

Abstract
The relentless efforts of thousands of researchers have allowed deciphering the molecular machinery that regulates and executes autophagy, thus identifying multiple molecular targets to enhance or block the process, rendering autophagy “druggable”. Autophagy inhibition may be useful for preserving the life of cells that otherwise would succumb to excessive self-digestion. Moreover, autophagy blockade may reduce the fitness of cancer cells or interrupt metabolic circuitries required for their growth. Autophagy stimulation is probably useful for the prevention or treatment of aging, cancer (when stimulation of immunosurveillance is the therapeutic goal), cardiovascular disease, cystic fibrosis, infection by intracellular pathogens, obesity, and intoxication by heavy metals, just to mention a few examples. Epidemiological evidence suggests broad health-improving effects for lifestyles, micronutrients, and drugs that favor autophagy. In this review, we discuss the role of autophagy in disease pathogenesis while focusing on the question, which disease will become the first clinically approved indication for therapeutic autophagy modulation.
The induction and inhibition of autophagy may provide new strategies for intervening on major and specific diseases, respectively. The scheme provides an overview on pathologies that might be targeted by the manipulation of autophagy.
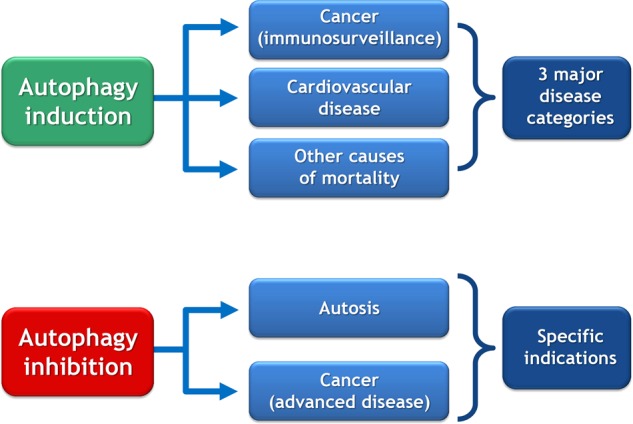
Facts
Autophagy is one of the best-studied phenomena in cell biology. Drugs for enhancing or inhibiting general or specific autophagy are being developed.
Acquired or genetically determined alterations in autophagic flux are involved in multiple pathologies across the entire spectrum of human diseases.
Autophagy inhibition might be useful for the avoidance of unwarranted autophagy-dependent cell death.
Chronic autophagy stimulation has a positive impact on preclinical models of aging and multiple distinct age-dependent diseases, including arteriosclerosis, cancer, and neurodegeneration. Acute autophagy stimulation also has organ-protective effects in models of ischemia or intoxication.
Open questions
Optimal pharmacological agents that modulate autophagy at the expense of acceptable side effects must be selected and characterized at the preclinical level.
For autophagy induction, it remains to be determined whether pharmacological agents are superior to life style interventions (quantity and quality of nutrition, physical exercise) for long-term benefits.
The question remains open, which particular disease will be the first-in-human indication for clinical trials that explore pharmacological autophagy modulators.
Introduction
The final fate of cells is death, while that of cytoplasmic organelles is macroautophagy [1, 2]. For this reason, many scientists that started their carrier on cell death switched the focus of their work to macroautophagy (hitherto “autophagy”). This move from one area of research to another has also been motivated by the ambiguous effect of autophagy on cell death.
On one hand, autophagy constitutes a formidable process allowing cells to adapt to changing and stressful conditions by removing damaged subcellular structures and by digesting macromolecules to small molecules that then can either be used to fuel bioenergetics or to rebuild new organelles, thus rejuvenating the cytoplasm [3] and perhaps even the nuclear envelope and nucleoli [4]. For this reason, autophagy is viewed as a major anti-aging mechanism that, if stimulated in an adequate fashion at the whole-body level, may enhance health span and longevity [5–7]. Regrettably, autophagy may also enhance the fitness of cancer cells that attempt to strive in a hostile microenvironment, thus resisting endogenous stressors (absence of trophic support, hypoxia, and attack by the immune system) or therapeutic measures (chemotherapy, radiotherapy, or targeted therapy) [8, 9]. Moreover, autophagy may be important for maintaining the pool of cancer stem cells [10]. Given the cytoprotective action of autophagy, scientists have been reasoning that autophagy should be stimulated when the goal is the preservation of normal cellular or organismal functions [5, 6], but inhibited when the goal is the treatment of cancer [8, 9].
On the other hand, deregulated autophagy may participate in the destruction of cells, be it during normal development (to get rid of superfluous cells, especially in model organisms) [11] or in response to environmental stress [12, 13]. Although nowadays the first idea (autophagy improving cellular fitness) largely dominates over the second one (autophagy as a cell death mechanism), there are indeed instances in mammalian pathophysiology in which excessive autophagy may destroy neurons (for instance in neonatal ischemia) [14, 15] or cardiomyocytes (in hypoxia–ischemia) [16, 17]. In this context, suppression of autophagy may protect specific cell types against death, calling for the development of cytoprotective autophagy inhibitors.
