

Abstract
The cannabinoid type-1 (CB1) receptor, a G-protein-coupled receptor, is an attractive target for drug discovery due to its involvement in many physiological processes. Historically, drug discovery efforts targeting the CB1 receptor have focused on the development of orthosteric ligands that interact with the active site to which endogenous cannabinoids bind. Research performed over the last several decades has revealed substantial difficulties in translating CB1 orthosteric ligands into druggable candidates. The difficulty is mainly due to the adverse effects associated with orthosteric CB1 ligands. Recent discoveries of allosteric CB1 modulators provide tremendous opportunities to develop CB1 ligands with novel mechanisms of action; these ligands may potentially improve the pharmacological effects and enhance drug safety in treating the disorders by regulating the functions of the CB1 receptor. In this paper, we review and summarize the complex pharmacological profiles of each class of CB1 allosteric modulators, the development of new classes of CB1 allosteric modulators and the results from in vivo assessments of their therapeutic value.
Keywords: allosteric modulator, cannabinoid CB1 receptor, G-protein-coupled receptor, biased signaling, functional selectivity, therapeutic potential, drug discovery
Introduction
The cannabinoid CB1 receptor is a member of the endocannabinoid system that comprises at least two G-protein-coupled receptors (GPCRs), the cannabinoid type-1 receptor (CB1) and the cannabinoid type-2 receptor (CB2). The CB1 receptor is the most abundant GPCR expressed in the central nervous system (CNS) and is widely distributed at lower concentrations in a variety of peripheral tissues [1, 2]. In contrast, the CB2 receptor is primarily located in the periphery, with high concentrations in the tonsil, spleen and immune-related cells [3]. Similar to ∆9-tetrahydrocannabinol (∆9-THC), the main active ingredient of cannabis, a group of lipid-derived molecules represented by N-arachidonoyl ethanolamide (AEA) and 2-arachidonoyl glycerol (2-AG) functions as endogenous agonists of the two cannabinoid receptors.
Several catabolic enzymes, including fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MGL), are responsible for promptly degrading the endogenous cannabinoid ligands to prevent hyperactivity of the cannabinoid receptors under physiological conditions [1–3]. The CB1 receptor is linked to several disease states, including chemotherapy-induced nausea, wasting syndromes associated with cancer and AIDS, obesity, neurodegenerative disorders, pain, and substance abuse disorders [1, 4]. Traditionally, drug discovery efforts aimed at the CB1 receptors have focused on ligands targeting the CB1 orthosteric sites to which the endogenous cannabinoids bind. However, ligands targeting the orthosteric sites of CB1 receptors exert either psychotropic side effects, such as CB1 agonists [5], or psychiatric side effects, such as globally active CB1 antagonists/inverse agonists [6]. These untoward side effects have hampered drug discovery efforts aimed at regulating CB1 receptors for therapeutic benefits. On the other hand, the CB2 receptor has attracted considerable attention as a therapeutic target for pain, cancer, inflammation-derived neurodegenerative diseases, and osteoporosis [7, 8]. A large number of preclinical models have been developed and employed to evaluate CB2-selective agonists [7–10]. Some CB2-selective agonists were assessed in clinical trials. Unfortunately, the outcomes were disappointing and failed to meet the primary endpoints. The possible causes for the disappointing clinical outcomes were recently discussed by other groups [7, 8]. Briefly, the failures are likely due to several factors: (1) inadequate preclinical studies to precisely predict clinical efficacies in humans; (2) the lack of CB2 specific ligands to validate the target in preclinical investigations; and (3) insufficient engagement of the drug with its target and possible off-target side effects. Therefore, additional in-depth investigations are needed to address these issues before CB2 selective ligands are translated into efficacious therapeutics.
The clinical failures of centrally active CB1 inverse agonists and the psychoactive liability of CB1 agonists have promoted the pursuit of new approaches to develop safer molecules targeting the cannabinoid CB1 receptor for drug discovery. These approaches include (1) neutral antagonists of the CB1 receptors, which spare the drugs from antagonizing the constitutive activity of CB1 receptor while antagonizing the effects of the endocannabinoids [11, 12], (2) peripherally acting CB1 ligands that prevent the ligands from interacting with central CB1 receptors to abolish CNS side effects [13], and (3) allosteric modulators of the CB1 receptor that display a novel mechanism of action to improve the drug safety profile of CB1 ligands [14, 15]. To date, approximately nine types of small molecules that allosterically modulate the CB1 receptors have been reported. These molecules offer researchers new opportunities to intervene in the actions of physiologically and pathologically important CB1 receptors. This review provides basic knowledge to improve our understanding of promising CB1 allosteric modulators and their potential therapeutic utility.
The cannabinoid CB1 receptor and its complex signaling network
The cannabinoid CB1 receptor was identified in the early 1990s in a study that revealed the mechanism of cannabis addiction and characterized a class 1A rhodopsin-like GPCR [3]. CB1 receptors are widely distributed in the human body, and are primarily expressed in the CNS and to a lesser but functional extent in the periphery [16, 17]. The CB1 receptor is the most abundant GPCR in the brain, with particularly high levels observed in the neocortex, hippocampus, basal ganglia, cerebellum, and brainstem [18]. CB1 receptors play important roles in a variety of physiological conditions, including neuronal development, neuronal plasticity, food intake and energy balance, perception processes, immunomodulation, cell apoptosis, and cardiovascular and reproductive functions [1, 19, 20]. The multiple functions of the CB1 receptor are apparently due to its complex cellular signaling pathways. Traditionally, the functions of CB1 receptor are regulated by (1) endogenous cannabinoid agonists (e.g., 1, AEA and 2, 2-AG, Fig. 1), (2) exogenous agonists from phytocannabinoids (e.g., 3, Δ9-THC, Fig. 1) and synthetic cannabimimetics (e.g., 4, HU210; 5, CP55,940; and 6, WIN55,212-2; Fig. 1), and (3) synthetic inverse agonists/antagonists (e.g., 7, SR141716A, and 8, AM251, Fig. 1) and neutral antagonists (e.g., 9, AM6545, Fig. 1). Typically, a CB1 orthosteric inverse agonist binds to the same receptor site as an orthosteric agonist but induces a biological response opposite to an agonist. In contrast, a neutral antagonist binds to the receptor in the orthosteric site, does not change the equilibrium of the receptor, and only antagonizes the endogenously released endocannabinoids, but not by modulating the constitutive activity of the CB1 receptor [12, 21].
Fig. 1.

Structures of representative orthosteric ligands of the CB1 receptor
Upon activation, the CB1 receptor transduces signals through its interaction with Gi/o-, Gs-, and Gq/11-proteins, of which Gi/o is preferentially coupled. Stimulation of the CB1 receptor typically results in activation and dissociation of the coupled Gi/o-protein heterotrimers (i.e., α and βγ subunits). The released Giα subunits interact with adenylyl cyclase (AC) and inhibit its catalysis of cAMP production. The reduction in cAMP production downregulates protein kinase A (PKA), which in turn suppresses PKA-mediated signaling events. The dissociated βγ subunits stimulate the phosphatidylinositide 3-kinase (PI3K) and protein kinase B (PKB) pathways, which induces the phosphorylation of mitogen-activated protein kinases (MAPKs). Along with the G-protein-mediated signal transduction, the CB1 receptor also interacts with a variety of non-G-protein partners, including β-arrestins, the adaptor protein AP-3, GPCR-associated sorting proteins (GASP), the factor associated with neutral sphingomyelinase (FAN) and the cannabinoid receptor-interacting protein 1a (CRIP1a), to regulate its downstream effectors that include some MAPKs, multiple receptor tyrosine kinases and extracellular signal-regulated kinases (ERK) [22–25]. Of these non-G-protein partners, CRIP1a is interesting since it is involved in CB1 desensitization and intracellular trafficking processes in which β-arrestins play critical roles. CRIP1a expression modulates ERK1/2 phosphorylation and the coupling selectivity among different G-protein α subunits [26], which are related to the functional selectivity of CB1 ligands.
CB1 receptors also transduce signals through other G-protein independent pathways that employ ceramide as the second messenger [27]. In addition to regulating the activities of the protein kinases, CB1 activation inhibits voltage-gated N- and P/Q-type calcium channels and activates A-type, and G-protein-coupled inwardly rectifying potassium channels (GIRK) [2]. Figure 2 illustrates the signaling network of the CB1 receptor. Signal transduction mediated by the CB1 receptor exhibits pluridimensional and spatiotemporal features.
