

Abstract
Human IgG comprises four subclasses with different biological functions. The IgG3 subclass has a unique character, exhibiting high effector function and Fab arm flexibility. However, it is not used as a therapeutic drug owing to an enhanced susceptibility to proteolysis. Antibody aggregation control is also important for therapeutic antibody development. To date, there have been few reports of IgG3 aggregation during protein expression and the low pH conditions needed for purification and virus inactivation. This study explored the potential of IgG3 antibody for therapeutics using anti‐CD20 IgG3 as a model to investigate aggregate formation. Initially, anti‐CD20 IgG3 antibody showed substantial aggregate formation during expression and low pH treatment. To circumvent this phenomenon, we systematically exchanged IgG3 constant domains with those of IgG1, a stable IgG. IgG3 antibody with the IgG1 CH3 domain exhibited reduced aggregate formation during expression. Differential scanning calorimetric analysis of individual amino acid substitutions revealed that two amino acid mutations in the CH3 domain, N392K and M397V, reduced aggregation and increased CH3 transition temperature. The engineered human IgG3 antibody was further improved by additional mutations of R435H to obtain IgG3KVH to achieve protein A binding and showed similar antigen binding as wild‐type IgG3. IgG3KVH also exhibited high binding activity for FcγRIIIa and C1q. In summary, we have successfully established an engineered human IgG3 antibody with reduced aggregation during bioprocessing, which will contribute to the better design of therapeutic antibodies with high effector function and Fab arm flexibility.
Keywords: antibody, IgG3, aggregation, subclass change, differential scanning calorimetry, thermodynamic stability, pH stability, ADCC, CDC
Abbreviations
- ADCC
antibody‐dependent cellular cytotoxicity
- CDC
complement‐dependent cytotoxicity
- CH
constant heavy chain
- DSC
differential scanning calorimetry
- HMWS
high‐molecular‐weight species
- LMWS
low‐molecular‐weight species
- UHP‐SEC
ultrahigh‐pressure size exclusion chromatography
- VH
variable regions of heavy chains
- VL
variable regions of light chains
Introduction
Monoclonal antibodies comprise extremely versatile agents that are extensively used as targeted treatments for many diseases. Therapeutic antibodies have been approved for clinical use against a variety of disorders including cancer and chronic and autoimmune diseases. Furthermore, several novel antibodies are under development by pharmaceutical companies.1 There are four classes of human IgG antibodies (IgG1–4), each having a different biological function in the body.2, 3 For example, IgG1 and IgG3 subclass antibodies exhibit high antibody‐dependent cellular cytotoxicity (ADCC) and complement‐dependent cytotoxicity (CDC) compared with IgG2 and IgG4.3 Therefore, IgG1 and IgG3 subclass antibodies are more suitable if the effector function is required to eliminate target cells. The unique character of IgG3 arises from a 62‐amino acid long hinge region that confers a high flexibility on the Fab arm, which in turn influences the antigen‐binding ability.3 For example, human IgG3 monoclonal antibodies targeting HIV have a higher neutralizing ability than the IgG1 subclass.4 Some studies have also reported that IgG3 correlates with a lower risk of HIV‐1 infection and other infectious diseases.5, 6 IgG3 antibodies efficiently opsonize red blood cells or microorganisms, and induce superior phagocytosis.7, 8, 9 These unique characteristics indicate that human IgG3 provides a new platform for developing therapeutic antibodies against several diseases.
However, the IgG3 subclass has received little attention for therapy development because of its high allotypic polymorphism, susceptibility to proteolysis owing to the long hinge region, and short half‐life in the body.2, 10, 11 In addition, aggregate formation during production remains an issue. Several therapeutic IgG1 subclass antibody studies have also shown that aggregation can cause immunogenicity, posing a safety issue.12 Furthermore, IgG1 aggregation can occur at various steps of the manufacturing process including expression, purification, formulation, and storage.13 For example, protein aggregates are formed during expression and in the cell culture medium, and also induced during the purification step by the low pH treatments used for protein A/G elution and virus inactivation.14, 15 However, to date, little information exists regarding IgG3 aggregate formation during expression and low pH treatment because of a lack of interest in using the IgG3 subclass for drug development.
Here, we used anti‐CD20 IgG1 and IgG3 antibodies to compare and evaluate aggregate formation during expression and assessed their resistance to aggregate formation under low pH stress conditions. Wild‐type anti‐CD20‐IgG3 showed high aggregation during expression and was less stable than anti‐CD20‐IgG1 under low pH conditions. To stabilize the human IgG3 antibody, we engineered important amino acids in the IgG3 CH3 domain, which reduced aggregate formation. Our study demonstrated a method to express and purify human IgG3 antibodies with low aggregate formation, contributing to the development of therapeutic IgG3 antibodies with high‐effector function and increased antigen binding flexibility.
Results
Comparison of anti‐CD20 IgG1 and IgG3 aggregate formation during recombinant expression
To investigate the aggregation profile of human IgG3 during expression, we designed anti‐CD20 IgG1 and IgG3 model antibodies (Fig. 1). The heavy and light chain constant regions were expressed with an identical VH domain and light chain derived from rituximab, a therapeutic anti‐CD20 antibody.16 Ultrahigh‐pressure size‐exclusion chromatography (UHP‐SEC) analysis of IgG1 showed high‐molecular‐weight species (HMWS) of less than 5%. In contrast, the HMWS of IgG3 was around 40% [Fig. 2(A)]. Anti‐CD20 IgG3 aggregation content was higher than that of anti‐CD20 IgG1, which is consistent with a previous report.17
Figure 1.
