

Abstract
Iron and copper have similar physiochemical properties; thus, physiologically relevant interactions seem likely. Indeed, points of intersection between these two essential trace minerals have been recognized for many decades, but mechanistic details have been lacking. Investigations in recent years have revealed that copper may positively influence iron homeostasis, and also that iron may antagonize copper metabolism. For example, when body iron stores are low, copper is apparently redistributed to tissues important for regulating iron balance, including enterocytes of upper small bowel, the liver, and blood. Copper in enterocytes may positively influence iron transport, and hepatic copper may enhance biosynthesis of a circulating ferroxidase, ceruloplasmin, which potentiates iron release from stores. Moreover, many intestinal genes related to iron absorption are transactivated by a hypoxia-inducible transcription factor, hypoxia-inducible factor-2α (HlF2α), during iron deficiency. Interestingly, copper influences the DNA-binding activity of the HIF factors, thus further exemplifying how copper may modulate intestinal iron homeostasis. Copper may also alter the activity of the iron-regulatory hormone hepcidin. Furthermore, copper depletion has been noted in iron-loading disorders, such as hereditary hemochromatosis. Copper depletion may also be caused by high-dose iron supplementation, raising concerns particularly in pregnancy when iron supplementation is widely recommended. This review will cover the basic physiology of intestinal iron and copper absorption as well as the metabolism of these minerals in the liver. Also considered in detail will be current experimental work in this field, with a focus on molecular aspects of intestinal and hepatic iron-copper interplay and how this relates to various disease states.
Introduction
Among the essential trace minerals, iron and copper are unique as they exist in two oxidation states in biological systems and can potentiate the formation of damaging oxygen free radicals when in excess. Deficiencies of both nutrients are also associated with significant physiological perturbations. Given the potential adverse effects of too much or too little iron or copper, their homeostasis is tightly controlled at the cellular and organismal levels by local and systemic mediators. The reactive nature of these metal ions underlies important biological functions related to electron transfer (i.e., redox) reactions, in which both metals function as enzyme cofactors. Moreover, given their similar physiochemical properties, including comparable atomic radii and electrical charges, it is not surprising that biologically-relevant interactions between iron and copper have been frequently noted in mammals (54, 87, 105).
Iron extraction from the diet in the proximal small intestine is tightly controlled since no active, regulated mechanisms exist in humans to excrete excess iron (although rodents do have a limited capacity to excrete iron in bile). Iron homeostasis is regulated at the whole-body level by the hepatic, peptide hormone hepcidin (HEPC). HEPC is released when body iron stores increase and during infection and inflammation, and it functions to reduce serum iron concentrations. It accomplishes this by binding to the iron exporter, ferroportin 1 (FPN1), which is expressed on the surface of cells that absorb and store iron, causing its internalization and degradation (208). Additional transcriptional and posttranscriptional mechanisms also exist at the cellular level to locally regulate iron homeostasis. Collectively, these homeostatic loops modulate the expression of genes encoding iron metabolism-related proteins, including iron transporters and an iron reductase (i.e., a “ferrireductase”). One such mechanism involves the transactivation of genes in enterocytes by a hypoxia-inducible factor-2α (HIF2α) during iron deprivation (with concurrent hypoxia). Another regulatory mechanism acts posttranscriptionally to control mRNA levels within many cells via interaction of a stem-loop structure within the transcripts [i.e., iron-responsive elements (IREs)] with cytosolic, iron-sensing proteins [called iron-regulatory proteins (IRPs)]. These interactions can either inhibit translation of a message or increase its stability, leading to the production of more protein. Intracellular modulation of “free,” or unbound, iron levels also occurs via interaction with ferritin, which sequesters excess iron, thus rendering it unreactive.
Copper metabolism is also regulated according to physiologic demand, but the mechanisms involved have not been elucidated to date. Modulation of copper homeostasis by a copper-regulatory hormone was proposed in mice (145), but more recent, confirmatory studies have not been reported. The purported factor was released from the heart in response to low copper levels, and it supposedly increased intestinal copper absorption and hepatic copper release by upregulating expression of a copper exporter [copper-transporting ATPase 1 (ATP7A)]. Furthermore, cellular copper metabolism is modulated within cells by a host of cytosolic chaperones, which control copper trafficking. Copper may also be sequestered within cells by metallothionein (MT), which is a copper- and zinc-binding protein (but it has a higher affinity for copper) (144). Whole-body copper concentrations are controlled by excretion into the bile; biliary copper is complexed with bile salts and thus cannot be reabsorbed in the gut.
