

Abstract
Herein we report a convenient and broadly applicable strategy for the difluoromethylation of aryl bromides via metallaphotoredox catalysis. Bromodifluoromethane, a simple and commercial alkyl halide, is harnessed as an effective source of difluoromethyl radical via silyl radical-mediated halogen abstraction. The merger of this fluoroalkyl electrophile activation pathway with a dual nickel/photoredox catalytic platform enables the difluoromethylation of a diverse array of aryl and heteroaryl bromides under mild conditions. The utility of this protocol is showcased in the late-stage functionalization of several drug analogues.
Keywords: difluoromethylation, photoredox catalysis, heterocycles, nickel, fluorine
Graphical Abstract
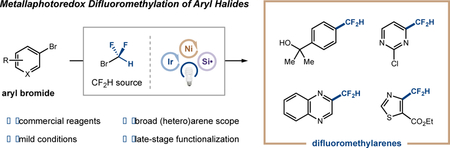
Bromodifluoromethane, a commercial alkyl halide, is harnessed as an effective source of difluoromethyl radical via silyl radical-mediated halogen abstraction. The merger of this fluoroalkyl electrophile activation pathway with a dual nickel/photoredox catalytic platform enables the difluoromethylation of a diverse array of aryl and heteroaryl bromides under mild conditions. The utility of this protocol is showcased in the late-stage functionalization of several drug analogues.
Within the realm of drug design, the chemoselective incorporation of fluorine or polyfluorinated alkyl substituents is a powerful and widely employed tactic to enhance binding selectivity, elevate lipophilicity, and/or circumvent metabolism issues arising from in vivo C–H bond oxidation.[1,2] While the implementation of the trifluoromethyl group (–CF3) has been widely studied in medicinal chemistry, the relatively underexplored difluoromethyl group (–CF2H) has recently garnered significant attention by virtue of its capacity to serve as a lipophilic hydrogen bond donor and to act as a bioisostere for thiol and alcohol functionalities.[3,4] Modern approaches to the direct and selective introduction of the difluoromethyl group into aromatic rings typically rely on the metal-catalyzed cross-coupling of aryl electrophiles or nucleophiles with an appropriate CF2HR reagent, often designed for facile transmetallation or formal oxidative addition by the metal catalyst.[5,6] Given the pronounced practical significance of this emerging area, one of the greatest challenges is the rendering of readily available CF2H sources as effective difluoromethylating agents in cross-coupling. Crucially, this coupling platform must display high functional tolerance and amenability towards medicinally relevant scaffolds. As such, the development of novel, operationally convenient, yet general routes to difluoromethylarenes and heteroarenes remains of high interest.
Metallaphotoredox catalysis has emerged in recent years as a valuable platform for the production of previously elusive C(sp3)–C(sp2) bonds, thereby enabling access to novel constructs of importance in medicinal chemistry.[7] One example from our laboratory involves a dual nickel/photoredox-catalyzed cross-electrophile coupling protocol, wherein the union of a broad range of aryl and alkyl halides is accomplished at room temperature using visible light irradiation.[8] A unique design feature of this mechanism is the implementation of silyl radical-mediated abstraction of bromine atoms from C(sp3)–Br bonds,[9] a pathway that allows alkyl halides to readily participate in metal-catalyzed cross-couplings. Most notably, the scope of these silane-mediated cross-electrophile couplings has been determined to be extensive with respect to both arene substitution pattern and functional group tolerance, a characteristic that has led to widescale adoption by medicinal chemistry groups within the pharmaceutical sector.[11]
Inspired by the success of this silyl radical-mediated cross-electrophile coupling, we wondered if an analogous strategy could serve as the basis for a general and direct synthesis of difluoromethylarenes. Specifically, we considered bromodifluoromethane, a simple and commercial alkyl halide, as a potential source of CF2H radical via a previously unexplored halogen abstraction step. Crucially, this pathway would be thermodynamically feasible given that the HF2C–Br bond (bond dissociation energy of 69 kcal/mol) is far weaker than the Si–Br bond in a typical abstraction product (e.g., 96 kcal/mol for Me3Si–Br).[12] Moreover, given the electron-rich character of the silyl radical, we surmised that halogen abstraction from bromodifluoromethane would be polarity matched and hence kinetically faster than from previously utilized alkyl bromide substrates.[13] Herein we disclose the successful implementation of these ideals and present a mild, convenient and broadly applicable metallaphotoredox-catalyzed difluoromethylation of a wide array of aryl and heteroaryl halides.