In view of the disease-modulatory potential of autophagy, scientists working in academia, biotechnology, or pharmaceutical industry are developing pharmacological autophagy inhibitors and inducers [18, 19]. This effort is encouraged by the increasingly accepted notion that the disease-preventive or therapeutic effects of some widely used drugs and food components can be explained by autophagy stimulation, as exemplified for aspirin [20], resveratrol [21], and spermidine [22, 23]. In theory, there are multiple molecularly defined targets to suppress autophagy (each of the essential autophage gene [ATG]-encoded proteins) and to enhance autophagy (by targeting each of the numerous endogenous inhibitors of autophagy). Prominent pharmacological targets for autophagy inhibition include proteases (such as the ATG4 isoenzymes required for the proteolytic maturation of LC3 family proteins), lipid kinases (in particular, phosphatidylinositol 3-kinase catalytic subunit type 3, PIK3C3, which acts within the Beclin 1, BECN1, complex) and protein kinases (especially, Unc-51-like autophagy activating kinase 1, ULK1) [19, 24]. Druggable targets to enhance autophagic flux include a series of negative regulators of autophagy such as multidomain proteins of the BCL-2 family (which can be targeted by so-called BH-3 mimetics and perhaps other agents that disrupt their inhibitory interaction with the Beclin 1/PIK3C3 complex) [25–28], the acetyltransferase E1A binding protein P300 (EP300, which is already known as the target of aspirin and spermidine) [20, 29], the mammalian target of rapamycin complex 1 (MTORC1, which is inhibited by rapamycin and rapalogs), as well as kinases acting upstream of MTORC1 such as the members of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) and protein kinase B (PKB) families [19, 30]. However, this list is not exhaustive and other potential targets for autophagy modulation must be considered.
In this article, we will provide an opinionated overview on the diseases that might be treated by autophagy-modulatory drugs, while polemically centering our focus on the following question. Which will be the first disease(s) to be treated with new drugs designed to enhance or reduce autophagy?
Prime indications for autophagy inhibition
As mentioned above in the Introduction, there are two possible indications for inhibiting autophagy: (i) autophagy-dependent cell death and (ii) cancer (Fig. 1a).
Fig. 1.
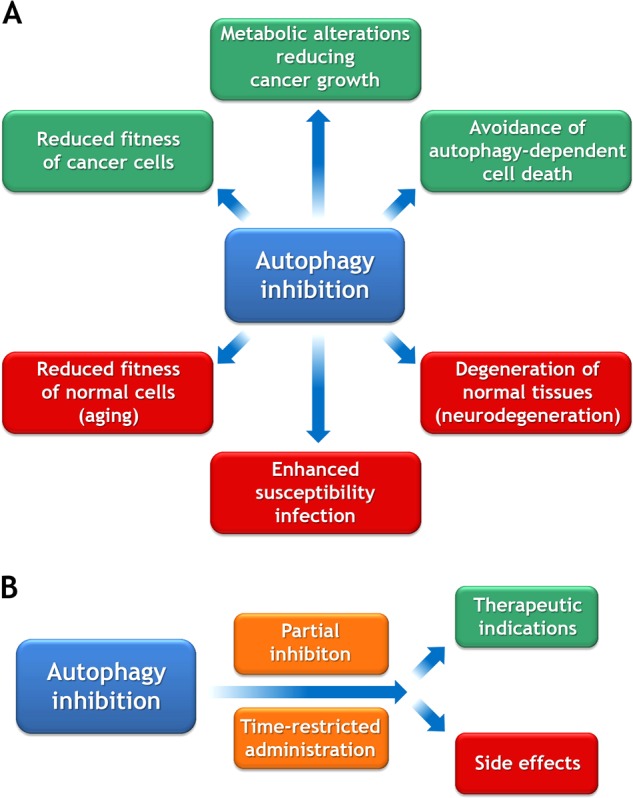
Indications for autophagy inhibition. a Hypothetical desirable and side effects of autophagy inhibition are shown in green and red, respectively. b For reducing unacceptable side effects, it may be necessary to administer autophagy inhibitors at a reduced dose and in short-term or intermittent schedules
Autophagy-dependent cell death
Cell death is often preceded by signs of autophagy, an observation that led to the common belief that autophagy would constitute a major cell death modality (“type-2 cell death”) [31, 32]. However, when autophagy is inhibited by specific (i.e., genetic) methods, it turned out that, in most cases, autophagy is not necessary for lethality but rather acts as a cytoprotective mechanisms, hence increasing the resistance of cells to fatal stimuli and avoiding or postponing their death [33, 34]. There are several notable exceptions to this rule. In model organisms, abolition of autophagy can affect the developmental death of some cell types, contrasting though with the observation that complete invalidation of conventional autophagy does not cause any developmental perturbations in mice [11]. Moreover, in human or mouse cell cultures responding to specific stimuli, cell death may be favored by autophagy (as a process) or by the expression of ATG proteins, meaning that autophagy inhibition or depletion/deletion of ATG gene products increases the resistance of the cells to potentially lethal insults [12, 16]. Thus, in mouse models, genetic inhibition of autophagy may reduce motorneuron loss in amyotrophic lateral sclerosis [35], as well as cerebral damage after neonatal asphyxiation [14]. Moreover, cardiac glycosides (CGs) can inhibit “autosis”, a peculiar autophagy-dependent cell death modality characterized by cytoplasmic vacuolization and separation of the nuclear envelope from the nucleus [36]. The precise molecular mechanisms through which autosis is inhibited by CG remains elusive, although it can be recapitulated by genetic interventions on the Na+, K+-ATPase, the molecular target of CG [16]. Notwithstanding this limitation, it is tempting to speculate that the well-known cardioprotective effects of CG are not only due to a positive inotropic action but may also involve cytoprotection through the suppression of excessive autophagy. Indeed, administration of CG can prevent ischemic lesions to the myocardium in mice [16]. Nonetheless, during recent years CGs have been largely replaced by other drugs, mainly due to safety concerns. Hence, it remains to be seen whether their recently discovered capacity to inhibit autosis will favor their reintroduction into the clinics.