Adequate iron and copper intake is critical for humans and other mammals, especially during the rapid postnatal growth period. This fact is exemplified by the pathophysiological consequences of deficiency of iron or copper in humans. Iron deficiency (ID) is the most common nutrient deficiency worldwide, according to the World Health Organization (www.who.int). Infants and children that lack adequate dietary iron during critical developmental periods develop irreversible cognitive deficits (54). ID is also common in developed countries like the United States (171), occurring in individuals that are unable to assimilate necessary amounts of dietary iron to meet demands. This occurs frequently in children and adolescents (who are rapidly growing), women of child-bearing age (who lose menstrual blood), and during pregnancy and lactation (when iron demands are elevated). ID may also occur in individuals that have malab-sorptive disorders (e.g., inflammatory bowel diseases, IBDs) or as a consequence of gastric bypass surgery for morbid obesity, which effectively increase dietary iron requirements. Dietary studies have shown that average iron intake is below the RDA for many Americans, particularly amongst infants, young children, teenaged girls, pregnant women, and premenopausal women (https://ods.od.nih.gov/factsheets/Iron-HealthProfessional/#h4) (21, 22, 115). ID is most commonly treated with either oral or intravenous iron supplementation. Furthermore, iron excess is also a common condition in humans, most commonly associated with a group of genetic disorders, collectively referred to as hereditary hemochromatosis (HH). Individuals with HH hyperabsorb dietary iron, and over time, excess iron accumulates in various tissues and eventually causes damage due to oxidative stress. HH is caused by mutations in genes that encode proteins that regulate HAMP (the gene encoding HEPC) transcription in hepatocytes, which effectively causes HEPC insufficiency. HH can be treated by iron chelators or by phlebotomy, which may decrease body iron burden over time.
Copper deficiency, conversely, occurs less frequently. It is most often occurs in patients with Menkes disease (MD), a genetic disorder of impaired copper homeostasis. MD results from mutations in the gene encoding ATP7A, which leads to a defective protein, resulting in impaired intestinal copper absorption and consequent severe systemic copper deficiency. The pathophysiologic outcomes of such are devastating, particularly with regard to brain development. If detected early enough, affected individuals can be treated with supplemental copper, which may lessen the severity of the disease. Excess copper has also been reported in humans, most often being associated with another, rare genetic disorder, Wilson’s disease (WD). This disorder is caused by impaired biliary copper excretion, due to mutations in the gene encoding copper-transporting ATPase 2 (ATP7B). As a result, copper accumulates in the liver and other tissues that require ATP7B for copper export, eventually resulting in pathologies related to copper accumulation (i.e., oxidative stress and consequent tissue damage). WD can be treated with copper chelators or by high zinc intake, which blocks intestinal copper absorption.
This review will focus on synergistic and antagonistic interactions between iron and copper at the level of the intestinal mucosa. This is an important, active area of research, as accumulating evidence supports the postulate that copper promotes iron absorption, especially during ID. Moreover, recent evidence suggests that high dietary and body iron levels can perturb copper homeostasis. A detailed description of mechanisms of intestinal iron absorption will be provided, and how this process is influenced by copper will be considered. Mechanisms of intestinal copper absorption will also be considered in detail, although less is known about this process (at least in comparison to what is known about intestinal iron absorption). How iron may influence copper absorption will also be covered. Also pertinent to this topic is the metabolism of iron and copper in the liver, given that the liver plays an important role in regulating intestinal iron transport (by producing and releasing HEPC), and this process may be influenced by copper. Therefore, this review will not only outline how these metals interact in the gut, but will also consider hepatic metabolism as well. The overall goal of this review is thus to provide updated information on mechanisms of iron and copper absorption and then to discuss in detail how these essential trace minerals intersect at the subcellular, cellular, and tissue levels in humans and other mammals.