Fig. 2.

Signaling network of the CB1 receptor. Upon activation, the CB1 receptor elicits G-protein-dependent signal transduction, mainly through Gαi/o-dependent inhibition of adenylyl cyclase (AC), Gβγ-dependent activation of PLC and PI3K, and Gβγ-dependent regulation of K+ and Ca2+ channels. Following the CB1-induced phosphorylation of GPCR kinases (GRKs), β-arrestins bind to the CB1 receptor and mediate G-protein independent signal transduction. CB1 also signals through other non-G protein partners, such as the adaptor protein, the factor associated with neutral sphingomyelinase (FAN). The CB1 receptor may induce ERK activation through several different routes, including cAMP-dependent pathways, β-arrestin-dependent pathways, the activation of PI3K/PKB, transactivation of tyrosine receptor kinases, or the use of ceramide as the second messenger
Based on published data, researchers have postulated that CB1-mediated signal transduction may occur in three different waves. The first is transient and mediated by heterotrimeric G-proteins (Gαβγ). Afterwards, the second wave is mediated by β-arrestins. The third and final wave occurs in intracellular compartments and is elicited either by G-proteins or β-arrestins [28]. Without activation by agonists, the CB1 receptor induces certain cellular responses through its constitutive activity, which is attenuated by CB1 inverse agonists [12]. Under certain circumstances, CB1 receptors exhibit functional selectivity between different cellular signaling pathways [29–32]. Functional selectivity (also known as biased agonism) refers to the ability of a ligand to stabilize a GPCR in a confirmation that preferentially binds to one or a subset of intracellular signaling proteins and selectively evokes certain response pathways over others [33–35]. For instance, the CB1 ligands HU210, CP55,940, WIN55, 212-2, 2-AG and AEA elicit preferential interactions with different subtypes of G-proteins (i.e., Gi, Go, Gs and Gq/11, respectively) with varying efficacies and produce ligand-dependent functional selectivity [36–38]. Additionally, the CB1 partial agonist Δ9-THC and full agonist CP55,940 induce the internalization of CB1 receptors through biased β-arrestin-2 recruitment [37]. Collectively, this evidence suggested that different CB1 agonists stabilize different receptor conformations to induce functional selectivity in cellular responses. The complex signaling network of the CB1 receptor indicates a necessity for special ligands that selectively transduce the signals required for the desired therapeutic effects while sparing the pathways linked to side effects. Although some orthosteric ligands have achieved ligand-dependent functional selectivity, allosteric ligands have been widely recognized and accepted as offering greater opportunities to produce functional selectivity than orthosteric ligands [39, 40]. Allosteric modulators of a given receptor induce various receptor conformations that are distinct from the conformations stabilized by orthosteric ligands [35, 41]. Different conformations of a receptor impact the ability of the receptor to interact with its downstream effectors, leading to functional selectivity. Recently, the X-ray crystallographic structures of CB1 receptors bound to agonists [42] and antagonists [43, 44] have been revealed. These ligand-bound CB1 receptor structures are critical in providing insights into the molecular conformation of the active and inactive states of the CB1 receptor while assisting the rational design of new CB1 ligands.
Allosteric modulators of the cannabinoid CB1 receptors
Historically, drug discovery programs aimed at regulating GPCR functions have been dominated by the identification of ligands to compete with endogenous ligands at the orthosteric sites. Recently, tremendous advances have been achieved in the discovery of ligands that regulate GPCR functions by binding to receptor sites that are topographically distinct from orthosteric sites, defining these compounds as allosteric modulators. Allosteric ligands of GPCRs induce and stabilize unique conformations of GPCRs and therefore provide fundamentally different receptors that are capable of exerting novel pharmacological effects. During the last 12 years, several structurally distinct molecules serving as allosteric modulators of the CB1 receptor have been discovered. These compounds (shown in Fig. 3) include the small molecules 5-chloro-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (10, Org27569) [45], 1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea (11, PSNCBAM-1) [46], 3-(4-chlorophenyl)-5-(8-methyl-3-p-tolyl-8-azabicyclo[3.2.1]octan-2-yl)-isoxazole (12, RTI-371) [47], 6-methyl-3-(2-nitro-1-(thiophen-2-yl)ethyl)-2-phenyl-1H-indole (13, ZCZ011) [48], cannabidiol (14), and fenofibrate (15), as well as the endogenous molecules (5 S,6 R,9E,11Z,13E,15 S)-5,6,15-trihydroxyicosa-9,11,13-trienoic acid (16, lipoxin A4) [49], pregnenolone (17) [50], and pepcan-12 (18) [51].
Fig. 3.

Structures of some representative CB1 allosteric modulators
Representative CB1 allosteric modulators and their allosteric properties at the cellular level
Exogenous allosteric modulators of the CB1 receptor
Org27569
Org27569 (10, Fig. 3) is the first-in-its-class of CB1 allosteric modulators. Ross and colleagues described its allosteric modulatory properties in 2005 [45]. In equilibrium binding assays, Org27569 significantly increased the binding of the CB1 receptor agonist [3H]CP55,940, suggesting positive binding cooperativity with the orthosteric ligand CP55,940. Simultaneously, Org27569 significantly, but not completely, reduced the specific binding of the CB1 receptor inverse agonist [3H]SR141716A, indicating a limited negative binding cooperativity. Its allosteric nature was further verified by studies of [3H]CP55,940 dissociation kinetics. Paradoxically, its potentiation of agonist binding did not lead to an augmentation of agonistic effects induced by the same agonist. In contrast, Org27569 behaved as an insurmountable inhibitor of some CB1 receptor functions and induced a significant reduction in the Emax values for some properties of CB1 agonists (i.e., CP55,940 and WIN55,212-2). According to the results from several functional assays, including the reporter gene assay (i.e., the luciferase reporter assay, which determines the increase in luciferase expression induced by CB1 agonist), the GTPγS binding assay and the mouse vas deferens assay, Org27569 functions as an inhibitor of some activities, and these findings identified the presence of allosteric sites on the CB1 receptor for the first time [45]. One significant finding in the study of Org27569 is that this molecule induces a CB1 conformation that promotes functional selectivity. Org27569 suppressed the CB1 agonist (CP55,940)-induced G-protein-mediated phosphorylation of JNK while it promoted β-arrestin-1-mediated phosphorylation of ERK1/2 in HEK-hCB1 cells either alone or after cotreatment with CP55,940 [52, 53]. In other words, Org27569 is a positive allosteric modulator (PAM) of certain β-arrestin-mediated CB1 signal transduction pathways, and inhibits G-protein-mediated CB1 signal transduction.
PSNCBAM-1
Soon after the discovery of Org27569, Prosidon Ltd. (Oxford, UK) identified another CB1 receptor allosteric modulator, PSNCBAM-1 (11, Fig. 3). PSNCBAM-1 is structurally unrelated to Org27569 and exhibits a diaryl urea scaffold [46]. The initial profile of this compound revealed that PSNCBAM-1 dose-dependently increased the binding of the CB1 agonist [3H]CP55,940 and significantly, but incompletely, decreased the binding of the CB1 inverse agonist [3H]SR141716A. Similar to Org27569, PSNCBAM-1 inhibited [35S]GTPγS binding induced by the CB1 agonist [3H]CP55,940 [53]. It also inhibited Δ9-THC-induced [35S]-GTPγS binding [54]. In yeast reporter assays using the CB1 receptor, PSNCBAM-1 blocked the agonistic effects of several CB1 orthosteric agonists, including CP55,940, WIN55,212-2, AEA and 2-AG. In the constitutive yeast reporter assay, PSNCBAM-1 exhibited no intrinsic negative regulation of the constitutive activity of CB1 receptor and did not behave as an inverse agonist like SR141716A. Similar to Org27569, PSNCBAM-1 promotes functional selectivity via the β-arrestin-1 mediated pathway, as evidenced in the study of its analogs in the ERK1/2 phosphorylation assay [55].