Cancer
Malignant cells can be considered as abnormally fit cells that have escaped from cell-autonomous tumor-suppressive mechanisms, as well as from immunosurveillance. Although the inhibition of autophagy can stimulate oncogenesis, especially at the initial steps [37–39], it has been observed in several instances that autophagy inhibition reduces the growth of advanced cancers [9, 40]. Thus, inducible knockout of Atg7 specifically within cancer cells (as opposed to stromal cells) reduces the growth of lung and pancreas cancers induced by oncogenic KRAS and inactivation of the tumor-suppressor TP53 [41, 42]. There are multiple reports showing that inhibition of autophagy using chloroquine or hydroxychloroquine may sensitize tumors to chemotherapy either in vitro or in vivo [43, 44]. However, chloroquine and its derivatives cannot be considered as specific autophagy inhibitors (they actually mediate autophagy-independent cytotoxicity due to their lysosome-disrupting effects), shedding doubts on the question whether these anticancer effects are truly mediated by autophagy suppression [45, 46].
Beyond the cancer cells themselves, autophagy in other cell types may also contribute to tumor progression. Thus, knockout of ATG genes in endothelial cells can retard tumor angiogenesis [47], whereas inhibition of autophagy in cancer-associated fibroblasts interrupts trophic support to malignant cells [48]. One recent report shows that an inducible total-body knockout of Atg7 triggers major changes in metabolism including an increase in the hepatic release of arginase 1 (ARG1), causing a reduction in extracellular arginine levels and consequent growth reduction of arginine auxotroph cancers [49]. This suggests that a global (rather than cancer cell-specific) inhibition of autophagy may be used for the treatment of neoplasia. That said, the inducible deletion of Atg7 from the mouse genome causes a general tissue-degenerative phenotype affecting all organs and causing death due to brain dysfunction within approximately 6 weeks [41]. It remains to be determined whether temporary, partial, or intermittent inhibition of autophagy obtained by pharmacological agents can avoid the manifestation of such an accelerated aging phenotype, yet conserve antitumor efficacy in vivo (Fig. 1b).
Prime indications for autophagy induction
Autophagy induction can be most easily obtained by reduced caloric intake and exercise, yet can also be stimulated by “caloric restriction mimetics (CRMs)” and other pharmacological agents [19, 50]. There are a number of pathological conditions that favorably respond to autophagy enhancement (Fig. 2).
Fig. 2.
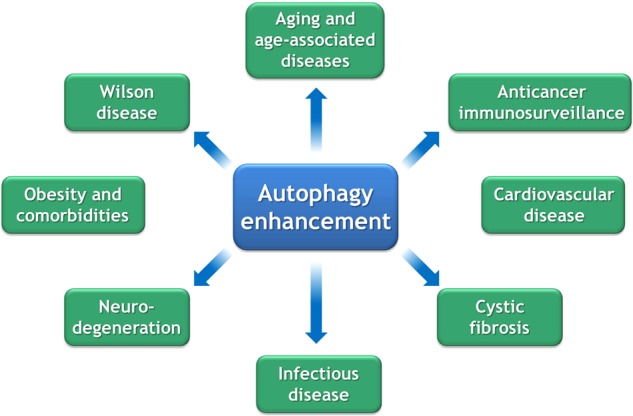
Indications for autophagy enhancement. For a detailed discussion, consult main text
Aging
In model organisms like yeast, nematodes, and fruit flies, there is overwhelming evidence that any kind of behavioral, nutritional, pharmacological, and genetic manipulation capable of extending longevity induces an increase in autophagic flux and actually requires autophagy to be efficient [51, 52]. Similarly, in mice, it is sufficient to overexpress ATG5 as a transgene [53] or to introduce a gain-of-function mutant of BECN1 (encoding a BECN1 protein that does not interact any more with the autophagy-inhibitory protein BCL-2) [54] to induce an increase in health span and lifespan. Treatment of mice with autophagy inducers including rapamycin (an inhibitor of MTORC1) or spermidine (an inhibitor of EP300) has a positive impact on life expectancy as well [22, 55]. Epidemiological observations in humans confirm that high nutritional spermidine uptake reduces mortality from cancer, cardiovascular disease, and other causes [56, 57]. Hence, spermidine might be used as a safe food supplement to reduce aging and to postpone the manifestations of age-related diseases. At this stage, however, it remains to be seen whether this type of intervention would be sufficiently lucrative for pharmaceutical companies to stimulate prospective clinical trials in this area. Moreover, aging is not yet considered as a disease, meaning that neither the Federal Drug Administration (FDA) nor the European Medical Agency (EMA) would authorize this kind of clinical trial to be launched, unless it would deal with the prevention or treatment of a particular pathology.