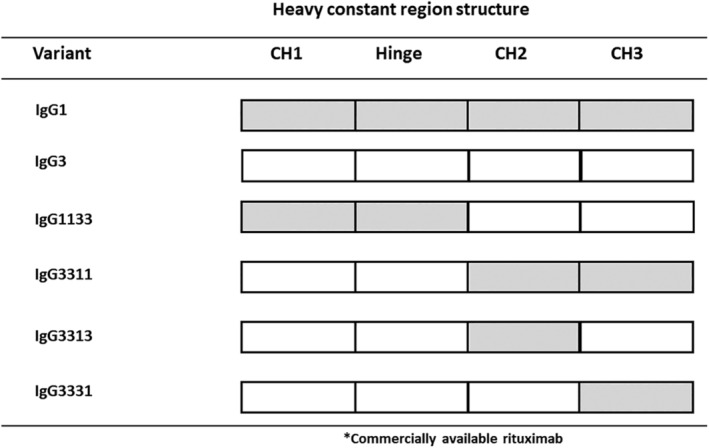
Summary of the domain‐exchanged antibodies constructed in this study. The closed and open squares represent domains derived from human IgG1 or IgG3, respectively. All antibodies shared variable regions from rituximab and the light chain constant region of Ck isotype.
Figure 2.
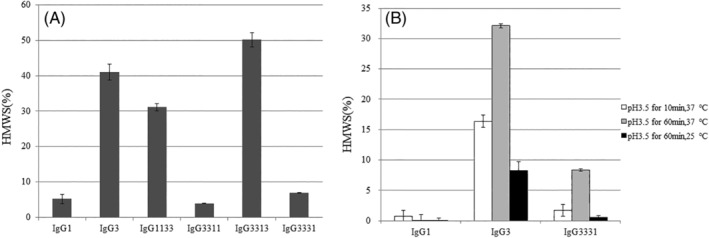
(A) Ultrahigh‐pressure size‐exclusion chromatography analysis of the proportion of high‐molecular‐weight species (HMWS) in preparations of domain‐exchanged antibodies. (B) Proportion of HMWS in preparations of pH‐stressed antibodies assessed using size‐exclusion HPLC. The pH stresses used were pH 3.5 for 10 min or 60 min at 25 or 37°C. Data are presented as the means ± SD and all experiments were performed in triplicate.
Design and generation of IgG1/IgG3 domain‐exchanged antibodies
To identify the important domain for aggregate formation during expression, we performed a systematic exchange of IgG3 and IgG1 domains within the CH region of each subclass (Fig. 1). We selected the IgG1 subclass for domain exchange, as it possesses particularly stable physicochemical properties.16, 18 During our analysis, we exchanged the IgG3 CH1, hinge, and Fc domains with the corresponding IgG1 domains. Specifically, we generated the 1133, 3311, 3313, and 3331 domain‐exchanged antibodies. The numbers in the names correspond to the IgG subclass of each domain, with the order of CH1‐hinge‐CH2‐CH3.
IgG3/IgG1 domain‐exchanged antibody aggregate formation during expression and low pH treatment
We next investigated the aggregate formation of domain‐exchanged antibodies using UHP‐SEC. Representative UHP‐SEC chromatograms of IgG3 are shown in Supporting Information Figure S1. The proportion of HMWS in the IgG3 and IgG1133 antibody with both the CH1 and Hinge domains exchanged was >30% [Fig. 2(A)]. In contrast, the IgG3311 antibody with both the CH2 and CH3 domains exchanged exhibited <10% HMWS. To determine the critical domain(s) for aggregate formation, we generated each individual CH2 or CH3 domain‐exchanged antibody, IgG3313 or IgG3331. The CH2 domain‐exchanged IgG3313 antibody showed >30% HMWS. In contrast, IgG3331, with only the CH3 domain exchanged, exhibited <10% HMWS. This indicated that the CH3 domain is important for IgG3 aggregate formation; although exchanging both the CH2 and CH3 domains between IgG3 and IgG1 (from IgG3 to IgG1) was most effective.
We also investigated IgG1, IgG3, and IgG3331 antibody stability under low pH stress. Initially, the proportion of HMWS in the three IgG purified samples was <5%. IgG1 exhibited a strong stability under low pH conditions with only a small increase in HMWS [Fig. 2(B)]. However, IgG3 showed marked HMWS formation after low pH treatment. In contrast, IgG3331 with IgG1 CH3 domain showed much less HMWS formation than IgG3.
Identification of amino acid residues in the CH3 domain critical for aggregation of IgG3
To identify important amino acid residues within the CH3 domain that contribute to IgG3 aggregate formation, we compared the amino acid sequences of the IgG1 and IgG3 CH3 domains using the IGHG1*01 and IGHG3*14 alleles [Fig. 3(A)]. These domains differed in six amino acids (positions 356, 358, 392, 397, 422, and 435 using Kabat numbering). Next, we converted the specific positions in the IgG3 construct to match the IgG1 sequence [Fig. 3(B)]. Among the six amino acid mutations, substituting N392K or M397V in IgG3 decreased the HMWS formation relative to that when wild‐type IgG3 is expressed. To determine the effect of the mutations in combination, we also generated an IgG3 variant carrying the N392K and M397V double substitution (IgG3KV). IgG3KV showed a further reduction of HMWS formation compared with that of individual N392K and M397V mutations. IgG3KV HMWS formation was similar to that of wild‐type IgG1 [Fig. 4(A)].
Figure 3.

(A) Amino acid alignment of the heavy chain CH3 domain of human IgG1(IGHG1*01) and IgG3(IGHG3*14). (B) Summary of the amino acids substitution antibodies constructed in this study. The gray bars indicate the substitution sites and open squares indicate the human IgG3 domains. All antibodies share variable regions from rituximab and the light chain constant region of Ck isotype. The substitutions made and positions are indicated.
Figure 4.