The proposed mechanism for this silane-mediated difluoromethylation is shown in Scheme 1. Visible-light excitation of IrIII photocatalyst [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (1)[14] is known to generate the excited-state *IrIII complex 2 which can readily oxidize bromide anion (3) (E1/2red [*IrIII/IrII] = +1.21 V vs. saturated calomel electrode (SCE) in MeCN; E1/2red [Br•/Br–] = +0.80 V vs. SCE in DME).[8,15] The resulting bromine radical (5) can participate in hydrogen atom transfer with (TMS)3SiH to yield the nucleophilic silyl radical 6.[13] Bromine abstraction from bromodifluoromethane (7) by open shell silyl species 6 would then afford the key difluoromethyl radical (8). Concurrently with the photoredox catalytic cycle, Ni0 catalyst 9[16] is expected to undergo facile oxidative addition into aryl bromide 10 to generate NiII–aryl intermediate 11. Trapping of difluoromethyl radical (8) would then generate the corresponding aryl–NiIII–CF2H complex 12, which upon reductive elimination should afford the desired difluoromethylarene product 13 and NiI species 14. Finally, single electron transfer between 14 and reduced photocatalyst 4 would simultaneously regenerate low-valent nickel catalyst 9 and ground-state photocatalyst 1.
Scheme 1.
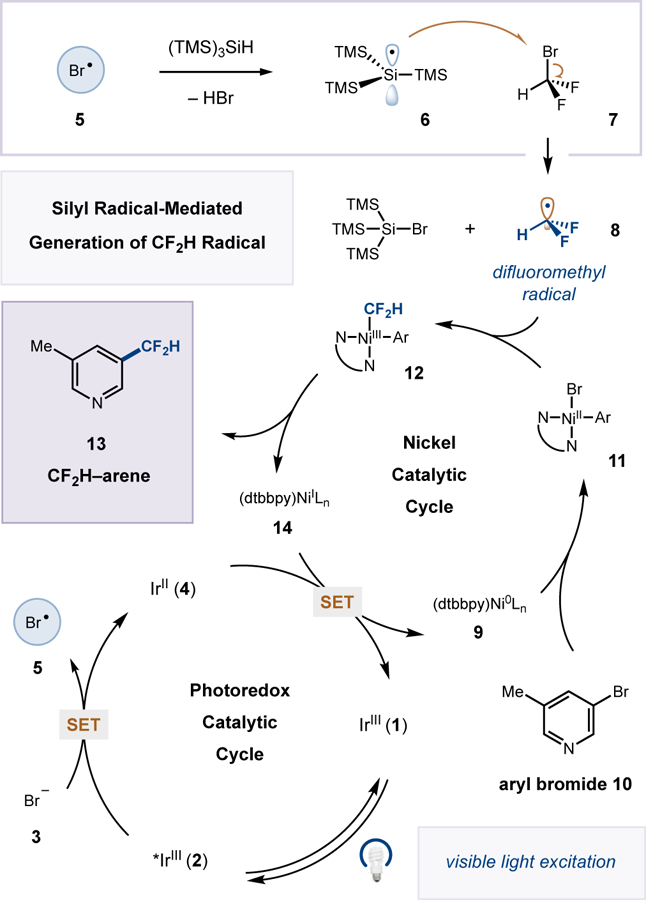
Proposed mechanism for metallaphotoredox difluoromethylation.