Anticancer immunosurveillance
We have given large room to the hypothesis that autophagy should be inhibited for the treatment of cancer (see above). However, there are also arguments in favor of the contrary, i.e., autophagy stimulation for cancer treatment. This debate—should we inhibit or enhance autophagy for cancer treatment?—is fueled by rather distinct visions of tumor pathogenesis. Indeed, most cell biologists have been adhering to the idea that malignant disease arises from genetic and epigenetic alterations within cancer cells [58]. This vision, has recently been revised when it was understood that cancers only manifest when immunosurveillance fails [59] and that antineoplastic therapies (such as cytotoxic chemotherapeutics and radiotherapy) only confer long-term benefits if they are capable of restoring immunosurveillance [60, 61]. Accordingly, the (re)activation of anticancer immune responses using so-called immune checkpoint blockers has become a close-to-universal treatment of neoplastic disease [62, 63]. In this context, it appears that the final outcome of chemotherapies and radiotherapies is not so much determined by the direct induction of cancer cell death but rather is dictated by the vigor of the antitumor immune response. Autophagy induction may favor the release of ATP from stressed and dying cancer cells, thus favoring the recruitment of myeloid cells including that of dendritic cell precursors into the tumor bed [64, 65]. For this reason, pharmacological autophagy inducers may act as immunostimulators in the context of anticancer chemotherapies, provided that the cancer cells are autophagy competent [66]. Accordingly, autophagy competence of malignant cells correlates with local immune responses and treatment outcome in human cancers [67–69]. Based on these results, it is tempting to speculate that autophagy induction by nutritional interventions (such as fasting or caloric restriction) or pharmacological agents might be introduced into the oncological armamentarium [70]. It appears plausible, yet remains to be clinically demonstrated, that short pulses of autophagy induction (as opposed to a protracted enhancement) would be sufficient to ignite durable anticancer immune responses, if appropriately combined with chemotherapy, radiotherapy, or immunotherapy.
Cardiovascular disease
Accumulating evidence suggests that repeated or continuous autophagy stimulation can protect against most if not all signs of cardiovascular aging including arteriosclerosis, increased arterial stiffness, and cardiac failure [71]. For this reason, dietary and pharmacological measures for enhancing autophagy might be considered for the prevention of cardiovascular diseases, perhaps in the context of a general strategy to promote healthy aging. Moreover, with the exception of the (controversial) maladaptive autophagy occurring during pressure overload [17], it appears that autophagy induction reduces myocardial damage after acute insult by ischemia. Preclinical evidence suggests that a variety of autophagy inducers including trehalose can improve the outcome of myocardial infarction [72]. Of note, coronary stents releasing rapamycin are FDA/EMA approved because of their superior clinical efficacy [73]. Although the official indication for these stents is the avoidance of restenosis, it can be speculated that they act through local autophagy induction. Pending proof of this concept, it can be envisaged that other (more potent and less immunosuppressive) autophagy inducers will enter the clinics for minimizing tissue damage and improving local repair.
Cystic fibrosis
Cystic fibrosis (CF) is the most frequent monogenetic lethal human disease, caused by loss-of-function mutations of CF transmembrane conductance regulator (CFTR), an ATP-gated chloride channel whose malfunction causes an ionic disequilibrium, both in mucus (due to lack or reabsorption of chloride by epithelial cells) and within cells. The most frequent mutation causing CF (responsible for 70–90% of all cases) consists in the deletion of amino-acid 506 (CFTRdel506). It has been thought for long that the cause of CF pathogenesis was the production of thick mucus, leading to infections of the respiratory tract and pancreatic/gastrointestinal dysfunction [74]. However, recently it has been understood that CFTR malfunction also affects intracellular proteostasis due to the overactivation of transglutaminase-2 (TGM2) and the depletion of the pro-autophagic BECN1 complex [75–78]. Feedforward loops amplifying the phenomenon (because TGM2 activation and BECN1 inhibition ultimately cause the disposal of CFTRdel506 protein, totally abolishing its residual function) then lock epithelial cells in a pro-inflammatory state and hinder them to return to homeostasis [77, 79]. Of note, preclinical work in mice and two independent phase 2 clinical trials have shown that TGM2 inhibition with cysteamine combined with epigallocatechin gallate (EGCG, a green tea flavoid that can stimulate autophagy, presumably by inhibiting EP300) [80] efficiently reverse the disease caused by CFTRdel506 [81, 82]. In CFTRdel506/del506 mice that are partially autophagy deficient due to BECN1 haploinsuffiency (BECN1+/-), the combination therapy with cysteamine plus EGCG fails, suggesting that this treatment indeed requires the induction of autophagy [82]. Moreover, respiratory epithelia from CFTRdel506 patients treated with cysteamine plus EGCG exhibit signs of improved autophagy (with increased expression of BECN1 and lipidation of LC3) [82]. Similarly, thymosin-α might mediate its beneficial effects on CF through autophagy induction [83]. Altogether, the clinical results suggest that autophagy enhancement in CF constitutes a safe and efficacious treatment. However, phase 3 clinical trials must be performed to validate this concept and to clear the path to clinical approval of autophagy enhancers.
Infectious disease
Autophagy and ATG proteins are not only involved in the sequestration and destruction of cytoplasmic structures of the cell; they also mediate the clearance of intracellular viruses and bacteria. This process, which then is called “xenophagy” constitutes the most primitive “immune” response, allowing single-cell eukaryotes and individual cells within multicellular organisms to rid themselves from intracellular pathogens. There is strong evidence that this xenophagy plays a major role in human physiology. Inherited defects in autophagy-relevant genes predispose to infectious disease [84], and some viruses have developed strategies to subvert their xenophagic destruction [85]. Moreover, pharmacological stimulation of autophagy is sufficient to reduce the mortality of mice infected with chikungunya or West Nile virus [86]. Similarly, cysteamine-mediated stimulation of autophagy in macrophages from CF patients (with the CFTRdel506 mutation) can improve their capacity to clear Pseudomonas aerginosa, a bacterium that frequently infects the CF lung [87]. Host resistance against infection by Mycobacterium tuberculosis is determined by xenophagy (mostly in macrophages) [88], and pharmacological stimulation of autophagy can actually help the host to clear M. tuberculosis [89]. These examples suggest that autophagy stimulation might constitute a valid host-directed therapeutic strategy for treating antibiotic-resistant infections [90]. Given the surge of multiresistant bacteria and the absolute urgency to control them, it would not be surprising that therapeutic autophagy inducers would find their way into medical routine practice for fighting against antibiotic-resistant infections.