Metabolic Intersection of Iron and Copper: History and Background
Iron-copper interactions in humans were perhaps first described in the middle of the nineteenth century in industrialized Europe. As described in the early literature, a disease referred to as the “greening sickness” or “chlorosis” was common among factory workers at the time (87). Young women, in particular, were most likely to suffer from this disorder. Common symptoms included lethargy and decreased work capacity, paleness, and amenorrhea (87). Based upon descriptions from publications at the time, it is a logical prediction that chlorosis was in actuality iron-deficiency anemia, which commonly afflicts young women of childbearing age even today. Although this pathological condition was common in the general population, young women working in copper factories did not develop chlorosis, suggesting that copper exposure was somehow protective. There were reports of young women breastfeeding their infants with copper salts splashed across their bodies. These decades’ old observations provide the earliest examples of possible interactions between iron and copper. Based upon the current state of knowledge in this area of scientific research, it is a logical postulate that copper exposure enhanced absorption of dietary iron or potentiated iron utilization by developing erythrocytes in the bone marrow in these female factory workers (thus preventing the development of anemia). These possibilities seem most likely given that intestinal iron absorption ultimately determines overall body iron levels (since no excretory mechanism exists in humans), and that most iron is utilized for hemoglobin production in red blood cells. Another possibility is that copper depletion caused chlorosis. This seems plausible since copper deficiency causes an anemia that is indistinguishable from the anemia associated with ID (31). Although these observational reports do not clarify the specific underlying cause of chlorosis, they nonetheless nicely exemplify the longstanding historical appreciation of the intersection of iron and copper metabolism as it relates to human physiology and pathophysiology.
Figure 1 highlights points of intersection between iron and copper metabolism from absorption in the gut, to utilization by body cells and tissues, to regulated (for copper) and unregulated losses (for both minerals). Both minerals are absorbed from the diet in the duodenum, with efficiency of absorption being matched to physiologic demand. Hormonal control of iron absorption has been described, but no copper-regulatory hormones have been identified to date. The liver-derived, peptide hormone HEPC modulates iron efflux from duodenal enterocytes by blocking iron export via FPN1. The mechanism involves HEPC binding to FPN1, which leads to endocytosis and eventual degradation of the FPN1 protein in the lysosome (208). HEPC also blocks iron release from stores in reticuloendothelial (RE) macrophages of the bone marrow, spleen, and liver (Kupffer cells), hepatocytes, and placental trophoblasts, by a similar mechanism. The overall effect of HEPC then is to lower serum iron concentrations, which occurs when body iron levels are high, and during infection and inflammation (as part of the acute-phase response). Subsequent to passage through enterocytes into the interstitial fluids, iron is bound by transferrin (TF) and copper by mainly albumin, which facilitates delivery of both minerals to the liver via the portal blood circulation. Diet-derived, hepatic iron may be utilized for metabolic purposes, stored in hepatocytes (in ferritin), or released into the blood where it again is bound by TF (Fig. 1). Similarly, hepatic copper may be utilized by liver cells, stored in hepatocytes (in MT), or biosynthetically incorporated into ceruloplasmin (CP). Most copper exits the liver as CP-copper, but other copper exporters release free copper into the blood (which binds to serum proteins, such as albumin). Iron and copper are then widely distributed throughout the body, as all body cells require these minerals for metabolic purposes. The bone marrow is an “iron sink” as most body iron is utilized for Hb production in developing erythrocytes. Copper, however, does not concentrate into one particular tissue like iron.
Figure 1.