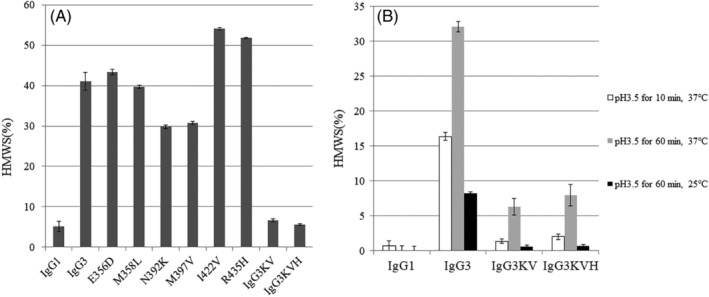
(A) Proportion of high‐molecular‐weight species (HMWS) in preparations of multipoint substitution antibodies. [IgG1 and IgG3 data are the same as in Fig. 2(A)]. (B) Proportion of HMWS in preparations of pH‐stressed antibodies using ultrahigh‐pressure size‐exclusion chromatography. The pH stresses used were pH 3.5 for 10 min or 60 min at 25 or 37°C. The antibodies examined were IgG1, IgG3, IgG3KV, and IgG3 KVH [IgG1 and IgG3 data are the same as in Fig. 2(B)]. Data are presented as the means ± SD and all experiments were performed in triplicate.
Development of an IgG3 antibody with reduced aggregate formation and improved protein A binding
We further modified IgG3KV by introducing an R435H substitution that was previously shown to be important for protein A binding.19 R435H substitution led to the creation of IgG3KVH, with three amino acid mutations introduced into the IgG3 antibody (N392K, M397V, and R435H). IgG3KVH aggregate formation during expression was <10%, similar to that of IgG3KV [Fig. 4(A)]. Next, IgG3KV and IgG3KVH antibodies were treated with different low pH conditions (pH 3.5 at 37°C for 10 or 60 min and at 25°C for 60 min), which are commonly used for elution from protein A and viral inactivation. Both IgG3KV and IgG3KVH showed less aggregate formation than IgG3 under low pH conditions (pH 3.5 at 37°C for 10 min and at 25°C for 60 min). Even under the most severe low pH conditions (pH 3.5 at 37°C for 60 min), the degree of aggregation by IgG3KV and IgG3KVH was <10% [Fig. 4(B)].
Biological activity of IgG3 antibody variants
Next, the biological functions of anti‐CD20 IgG3 variants were investigated. Flow cytometric experiments showed that the anti‐CD20 IgG3 variants IgG3331, IgG3KV, and IgG3KVH bound to CD20‐expressing cells similarly to wild‐type anti‐CD20 IgG3 (Fig. 5). This indicated that our stabilized antibodies do not lose their antigen binding ability.
Figure 5.
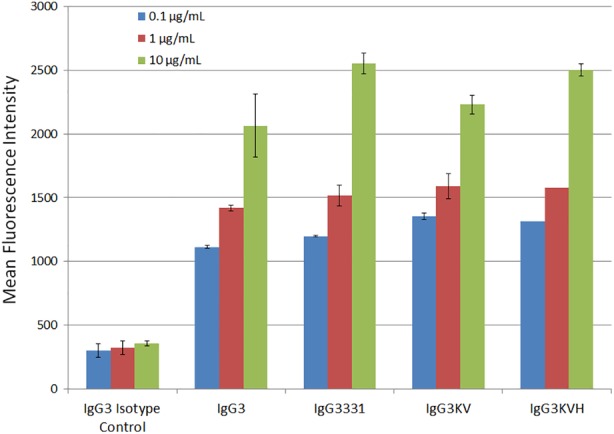
Flow cytometry antibody binding analysis of IgG3 rituximab wild‐type and variant antibodies to CD20 expressed by mouse EL‐4 cells. The antibody concentrations examined were 0.1, 1, and 10 μg/mL and the binding of each was measured using a FITC‐conjugated anti human IgG (H + L) secondary antibody.
The binding profiles of the anti‐CD20 IgG3 variants to human FcγRIIIa‐158V (high affinity) and FcγRIIIa‐158F (low affinity) allotypes that mediate ADCC activity were also examined by ELISA.20, 21, 22 Human IgG3 has a strong affinity for FcγRIIIa compared with IgG1.23 Consistent with these studies, the wild‐type IgG3 showed a high binding activity for FcγRIIIa than did IgG1, for both FcγRIIIa allotypes [Fig. 6(A,B)]. Although the stabilized CH3 variants of IgG3 (IgG3331 and IgG3KVH) showed a slightly reduced binding profile compared with wild‐type IgG3, they had higher binding activity for FcγRIIIa than did IgG1.
Figure 6.
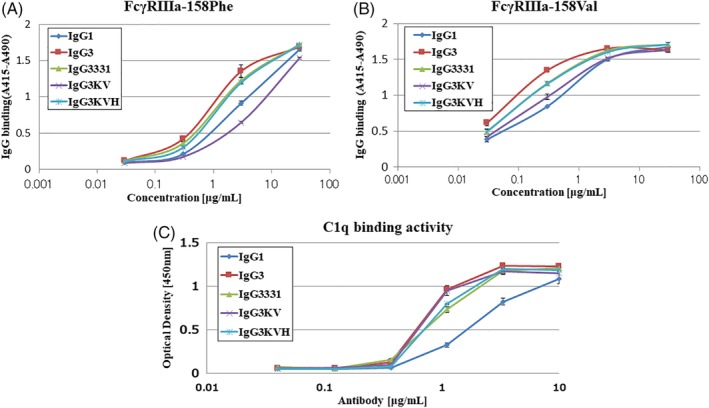
Effect of IgG3 CH3 domain amino acid substitutions on FcγRIIIa and C1q binding as assessed by ELISA. (A,B) two alleles of FcγRIIIa, FcγRIIIa‐158F and FcγRIIIa‐158 V, were individually coated on 96 well immunoplates using anti‐tetra His antibodies and then incubated with the indicated concentration of IgG1, IgG3, IgG3331 IgG3KV, or IgG3KVH. Binding was assessed using a peroxidase‐labeled secondary antibody. Data are presented as the means ± S.D. and all experiments were performed in triplicate. (C) C1q protein was coated on 96 well immunoplates and incubated with the indicated concentration of IgG1, IgG3, IgG3331, or IgG3KVH. Binding was assessed using anti C1q peroxidase‐labeled secondary antibody. Data are presented as the means ± S.D. and all experiments were performed in triplicate.