We began our investigation by examining three separate aryl halide precursors in the proposed difluoromethylation protocol: namely, cyanopyridine 16, as well as trifluoromethyl-and fluoro-substituted bromobenzenes (17 and 18, respectively; see Table 1). For each substrate, we employed photocatalyst 1, nickel catalyst NiBr2•dtbbpy (15), commercially available (TMS)3SiH, 2,6-lutidine as the base, DME as solvent, and blue LEDs as the photon source. In the case of the electron-deficient heteroaryl bromide 16, optimal levels of reaction efficiency were observed using excess CF2HBr (4 equiv., 77% yield). Remarkably, however, with the less electron-deficient CF3-arene 17, the use of 2 equivalents of bromodifluoromethane gave a superior outcome, while electron-neutral para-fluoro substrate 18 achieved the highest yield with a 1:1 stoichiometry of arene to CF2HBr.[17] Perhaps more surprising, the use of a large excess of bromodifluoromethane (4 equiv.) led to dramatically diminished yields in the latter two cases.[18,19]
Table 1.
Effect of stoichiometry of bromodifluoromethane.[a]
 |
To rationalize these trends, we propose that when less electron-deficient arenes are employed, the catalytic nickel species can undergo oxidative addition with the electron-deficient CF2HBr reagent at a rate competitive with the aryl bromide insertion step.[20] Moreover, we believe that such a pathway would be deleterious, given that the resultant NiII–CF2H complex would be unlikely to participate in further oxidative addition steps with the aryl bromide.[21] As such, for more electron-rich or hindered aryl halides that undergo relatively slow oxidative addition with nickel, the issue of competitive CF2HBr insertion is mitigated by employing lower concentrations of the CF2H source. However, at the other end of the electronic spectrum, higher stoichiometry of CF2HBr ensures that the silane-mediated generation of CF2H radical occurs in synchronicity with the rapid oxidative addition of the nickel catalyst into highly electron-deficient arenes (e.g., 16).[22]