Following the identification of Org27569 and PSNCBAM-1, some functional assays have been employed to characterize the two compounds at molecular, receptor and cellular levels [53, 54, 56–60]. The assays employed in the characterization of these compounds include equilibrium binding, CB1 agonist binding kinetics, CB1 agonist-induced [35S]GTPγS binding, the Gi-mediated inhibition of cAMP production and Gs-mediated stimulation of cAMP production, phosphorylation of ERK1/2, and β-arrestin recruitment, cellular hyperpolarization and receptor-internalization and desensitization. The allosteric modulation of the CB1 receptor by Org27569 and PSNCBAM-1 showed a certain dependence on the orthosteric ligands, receptor sources, cell types, pathways and time frame over which the signaling responses occurred. Generally, Org27569 and PSNCBAM-1 show a similar pharmacological profile and behave as PAMs in potentiating the binding of the orthosteric CB1 agonist (i.e., CP55,940) and function as inhibitors by antagonizing the CB1 activity induced by various CB1 agonists. Recent reviews have reported the multifaceted biochemical and pharmacological effects of these CB1 allosteric modulators [61]. The data from those investigations confirmed that CB1 allosteric modulators promote functional selectivity, an effect that is highly relevant for the discovery of new drugs with more defined pharmacology and improved drug safety.
ZCZ011
Immediately after the discovery of Org27569 and PSNCBAM-1, ZCZ011 was identified as a CB1 allosteric modulator that behaves differently from Org27569 and PSNCBAM-1 [48]. This compound not only potentiates the binding of the CB1 agonist CP55,940 but also enhances the binding of WIN55,212-2, a CB1 orthosteric agonist with which Org27569 and PSNCBAM-1 did not show any binding cooperativity. ZCZ011 decreased the binding of the CB1 inverse agonist SR141716A, similar to Org27569 and PSNCBAM-1. Unlike the Org27569- and PSNCBAM-1-mediated inhibition of CP55,940-induced [35S]GTPγS binding to the CB1 receptor, ZCZ011 enhanced [35S]GTPγS binding to the CB1 receptors stimulated by the CB1 agonists CP55,940 and AEA in mouse brain membranes. This compound also increased AEA-induced β-arrestin recruitment and ERK1/2 phosphorylation induced by AEA and CP55,940 in CHO-hCB1 cells. These properties established that ZCZ011 is a PAM of the CB1 receptor at a functional level. Notably, ZCZ011 alone acted as an agonist and inhibited forskolin-stimulated cAMP production, while it did not significantly enhance the CP55,940- and AEA-induced inhibition of cAMP production at a test concentration of 1 μM.
Endogenous allosteric modulators of the CB1 receptor
Following the discovery of small synthetic molecules that allosterically regulate the CB1 receptor, several endogenous molecules were identified as CB1 allosteric modulators. These molecules include pepcan-12 [51], lipoxin A4 [49] and pregnenolone [50].
Pepcan-12
Pepcan-12 is an α-hemoglobin-derived peptide (pepcan) [51]. Within this group, pepcan-12 (a dodecapeptide) exhibits the properties of a negative allosteric modulator (NAM) of the CB1 receptor, although previous reports indicated that it is a partial agonist of the CB1 receptor [62]. In equilibrium binding studies, pepcan-12 showed saturable but incomplete displacement of the CB1 orthosteric agonists [3H]CP55,950 and [3H]WIN55,212–2. In dissociation kinetic studies, pepcan-12 increased the dissociation constant of the CB1 orthosteric agonist CP55,940. These results are consistent with the negative allosteric modulation of orthosteric agonist binding. Compared with the synthetic CB1 allosteric modulators, endogenous pepcan-12 exhibited relatively high binding affinity for the CB1 receptor (Ki < 29 nM) [51, 62]. In functional assays, pepcan-12 reduced the efficacy of CB1 receptor agonist-induced cAMP accumulation, [35S]GTPγS binding, and CB1 receptor internalization. The employed CB1 agonists included WIN55,212–2, 2-AG and HU210. The negative modulation induced by pepcan-12 did not depend on the probe. Probe-dependence is one of the benefits of allosteric modulation, which allows a GPCR such as the CB1 receptor to respond differently to the same allosteric modulator when different orthosteric ligands are bound. Pepcan-12 strongly decreases the efficacy, but not potency, of the endogenous CB1 agonist 2-AG.
Lipoxin A4
Following the identification of an endogenous NAM of the CB1 receptor, an endogenous PAM of the CB1 receptor (i.e., lipoxin A4) was reported [49]. Lipoxin A4 enhances the binding affinity of the CB1 agonists [3H]CP55,940, AEA and [3H]WIN55,212–2. Lipoxin A4 increased [3H]CP55,940 binding by 100% and [3H]WIN55,212–2 binding by approximately 30%, suggesting a probe dependence of the lipoxin A4 effects. Based on the results of kinetic dissociation-binding studies, lipoxin A4 slowed the rate of [3H]CP55,940 displacement by competing with WIN55,212–2. This finding confirms the nature of lipoxin A4 as an allosteric modulator. In CB1 functional assays, lipoxin A4 reduced [35S]GTPγS binding induced by the CB1 agonist AEA and yet strongly augmented the AEA-elicited inhibition of forskolin-stimulated cAMP production. Additionally, lipoxin A4 potentiated the cannabinergic effects of AEA on mice. Lipoxin A4 increased the AEA-induced cannabinergic effects (e.g., catalepsy) on wild-type (CB1+/+) mice, but not CB1 knockout mice, indicating a CB1-dependent mechanism of allosteric modulation. Thus, Lipoxin A4 was characterized as a PAM of CB1 at the molecular, receptor, cellular and whole animal levels.
Pregnenolone
In a study of the impact of the major classes of drugs of abuse on the production of neurosteroids in the rat and mouse brains, the production of pregnenolone was upregulated by ∆9-THC. Moreover, pregnenolone functions as a signaling-specific CB1 NAM [50]. Pregnenolone (at a concentration up to 100 µM) did not alter the equilibrium binding of the CB1 agonists [3H]CP55,940 and [3H]WIN55,212–2, indicating a lack of binding cooperativity with orthosteric agonists. Pregnenolone binds to a distinct site located at the lipid face of the CB1 receptor in the transmembrane TMH1/TMH7/Hx8 region. Binding to this region was validated by a mutant CB1 receptor. Additionally, the binding of pregnenolone to the CB1 receptor was supported by a concentration-dependent but incomplete displacement of [3H]SR141716A. The TMH7/Hx8 region is located in the C-terminal region of the CB1 receptor, the site at which CRIP1a has been proposed to interact with the CB1 receptor and β-arrestins [26]. Pregnenolone may modulate the CB1 receptor by interacting with CRIP1a. In CHO-CB1 cells, pregnenolone decreased Δ9-THC-induced ERK1/2 phosphorylation. Based on these findings, pregnenolone is characterized as an endogenous NAM of the CB1 receptor. Its negative allosteric modulation was further confirmed through in vivo studies [50].
Recently, a follow-up investigation of pregnenolone and lipoxin A4 was unable to validate their allosteric natures, except that the partial displacement of [3H]SR141716A by pregnenolone was reproducible [57]. Therefore, the in vitro allosteric effects of pregnenolone and lipoxin A4 require further investigation.
Miscellaneous allosteric modulators of the CB1 receptor
Cannabidiol
Cannabidiol, a major ingredient of cannabis without psychoactive properties, exerts a variety of pharmacological effects [63, 64]. Unlike Δ9-THC, cannabidiol does not bind to the orthosteric binding sites of the cannabinoid CB1 and CB2 receptors. However, it possesses receptor-dependent and receptor-independent pharmacological effects [63, 65, 66]. Recently, cannabidiol was shown to behave as a non‐competitive NAM of CB1 receptors [67]. It reduced the efficacy and potency of CB1 agonists 2‐AG and Δ9‐THC at inducing the phosphorylation of PLCβ3‐ and ERK1/2 in HEK293A cells (heterologously expressing CB1) and in STHdhQ7/Q7 cells (endogenously expressing CB1). Cannabidiol reduced β-arrestin-2 recruitment, leading to a reduced efficacy and potency of Δ9‐THC and 2‐AG in stimulating the internalization of CB1 receptors. These data supported the hypothesis that cannabidiol functions as a NAM of the CB1 receptor.
Fenofibrate
Cannabinoid compounds have been shown to interact with peroxisome proliferator-activated receptors (PPARs), members of the nuclear hormone receptor family [68]. For instance, AEA, Δ9-THC, and WIN55,212–2 all function as PPAR-α agonists in vitro at concentrations higher than those required to activate cannabinoid receptors. Interestingly, one of the known PPAR-α receptor agonists, fenofibrate, exhibits allosteric modulation properties at high concentrations. It displaced the binding of the CB1 agonist [3H]-CP55,940 in an incomplete manner in CHO-hCB1 cells. In functional assays, fenofibrate (at concentrations greater than 3.1 µM) dose-dependently reduces [35S]GTPγS binding, ERK1/2 phosphorylation, and β-arrestin recruitment induced by the CB1 agonist CP55,940, whereas it potentiates these effects at a concentration less than 3.1 µM [69]. Thus, fenofibrate functions as a bitopic ligand of the CB1 receptor. At low concentrations, it functions as a partial agonist, whereas it acts as a NAM at high concentration.