To confirm that the structure of the N‐linked oligosaccharide chains in the CH2 domains of the Fc region was the same in all the antibodies and had no effect on the binding affinity for FcγRIIIa, oligosaccharide analysis was performed with LC–MS, because human IgG1 antibodies that lack core fucose have previously been shown to have a high binding affinity for human FcγRIIIa.23, 24 The Fc oligosaccharide structures of anti‐CD20 antibodies were almost the same, comprising complex biantennary type and fully core‐fucosylated (Supporting Information Fig. S2).
Next, C1q binding activity was assessed using ELISA, because C1q binding activity is known to correlate with antibody CDC activity [Fig. 6(c)].25 IgG3 possessed higher C1q binding activity than IgG1, consistent with a previous report.26 The stabilized IgG3KV variant was found to bind C1q similar to IgG3. In contrast, IgG3331 and IgG3KVH exhibited reduced C1q binding compared to IgG3. However, the binding of both IgG3KV and IgG3KVH to C1q was still higher than that of IgG1.
Thermodynamic stabilities of IgG3 antibody variants
Finally, the thermodynamic stabilities of IgG3KV and IgG3KVH were examined using differential scanning calorimetry (DSC) (Fig. 7). For IgG1, Tm values of CH2 (Tm1), Fab (Tm2), and CH3 (Tm3) in PBS were approximately 72.2°C, 75.8°C, and 83.0°C, respectively. These values were ascribed to the unfolding of the CH2, Fab, and CH3 domain, respectively, and are in close agreement with previous results.27, 28 In relation to the Fab domain, Tm2 values among IgG3, IgG3KV, and IgG3KVH (ΔTm < 0.5) were almost identical. This is likely because the antibodies have the same variable region. Notably, the Tm1 values (CH2 domain) of IgG3, IgG3KV, and IgG3KVH differed (65.9°C, 64.9°C, and 68.8°C, respectively), even though all amino acids substitutions were within the CH3 domain. The Tm1 value of IgG3KV was lower than that of IgG3. In contrast, IgG3KVH had a 2.9°C higher Tm1 value than IgG3, even though it differed from IgG3KV with only one substitution. A similar trend was observed with the onset temperature (Tonset) values of each antibody [IgG3KVH (64.0°C) > IgG3 (61.8°C) > IgG3KV (60.4°C)]. Regarding the CH3 domain, Tm3 values were increased as predicted. Although we could not measure IgG3 Tm3 value, those of IgG3KV and IgG3KVH were measured as 82.7°C and 82.3°C, respectively. The Tm3 (CH3 domain) value of IgG1 was found to be 83.0°C which is almost the same as those of IgG3KV and IgG3KVH. Our data suggested that IgG3KV and IgG3KVH have an altered CH3 domain and Tm3 value that is comparable to those of IgG1.
Figure 7.
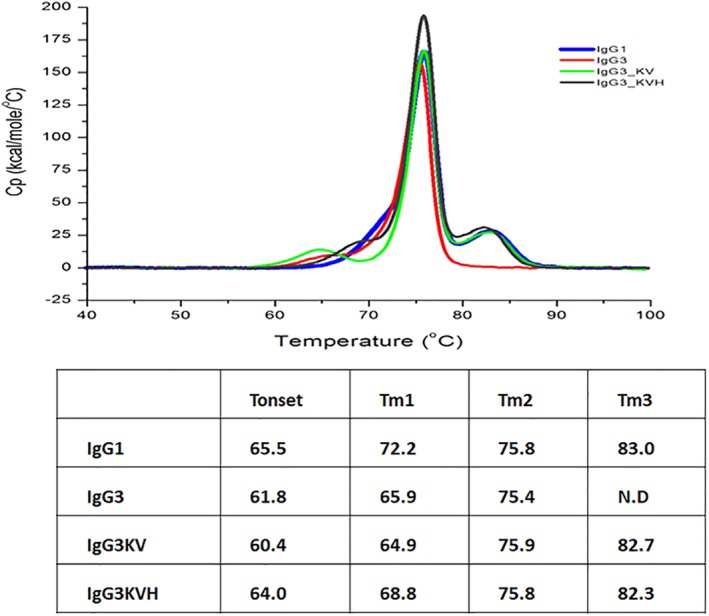
Analysis of the thermodynamic stabilities of antibody domains in PBS using differential scanning calorimetry (DSC). (A) DSC charts of IgG1, IgG3 wild‐type, and IgG3 variant antibodies. (B) Melting transition (Tm) and onset temperature (Tonset) of IgG3 variant antibodies determined from the DSC analysis.
Discussion
In this study, an engineered IgG3 antibody with reduced aggregation during expression and low pH treatment was explored. A previous study reported that the CH3 region is the most important domain contributing to antibody aggregation under acidic conditions.29 However, a second study suggested that the CH2 domain of both IgG1 and IgG2 subclasses differentially affect aggregation and stability.30 Our experiments appeared to support the former study in that the N392K and M397V substitutions, which are within the CH3 region, were responsible for controlling aggregate formation during expression and low pH treatments. These two residues are located within the CH3:CH3 interface and are involved in CH3‐CH3 interactions (Fig. 8).31, 32 It has also been reported that the Val397 residue is part of an aggregation‐prone motif found in all IgGs.33 In the present study, the introduction of an M397V substitution decreased HMWS formation, suggesting that this modification increases CH3‐CH3 interactions in IgG3 antibodies. Moreover, thermal stability of the IgG3 KVH CH3 domain was increased. Although we did not measure DSC at low pH, for example, pH 4.0/5.0/6.0, a prior study reported that a decrease in the Tm of the CH2 domain of IgG1, IgG2, and IgG4 subclass antibodies occurs with pH decrease.27 We hypothesized that a Tm decrease of the CH2 domain would also occur in IgG3 and IgG3KVH, which correlates with the observed formation of aggregates by both antibodies at low pH.
Figure 8.