With optimized conditions in hand, we next evaluated the scope of the aryl bromide component (Table 2). Notably, substrates bearing electron-withdrawing groups, such as esters, ketones, nitriles, and sulfones, generated the respective difluoromethyl adducts in high yields (19–22, 75–83% yield). In accord with our optimization studies, electron-neutral and electron-rich bromoarenes performed well using lower loadings of CF2HBr (23–26, 55–80% yield). As a useful demonstration of the mild conditions and functional group tolerance of this new coupling protocol, we found that aryl electrophiles bearing chloride and boronate ester groups can be readily implemented (27 and 28, 80% and 85% yield, respectively). This characteristic was further underscored by the performance of substrates containing alcohol and silylalkyne moieties (29 and 30, 71% and 75% yield, respectively). Meta-and ortho-substituted aryl bromides were also shown to be competent electrophiles in this transformation (31–34, 60–78% yield).
Table 2.
Scope of aryl halide electrophiles in silyl radical-mediated difluoromethylation using bromodifluoromethane as a CF2H source[a]
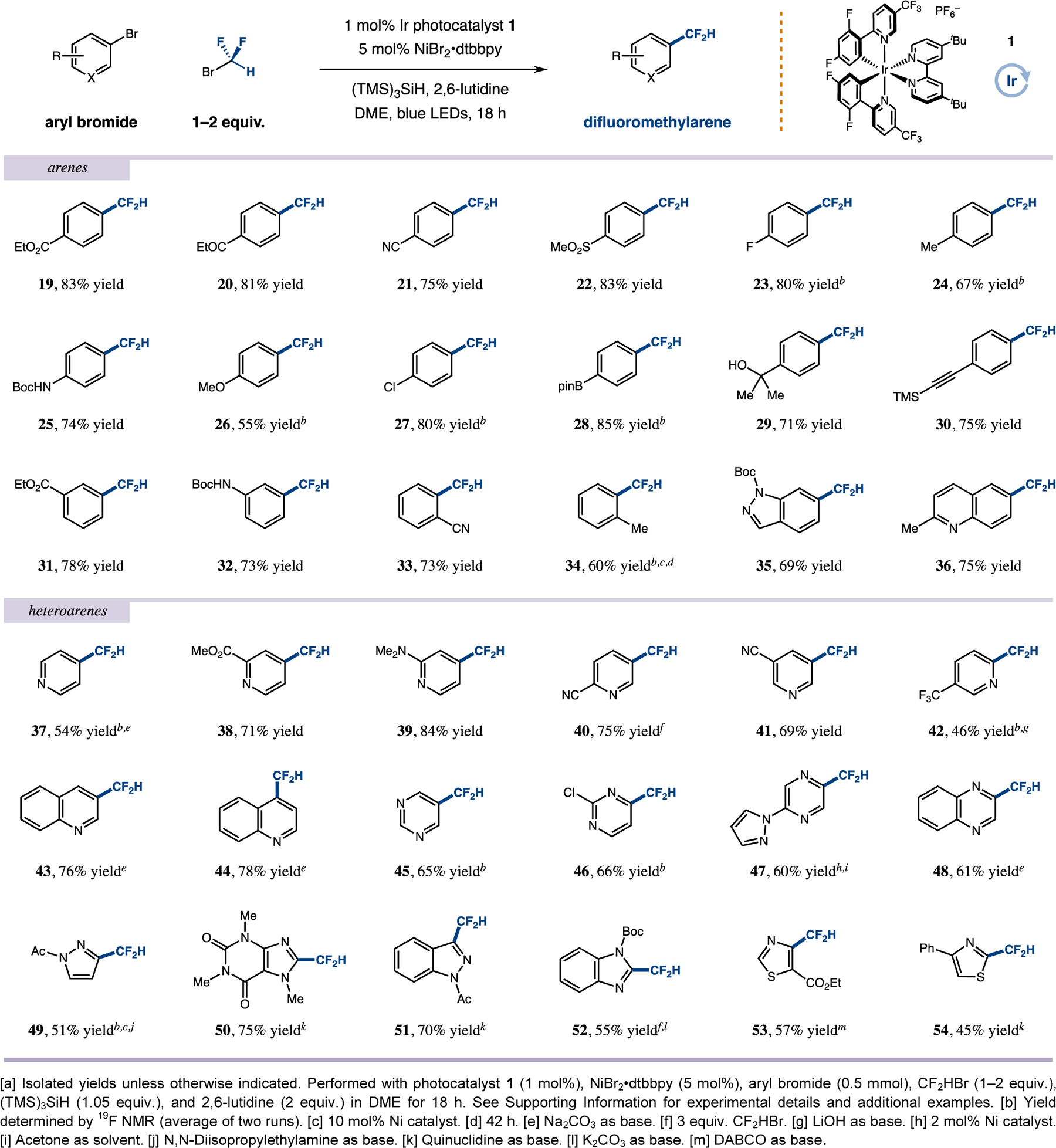 |
We next turned our attention to the scope of heteroaryl halides, a critical group of substrates with respect to the utility of this protocol in the medicinal chemistry sector. As shown in Table 2, a broad range of 2-, 3-, and 4-bromopyridines were found to be suitable coupling partners (37–42, 46–84% yield). Moreover, bromoquinolines afforded the desired products with good efficiency (43 and 44, 76% and 78% yield, respectively). Multi-nitrogen-bearing heteroaryls, such as pyrimidines, pyrazines and quinoxalines, have long been viewed as problematic substrates for a range of cross-coupling reactions. As such, it was notable that all of these heteroaryl ring classes were readily converted to their corresponding difluoromethylarenes (45–48, 60–66% yield). In the same context, five-membered bromoarenes were also found to be competent electrophiles. In particular, difluoromethyl derivatives of pyrazole, indazole, benzimidazole, and caffeine were obtained in good to high yields (49–52, 51–75% yield). Perhaps most notable, bromothiazoles, a traditionally difficult cross-coupling class,[23] were readily transformed to their corresponding CF2H adducts (53 and 54, 57% and 45% yield, respectively).
