS using Canu31. If a single large contig was produced after the first original assembly, this selection step was not performed.
Successful final assemblies should produce a single large contig with the beginning and end of the genome assembly containing a duplicate sequence. To create a circularized genome, a “break” was introduced in the single contig and minimus2 (modified version of the minimus pipeline34) was used to detect overlaps and join the ends of the two contigs (Fig. 2). The final step was error correction of the draft. The CCS reads were once again mapped against the produced circularized genome using ngmlr33. Tests of polishing were performed to optimize the consensus sequence calling. Two methods were used: one using pilon35 in order to identify misassemblies and detect variants and a second using SAMtools and BCFtools (the parameter was tuned to reduce the effect of reads with excessive mismatches) (http://samtools.sourceforge.net/mpileup.shtml) (Fig. 2). Assembly statistics were evaluated using QUAST36. To analyze the polishing, the produced drafts and the genome reference were aligned using progressiveMauve37. PCR primers were designed to confirm the sites of circularization of the single contigs, as well as any sequences containing potential polymorphisms observed between the produced genome sequence and database references, when available (Table S5).
Evaluation of the enrichment was established by mapping each CCS against genome references and produced genomes using ngmlr33 with the PacBio preset settings (Fig. 2). In the case of B. malayi, the available wBm complete genome ASM838v1 (NC_006833)19 was used. In the case of A. albopictus, mapping was tested with the available different drafts: wAlbB ASM24241v3 (156 contigs, N50 = 12,719; 1,162,431 bp), ASM237914v1 (131 contigs, N50 = 12,474; 1,176,060 bp) and ASM237484v1 (177 contigs, N50 = 11,063; 1,517,743 bp) (direct NCBI submission). Unlike the two other samples, the enrichment for D. mauritiana was established mapping with the assembly of the current study because no reference was available. The coverage of the assembly was evaluated with SAMtools (samtools depth)38. In order to study the limitations of the assemblies, the processed genomes sequence or drafts were analyzed using RAST39. Transposase elements were identified: insertion sequences (ISs) using ISSAGA40 and group II introns were annotated by the RAST algorithm.
Supplementary information
Acknowledgements
We thank Stephen L. Dobson and Jason L. Rasgon for providing the mosquitoes infected by wAlbB. We thank Seth Bordenstein for original work in this area. We thank Rick Morgan, Lise Raleigh, Tom Evans, Eileen Dimalanta, Andy Gardner, Rich Roberts and Don Comb from New England Biolabs for helpful comments, suggestions and support. We thank Michael Weiand, Roberto Lleras and Cheryl Heiner from Pacific Biosciences and Roche Sequencing for their expertise. Supported by internal funding from NEB.
Author Contributions
E.L., L.S. and B.E.S. conceived and designed the experiments. E.L., L.V. and N.V. performed the experiments. E.L. analyzed the data. H.F. contributed materials. E.L., J.M.F. and B.E.S. wrote the main manuscript text. All authors reviewed the manuscript.
Data Availability
Data generated are available in GenBank: BioProject PRJNA508212; BioSample SAMN10519683 for Wolbachia endosymbiont strain TRS of Brugia malayi (genome: CP034333); BioSample SAMN10519763 and SAMN10519765 for Wolbachia endosymbiont of Drosophila mauritiana strain (respectively wMau lineages 177 and 181) (genome: CP034334 and CP034335); BioSample SAMN10519629 for Wolbachia endosymbiont of Aedes albopictus wAlbB (genome: RWIK00000000). The raw data are available in GenBank as Sequence Read Archive (SRA): SRR8283319 to SRR8283325.
Competing Interests
E.L., L.S., J.F., L.V. and B.S. are employed by New England Biolabs, Inc., who provided funding for this project.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
12/24/2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-42454-w.