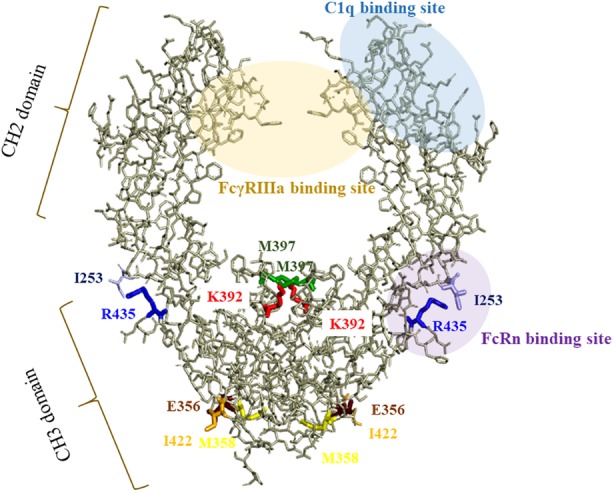
Structure of the Fc region in human IgG3. The model was built from published structural data of human IgG3 Fc (N392K) (Protein Data Bank accession code: 5W38). The six amino acids of IgG3 substitution sites (253, 356, 392, 397, 422, 435) within the CH3 domain and I253 within the CH2 domain are highlighted in color.
R435H substitution into the recombinant antibody with low aggregate formation (IgG3KVH) was successfully performed. Only two (G3 m[s,t]/G3 m15,16) of 13 allotypes, which are often found in the Mongoloid population, contained H435.11 This allows purification of the antibodies with both proteins G and A. Protein A is widely used for the purification of IgG1, IgG2, and IgG4 antibodies but not IgG3. Although R435H is within the CH3 domain of IgG3, the thermal stability of the IgG3KVH CH2 domain was higher than that of the IgG3KV CH2 domain. Previously, it was shown that the R435 side‐chain of the IgG3 CH3 domain is close to the I253 side chain of the CH2 domain. In contrast, the H435 residue is located some distance away from the I253 in the IgG1 CH2 domain. Therefore, the R435 and I253 residues may actually bring the CH2 domain closer.19 We hypothesized that the R435H substitution affects the CH2 pivot, increasing the thermal stability of the CH2 domain. However, our study only analyzed IgG3 aggregation during expression and low pH treatments. As aggregation can also occur during storage, long‐term stability studies should also be conducted with IgG3KVH to fully understand the true effectiveness of IgG3KVH as a potential therapeutic antibody. In addition, it has been reported that some excipients affect the IgG1 antibody aggregation behavior.34 For example, sucrose stabilizes IgG1‐type antibodies against aggregate formation and enhances the first conformation transition (i.e., CH2 domain for IgG1). In contrast, arginine destabilizes some antibodies in a concentration‐dependent manner. These phenomena suggest that an appropriate formulation may stabilize the IgG3KVH antibody and decrease the aggregation during the storage step.
To examine the biological activity of IgG3KVH, FcγRIIIa, C1q, and antigen binding were investigated. Previously, a strong correlation was found between ADCC activity and FcγRIIIa binding.22 The FcγRIIIa‐158V allele exhibits a high binding affinity for IgG Fc regions, whereas FcγRIIIa‐158F has low affinity. Although both IgG3KVH and IgG3331 more strongly bound to FcγRIIIa‐158V and FcγRIIIa‐158F than IgG1, FcγRIIIa binding activity was slightly lower than that for IgG3. For the IgG3KV antibody, FcγRIIIa binding activity was drastically decreased. It has also been shown that IgG1 binding to FcγRIIIa is mediated through the hinge and CH2 regions and that the CH3 domain does not directly affect binding.35, 36 A previous study indicated that the CH3 and CH2 domains of the IgG2 antibody interacted via L251 in the CH2 region and residues M428, H429, E430, and H435 in the CH3 domain.37 This is supported by another study that suggested that the R435 residue in the IgG3 CH3 domain may influence the CH2 domain pivot.19 Based on these data and our own results, we hypothesized that the substitutions made in the IgG3 CH3 domain affected the CH2 domain structure allosterically, impacting FcγRIIIa binding. In addition, it has been suggested that the FcγRIIIa binding, as detected by solid phase ELISA assay alone, does not fully predict the actual cellular ADCC.23 This suggested that further ADCC activity analysis is required to fully understand the potential of the stabilized IgG3 antibodies we have established.
Our recombinant antibody (IgG3KVH) showed stronger binding to C1q than IgG1. Previously, it was reported that residues D270, K322, P329, and P331 in the CH2 domain constitute the human C1q binding site of the Fc region.25, 36, 38 However, the substitutions introduced into IgG3KVH were all within the CH3 region. We hypothesized that the CH2 domain structure of IgG3KVH, IgG3331, and IgG3KV is different owing to interaction with the CH3 region, which affects the C1q binding domain allosterically.
It has been demonstrated that a R435H substitution in IgG3 can increase FcRn binding, thereby impacting the overall pharmacokinetics (PK).39 Therefore, it is possible that IgG3KVH has an increased PK profile compared to IgG3. Further studies are needed to better understand the FcRn binding and PK profiles of IgG3KVH.
Our stabilized IgG3 antibody addresses the aggregation issue during expression and low pH treatment. However, the issue of sensitivity to proteolysis remains and must be addressed before IgG3 can be used as a therapeutic antibody. We investigated the degradation of the IgG3 antibodies under low pH treatment by monitoring the LMWS using UPLC‐SEC. The results indicated that IgG3 LMWS formation was well correlated with HMWS formation tendency (Supporting Information Fig. S3). Moreover, a previous study suggested that the hinge region of human serum IgG3 is O‐glycosylated, indicating that O‐glycans may be able to shield the hinge region from proteolytic cleavage.40 Increasing O‐glycosylation in the IgG3 hinge region may therefore be an effective way to prevent degradation and proteolysis. Mutation study of the hinge region may also be able to resolve the issue of susceptibility to proteolysis. Nevertheless, further studies including in vivo analysis of proteolysis will be needed prior to IgG3 antibody use as a therapeutic drug.