Finally, given the pharmaceutical relevance of the CF2H group, we sought to showcase the utility of our protocol in the late-stage difluoromethylation of analogues of several known medicinal agents (Scheme 2). Specifically, difluoromethyl-containing derivatives of sulfadimethoxine, celecoxib, indometacin, and pomalidomide were obtained in good to high yields from the aryl bromide precursors (55–58, 64–82% yield). These results further highlight the real-world utility of this metallaphotoredox technology with respect to tolerance of medicinally relevant functionality, such as sulfonamides, imides, electron-rich pyrimidines, pyrazoles, and indoles.
Scheme 2.
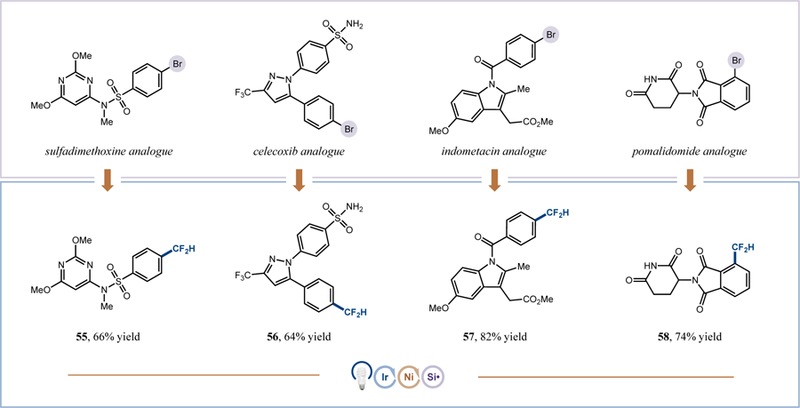
Application of difluoromethylation: late-stage functionalization for the expedient synthesis of difluoromethyl analogues of pharmaceutical agents.
In conclusion, we have developed a novel metallaphotoredox platform for the difluoromethylation of a broad range of aryl and heteroaryl halides. This protocol employs commercially available bromodifluoromethane as a direct source of CF2H radical via a silyl radical-mediated halogen abstraction pathway previously unexplored for fluoroalkyl electrophiles within the realm of cross-coupling. Given its distinct convenience and broad applicability to pharmaceutically relevant scaffolds, we expect this method to be widely adopted within the synthetic and medicinal chemistry community.
Supplementary Material
Figure 1.
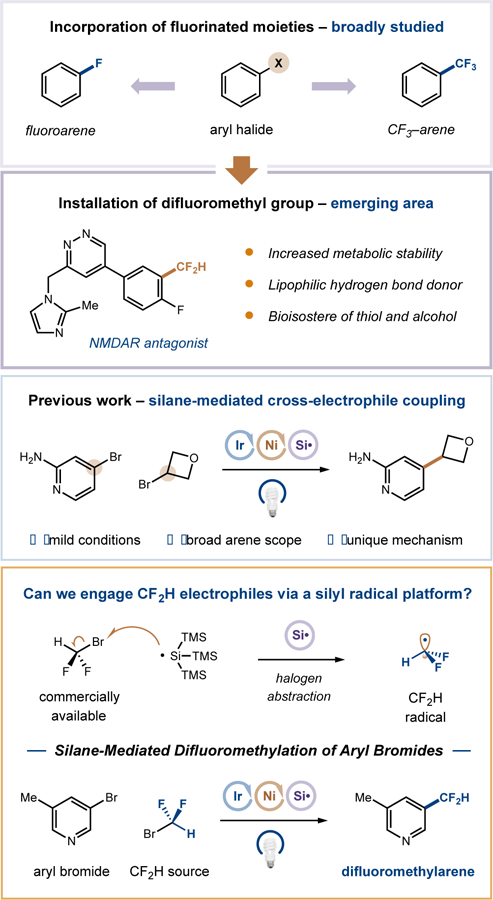
Silane-mediated difluoromethylation of aryl bromides.[10]
Acknowledgements
Financial support provided by the NIHGMS (R01 GM103558–05), and kind gifts from Merck, BMS, Pfizer, Janssen, and Eli Lilly. S.C. gratefully thanks the Fonds de Recherche du Québec – Nature et Technologies (FRQNT) for a postdoctoral fellowship (#198859). M.Y. thanks Daiichi Sankyo Co., Ltd. for funding. M.K. thanks the Japan Society for the Promotion of Science for Grant-in-Aid for JSPS Fellows. D.F.F. thanks the MINECO for a fellowship (BES-2014–068776 and EEBB-I-17–11925). R.R. thanks the Swiss National Science Foundation for a postdoctoral fellowship (P2FRP2–178091). The authors acknowledge C. Liu for assistance in preparing the manuscript.