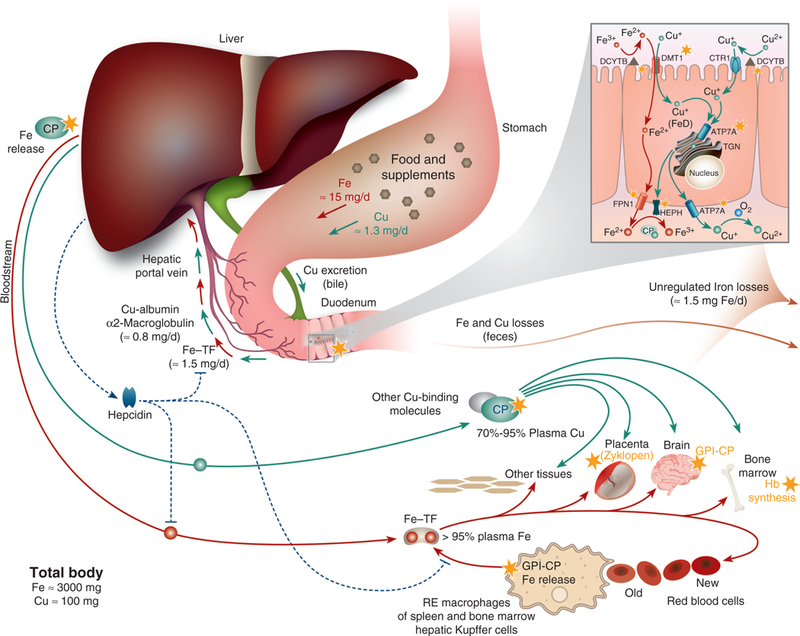
Iron and copper metabolism in mammals, highlighting points of intersection between these two essential trace minerals. Iron and copper homeostasis during physiological conditions is displayed with points of iron-copper intersection demarcated by yellow stars. Copper movement is indicated with green lines and iron flux in a rust color. Both minerals are absorbed in the duodenum. The inset shows points of iron-copper intersection in a duodenal enterocyte; more details are provided in Figure 2. Copper is mainly incorporated into ceruloplasmin (CP) in hepatocytes, which is secreted into the blood where it functions predominantly in iron metabolism, facilitating iron release from some tissues. A membrane-anchored form of CP, GPI-CP, has a similar function in some tissues. Excess body copper is excreted in bile. Ferric iron binds transferrin (TF) in the portal blood, and after reduction and import into the liver, it is utilized for metabolic purposes or stored in hepatocytes within ferritin. Ferrous iron is then exported into the serum by FPN1, where it is oxidized by CP and then binds to TF for distribution in the blood. Most diferric-TF is taken up by immature red blood cells in the bone marrow and utilized predominantly for hemoglobin synthesis. Iron utilization by developing erythrocytes is copper dependent, although the mechanism by which this occurs is unclear. Iron is also taken up into other tissues, including the brain, where iron release requires GPI-CP. The FOX zyklopen, a copper-dependent protein, may be required for proper iron flux in the placenta. Iron within hemoglobin of senescent red blood cells is recovered and stored by RE macrophages in spleen, bone marrow, and liver (i.e., Kupffer cells). Iron release from these macrophages requires CP or possibly GPI-CP. Iron homeostasis is regulated by hepcidin, which modulates iron flux by inhibiting intestinal iron absorption and iron release from stores in RE macrophages and hepatocytes. Hepcidin may be stabilized by copper, exemplifying another point of iron-copper intersection. Iron is lost from the body predominantly by desquamation of skin cells and exfoliation of enterocytes, and by blood loss, since no active, regulatory excretory system for iron has evolved in humans.
Known points of intersection between iron and copper have been identified in enterocytes of the proximal small bowel (Fig. 2), which mediate assimilation of both minerals from the diet. Several proteins expressed in these cells may impact iron and copper metabolism, as detailed in subsequent sections of this review. These include the major iron importer divalent metal-ion transporter 1 (DMT1), a brush-border membrane (BBM) ferric iron reductase duodenal cytochrome B (DCYTB), the iron exporter FPN1 and a ferrous iron oxidase hephaestin (HEPH). DMT1 may transport iron and copper; DCTYB may reduce both metals; FPN1 expression/activity may be influenced by copper; and HEPH is a copper-containing protein that functions in iron metabolism. Also highlighted in Figure 2 is ATP7A, which is strongly induced in the duodenum of iron-depleted rodents. Based upon this and its coregulation with iron transporters during ID (317), it was hypothesized that ATP7A (and/or copper) positively influences iron transport in enterocytes.
Figure 2.