Conclusion
In summary, we have engineered an IgG3 antibody with a triple substitution (N392K, M397 V, and R435H) that displays reduced aggregation during expression and increased resistance to acid‐induced stress (IgG3KVH). Notably, IgG3KVH retained the unique high binding to FcγRIIIa and C1q, and displayed antigen binding similar to that of the wild‐type IgG3. Our design therefore contributes to the engineering of therapeutic IgG3 antibodies with high effector function and Fab flexibility.
Materials and Methods
Materials
IgG antibodies were manufactured by Kyowa Hakko Kirin (Tokyo, Japan) and expressed in Expi293F cells under identical conditions. Secreted antibodies were recovered from culture medium and purified using protein G chromatography. SEC chromatography was used to isolate monomer fractions.
Construction, expression, and purification of antibodies
Expression vectors for the anti‐CD20 IgG3 antibody were constructed by cloning the VH and VL coding regions of C2B8 (VH: GenBank accession no. AR000013, VL: GenBank accession no. AR015962) along with the IgG3 wild‐type (IgG3) constant region into the pCI plasmid (Biogen IDEC, Cambridge, MA, USA). The constructs were then transfected into Expi293F cells (Invitrogen, Carlsbad, CA, USA), and purified from supernatants with Protein G‐conjugated sepharose columns (GE Healthcare, Chicago, IL, USA). We also constructed antibody expression vectors encoding exchanged and/or substituted IgG1 subclass domains including IgG1133, IgG3311, IgG3313, IgG3331, IgG3_Glu356Asp(E356D), IgG3_Met358Leu(M358 L), IgG3_Asn392Lys(N392K), IgG3_Ile422Val(I422V), and IgG3_Arg435His(R435H), where each numeral indicates EU numbering. All antibodies were purified using a Protein G sepharose 4 Fast Flow (GE Healthcare), according to the manufacturer's instructions. Purified antibodies were buffer‐exchanged with D‐PBS (Gibco, Gaithersburg, MD, USA) using an NAP25 desalting column (GE Healthcare). Finally, antibody concentrations were adjusted to 1.0 mg/mL and the purity of prepared samples analyzed by SDS‐PAGE using a previously published method.41
Ultrahigh‐pressure size‐exclusion chromatography (UHP‐SEC)
The proportions of monomers, HMWS and LMWS, were measured using UHP‐SEC on an ACQUITY UPLC BEH200 SEC (200 Å, 1.7 μm, 4.6 × 150 mm; Waters Corp., Milford, MA USA). The mobile phase contained 20 mM sodium phosphate (pH 7.0) and 500 mM NaCl. The experimental conditions were: flow rate of 0.5 mL/min, detection wavelength of 215 nm, and analysis time of 5.5 min. Samples (5 μL) from 1 mg/mL MAb solutions were injected into each column, corresponding to 5 μg total protein. Peaks of antibody protein monomers, HMWS and LMWS, were analyzed by integrating the area under each eluting peak using Empower 2 chromatography Data Software (Waters Corp.) and recorded as percentages of HMWS and LMWS.
Low pH stress assays
Antibodies were treated with 0.1 M citric acid buffer (pH 2.7) adjusted to pH 3.5, at 37°C for 10 or 60 min and 25°C for 60 min using a PCR Thermocycler (Applied Systems, Foster City, CA, USA). For neutralization, the samples were treated with 500 mM phosphate buffer (pH 8.0) at 4°C. The degree of aggregation was analyzed by UHP‐SEC, as previously described.
Measurement of CD20 binding activity
CD20‐binding analysis was performed by flow cytometry. Briefly, CD20/EL4 cells (106) were stained with 10, 1, or 0.1 μg/mL anti‐CD20 IgG for 60 min on ice.24 Fluorescein Isothiocyanate (FITC)‐conjugated anti‐human IgG (H + L) was used as the secondary reagent. Stained cells were analyzed using a Canto2 flow cytometer (Beckman Coulter, Tokyo, Japan). Anti‐Human IgG3 antibody was used as a control.
Preparation of hexa‐His‐tagged recombinant soluble human Fc receptors
FcγRIIIa‐158F and FcγRIIIa‐158 V were prepared as described previously.23 For both FcγRIIIa receptors, the transmembrane and intracellular domains were replaced by a hexa‐His tag. The concentrations of purified proteins were measured by assessing absorbance at 280 nm and their purities and molecular weights confirmed by SDS‐PAGE.
FcγRIIIa binding assay
Assessment of IgG binding to recombinant FcγRIIIa was performed by ELISA as previously described.42 Both polymorphic variants of recombinant FcγRIIIa (FcγRIIIa‐158F and FcγRIIIa‐158 V) were used in the assay. Human Anti IgG3‐isotype control antibody was used as the control.
C1q binding assay
The binding of human C1q (Quidel, San Diego, CA) to rituximab and recombinant antibodies was assessed by ELISA using a previously published method.25
Differential scanning calorimetry (DSC)
The thermal stability of individual domains was evaluated by DSC. Measurements were performed on 0.5 mg/mL IgG in PBS buffer (pH 7.4) using a Micro Cal VP‐Capillary DSC system (Malvern Instruments Ltd., Worcestershire, UK). Temperature scans were performed from 25 to 100°C at a scan rate of 1°C/min. A buffer‐buffer reference scan was subtracted from each sample scan before concentration normalization. Baselines were established using Origin7.0 (Origin Lab, Northampton, MA, USA) and cubic interpolation of the pre‐ and post‐transition baselines.
Oligosaccharide analysis using LC‐MS
N297‐attached oligosaccharides were analyzed with LC‐MS as previously described.43 Each glycoform was distinguished by its specific mass and the intensities estimated using total ion chromatograms generated by extracted ion chromatograms. The extracted ion chromatograms were created by extracting the ion signal for the mass of a particular peptide from the total ion chromatograms acquired on the mass spectrometer. All glycoform peak intensities were summed and the ratio of each glycoform calculated.
Supporting information
Supplementary Fig. S1 UHP‐SEC analysis of IgG3.
Supplementary Fig. S2 LC–MS analysis of Asn297‐attached oligosaccharide.
Supplementary Fig. S3 Ultrahigh‐pressure size‐exclusion chromatography analysis of the proportion of LMWS in preparations of domain‐exchanged antibodies.