References
- [1].a) Hagmann WK, J. Med. Chem 2008, 51, 4359. [DOI] [PubMed] [Google Scholar]; b) Purser S, Moore PR, Swallow S, Gouverneur V, Chem. Soc. Rev 2008, 37, 320. [DOI] [PubMed] [Google Scholar]; c) Zhuo Y, Wang J, Gu Z, Wang S, Zhu W, Aceña JL, Soloshonok VA, Izawa K, Liu H, Chem. Rev 2016, 116, 422. [DOI] [PubMed] [Google Scholar]
- [2].a) Liang T, Neumann CN, Ritter T, Angew. Chem. Int. Ed 2013, 52, 8214. [DOI] [PubMed] [Google Scholar]; b) Campbell MG, Ritter T, Chem. Rev 2015, 115, 612. [DOI] [PubMed] [Google Scholar]; c) Tomashenko OA, Grushin VV, Chem. Rev 2011, 111, 4475. [DOI] [PubMed] [Google Scholar]; d) Alonso C, de Marigorta EM, Rubiales G, Palacios F, Chem. Rev 2015, 115, 1847. [DOI] [PubMed] [Google Scholar]
- [3].a) Erickson JA, McLoughlin JI, J. Org. Chem 1995, 60, 1626 [Google Scholar]; b) Zafrani Y, Yeffet D, Sod-Moriah G, Berliner A, Amir D, Marciano D, Gershonov E, Saphier S, J. Med. Chem 2017, 60, 797. [DOI] [PubMed] [Google Scholar]; c) Sessler CD, Rahm M, Becker S, Goldberg JM, Wang F, Lippard SJ, J. Am. Chem. Soc 2017, 139, 9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Meanwell NA, J. Med. Chem 2011, 54, 2529. [DOI] [PubMed] [Google Scholar]; b) Meanwell NA, J. Med. Chem 2018. Article ASAP. DOI: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- [5].For recent reviews on difluoromethylation reactions, see: a) Chen B, Vicic DA, Top. Organomet. Chem 2015, 52, 113 [Google Scholar]; b) Yerien DE, Barata-Vallejo S, Postigo A, Chem. Eur. J 2017, 23, 14676. [DOI] [PubMed] [Google Scholar]; c) Rong J, Ni C, Hu J, Asian J Org. Chem 2017, 6, 139. [Google Scholar]
- [6].Selected examples: a) Fier PS, Hartwig JF, J. Am. Chem. Soc 2012, 134, 5524. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gu Y, Leng X, Shen Q, Nat. Commun 2014, 5, 5405. [DOI] [PubMed] [Google Scholar]; c) Xu L, Vicic DA, J. Am. Chem. Soc 2016, 138, 2536. [DOI] [PubMed] [Google Scholar]; d) Lu C, Gu Y, Wu J, Gu Y, Shen Q, Chem. Sci 2017, 8, 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Feng Z, Min Q-Q, Fu X-P, An L, Zhang X, Nat. Chem 2017, 9, 918. [DOI] [PubMed] [Google Scholar]; f) Lu C, Lu H, Wu J, Shen HC, Hu T, Gu Y, Shen Q, J. Org. Chem 2018, 83, 1077. [DOI] [PubMed] [Google Scholar]; g) Miao W, Zhao Y, Ni C, Gao B, Zhang W, Hu J, J. Am. Chem. Soc 2018, 140, 880. [DOI] [PubMed] [Google Scholar]; h) Merchant RR, Edwards JT, Qin T, Kruszyk MM, Bi C, Che G, Bao D-H, Qiao W, Sun L, Collins MR, Fadeyi OO, Gallego GM, Mousseau JJ, Nuhant P, Baran PS, Science 2018, 360, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Xu C, Guo W-H, He X, Guo Y-L, Zhang X-Y, Zhang X, Nat. Commun 2018, 9, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].For reviews on metallaphotoredox catalysis, see: a) Skubi KL, Blum TR, Yoon TP, Chem. Rev 2016, 116, 10035. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Levin MD, Kim S, Toste FD, ACS Cent. Sci 2016, 2, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Twilton J, Le CC, Zhang P, Shaw MH, Evans RW, MacMillan DWC, Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]