RTI-371
In a study of a group of dopamine transporter inhibitors derived from the tropane scaffold, several compounds represented by RTI-371 showed the capability to potentiate induced calcium mobilization in CHO-hCB1 cells induced by the CB1 agonist CP55,940. Hence, RTI-371 was categorized as a PAM of CB1 receptor. However, no other evidence was provided to support the claim and characterization of this compound as a CB1 allosteric modulator.
Recent developments in CB1 allosteric modulators
To date, approximately nine classes of CB1 allosteric modulators have been identified. By far the best-characterized allosteric modulators of CB1 receptors include the NAMs Org27569 and PSNCBAM-1, as well as the PAMs ZCZ011 and lipoxin A4. Since the discovery of the nine scaffolds of CB1 allosteric modulators (Fig. 2), studies in medicinal chemistry attempting to optimize these leads have mainly focused on the scaffolds of Org27569 and PSNCBAM-1. The results of their structure–activity-relationship (SAR) studies are summarized below, and representative molecules are illustrated.
SARs of indole-2-carboxamides represented by Org27569
Since the discovery of Org27569, several SAR studies have been performed on the indole-2-carboxamide scaffold [70–76]. The identified key structural requirements of indole-2-carboxamides for allosteric modulation of CB1 receptors include several factors, as listed below. (1) The carboxamide functional group is required. When replaced by an ester group, the allosteric modulation of agonist binding is drastically altered, or the molecule becomes inhibitory rather than potentiating [71]. (2) The indole ring is critical. The replacement of this ring with other heteroaromatic rings, such as benzofuran or benzimidazole, abolishes or reduces the allosteric activity [72, 77]. (3) The length of the linker between the amide group and the aminophenyl ring is instrumental. Only an ethylene linker is tolerated, and any alteration in the linker length results in a complete loss of allosteric activity [74]. (4) The C3 alkyl group on the indole ring has a fairly substantial influence on allostery, with a lower linear alkyl group preferred [72, 74]. (5) The NH of indole ring must be unsubstituted [70]. (6) The amino substituent on the phenyl ring impacts both the affinity and efficacy of allosteric modulation, with an N,N-dimethyl amino group showing improved allostery compared to a piperidinyl moiety [71, 74]. (7) The 5-position of the indole ring favors an electron-withdrawing group, with a preference for a halogen or an isothiocyanate group [71, 74, 76]. Figure 4 summarizes the SARs of this class of compounds. The representative members that emerged from SAR studies are presented in Fig. 5. These novel allosteric modulators include compounds 19 [73], 20 [72], 21 [73], and 22 [76].
Fig. 4.

Summary of SARs for indole-2-carboxamides represented by Org27569
Fig. 5.
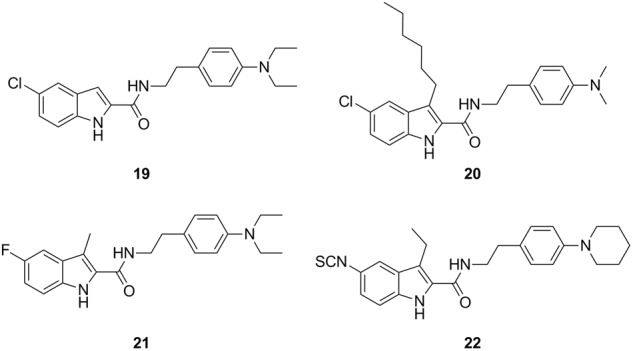
Representative indole-2-carboxamide analogs of Org27569 with improved or comparable activities
SARs of diaryl ureas represented by PSNCBAM-1
Several SAR investigations have focused on the substituents, the major linker bond and the heteroaromatic ring of the molecule to optimize the scaffold of diaryl ureas represented by PSNCBAM-1 [55, 76, 78, 79]. The key findings are listed below. (1) An electron-withdrawing substituent is optimal at the 4-chlorophenyl position, with a cyano group being more potent than other substituents. (2) The urea skeleton is instrumental, and its replacement with a carbamate, methylated urea, or amide group abolishes the allosteric properties. (3) The pyridine ring is not necessary and can be replaced by a phenyl ring. (4) The pyrrolidinyl ring position can tolerate replacement by tertiary amino groups, such as dialkylamino, piperidinyl groups or other cyclic rings, with some size limitation. The SARs of this class of compounds are summarized in Fig. 6. The representative members that emerged from SAR studies are presented in Fig. 7. These novel allosteric modulators include compounds 23 [78], 24 [79], 25 [55], and 26 [55].
Fig. 6.

Summary of SARs for diaryl ureas represented by PSNCBAM-1
Fig. 7.
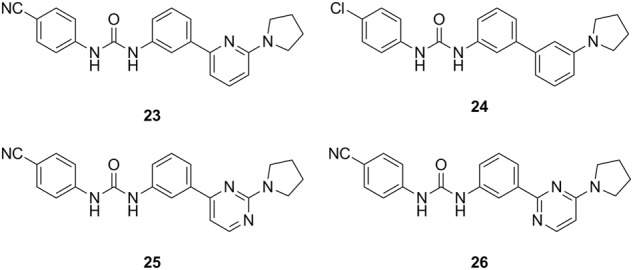
Representative diaryl urea analogs of PSNCBAM-1 with improved or comparable activities
Development of new ZCZ011 analogs
Recently, a close analog of ZCZ011, GAT211 (27, Fig. 8), was synthesized and reported to exert pharmacologic effects similar to ZCZ011 at the molecular, receptor and cellular levels [80]. ZCZ011 and GAT211 are racemates. Chiral resolution of GAT211 led to a pair of optically pure enantiomers of GAT211. Interestingly, the (R)-enantiomer GAT228 (28, Fig. 8) behaves as an allosteric partial agonist of the CB1 receptor, while the (S)-enantiomer GAT229 (29, Fig. 8) functions as a pure PAM [80, 81]. GAT229 functions as a biphasic ligand: at high concentrations, it competes with orthosteric agonists, while at low concentrations, it behaves as an allosteric modulator [80]. A recent communication describing the binding sites of this class of compounds reported a similar biphasic mechanism for the allosteric ligands GAT228 and ZCZ011 [82]. Based on these data, 2-phenylindole analogs are likely able to bind to multiple sites on the CB1 receptor. The mechanism of allosteric modulation may depend on the drug concentration and the presence of an orthosteric agonist. When an orthosteric agonist is not bound to the receptor, these compounds function as an orthosteric ligand, while they function as allosteric modulators by binding to a distinct site when an orthosteric agonist binds to the receptor [82].
Fig. 8.

Representative analogs of the CB1 allosteric modulator ZCZ011
Currently, the reports of ZCZ011 analogs are fairly limited [83, 84]. However, a preliminary SAR can be inferred. The SARs of ZCZ011 analogs are summarized below and illustrated in Fig. 9. The key SAR includes the factors listed below. (1) The 2-aryl indole skeleton is essential for allosteric modulation effects. (2) The 6-methyl group is influential, and its replacement with a chloro group or a hydrogen is tolerated, but the compound exhibits reduced activity. (3) The disubstituted ethyl group at the 3-position of the indole ring is instrumental. Its thiophene group can be replaced by a pyridine ring, while its replacement with a phenyl ring reduces the activity. The 2-nitro group is fairly resistant to modification. Only a cyano group (CN) or a trifluoromethyl group (CF3) is tolerated. The replacement of the nitro group with a carboxylic acid (COOH), an ester (COOR), an amide (CONH2) or an amino group (NH2) abolishes the activity. (4) The 2-phenyl moiety can be substituted at the para-position with halogens. The racemic nature of this class of compounds becomes challenging for SAR investigations since the (R)- and (S)-isomers of GAT211 function differently in allosterically modulating the CB1 receptor [80, 81].
Fig. 9.