Acknowledgments
We would like to thank Dr. Rinpei Niwa for valuable discussion and suggestions. We are also grateful to Mrs. Machi Kusunoki for ELISA analysis. We thank Editage (www.editage.jp) for English language editing.
Statement of importance: Antibody‐based therapies are used for many diseases; although few utilize immunoglobin‐G3, as such antibodies are susceptible to degradation and aggregation. However, this subclass possesses features that are ideal for therapeutic applications, such as high effector function. To encourage development of immunoglobin‐G3‐based therapies, we created variants that exhibit less aggregation and are easier to purify. This work contributes to the development of a wider range of antibody therapies that offer distinct advantages with fewer disadvantages.
References
- 1. Kaplon H, Reichert JM (2018) Antibodies to watch in 2018. MAbs 10:183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salfeld JG (2007) Isotype selection in antibody engineering. Nat Biotechnol 25:1369–1372. [DOI] [PubMed] [Google Scholar]
- 3. Vidarsson G, Dekkers G, Rispens T (2014) IgG subclasses and allotypes: from structure to effector functions. Front Immunol 5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scharf O, Golding H, King LR, Eller N, Frazier D, Golding B, Scott DE (2001) Immunoglobulin G3 from polyclonal human immunodeficiency virus (HIV) immune globulin is more potent than other subclasses in neutralizing HIV type 1. J Virol 75:6558–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Irani V, Guy AJ, Andrew D, Beeson JG, Ramsland PA, Richards JS (2015) Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol Immunol 67:171–182. [DOI] [PubMed] [Google Scholar]
- 6. Yates NL, Liao HX, Fong Y, deCamp A, Vandergrift NA, Williams WT, Alam SM, Ferrari G, Yang ZY, Seaton KE, Berman PW, Alpert MD, Evans DT, O'Connell RJ, Francis D, Sinangil F, Lee C, Nitayaphan S, Rerks‐Ngarm S, Kaewkungwal J, Pitisuttithum P, Tartaglia J, Pinter A, Zolla‐Pazner S, Gilbert PB, Nabel GJ, Michael NL, Kim JH, Montefiori DC, Haynes BF, Tomaras GD (2014) Vaccine‐induced Env V1‐V2 IgG3 correlates with lower HIV‐1 infection risk and declines soon after vaccination. Sci Transl Med 6:228ra239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tebo AE, Kremsner PG, Luty AJ (2002) Fcgamma receptor‐mediated phagocytosis of plasmodium falciparum‐infected erythrocytes in vitro. Clin Exp Immunol 130:300–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wiener E, Atwal A, Thompson KM, Melamed MD, Gorick B, Hughes‐Jones NC (1987) Differences between the activities of human monoclonal IgG1 and IgG3 subclasses of anti‐D(Rh) antibody in their ability to mediate red cell‐binding to macrophages. Immunology 62:401–404. [PMC free article] [PubMed] [Google Scholar]
- 9. Braster R, Grewal S, Visser R, Einarsdottir HK, van Egmond M, Vidarsson G, Bögels M (2017) Human IgG3 with extended half‐life does not improve fc‐gamma receptor‐mediated cancer antibody therapies in mice. PLoS One 12:e0177736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carter PJ (2006) Potent antibody therapeutics by design. Nat Rev Immunol 6:343–357. [DOI] [PubMed] [Google Scholar]
- 11. Lefranc MP, Lefranc G (2012) Human Gm, km, and am allotypes and their molecular characterization: a remarkable demonstration of polymorphism. Methods Mol Biol 882:635–680. [DOI] [PubMed] [Google Scholar]
- 12. Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, Topp EM (2016) Immunogenicity of therapeutic protein aggregates. J Pharm Sci 105:417–430. [DOI] [PubMed] [Google Scholar]
- 13. Mahler HC, Friess W, Grauschopf U, Kiese S (2009) Protein aggregation: pathways, induction factors and analysis. J Pharm Sci 98:2909–2934. [DOI] [PubMed] [Google Scholar]
- 14. Cromwell ME, Hilario E, Jacobson F (2006) Protein aggregation and bioprocessing. AAPS J 8:E572–E579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ejima D, Tsumoto K, Fukada H, Yumioka R, Nagase K, Arakawa T, Philo JS (2007) Effects of acid exposure on the conformation, stability, and aggregation of monoclonal antibodies. Proteins 66:954–962. [DOI] [PubMed] [Google Scholar]
- 16. McLaughlin P, Grillo‐López AJ, Link BK, Levy R, Czuczman MS, Williams ME, Heyman MR, Bence‐Bruckler I, White CA, Cabanillas F, Jain V, Ho AD, Lister J, Wey K, Shen D, Dallaire BK (1998) Rituximab chimeric anti‐CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four‐dose treatment program. J Clin Oncol 16:2825–2833. [DOI] [PubMed] [Google Scholar]
- 17. Rösner T, Derer S, Kellner C, Dechant M, Lohse S, Vidarsson G, Peipp M, Valerius T (2013) An IgG3 switch variant of rituximab mediates enhanced complement‐dependent cytotoxicity against tumour cells with low CD20 expression levels. Br J Haematol 161:282–286. [DOI] [PubMed] [Google Scholar]
- 18. Rouet R, Lowe D, Christ D (2014) Stability engineering of the human antibody repertoire. FEBS Lett 588:269–277. [DOI] [PubMed] [Google Scholar]
- 19. Shah IS, Lovell S, Mehzabeen N, Battaile KP, Tolbert TJ (2017) Structural characterization of the Man5 glycoform of human IgG3 Fc. Mol Immunol 92:28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cartron G, Dacheux L, Salles G, Solal‐Celigny P, Bardos P, Colombat P, Watier H (2002) Therapeutic activity of humanized anti‐CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood 99:754–758. [DOI] [PubMed] [Google Scholar]
- 21. Weng WK, Levy R (2003) Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol 21:3940–3947. [DOI] [PubMed] [Google Scholar]