- [8].Zhang P, Le CC, MacMillan DWC, J. Am. Chem. Soc 2016, 138, 8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Ballestri M, Chatgilialoglu C, Clark KB, Griller D, Giese B, Kopping B, J. Org. Chem 1991, 56, 678 [Google Scholar]; b) Chatgilialoglu C, Acc. Chem. Res 1992, 25, 188 [Google Scholar]; c) Chatgilialoglu C, Lalevée J, Molecules 2012, 17, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF, Liotta DC, Expert Opin. Ther. Pat 2012, 22, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Blakemore DC, Castro L, Chucher I, Rees DC, Thomas AW, Wilson DM, Wood A, Nat. Chem 2018, 10, 383. [DOI] [PubMed] [Google Scholar]; b) Zhang R, Li G, Wismer M, Vachal P, Colletti SL, Shi Z-C, ACS Med. Chem. Lett Article ASAP. DOI: 10.1021/acsmedchemlett.8b00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo Y-R, Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- [13].For a study on the nucleophilicity of silyl radicals and the kinetic effect of polarity matching in halogen abstraction from benzyl bromides, see: Jiang X-K, Ding WF-X, Zhang Y-H, Tetrahedron 1997, 53, 8479 and references therein. [Google Scholar]
- [14]. dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-trifluoromethylpyridine; dtbbpy = 4,4′-di-t-Bu-2,2′-bipyridine.
- [15].Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S, Chem. Mater 2005, 17, 5712. [Google Scholar]
- [16].Ni0 catalyst 9 can likely be generated in situ from a NiII precatalyst via two single electron reductions performed by reduced photocatalyst 4. A sacrificial amount of bromide anion could turn over the photocatalytic cycle in this case. See: Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC, Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].The fluoro substituent has a Hammett σp value of –0.06. See: Hansch C, Leo A, Taft RW, Chem. Rev 1991, 91, 165. [Google Scholar]
- [18]. Control experiments with aryl halide 17 revealed that light, photocatalyst, nickel, and silane are all required for the success of the transformation (<1% yield in the absence of at least one component). Reduction of CF2HBr to CF2H2 was observed when the nickel catalyst was excluded, an observation that supports the capacity of the silyl radical to engage in halide abstraction with CF2HBr. See Supporting Information for details.
- [19]. The implementation of commercially available (bromodifluoromethyl)trimethylsilane as a CF2H source was also possible, but not pursued due to lower reaction efficiency (Table 1).
- [20].For reports of proposed NiI catalytic intermediates undergoing formal oxidative addition into bromodifluoromethane and chlorodifluoromethane, see: a) Fu X-P, Xiao Y-L, Zhang X, Chin. J. Chem 2018, 36, 143 [Google Scholar]; b) ref 6i. [Google Scholar]
- [21]. For experimental observations consistent with the deactivation of the nickel catalyst (i.e., poor conversion of starting material), see Supporting Information.
- [22]. In support of the dramatic differences among rates of oxidative addition, competition experiments between 16 and 18 revealed that at early reaction time points the difluoromethyl product from electron-deficient 18 is obtained in up to 45% yield, whereas electron-neutral bromoarene 16 remains entirely unreacted. See Supporting Information.
- [23].Su M, Buchwald SL, Angew. Chem. Int. Ed 2012, 51, 4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.