Neurodegeneration
There is overwhelming evidence that loss-of-function variants of genes involved in autophagy including in selective autophagy (such as mitophagy) favor the manifestation of an entire panoply of distinct neurodegenerative diseases ranging from Alzheimer’s to Parkinson’s, Huntington’s and less frequent, though equally terrifying conditions. The precise molecular etiology of these (genetically determined and environmentally modulated) diseases are rather disparate, ranging from defects in cargo detection/sequestration to its degradation along the entire autophagic pathway with its ramifications. As a general scheme, the production of neurotoxic proteins exceeds their proteostatic clearance, meaning that even subtle defects in autophagy cause age-dependent neurodegeneration [30]. Based on this consideration, novel safe autophagy inducers have been identified and are ongoing initial clinical evaluation, for instance for the treatment of Huntington’s disease [91]. Nonetheless, it remains to be demonstrated that therapeutic interventions on even incipient neurodegenerative disease can disrupt their pathogenesis or whether prophylactic treatments (well before the disease becomes clinically manifest) would be the option of choice. On a pessimistic note, it is possible that close-to-etiological treatments like autophagy induction would be administered at a stage that is too late to arrest—let alone, to revert—the process of neurodegeneration.
Obesity
Obesity, the most frequent pathological condition that afflicts humanity, inhibits autophagy due to multiple reasons including enhanced circulating nutrients (glucose, free fatty acids, triglycerides etc.), and elevated levels of autophagy-inhibitory hormones (insulin, insulin-like growth factor and a series of adiponectins such as leptin) [92]. In aggregate, these factors cause an elevation of intracellular acetyl coenzyme A (CoA) levels coupled to hyperactivation of the PKB/PI3K/MTORC1 pathway, thus inhibiting autophagy [93, 94]. Although formal proof for this assertion is missing, it appears logical that obesity acts as a major accelerator of aging and age-related diseases (including cancer and cardiovascular diseases) due to the chronic inhibition of autophagy [95]. In mice, partial autophagy defects mediated by knockout of Atg4b (genotype: Atg4b-/-) or haploinsufficiency of Atg7 (genotype: Atg5b-/-) predispose to the induction of obesity by high sucrose or high-fat diets, respectively [96, 97]. Logically, it may be attempted to increase autophagic flux to increase energy expenditure and to interrupt the obesity-associated positive feedback loops resulting in ever-more increased appetite and adipogenesis to avoid metabolic syndrome and to retard the manifestation of comorbidities such as diabetes and hepatosteatosis. This is supported by promising evidence in rodent models [98, 99], yet awaits clinical validation.
Wilson disease
Wilson disease (WD) is the second most frequent lethal monogenetic human disease (after CF). It is caused by loss-of-function mutations in ATP7B, an ATPase that is expressed in the plasma membrane of hepatocytes and other cell types to extrude excessive copper ions (Cu2+). Excessive intracellular (and in particular intramitochondrial) accumulation of Cu2+ over time results into the progressive destruction of mitochondria, culminating in hepatocyte death [100]. In WD patients, as well as in Atb7b-/- rats, autophagy is induced in hepatocytes. In vitro experiments suggest that autophagy induction can protect hepatocytes against Cu2+-induced toxicity [101] Thus, beyond the chelation of Cu2+[102], it appears possible that autophagy induction might be useful for the treatment of WD and perhaps other intoxications with heavy metals. It remains to be seen whether the acute management of recently diagnosed, hepato-insufficient WD will involve sessions of autophagy induction to preserve organ function.
Conclusions and speculations
As autophagy is deciphered in its detailed molecular mechanisms, the process has become “druggable”, with the promise to enhance or to inhibit the phenomenon at will. Which autophagy-modulatory drug will then win the race to become the first of its class to reach FDA/EMA approval? And for which indication?
General versus selective autophagy modulation
At this stage, it remains to be seen whether the first drug(s) that will reach the clinics will target the core machinery of autophagy or rather influence the recognition/sequestration of specific autophagic cargo (selective autophagy). Indeed, inhibiting the core machinery of autophagy might have major side effects such as acute infectious disease or accelerated tissue aging, whereas excessive stimulation of the phenomenon might trigger autosis in vulnerable cell types. Moreover, many of the core processes of autophagy (such as the action of the BECN1/PIK3C3 complex leading to the generation of phosphatidylinositol 3-phosphate) are not exclusively involved in autophagy [84], meaning that their manipulation may affect other cellular processes with the consequent risk of side effects. Thus, yet-to-be-developed strategies for improving or reducing selective autophagy pathways to cause the specific removal of intracellular pathogens, organelles, and protein aggregates might be preferable over the global manipulation of general autophagy.