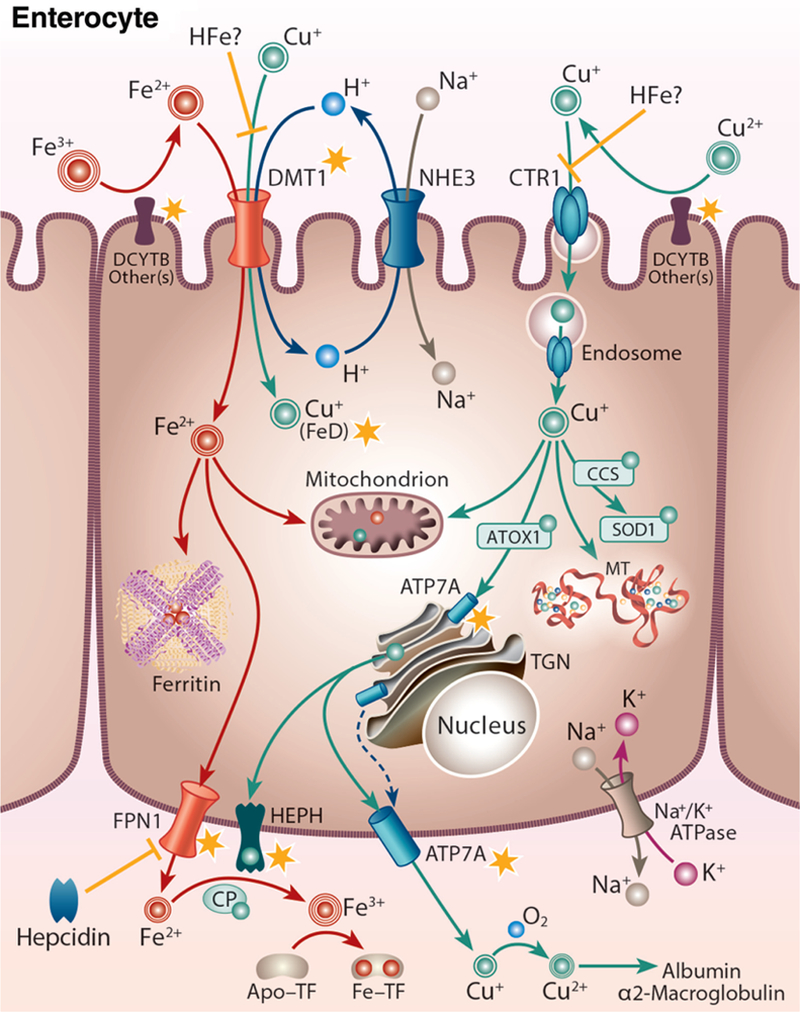
Iron-copper metabolism in a duodenal enterocyte, highlighting points of intersection between these two essential trace minerals. A duodenal enterocyte is depicted along with the proteins which mediate iron and copper absorption. Points where iron and copper metabolism intersect are demarcated by yellow stars. Both metals require reduction prior to absorption, which may be mediated by DCYTB and/or other reductases. Subsequently, iron is transported along with protons across the BBM by DMT1. The electrochemical proton gradient across the BBM that provides the driving force for ferrous iron transport is maintained via the action of a sodium-hydrogen antiporter (NHE3) and the Na+/K+ ATPase on the BLM. DMT1 may also transport copper during iron deficiency (FeD). High-iron (HFe) intake may block copper transport by DMT1 and/or CTR1, eventually leading to copper depletion. Cytosolic iron may be transported into mitochondria for metabolic use, stored in ferritin, or exported across the BLM by FPN1. FPN1 activity may be impacted by copper. Ferrous iron must then be oxidized by HEPH, CP, or other FOXs (not shown) to enable binding to TF in the interstitial fluids. After reduction, dietary copper is transported into enterocytes by CTR1 and is then distributed to various cellular locations by intracellular copper-binding proteins (i.e. chaperones). Excess copper may be stored in the cell by MT. Copper is pumped into the TGN by ATP7A, supporting cuproenzyme synthesis, or exported from the cell by ATP7A, which moves to the BLM when copper is in excess. ATP7A expression is strongly upregulated by iron depletion, suggesting that it (or copper) may positively influence iron metabolism in enterocytes. Copper is spontaneously oxidized by dissolved oxygen in the blood and then bound to mainly albumin and α2-macrogloubuoin in the portal blood and delivered to the liver.