Summary of SARs for 2-phenyl indoles represented by ZCZ011
Therapeutic relevance of CB1 allosteric modulators
While CB1 orthosteric agonists and antagonists have traditionally been pursued to target cannabinoid receptors, findings show untoward side effects that are unacceptable for chronic applications in clinical settings. Generally, allosteric modulators provide several mechanism-based advantages to overcome the on- and off-target side effects and increase drug safety and specificity. First, they offer the potential for better receptor subtype selectivity because of the greater structural variance in allosteric sites than the orthosteric sites, which generally are highly conserved. Second, allosteric modulators without intrinsic activity selectively exert biological responses only in tissues where the endogenous ligands are present and function. Third, once completely occupied, the allosteric sites produce a saturation effect and subsequently limit the effect of the allosteric modulator on the response induced by the orthosteric ligand. This “ceiling” action prevents drug overdose. More importantly, allosteric modulators enable the fine-tuning of receptor pharmacology through ligand-dependent signaling and functional selectivity. This approach may facilitate signal transduction through the pathways that are more therapeutically relevant while failing to activate pathways involved in the untoward effects. The discovery of CB1 allosteric modulators has increased the number of approaches by which the functions of the CB1 receptor are manipulated for potential therapeutic benefits with the aim of improving the pharmacology and drug safety compared with traditional orthosteric CB1 ligands. Figure 10 illustrates the possible allostery-engendered functional selectivity of the CB1 receptor. The in vitro characterization of CB1 allosteric modulators has provided the foundation for the investigation of the therapeutic values of positive and negative allosteric modulators of the CB1 receptor.
Fig. 10.

Schematic illustrating GPCR allosteric modulation and functional selectivity. Allosteric modulators alter the binding affinity (a: pink arrow) and/or efficacy (b: cyan arrow) of orthosteric ligands in a positive (PAM) or negative manner (NAM). Allosteric modulation often shows ligand dependence (i.e., different orthosteric ligands induce different forms of allostery). An allosteric modulator induces the receptor to adopt various conformations that preferentially activate either G-protein-mediated or a β-arrestin-mediated signal transduction. Other downstream effectors may also be preferentially involved in signaling pathways at the expense of others (functional selectivity). The resulting functional selectivity might generate a pharmacologically improved therapeutic effect, with reduced on-target adverse effects compared to the orthosteric ligand
The evidence from CB1 positive allosteric modulators
Theoretically, PAMs function through permissive augmentation of the tone from the endogenous agonists and/or by stabilization of novel receptor conformations that have a preference for certain endogenous ligands. These effects may lead to biased signaling toward the upregulation of the preferred response induced by the endogenous agonists that cooperate with the allosteric modulator.
The first evidence of some therapeutic value of a CB1 allosteric modulator was obtained from the in vivo studies of the CB1 PAM lipoxin A4. Intracerebroventricular (i.c.v.) injections of lipoxin A4 in wild-type mice potentiate the cataleptic effects of the endogenous CB1 agonist AEA. This potentiation was not observed in CB1 knockout mice. Hence, the response is a CB1-dependent allosteric modulatory effect. Because AEA exerts protective effects against pathogenic amyloid-β induced neurotoxicity [85, 86], the neuroprotective effects of lipoxin A4 on amyloid-β induced spatial memory impairments were assessed using the Morris water maze test. Lipoxin A4 provides the CB1 receptor-dependent protection against pathogenic β-amyloid-induced memory impairments [49].
More convincing evidence was provided by the investigation of the effects of the CB1 PAM ZCZ011 on wild-type and CB1 knockout mice [48]. The administration of ZCZ011 alone does not produce any cannabinergic effects, such as catalepsy, hypothermia, antinociception and decreased locomotion. In contrast, ZCZ011 significantly potentiated the antinociceptive, cataleptic and hypothermic effects elicited by the CB1 orthosteric agonist CP55,940. It also augmented the AEA-induced hypothermia, but did not alter the AEA-induced antinociceptive and cataleptic effects on FAAH knockout mice. These findings suggested a functional selectivity mediated by the allosteric modulator. In a drug discrimination assay, ZCZ011 significantly enhanced the potency of the discriminative stimulus effects of AEA on FAAH knockout mice. Additionally, ZCZ011 reverses nociceptive behaviors in well-established murine models of neuropathic and inflammatory pain. ZCZ011 blocks mechanical and cold allodynia for a fairly long period, without the development of tolerance. The compound does not produce conditioned place preference or aversion. The antiallodynic effects of ZCZ011 depend on the CB1 receptor.
Recently, another PAM of the CB1 receptor, GAT211, was shown to suppress the inflammatory nociception induced by complete Freund’s adjuvant (CFA) and the neuropathic pain evoked by the cancer chemotherapeutic agent paclitaxel [87]. Antiallodynic effects of GAT211 were observed on wild-type but not CB1 knockout mice, suggesting that the allosteric modulation depended on the CB1 receptor. GAT211 produced synergistic antiallodynic effects with the inhibitors of FAAH and MGL on paclitaxel-treated mice. It also synergized with the orthosteric agonist WIN55,212–2 to reduce CFA-induced mechanical allodynia. Its therapeutic efficacy persisted for 19 days of chronic dosing with GAT211, whereas it was not preserved with the MGL inhibitor tested in the same study. Cannabimimetic withdrawal precipitated by the CB1 antagonist SR141716A was observed in mice chronically treated with the orthosteric agonist WIN55,212–2, but not in mice treated with GAT211. The PAM GAT211 alone did not produce cannabinergic effects or other cardinal signs of direct CB1 activation in the presence or absence of pathological pain. Similar to ZCZ011, GAT211 did not induce conditioned place preference or aversion in the test animals.
The positive CB1 allosteric modulator GAT229 was effective at decreasing intraocular pressure (IOP) in ocular normotensive and ocular hypertensive nee mice [88]. In normotensive mice, the topical administration of GAT229 alone did not appear to exert any effect on reducing the IOP. This finding probably was due to insufficient local endocannabinoid concentrations at the site of action. However, the effect of a subthreshold concentration of WIN55,212–2 on decreasing IOP was potentiated by GAT229. In contrast, the topical administration of GAT229 alone in nee mice with ocular hypertension was sufficient to reduce the IOP. The authors did not clearly determine whether the levels of endocannabinoids at the site of action in nee mice were due to pathological conditions.
The results from recent in vivo studies of the effects of lipoxin A4, ZCZ011, GAT211, and GAT229 provided compelling evidence that CB1 PAMs show promise as potential therapeutics for neurodegenerative diseases, neuropathic and inflammatory pain, and glaucoma. In particular, the outcomes from studies of ZCZ011 and GAT211 suggested that CB1 PAMs may lack significant abuse liability and other CNS side effects associated with orthosteric CB1 agonists.
The evidence from CB1 NAMs
Theoretically, the NAMs of the CB1 receptor should be capable of downregulating the endocannabinoid tone or inducing a conformation that decreases or abolishes the constitutive activity of the CB1 receptor to produce pharmacological effects. CB1 antagonism or inverse agonism shows promising therapeutic effects on obesity, obesity-related metabolic syndromes, and substance abuse [11, 89]. Several preclinical studies have assessed the therapeutic potential of the CB1 NAMs Org27569, PSNCBAM-1, and pregnenolone.
As expected for a negative CB1 allosteric modulator, the administration of Org27569 alone does not elicit cannabimimetic effects [90]. However, this compound does not exhibit efficacy in attenuating the antinociceptive, cataleptic, and hypothermic effects produced by the orthosteric CB1 agonists AEA, CP55,940, and Δ9-THC, indicating a lack of negative allosteric modulation. In the drug discrimination paradigm, Org27569 was not able to substitute for either CB1 agonist AEA or Δ9-THC and did not modify the discriminative stimulus effects of any of the two CB1 orthosteric agonists. Notably, Org27569 produced small but statistically significant increases in the potency of AEA-elicited catalepsy and antinociception in FAAH knockout mice [90], suggesting weak positive allosteric modulation. However, this potentiation was not observed for AEA-induced hypothermia. The aforementioned results indicated the ligand-dependent allostery and functional selectivity of a CB1 allosteric modulator. In the same study, Org27569 reduced food intake by both wild-type and CB1 knockout mice, while the orthosteric CB1 antagonist SR141716A (rimonabant) only reduced food intake by wild-type mice [90]. Thus, the anorectic effects of Org27569 most likely were not mediated by a CB1-dependent mechanism. The results from this array of studies implied that the well-characterized CB1 negative allosteric properties of Org27569 obtained from in vitro studies were not translated into the pharmacological effects on the downregulation of the CB1 receptors in mice.