Acute versus protracted treatments
There are therapeutic indications for autophagy modulation that may require acute short-term treatments such as inhibition of autophagy for preventing brain damage in neonatal asphyxiation or induction of autophagy for the treatment of infection by multiresistant bacteria or viral pathogens, as well as for the avoidance of organ failure after ischemia or intoxication. However, there are also indications that may require long-term interventions such as the treatment of cancer and, on an even longer scale, the treatment of obesity, neurodegeneration, and aging. Given safety considerations (which become particularly stringent when mild chronic diseases are treated but can be more relaxed in the context of acute life-threatening pathologies), as well as the duration of clinical trials (ranging from a few weeks for acute infection to decade-long interventions for the treatment of aging), it appears probable that the first clinically approved indication for autophagy modulation will be a severe, rapidly developing disease.
Natural versus drug treatments
The natural way of inducing autophagy is exercise, fasting, and dieting, and this is the most efficient way to keep our body young and disease free [95]. Moreover, there are naturally occurring food items containing elevated levels of CRMs, such as the polyamine spermidine or polyphenols, which may enhance autophagy, hence posponing the manifestation of age-associated pathologies. It appears fascinating that, for therapeutic purposes, folk medicine often prescribes fasting, exercise and CRM-containing plants (such as ashitaba containing dimethoxychalcone) [103]. In this sense, autophagy induction is already used in traditional medicine (Fig. 3). It will be interesting to see whether new pharmacological agents designed to modulate autophagy will favorably compare with such natural therapies, although there is little doubt that the economic interests linked to modern medical practice, as well as the attitude of the patient/consumer will ultimately favor the oral or parenteral use of fully synthetic compounds.
Fig. 3.
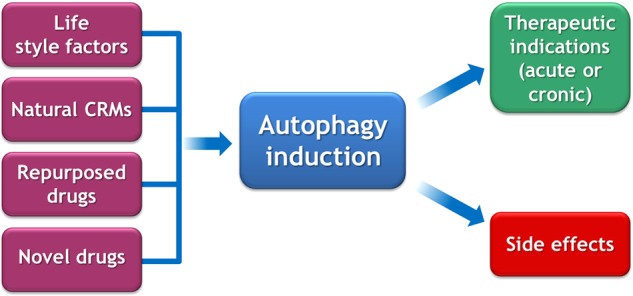
General considerations on strategies for autophagy induction. Life style modifications (exercise, fasting, diet), natural caloric restriction mimetics (CRMs), old drugs for which autophagy induction has been discovered, as well as completely new drugs may be used to stimulate autophagy for therapeutic purposes
Old versus new drugs
Some widely used drugs are efficient autophagy inducers. This applies to aspirin, a widely used (and relatively safe) drug that is reputed for its preventive effects on cardiovascular disease and solid tumors [104]. Other, “old” drugs that might be repurposed for autophagy induction include ambroxol [89], carbamazepine [105], cysteamine [81], rilmenidine [91], and valproic acid [105], just to mention a few. Although it appears logical to use such drugs with a known safety profile for new autophagy-related indications, the biotechnology and pharmaceutical industry is not interested in developing them for clinical use and hence to introduce them into phase 2 and phase 3 trials. Therefore, patients will have to wait for the development of new, more lucrative drugs that will have to undergo full toxicological evaluation before they can enter phase 1 studies in humans (Fig. 3). In sum, although some efficient (and safe) drugs for autophagy induction are already available for clinical use, it is almost certain that they will not reach FDA or EMA approval for new indications, unless public sponsors of their clinical evaluation step in.
Acknowledgements
GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Compliance with ethical standards
Conflict of interest
GK is one of the scientific co-founders of Samsara Therapeutics. MCM declares that she has no conflict of interest.
Footnotes
Edited by G. Melino
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Maria Chiara Maiuri, Email: chiara.maiuri@crc.jussieu.fr.
Guido Kroemer, Email: Kroemer@orange.fr.
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ. 2009;16:1–2. doi: 10.1038/cdd.2008.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papandreou ME, Tavernarakis N. Nucleophagy: from homeostasis to disease. Cell Death Differ. 2019; (in press). [DOI] [PMC free article] [PubMed]
- 5.Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19:579–93. doi: 10.1038/s41580-018-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyamoto S. Autophagy and cardiac aging. Cell Death Differ. 2019; (in press). [DOI] [PMC free article] [PubMed]
- 8.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest. 2015;125:47–54. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–43. doi: 10.1016/j.cmet.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019; (in press). [DOI] [PMC free article] [PubMed]
- 11.Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019; (in press). [DOI] [PMC free article] [PubMed]
- 12.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20:1110–7. doi: 10.1038/s41556-018-0201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie C, Ginet V, Sun Y, Koike M, Zhou K, Li T, et al. Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy. 2016;12:410–23. doi: 10.1080/15548627.2015.1132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17:467–84. doi: 10.1038/nrn.2016.51. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Shoji-Kawata S, Sumpter RM, Jr, Wei Y, Ginet V, Zhang L, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci USA. 2013;110:20364–71. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018;80:1–26. doi: 10.1146/annurev-physiol-021317-121427. [DOI] [PubMed] [Google Scholar]