References
- 1.Sachs JL, Skophammer RG, Bansal N, Stajich JE. Evolutionary origins and diversification of proteobacterial mutualists. Proceedings. Biological sciences/The Royal Society. 2014;281:20132146. doi: 10.1098/rspb.2013.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bandi C, Anderson TJ, Genchi C, Blaxter ML. Phylogeny of Wolbachia in filarial nematodes. Proceedings. Biological sciences/The Royal Society. 1998;265:2407–2413. doi: 10.1098/rspb.1998.0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sironi M, et al. Molecular evidence for a close relative of the arthropod endosymbiont Wolbachia in a filarial worm. Molecular and biochemical parasitology. 1995;74:223–227. doi: 10.1016/0166-6851(95)02494-8. [DOI] [PubMed] [Google Scholar]
- 4.Brown AM, et al. Genomic evidence for plant-parasitic nematodes as the earliest Wolbachia hosts. Sci Rep. 2016;6:34955. doi: 10.1038/srep34955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One. 2012;7:e38544. doi: 10.1371/journal.pone.0038544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nature reviews. Microbiology. 2008;6:741–751. doi: 10.1038/nrmicro1969. [DOI] [PubMed] [Google Scholar]
- 7.Bouchery T., Lefoulon E., Karadjian G., Nieguitsila A., Martin C. The symbiotic role of Wolbachia in Onchocercidae and its impact on filariasis. Clinical Microbiology and Infection. 2013;19(2):131–140. doi: 10.1111/1469-0691.12069. [DOI] [PubMed] [Google Scholar]
- 8.Slatko BE, Taylor MJ, Foster JM. The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis. 2010;51:55–65. doi: 10.1007/s13199-010-0067-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LePage DP, et al. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature. 2017;543:243–247. doi: 10.1038/nature21391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckmann JF, Ronau JA, Hochstrasser M. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat Microbiol. 2017;2:17007. doi: 10.1038/nmicrobiol.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newton IL, et al. Comparative Genomics of Two Closely Related Wolbachia with Different Reproductive Effects on Hosts. Genome biology and evolution. 2016;8:1526–1542. doi: 10.1093/gbe/evw096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunning Hotopp JC, et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317:1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- 13.Blaxter M. Symbiont genes in host genomes: fragments with a future? Cell host & microbe. 2007;2:211–213. doi: 10.1016/j.chom.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Dunning Hotopp JC, Klasson L. The Complexities and Nuances of Analyzing the Genome of Drosophila ananassae and Its Wolbachia Endosymbiont. G3 (Bethesda) 2018;8:373–374. doi: 10.1534/g3.117.300164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kent BN, et al. Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture. Genome biology and evolution. 2011;3:209–218. doi: 10.1093/gbe/evr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geniez S, et al. Targeted genome enrichment for efficient purification of endosymbiont DNA from host DNA. Symbiosis. 2012;58:201–207. doi: 10.1007/s13199-012-0215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nature reviews. Genetics. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brownlie JC, O’Neill SL. Wolbachia genomes: insights into an intracellular lifestyle. Current biology: CB. 2005;15:R507–509. doi: 10.1016/j.cub.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 19.Foster J, et al. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS biology. 2005;3:e121. doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comandatore F, et al. Supergroup C Wolbachia, mutualist symbionts of filarial nematodes, have a distinct genome structure. Open Biol. 2015;5:150099. doi: 10.1098/rsob.150099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma X, et al. Analysis of error profiles in deep next-generation sequencing data. Genome biology. 2019;20:50. doi: 10.1186/s13059-019-1659-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quail MA, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sohn JI, Nam JW. The present and future of de novo whole-genome assembly. Briefings in bioinformatics. 2018;19:23–40. doi: 10.1093/bib/bbw096. [DOI] [PubMed] [Google Scholar]
- 24.Eid J, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 25.Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic acids research. 2010;38:e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wenger, A. M. et al. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome. bioRxiv 10.1101/519025 (2019). [DOI] [PMC free article] [PubMed]
- 27.Giolai M, et al. Targeted capture and sequencing of gene-sized DNA molecules. Biotechniques. 2016;61:315–322. doi: 10.2144/000114484. [DOI] [PubMed] [Google Scholar]
- 28.Toomey ME, Panaram K, Fast EM, Beatty C, Frydman HM. Evolutionarily conserved Wolbachia-encoded factors control pattern of stem-cell niche tropism in Drosophila ovaries and favor infection. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:10788–10793. doi: 10.1073/pnas.1301524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xi Z, Dean JL, Khoo C, Dobson SL. Generation of a novel Wolbachia infection in Aedes albopictus (Asian tiger mosquito) via embryonic microinjection. Insect biochemistry and molecular biology. 2005;35:903–910. doi: 10.1016/j.ibmb.2005.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria 2017).
- 31.Koren S, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camacho C, et al. BLAST+: architecture and applications. BMC bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sedlazeck FJ, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature methods. 2018;15:461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darling AE, Mau B, Perna N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aziz RK, et al. The RAST Server: rapid annotations using subsystems technology. BMC genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome biology. 2011;12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated are available in GenBank: BioProject PRJNA508212; BioSample SAMN10519683 for Wolbachia endosymbiont strain TRS of Brugia malayi (genome: CP034333); BioSample SAMN10519763 and SAMN10519765 for Wolbachia endosymbiont of Drosophila mauritiana strain (respectively wMau lineages 177 and 181) (genome: CP034334 and CP034335); BioSample SAMN10519629 for Wolbachia endosymbiont of Aedes albopictus wAlbB (genome: RWIK00000000). The raw data are available in GenBank as Sequence Read Archive (SRA): SRR8283319 to SRR8283325.