In a preclinical study of Org27569 using rats, the compound showed mixed effects [91]. It did not markedly alter the body temperature alone, but significantly attenuated the hypothermic effect induced by the CB1 agonists CP55,940 and AEA, suggesting negative modulation. Pretreatment with Org27569 did not significantly alter the cataleptic and antinociceptive effects induced by CP55,940, whereas the CB1 antagonist/inverse agonist SR141716A significantly decreased CP55,940-induced catalepsy and nociception, suggesting a lack of modulation on these CB1 mediated cannabinergic effects. SR141716A precipitates Δ9-THC withdrawal in mice and rats [91]. Typical withdrawal behavior was observed, including an increase in paw tremors and head shakes, accompanied by a decrease in normal behaviors such as grooming and scratching. Org27569 alone did not elicit increased grooming and scratching behaviors and did not significantly alter the grooming and scratching behaviors induced by SR141716A, which was shown to significantly increase the frequency of scratching and grooming. Based on these results, Org27569 does not show the pharmacological effects associated with the downregulation of CB1 receptor, and it functions differently from the orthosteric CB1 antagonist/inverse agonist SR141716A (rimonabant). Surprisingly, Org27569 produced hypophagic effects similar to the CB1 antagonist SR141716A. It reduced food intake and body weight in rats, with outcomes similar to SR141716A. However, whether the hypophagic effects of Org27569 depend on the CB1 receptor mediation remains unknown due to lack of CB1 knockout rat models [91]. A follow-up investigation of Org27569 in rats found that pretreatment with Org27569 dose-dependently attenuated both cue- and drug-induced reinstatement of cocaine- and methamphetamine-seeking behaviors. SR141716A also exerted similar inhibitory effects on the reinstatement of drug-seeking behaviors [92]. Thus, Org27569 downregulates the CB1 receptor in rats by functioning as a CB1 inverse agonist. In the study described above, Org27569 was screened at a concentration of 10 µM to evaluate its binding selectivity toward more than 40 GPCRs, including some receptors associated with drug addiction. Org27569 showed high selectivity for CB1 compared with the screened panel. The highly selective binding profile of Org27569 suggested that its effects on relapse to psychostimulant seeking behaviors are likely mediated by negative modulation of CB1 receptors. The results warrant further investigations of the utility of CB1 NAMs to treat substance addiction.
Similarly, another allosteric modulator of the CB1 receptor, PSNCBAM-1, exerted anorectic effects on rats, without any obvious adverse effects on animal behaviors or signs of toxicity under the assessment conditions [46]. However, in a recent study, the anorectic effects of PSNCBAM-1 were not observed in wild-type mice [54]. In the same study, the compound was ineffective at attenuating the reduced locomotion, catalepsy, and hypothermia induced by CB1 agonist Δ9-THC. Similar to Org27569, PSNCBAM-1 exhibited a modest but statistically significant reduction in the antinociceptive effects induced by the CB1 agonist Δ9-THC [54]. The above results again revealed the functional selectivity of a CB1 NAM. In addition to the anorectic effects, other potential therapeutic effects of PSNCBAM-1 are emerging, such as possible neuroprotective effects mediated by the inhibition of 2-AG-mediated depolarization-induced suppression of excitation (DSE) [93], and its effects on altering neuronal excitability [58].
The unexpected anorectic efficacy of Org27569 in CB1 knockout mice should not discourage the investigation of the effects of CB1 allosteric modulators on feeding behaviors. A careful interpretation of the results is necessssary. First, the two compounds exhibited a relatively weaker ability to interact with CB1 receptors than the CB1 inverse agonist SR141716A, as reflected by their equilibrium dissociation constants (i.e., KB, 217 nM for Org27569 and 54 nM for PSNCBAM-1) [55, 72], and the binding affinity of SR141716A (i.e., Ki = 1.98 nM). Some CB1-independent anorectic response may be triggered in wild-type and CB1(-/-) mice at the tested doses. Second, the two compounds have very low solubility. Further studies are needed to determine whether the two CB1 allosteric modulators are able to be sufficiently delivered to their sites of action in animals. In in vivo experiments, the potency at which the drug binds the receptor, as well as the absorption, distribution, metabolism, and excretion (ADME), impact the pharmacological effects. The investigations of feeding behaviors in mice treated with Org27569 and PSNCBAM-1 indicated that more potent CB1 allosteric modulators and preclinical studies of their ADME properties are needed to validate the target.
Unlike Org27569 and PSNCBAM-1, which did not attenuate the complete spectrum of the cannabinergic effects induced by CB1 agonists, the CB1 NAM pregnenolone exhibited the capability to attenuate the locomotor suppression, hypothermia, catalepsy, and antinociception induced by Δ9-THC [50]. Furthermore, pregnenolone suppressed the Δ9-THC-induced increase in food consumption in rats and mice. This NAM ameliorates the Δ9-THC-induced memory impairments in mice and blocks the Δ9-THC-induced release of glutamate and dopamine. The compound was also capable of blocking and reinforcing the effects of cannabimimetic drugs [50]. Very recently, pregnenolone was shown to block a wide spectrum of Δ9-THC-induced endophenotypes in mice that are typically associated with psychotic-like states [94]. Collectively, the results from preclinical studies of pregnenolone suggested that pregnenolone-based NAMs of CB1 receptor represent promising new approaches for the treatment of disease states associated with overactive CB1 receptors.
The results from recent in vitro and in vivo studies of the allosteric modulators Org27569, PSNCBAM-1 and pregnenolone revealed new avenues to downregulate the endocannabinoid tone and achieve therapeutic benefits in the areas of substance addiction, neurological disorders and obesity-related diseases.
Closing remarks
Since the discovery of the first CB1 allosteric modulator Org27569 in 2005, nine classes of structurally diverse chemical entities have been identified as CB1 allosteric modulators. Accumulating evidence from in vitro and preclinical studies have confirmed their capabilities to either positively or negatively regulate the functions of the cannabinoid CB1 receptor. Some of the CB1 allosteric modulators selectively manipulate signaling pathways downstream of cannabinoid CB1 receptors through functional selectivity. These properties of allosteric modulators suggest that they possess tremendous potential to separate the therapy-relevant CB1 responses from CB1 responses that cause untoward side effects. This goal has been difficult or impossible to achieve using orthosteric CB1 ligands, but may be achievable with CB1 allosteric modulators, as evidenced for other GPCRs, such as the angiotensin II receptor [95]. However, the links between the specific pathways of the CB1 receptor and the untoward side effects have not yet been elucidated. Therefore, advancements in the discovery of novel and more potent CB1 allosteric modulators are needed; these molecules represent potentially powerful tools for studies aiming to identify the signaling factors that cause the side effects mediated by activation or downregulation of the CB1 receptor. By employing the advantages of the well-established therapeutic benefits obtained from CB1 orthosteric agonists and antagonists, positive and negative allosteric modulators hold stronger promise to deliver cannabinoid-based medications that exhibit improved specificity, efficacy and drug safety.