- 18.Rubinsztein DC, Bento CF, Deretic V. Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med. 2015;212:979–90. doi: 10.1084/jem.20150956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511. doi: 10.1038/nrd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietrocola F, Castoldi F, Markaki M, Lachkar S, Chen G, Enot DP, et al. Aspirin recapitulates features of caloric restriction. Cell Rep. 2018;22:2395–407. doi: 10.1016/j.celrep.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–38. doi: 10.1038/nm.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madeo F, Eisenberg T, Pietrocola F, Kroemer G. Spermidine in health and disease. Science. 2018;359:6374. doi: 10.1126/science.aan2788. [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G. Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Invest. 2015;125:1–4. doi: 10.1172/JCI78652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 27.Chiang WC, Wei Y, Kuo YC, Wei S, Zhou A, Zou Z, et al. High-throughput screens to identify autophagy inducers that function by disrupting beclin 1/Bcl-2 binding. ACS Chem Biol. 2018;13:2247–60. doi: 10.1021/acschembio.8b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill Sandra M., Wrobel Lidia, Rubinsztein David C. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death & Differentiation. 2018;26(4):617–629. doi: 10.1038/s41418-018-0254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pietrocola F, Lachkar S, Enot DP, Niso-Santano M, Bravo-San Pedro JM, Sica V, et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015;22:509–16. doi: 10.1038/cdd.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17:660–88. doi: 10.1038/nrd.2018.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–81. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 32.Golstein P, Kroemer G. Redundant cell death mechanisms as relics and backups. Cell Death Differ. 2005;12(Suppl 2):1490–6. doi: 10.1038/sj.cdd.4401607. [DOI] [PubMed] [Google Scholar]
- 33.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.González-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquère S, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 35.Nassif M, Valenzuela V, Rojas-Rivera D, Vidal R, Matus S, Castillo K, et al. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy. 2014;10:1256–71. doi: 10.4161/auto.28784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kriel J, Loos B. The good, the bad and the autophagosome: exploring unanswered questions of autophagy dependent cell death. Cell Death Differ. 2019; (in press). [DOI] [PMC free article] [PubMed]
- 37.Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 38.Park SM, Ou J, Chamberlain L, Simone TM, Yang H, Virbasius CM, et al. U2AF35(S34F) promotes transformation by directing aberrant ATG7 pre-mRNA 3’ end formation. Mol Cell. 2016;62:479–90. doi: 10.1016/j.molcel.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galluzzi L, Bravo-San Pedro JM, Kroemer G. Defective autophagy iinitiates malignant transformation. Mol Cell. 2016;62:473–4. doi: 10.1016/j.molcel.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 40.Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–85. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914–27. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 43.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–36. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: a systematic review and meta-analysis. Med (Baltim) 2018;97:e12912. doi: 10.1097/MD.0000000000012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med. 2003;197:1323–34. doi: 10.1084/jem.20021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nyfeler B, Eng CH. Revisiting autophagy addiction of tumor cells. Autophagy. 2016;12:1206–7. doi: 10.1080/15548627.2016.1170265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaaf M, Houbaert D, Meçe O, Agostinis P. Autophagy in endothelial cell and tumour angiogenesis. Cell Death Differ. 2019; (in press). [DOI] [PMC free article] [PubMed]
- 48.Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11:2285–302. doi: 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563:569–73. doi: 10.1038/s41586-018-0697-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madeo F, Pietrocola F, Eisenberg T, Kroemer G. Caloric restriction mimetics: towards a molecular definition. Nat Rev Drug Discov. 2014;13:727–40. doi: 10.1038/nrd4391. [DOI] [PubMed] [Google Scholar]
- 51.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 52.Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol. 2018;20:1338–48. doi: 10.1038/s41556-018-0235-8. [DOI] [PubMed] [Google Scholar]
- 53.Pyo JO, Yoo SM, Ahn HH, Nah J, Hong SH, Kam TI, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fernández AacuteF, Sebti S, Wei Y, Zou Z, Shi M, McMillan KL, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–40. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kiechl S, Pechlaner R, Willeit P, Notdurfter M, Paulweber B, Willeit K, et al. Higher spermidine intake is linked to lower mortality: a prospective population-based study. Am J Clin Nutr. 2018;108:371–80. doi: 10.1093/ajcn/nqy102. [DOI] [PubMed] [Google Scholar]
- 57.Pietrocola Federico, Castoldi Francesca, Kepp Oliver, Carmona-Gutierrez Didac, Madeo Frank, Kroemer Guido. Spermidine reduces cancer-related mortality in humans. Autophagy. 2018;15(2):362–365. doi: 10.1080/15548627.2018.1539592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 59.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 60.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 61.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 62.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. 2018;10:459. doi: 10.1126/scitranslmed.aat7807. [DOI] [PubMed] [Google Scholar]
- 64.Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–41. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 65.Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity. 2013;39:211–27. doi: 10.1016/j.immuni.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 66.Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–60. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. 2015;11:1878–90. doi: 10.1080/15548627.2015.1082022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K, et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. 2016;12:864–75. doi: 10.1080/15548627.2016.1154244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C, et al. Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell. 2018;173:624. doi: 10.1016/j.cell.2018.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat Rev Clin Oncol. 2017;14:247–58. doi: 10.1038/nrclinonc.2016.183. [DOI] [PubMed] [Google Scholar]
- 71.Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in cardiovascular aging. Circ Res. 2018;123:803–24. doi: 10.1161/CIRCRESAHA.118.312208. [DOI] [PubMed] [Google Scholar]
- 72.Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2018;71:1999–2010. doi: 10.1016/j.jacc.2018.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thompson CA. First drug-eluting coronary stent approved. Am J Health Syst Pharm. 2003;60:1212. doi: 10.1093/ajhp/60.12.1210. [DOI] [PubMed] [Google Scholar]