In addition to the intestine, recent evidence supports the metabolic intersection of iron and copper in the mammalian liver. Hepatocytes produce and secrete a soluble, circulating ferrous iron oxidase, CP, which has significant homology to HEPH. CP is a copper-containing protein, like HEPH, and it is necessary for iron oxidation after release from certain tissues (e.g., the liver, brain, etc.) (225). As mentioned earlier, most copper in the blood is associated with CP (65%−90% depending upon the species), but CP is not required for copper delivery to tissues (124). Other mechanisms of copper export from the liver and distribution in the blood must thus exist, although these have not been described in detail to date. CP activity is critical in humans, since mutations that decrease or abolish CP production (as seen in the rare, genetic disease aceruloplasminemia), lead to iron accumulation in some tissues (e.g., brain, liver, pancreas, and retina). Interestingly, copper depletion, which impairs the biosynthesis of CP in hepatocytes, leads to a similar iron-overload phenotype as in aceruloplasminemia, further exemplifying one important aspect of the reciprocal relationship between these minerals in the liver, whereby deficiency of one metal leads to the loading of the other. It is unclear how ID causes hepatic copper accumulation; however, the mechanism by which copper depletion causes hepatic iron loading is clear, presumably being due to decreased CP activity, which impairs iron release (98). Furthermore, iron and copper are required for hemoglobin synthesis in developing erythrocytes within the bone marrow, as exemplified by the almost indistinguishable anemia associated with deficiency of either mineral. Iron is obviously necessary for producing hemoglobin, which contains an iron atom at its active (i.e., oxygen-binding) site. Copper is also required for hemoglobin synthesis; the mechanism behind this observation is currently unknown, but it likely relates to iron import into or utilization within mitochondria. A third copper-dependent iron oxidase has also recently been identified in the mammalian placenta, named Zyklopen (41). It presumably potentiates iron release from maternal sources to support the developing fetus, but this remains to be experimentally verified.
The purpose of this review is to summarize and critically analyze current research that relates to the intersection of iron and copper metabolism in humans and other mammals. In many cases, much experimental work has been done in laboratory rodents, but wherever applicable, human correlates will be highlighted. Although interplay between these two essential trace minerals has been appreciated at a superficial level for many decades (87), it has only been within the past 20 years or so that molecular details have emerged. One proven area of interaction is the copper-dependent ferroxi-dases (FOXs), HEPH and CP. It is, however, likely that other proteins also either mediate homeostasis of iron and copper, or are somehow influenced by both minerals. Although important iron-copper interactions occur in many tissues, including erythroid cells, RE macrophages, and brain (54, 105), the focus of this review will be on duodenal enterocytes and also hepatocytes, since the liver plays important regulatory roles in iron and copper metabolism.
Overview of Intestinal Iron Absorption
Humans and other mammals require iron for a variety of biological functions. ID is thus associated with significant pathophysiologic perturbations. Iron-containing proteins mediate energy (i.e., ATP) production, gas transport in the blood, and regulation of cell growth and differentiation and gene expression. Moreover, iron, when in excess, is toxic. Body iron levels are therefore controlled to ensure that adequate iron is available, while preventing excess accumulation in tissues and cells. Regulatory mechanisms that govern iron absorption, and storage and recycling, have developed over evolutionary time in humans. Iron is required for the activity of numerous proteins, where it facilitates important functions (e.g., electron transfer in redox reactions). Additionally, some proteins bind iron but have no known enzymatic function. These proteins contain iron in heme, in iron-sulfur clusters, or in other chemical configurations (17). Examples of these proteins include hemoglobin and myoglobin, which function in oxygen transport. Some iron-sulfur cluster-containing proteins (e.g., cytochromes of the electron transport chain) mediate energy production, by transferring electrons. Heme-containing proteins, such as cytochrome P450 complexes, also mediate electron transfer reactions. Iron transporters, such as DMT1 and FPN1, transiently bind iron and facilitate its movement across cellular membranes. Given the importance of these example iron-dependent enzymes (and others not mentioned) in normal physiology, it is a logical postulate that ID will have dire consequences.