Acknowledgements
This work was partially supported by NIH Grant DA039942. The authors thank Mrs. Michelle Walbeck for assisting with the revision of this manuscript.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howlett A. Cannabinoid receptor signaling. In: Pertwee R, editor. Cannabinoids. Berlin, Heidelberg: Springer; 2005. pp. 53–79. [Google Scholar]
- 3.Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47:345–58. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 4.Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–22. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 5.Grotenhermen F, Müller-Vahl K. The therapeutic potential of cannabis and cannabinoids. Dtsch Arztebl Int. 2012;109:495–501. doi: 10.3238/arztebl.2012.0495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cridge BJ, Rosengren RJ. Critical appraisal of the potential use of cannabinoids in cancer management. Cancer Manag Res. 2013;5:301–13. doi: 10.2147/CMAR.S36105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dhopeshwarkar A, Mackie K. CB2 Cannabinoid receptors as a therapeutic target—what does the future hold? Mol Pharmacol. 2014;86:430–7. doi: 10.1124/mol.114.094649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atwood BK, Straiker A, Mackie K. CB2: therapeutic target-in-waiting. Prog Neuro-Psychopharmacol Biol Psychiatry. 2012;38:16–20. doi: 10.1016/j.pnpbp.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang P, Wang L, Xie XQ. Latest advances in novel cannabinoid CB2 ligands for drug abuse and their therapeutic potential. Future Med Chem. 2012;4:187–204. doi: 10.4155/fmc.11.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han S, Thatte J, Buzard DJ, Jones RM. Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J Med Chem. 2013;56:8224–56. doi: 10.1021/jm4005626. [DOI] [PubMed] [Google Scholar]
- 11.Janero DR, Makriyannis A. Cannabinoid receptor antagonists: pharmacological opportunities, clinical experience, and translational prognosis. Expert Opin Emerg Drugs. 2009;14:43–65. doi: 10.1517/14728210902736568. [DOI] [PubMed] [Google Scholar]
- 12.Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–24. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 13.Kunos G, Osei-Hyiaman D, Bátkai S, Sharkey KA, Makriyannis A. Should peripheral CB1 cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol Sci. 2009;30:1–7. doi: 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christopoulos A. Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat Rev Drug Discov. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 15.Ross R. Tuning the endocannabinoid system: allosteric modulators of the CB1 receptor. Br J Pharmacol. 2007;152:565–6. doi: 10.1038/sj.bjp.0707349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackie K Distribution of cannabinoid receptors in the central and peripheral nervous system. In: Cannabinoids. Springer, Berlin; 2005. p. 299–325. [DOI] [PubMed]
- 17.Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74:129–80. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- 18.Mackie K. Cannabinoid receptors: where they are and what they do. J Neuroendocrinol. 2008;20:10–4. doi: 10.1111/j.1365-2826.2008.01671.x. [DOI] [PubMed] [Google Scholar]
- 19.Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–84. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez de Fonseca F, Del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, Navarro M. The endocannabinoid system: physiology and pharmacology. Alcohol Alcohol. 2005;40:2–14. doi: 10.1093/alcalc/agh110. [DOI] [PubMed] [Google Scholar]
- 21.Cluny N, Vemuri V, Chambers A, Limebeer C, Bedard H, Wood J, et al. A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br J Pharmacol. 2010;161:629–42. doi: 10.1111/j.1476-5381.2010.00908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 23.Howlett AC, Blume LC, Dalton GD. CB1 cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17:1382–93. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith TH, Sim-Selley LJ, Selley DE. Cannabinoid CB1 receptor-interacting proteins: novel targets for central nervous system drug discovery? Br J Pharmacol. 2010;160:454–66. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blume LC, Patten T, Eldeeb K, Leone-Kabler S, Ilyasov AA, Keegan BM, et al. Cannabinoid Receptor Interacting Protein (CRIP) 1a competition with β-arrestin for CB1 receptor binding sites. Mol Pharmacol. 2017;91:75–86. doi: 10.1124/mol.116.104638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith TH, Blume LC, Straiker A, Cox JO, David BG, McVoy JS, et al. Cannabinoid receptor interacting protein 1a (CRIP1a) modulates CB1 receptor signaling and regulation. Mol Pharmacol. 2015;87:747–65. doi: 10.1124/mol.114.096495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velasco G, Galve-Roperh I, Sanchez C, Blazquez C, Haro A, Guzman M. Cannabinoids and ceramide: two lipids acting hand-by-hand. Life Sci. 2005;77:1723–31. doi: 10.1016/j.lfs.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Nogueras-Ortiz C, Yudowski GA. The multiple waves of cannabinoid 1 receptor signaling. Mol Pharmacol. 2016;90:620–6. doi: 10.1124/mol.116.104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ibsen MS, Connor M, Glass M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017;2:48–60. doi: 10.1089/can.2016.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosier B, Muccioli GG, Hermans E, Lambert DM. Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem Pharmacol. 2010;80:1–12. doi: 10.1016/j.bcp.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Varga E, Georgieva T, Tumati S, Alves I, Salamon Z, Tollin G, et al. Functional selectivity in cannabinoid signaling. Curr Mol Pharmacol. 2008;1:273–84. doi: 10.2174/1874467210801030273. [DOI] [PubMed] [Google Scholar]
- 32.Mallipeddi S, Janero DR, Zvonok N, Makriyannis A. Functional selectivity at G-protein coupled receptors: Advancing cannabinoid receptors as drug targets. Biochem Pharmacol. 2017;128:1–11. doi: 10.1016/j.bcp.2016.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- 34.Urban JD, Clarke WP, Von Zastrow M, Nichols DE, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 35.Vaidehi N, Kenakin T. The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Curr Opin Pharm. 2010;10:775–81. doi: 10.1016/j.coph.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1999;56:1362–9. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- 37.Laprairie RB, Bagher AM, Kelly ME, Dupré DJ, Denovan-Wright EM. Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J Biol Chem. 2014;289:24845–62. doi: 10.1074/jbc.M114.557025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci USA. 2005;102:19144–9. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khoury E, Clément S, Laporte SA. Allosteric and biased g protein-coupled receptor signaling regulation: potentials for new therapeutics. Front Endocrinol (Lausanne) 2014;5:68. doi: 10.3389/fendo.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grover AK. Use of allosteric targets in the discovery of safer drugs. Med Princ Pract. 2013;22:418–26. doi: 10.1159/000350417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foster DJ, Conn PJ. Allosteric modulation of GPCRs: new insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron. 2017;94:431–46. doi: 10.1016/j.neuron.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, et al. Crystal structures of agonist-bound human cannabinoid receptor CB 1. Nature. 2017;547:468–71. doi: 10.1038/nature23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, et al. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature. 2016;540:602–6. doi: 10.1038/nature20613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, et al. Crystal structure of the human cannabinoid receptor CB 1. Cell. 2016;167:750–62. doi: 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–95. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 46.Horswill J, Bali U, Shaaban S, Keily J, Jeevaratnam P, Babbs A, et al. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–14. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Navarro HA, Howard JL, Pollard GT, Carroll F. Positive allosteric modulation of the human cannabinoid (CB1) receptor by RTI-371, a selective inhibitor of the dopamine transporter. Br J Pharmacol. 2009;156:1178–84. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, et al. A cannabinoid CB1 receptor-positive allosteric modulator reduces neuropathic pain in the mouse with no psychoactive effects. Neuropsychopharmacology. 2015;40:2948–59. doi: 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pamplona FA, Ferreira J, de Lima OM, Duarte FS, Bento AF, Forner S, et al. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc Natl Acad Sci USA. 2012;109:21134–9. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vallée M, Vitiello S, Bellocchio L, Hébert-Chatelain E, Monlezun S, Martin-Garcia E, et al. Pregnenolone can protect the brain from cannabis intoxication. Science. 2014;343:94–8. doi: 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauer M, Chicca A, Tamborrini M, Eisen D, Lerner R, Lutz B, et al. Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J Biol Chem. 2012;287:36944–67. doi: 10.1074/jbc.M112.382481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1) J Biol Chem. 2013;288:9790–800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baillie GL, Horswill J, Anavi-Goffer S, Reggio PH, Abood ME, Bolognini D, et al. CB1 receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2012;83:322–38. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gamage TF, Farquhar CE, Lefever TW, Thomas BF, Nguyen T, Zhang Y, et al. The great divide: separation between in vitro and in vivo effects of PSNCBAM-based CB1 receptor allosteric modulators. Neuropharmacology. 2017;125:365–75. doi: 10.1016/j.neuropharm.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khurana L, Fu BQ, Duddupudi AL, Liao YH, Immadi SS, Kendall DA, et al. Pyrimidinyl biphenylureas: identification of new lead compounds as allosteric modulators of the cannabinoid receptor CB1. J Med Chem. 2017;60:1089–104. doi: 10.1021/acs.jmedchem.6b01448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–82. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khajehali E, Malone DT, Glass M, Sexton PM, Christopoulos A, Leach K. Biased agonism and biased allosteric modulation at the CB1 cannabinoid receptor. Mol Pharmacol. 2015;88:368–79. doi: 10.1124/mol.115.099192. [DOI] [PubMed] [Google Scholar]