- 74.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16:45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on DeltaF508 cystic fibrosis transmembrane conductance regulator. Autophagy. 2012;8:1657–72. doi: 10.4161/auto.21483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Villella VR, Esposito S, Bruscia EM, Vicinanza M, Cenci S, Guido S, et al. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ. 2013;20:1101–15. doi: 10.1038/cdd.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maiuri L, Raia V, Kroemer G. Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 2017;24:1825–44. doi: 10.1038/cdd.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang S, Stoll G, Bravo San Pedro JM, Sica V, Sauvat A, Obrist F, et al. Evaluation of autophagy inducers in epithelial cells carrying the DeltaF508 mutation of the cystic fibrosis transmembrane conductance regulator CFTR. Cell Death Dis. 2018;9:191. doi: 10.1038/s41419-017-0235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Villella VR, Venerando A, Cozza G, Esposito S, Ferrari E, Monzani R, et al. A pathogenic role for cystic fibrosis transmembrane conductance regulator in celiac disease. EMBO J. 2019;38:e100101. [DOI] [PMC free article] [PubMed]
- 80.Izzo V, Pietrocola F, Sica V, Durand S, Lachkar S, Enot D, et al. Metabolic interactions between cysteamine and epigallocatechin gallate. Cell Cycle. 2017;16:271–9. doi: 10.1080/15384101.2016.1249550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. 2014;10:2053–74. doi: 10.4161/15548627.2014.973737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, et al. A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016;23:1380–93. doi: 10.1038/cdd.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Romani L, Oikonomou V, Moretti S, Iannitti RG, D’Adamo MC, Villella VR, et al. Thymosin alpha1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat Med. 2017;23:590–600. doi: 10.1038/nm.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42. doi: 10.1016/j.cell.2018.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong X, Levine B. Autophagy and viruses: adversaries or allies? J Innate Immun. 2013;5:480–93. doi: 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494:201–6. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, et al. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 2017;8:e2544. doi: 10.1038/cddis.2016.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, Levine B. The ubiquitin ligase Smurf1 functions in selective autophagy of mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe. 2017;21:59–72. doi: 10.1016/j.chom.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi SW, Gu Y, Peters RS, Salgame P, Ellner JJ, Timmins GS, Deretic V. Ambroxol induces autophagy and potentiates rifampin antimycobacterial activity. Antimicrob Agents Chemother. 2018;62:e01019–18. doi: 10.1128/AAC.01019-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kolloli A, Subbian S. Host-directed therapeutic strategies for tuberculosis. Front Med (Lausanne) 2017;4:171. doi: 10.3389/fmed.2017.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Underwood BR, Green-Thompson ZW, Pugh PJ, Lazic SE, Mason SL, Griffin J, et al. An open-label study to assess the feasibility and tolerability of rilmenidine for the treatment of Huntington’s disease. J Neurol. 2017;264:2457–63. doi: 10.1007/s00415-017-8647-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018;14:356–76. doi: 10.1038/s41574-018-0009-1. [DOI] [PubMed] [Google Scholar]
- 93.Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53:710–25. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 94.Son SM, Park SJ, Lee H, Siddiqi F, Lee JE, Menzies FM, Rubinsztein DC. Leucine signals to mTORC1 via its metabolite acetyl-coenzyme A. Cell Metab. 2018;29:192–201. doi: 10.1016/j.cmet.2018.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lopez-Otin C, Galluzzi L, Freije JM, Madeo F, Kroemer G. Metabolic control of longevity. Cell. 2016;166:802–21. doi: 10.1016/j.cell.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 96.Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun. 2014;5:4934. doi: 10.1038/ncomms5934. [DOI] [PubMed] [Google Scholar]
- 97.Fernández AacuteF, Bárcena C, Martínez-García GG, Tamargo-Gómez I, Suárez MF, Pietrocola F, et al. Autophagy couteracts weight gain, lipotoxicity and pancreatic beta-cell death upon hypercaloric pro-diabetic regimens. Cell Death Dis. 2017;8:e2970. doi: 10.1038/cddis.2017.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang C, Niederstrasser H, Douglas PM, Lin R, Jaramillo J, Li Y, et al. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun. 2017;8:2270. doi: 10.1038/s41467-017-02332-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lim H, Lim YM, Kim KH, Jeon YE, Park K, Kim J, et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat Commun. 2018;9:1438. doi: 10.1038/s41467-018-03939-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zischka H, Lichtmannegger J, Schmitt S, Jägemann N, Schulz S, Wartini D, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011;121:1508–18. doi: 10.1172/JCI45401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, et al. Activation of autophagy, observed in liver tissues from patients with Wilson disease and from Atp7b-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology. 2018;S0016-5085:35280–6. doi: 10.1053/j.gastro.2018.11.032. [DOI] [PubMed] [Google Scholar]
- 102.Lichtmannegger J, Leitzinger C, Wimmer R, Schmitt S, Schulz S, Kabiri Y, et al. Methanobactin reverses acute liver failure in a rat model of Wilson disease. J Clin Invest. 2016;126:2721–35. doi: 10.1172/JCI85226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carmona-Gutierrez D, Zimmermann A, Kainz K, Pietrocola F, Chen G, Maglioni S, et al. The flavonoid 4,4’-dimethoxychalcone promotes autophagy-dependent,3 longevity across species. Nat Commun 2019; (in press). [DOI] [PMC free article] [PubMed]
- 104.Pietrocola Federico, Castoldi Francesca, Maiuri Maria Chiara, Kroemer Guido. Aspirin—another caloric-restriction mimetic. Autophagy. 2018;14(7):1162–1163. doi: 10.1080/15548627.2018.1454810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med. 2015;7:127–39. doi: 10.15252/emmm.201404137. [DOI] [PMC free article] [PubMed] [Google Scholar]