- 22. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M (1997) Fc gammaRIIIa‐158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa‐48L/R/H phenotype. Blood 90:1109–1114. [PubMed] [Google Scholar]
- 23. Niwa R, Natsume A, Uehara A, Wakitani M, Iida S, Uchida K, Satoh M, Shitara K (2005) IgG subclass‐independent improvement of antibody‐dependent cellular cytotoxicity by fucose removal from Asn297‐linked oligosaccharides. J Immunol Methods 306:151–160. [DOI] [PubMed] [Google Scholar]
- 24. Niwa R, Sakurada M, Kobayashi Y, Uehara A, Matsushima K, Ueda R, Nakamura K, Shitara K (2005) Enhanced natural killer cell binding and activation by low‐fucose IgG1 antibody results in potent antibody‐dependent cellular cytotoxicity induction at lower antigen density. Clin Cancer Res 11:2327–2336. [DOI] [PubMed] [Google Scholar]
- 25. Idusogie EE, Presta LG, Gazzano‐Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG (2000) Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol 164:4178–4184. [DOI] [PubMed] [Google Scholar]
- 26. Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, Wakitani M, Ohta S, Satoh M, Shitara K, Niwa R (2008) Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res 68:3863–3872. [DOI] [PubMed] [Google Scholar]
- 27. Ito T, Tsumoto K (2013) Effects of subclass change on the structural stability of chimeric, humanized, and human antibodies under thermal stress. Protein Sci 22:1542–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tischenko VM, Abramov VM, Zav'yalov VP (1998) Investigation of the cooperative structure of Fc fragments from myeloma immunoglobulin G. Biochemistry 37:5576–5581. [DOI] [PubMed] [Google Scholar]
- 29. Yageta S, Lauer TM, Trout BL, Honda S (2015) Conformational and colloidal stabilities of isolated constant domains of human immunoglobulin G and their impact on antibody aggregation under acidic conditions. Mol Pharm 12:1443–1455. [DOI] [PubMed] [Google Scholar]
- 30. Hari SB, Lau H, Razinkov VI, Chen S, Latypov RF (2010) Acid‐induced aggregation of human monoclonal IgG1 and IgG2: molecular mechanism and the effect of solution composition. Biochemistry 49:9328–9338. [DOI] [PubMed] [Google Scholar]
- 31. Ridgway JB, Presta LG, Carter P (1996) 'Knobs‐into‐holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng 9:617–621. [DOI] [PubMed] [Google Scholar]
- 32. Rispens T, Davies AM, Ooijevaar‐de Heer P, Absalah S, Bende O, Sutton BJ, Vidarsson G, Aalberse RC (2014) Dynamics of inter‐heavy chain interactions in human immunoglobulin G (IgG) subclasses studied by kinetic Fab arm exchange. J Biol Chem 289:6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chennamsetty N, Helk B, Voynov V, Kayser V, Trout BL (2009) Aggregation‐prone motifs in human immunoglobulin G. J Mol Biol 391:404–413. [DOI] [PubMed] [Google Scholar]
- 34. Thakkar SV, Joshi SB, Jones ME, Sathish HA, Bishop SM, Volkin DB, Middaugh CR (2012) Excipients differentially influence the conformational stability and pretransition dynamics of two IgG1 monoclonal antibodies. J Pharm Sci 101:3062–3077. [DOI] [PubMed] [Google Scholar]
- 35. Sondermann P, Oosthuizen V (2002) X‐ray crystallographic studies of IgG‐Fc gamma receptor interactions. Biochem Soc Trans 30:481–486. [DOI] [PubMed] [Google Scholar]
- 36. Leoh LS, Daniels‐Wells TR, Martinez‐Maza O, Penichet ML (2015) Insights into the effector functions of human IgG3 in the context of an antibody targeting transferrin receptor 1. Mol Immunol 67:407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Teplyakov A, Zhao Y, Malia TJ, Obmolova G, Gilliland GL (2013) IgG2 Fc structure and the dynamic features of the IgG CH2‐CH3 interface. Mol Immunol 56:131–139. [DOI] [PubMed] [Google Scholar]
- 38. Michaelsen TE, Sandlie I, Bratlie DB, Sandin RH, Ihle O (2009) Structural difference in the complement activation site of human IgG1 and IgG3. Scand J Immunol 70:553–564. [DOI] [PubMed] [Google Scholar]
- 39. Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, Zhao Y, Kleijer M, Sandlie I, de Haas M, Jonsdottir I, van der Schoot CE, Vidarsson G (2011) Competition for FcRn‐mediated transport gives rise to short half‐life of human IgG3 and offers therapeutic potential. Nat Commun 2:599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Plomp R, Dekkers G, Rombouts Y, Visser R, Koeleman CA, Kammeijer GS, Jansen BC, Rispens T, Hensbergen PJ, Vidarsson G, Wuhrer M (2015) Hinge‐region O‐glycosylation of human immunoglobulin G3 (IgG3). Mol Cell Proteomics 14:1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laemmli UK (1970) Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 227:680–685. [DOI] [PubMed] [Google Scholar]
- 42. Niwa R, Hatanaka S, Shoji‐Hosaka E, Sakurada M, Kobayashi Y, Uehara A, Yokoi H, Nakamura K, Shitara K (2004) Enhancement of the antibody‐dependent cellular cytotoxicity of low‐fucose IgG1 is independent of FcgammaRIIIa functional polymorphism. Clin Cancer Res 10:6248–6255. [DOI] [PubMed] [Google Scholar]
- 43. Sinha S, Pipes G, Topp EM, Bondarenko PV, Treuheit MJ, Gadgil HS (2008) Comparison of LC and LC/MS methods for quantifying N‐glycosylation in recombinant IgGs. J Am Soc Mass Spectrom 19:1643–1654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1 UHP‐SEC analysis of IgG3.
Supplementary Fig. S2 LC–MS analysis of Asn297‐attached oligosaccharide.
Supplementary Fig. S3 Ultrahigh‐pressure size‐exclusion chromatography analysis of the proportion of LMWS in preparations of domain‐exchanged antibodies.