- 58.Wang X, Horswill JG, Whalley BJ, Stephens GJ. Effects of the allosteric antagonist 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl) phenyl] urea (PSNCBAM-1) on CB1 receptor modulation in the cerebellum. Mol Pharmacol. 2011;79:758–67. doi: 10.1124/mol.110.068197. [DOI] [PubMed] [Google Scholar]
- 59.Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M. Real-time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol. 2013;170:893–907. doi: 10.1111/bph.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gamage TF, Anderson JC, Abood ME. CB1 allosteric modulator Org27569 is an antagonist/inverse agonist of ERK1/2 signaling. Cannabis Cannabinoid Res. 2016;1:272–80. doi: 10.1089/can.2016.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen T, Li JX, Thomas BF, Wiley JL, Kenakin TP, Zhang Y. Allosteric modulation: an alternate approach targeting the cannabinoid CB1 receptor. Med Res Rev. 2017;37:441–74. doi: 10.1002/med.21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gomes I, Grushko JS, Golebiewska U, Hoogendoorn S, Gupta A, Heimann AS, et al. Novel endogenous peptide agonists of cannabinoid receptors. FASEB J. 2009;23:3020–9. doi: 10.1096/fj.09-132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pisanti S, Malfitano AM, Ciaglia E, Lamberti A, Ranieri R, Cuomo G, et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol Ther. 2017;175:133–50. doi: 10.1016/j.pharmthera.2017.02.041. [DOI] [PubMed] [Google Scholar]
- 64.Morales P, Reggio PH, Jagerovic N. An overview on medicinal chemistry of synthetic and natural derivatives of cannabidiol. Front Pharmacol. 2017;8:422. doi: 10.3389/fphar.2017.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morales P, Hurst DP, Reggio PH. Molecular targets of the phytocannabinoids: a complex picture. In: Phytocannabinoids. Springer, Berlin; 2017. p. 103–31. [DOI] [PMC free article] [PubMed]
- 66.Turner SE, Williams CM, Iversen L, Whalley BJ. Molecular pharmacology of phytocannabinoids. In: Phytocannabinoids. (Springer, 2017), p 61–101. [DOI] [PubMed]
- 67.Laprairie R, Bagher A, Kelly M, Denovan-Wright E. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. 2015;172:4790–805. doi: 10.1111/bph.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pistis M, O’Sullivan SE. The role of nuclear hormone receptors in cannabinoid function. Adv Pharmacol. 2017; 80:291–328. [DOI] [PubMed]
- 69.Priestley RS, Nickolls SA, Alexander SP, Kendall DA. A potential role for cannabinoid receptors in the therapeutic action of fenofibrate. FASEB J. 2015;29:1446–55. doi: 10.1096/fj.14-263053. [DOI] [PubMed] [Google Scholar]
- 70.Ahn KH, Mahmoud MM, Samala S, Lu D, Kendall DA. Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. J Neurochem. 2013;124:584–9. doi: 10.1111/jnc.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Piscitelli F, Ligresti A, La Regina G, Coluccia A, Morera L, Allarà M, et al. Indole-2-carboxamides as allosteric modulators of the cannabinoid CB1 receptor. J Med Chem. 2012;55:5627–31. doi: 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- 72.Mahmoud MM, Ali HI, Ahn KH, Damaraju A, Samala S, Pulipati VK, et al. Structure–activity relationship study of indole-2-carboxamides identifies a potent allosteric modulator for the cannabinoid receptor 1 (CB1) J Med Chem. 2013;56:7965–75. doi: 10.1021/jm4009828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nguyen T, German N, Decker AM, Li JX, Wiley JL, Thomas BF, et al. Structure–activity relationships of substituted 1H-indole-2-carboxamides as CB1 receptor allosteric modulators. Biorg. Med Chem. 2015;23:2195–203. doi: 10.1016/j.bmc.2015.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khurana L, Ali HI, Olszewska T, Ahn KH, Damaraju A, Kendall DA, et al. Optimization of chemical functionalities of indole-2-carboxamides to improve allosteric parameters for the cannabinoid receptor 1 (CB1) J Med Chem. 2014;57:3040–52. doi: 10.1021/jm5000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cawston EE, Connor M, Di Marzo V, Silvestri R, Glass M. Distinct temporal fingerprint for cyclic adenosine monophosphate (cAMP) signaling of indole-2-carboxamides as allosteric modulators of the cannabinoid receptors. J Med Chem. 2015;58:5979–88. doi: 10.1021/acs.jmedchem.5b00579. [DOI] [PubMed] [Google Scholar]
- 76.Kulkarni PM, Kulkarni AR, Korde A, Tichkule RB, Laprairie RB, Denovan-Wright EM, et al. Novel electrophilic and photoaffinity covalent probes for mapping the cannabinoid 1 receptor allosteric site (s) J Med Chem. 2015;59:44–60. doi: 10.1021/acs.jmedchem.5b01303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hernandez-Folgado L, Stevenson LA, Morales P, Gómez-Cañas M, Pazos MR, Cascio MG, et al. Exploring the benzimidazole ring as a substitution for indole in cannabinoid allosteric modulators. cannabis and cannabinoid. Research. 2016;1:196–201. doi: 10.1089/can.2015.0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.German N, Decker AM, Gilmour BP, Gay EA, Wiley JL, Thomas BF, et al. Diarylureas as allosteric modulators of the cannabinoid CB1 receptor: structure–activity relationship studies on 1-(4-chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl) pyridin-2-yl] phenyl} urea (PSNCBAM-1) J Med Chem. 2014;57:7758–69. doi: 10.1021/jm501042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bertini S, Chicca A, Gado F, Arena C, Nieri D, Digiacomo M, et al. Novel analogs of PSNCBAM-1 as allosteric modulators of cannabinoid CB1 receptor. Biorg. Med Chem. 2017;25:6427–34. doi: 10.1016/j.bmc.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laprairie RB, Kulkarni PM, Deschamps JR, Kelly ME, Janero DR, Cascio MG, et al. Enantiospecific allosteric modulation of cannabinoid 1 receptor. ACS Chem Neurosci. 2017;8:1188–203. doi: 10.1021/acschemneuro.6b00310. [DOI] [PubMed] [Google Scholar]
- 81.Mitjavila J, Yin D, Kulkarni PM, Zanato C, Thakur GA, Ross R, et al. Enantiomer-specific positive allosteric modulation of CB1 signaling in autaptic hippocampal neurons. Pharmacol Res. 2017;129:475–81. doi: 10.1016/j.phrs.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saleh N, Hucke O, Kramer G, Schmidt E, Montel F, Lipinski R, et al. Multiple binding sites contribute to the mechanism of mixed agonistic and positive allosteric modulators of the cannabinoid CB1 receptor. Angew Chem. 2018;130:2610–5. doi: 10.1002/anie.201708764. [DOI] [PubMed] [Google Scholar]
- 83.Thakur GA. Kulkarni PM Allosteric modulators of CB1 cannabinoid receptors. Patent application WO2013103967, 2017.
- 84.Ruth R, Greig I, Zanda M, Tseng CC Cannabinoid type 1 receptor modulators. Patent application, WO2016029310, 2018.
- 85.Ramírez BG, Blázquez C, del Pulgar TG, Guzmán M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–13. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, et al. Endocannabinoids and β-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci CMLS. 2006;63:1410–24. doi: 10.1007/s00018-006-6037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA, et al. Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biol Psychiatry. 2017;17:31761–4. doi: 10.1016/j.biopsych.2017.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cairns EA, Szczesniak AM, Straiker AJ, Kulkarni PM, Pertwee RG, Thakur GA, et al. The in vivo effects of the CB1-positive allosteric modulator GAT229 on intraocular pressure in ocular normotensive and hypertensive mice. J Ocul Pharmacol Ther. 2017;33:582–90. doi: 10.1089/jop.2017.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lu D, Dopart R, Kendall DA. Controlled downregulation of the cannabinoid CB1 receptor provides a promising approach for the treatment of obesity and obesity-derived type 2 diabetes. Cell Stress Chaperon- 2016;21:1–7. doi: 10.1007/s12192-015-0653-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gamage TF, Ignatowska-Jankowska BM, Wiley JL, Abdelrahman M, Trembleau L, Greig IR, et al. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org27569. Behav Pharmacol. 2014;25:182–5. doi: 10.1097/FBP.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ding Y, Qiu Y, Jing L, Thorn DA, Zhang Y, Li JX. Behavioral effects of the cannabinoid CB1 receptor allosteric modulator ORG27569 in rats. Pharmacol Res Perspect. 2014;2:1–11. doi: 10.1002/prp2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jing L, Qiu Y, Zhang Y, Li JX. Effects of the cannabinoid CB1 receptor allosteric modulator ORG 27569 on reinstatement of cocaine-and methamphetamine-seeking behavior in rats. Drug Alcohol Depend. 2014;143:251–6. doi: 10.1016/j.drugalcdep.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Straiker A, Mitjavila J, Yin D, Gibson A, Mackie K. Aiming for allosterism: evaluation of allosteric modulators of CB1 in a neuronal model. Pharmacol Res. 2015;99:370–6. doi: 10.1016/j.phrs.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Busquets-Garcia A, Soria-Gómez E, Redon B, Mackenbach Y, Vallee M, Chaouloff F, et al. Pregnenolone blocks cannabinoid-induced acute psychotic-like states in mice. Mol Psychiatry. 2017;22:1594–603. doi: 10.1038/mp.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]