Overall body iron homeostasis is regulated by the liver-derived, peptide hormone HEPC (206). HEPC blocks absorption of dietary iron, and inhibits iron release from RE macrophages of the spleen, liver (Kupffer cells), and bone marrow, and hepatocytes. Hepcidin transcription increases during infection and inflammation and when body iron levels are high. Conversely, when erythropoiesis is stimulated, for example, during ID and hypoxia, hepcidin production decreases. Under these circumstances, assimilation of dietary iron is enhanced by additional regulatory mechanisms at the level of intestinal enterocytes. In a normal healthy individual, absorption of dietary iron reflects body iron requirements; however, dysregulation of iron absorption occurs in a variety of disease states, with pathological outcomes. Humans have no regulated means to excrete excess iron (308), so intestinal iron absorption ultimately controls whole-body iron levels. Iron absorption typically matches non-specific, unregulated losses in urine and bile, from desquamation of skin cells and exfoliation of intestinal epithelial cells (IECs), and as a result of menstrual blood loss (81). A typical human will absorb 25 to 50 g of dietary iron over their lifespan. Adult males absorb ~ 1 mg of iron/day, while adult females absorb more (~ 1.5–2 mg/day), to compensate for iron lost during menstruation and pregnancy. The same basic regulatory schemes that have been described in humans are likely also operative in other mammals; but unlike humans, commonly used laboratory rodents can excrete limited quantities of iron in bile.
Basic physiological aspects of intestinal iron absorption were elucidated in the mid-1900s, but with the development of molecular and genetic techniques in recent years, contemporary advances have led to a basic mechanistic understanding of how dietary iron is transferred across the duodenal mucosa. Anatomically, iron is absorbed mainly in the proximal small intestine. Absorption occurs in differentiated enterocytes on the upper half of duodenal villi. Dietary iron exists mainly in the highly insoluble ferric form (Fe3+), which must be reduced to the more soluble ferrous (Fe2+) form prior to uptake into IECs. DCYTB is one candidate BBM ferrire-ductase (194). Dietary (e.g., ascorbic acid) and endogenous (e.g., gastric acid) factors also promote ferrous iron formation. After reduction ferric iron, Fe2+ is imported into cells via DMT1 (84,107). How enterocytes handle newly absorbed iron depends upon whole-body iron status. If iron stores are adequate, iron may be stored within intracellular ferritin and then lost when enterocytes are exfoliated into the intestinal lumen. Conversely, if body iron stores are inadequate, newly absorbed iron will be exported from cells across the basolateral membrane (BLM) by FPN1 (1, 69, 195). The export (or transfer) step requires that ferrous iron be oxidized to enable binding to TF in the interstitial fluids. This is likely mediated by the FOX HEPH (297), which is expressed on the BLM of enterocytes. Iron export is regulated by HEPC (210), which binds to and targets FPN1 for internalization and subsequent degradation. HAMP (encoding hepcidin) transcription in hepatocytes is regulated by physiological signals that are relayed to the HAMP gene by the hemochromatosis (HFE) protein (26), transferrin receptor 2 (TFR2) (207), and hemojuvelin (223). Mutations in these genes impair HEPC production, leading to increased intestinal iron absorption and consequent systemic iron loading, with eventual tissue and organ damage. Each of these processes involved in intestinal iron absorption and regulatory mechanisms that control them will be considered in greater detail in subsequent sections of this review.
Unlike in mature, differentiated enterocytes of the upper villus, iron homeostasis in undifferentiated enterocytes of the lower villus and intestinal crypts is distinct. Immature, undifferentiated IECs are not specialized to absorb nutrients (including iron), and as such, they are more similar to a generic cell. These cells are rapidly proliferating, so they require a relatively large amount of iron, which is absorbed as diferric-TF from the serosal (or blood) side. This is different from mature, fully differentiated enterocytes which obtain iron predominantly from digested food in the gut lumen. Cryptal enterocytes may absorb iron from the circulation by TF-mediated and TF-independent mechanisms (6). As these cells migrate upward along the villus and mature, they begin actively absorbing nutrients from the luminal side and subsequently lose the ability to absorb diferric-TF from the blood.
Perturbations of intestinal iron absorption can result in dire pathophysiological outcomes. In humans, this occurs most frequently in those afflicted with HH, which is a group of genetic diseases in which HEPC production is dysregulated, causing unregulated (and inappropriately high) intestinal iron absorption, eventually causing tissue iron accumulation and consequent oxidative damage. HFE-related HH is the most common form, but other less common forms have been documented as well. Pathological impairments of iron absorption, though occurring less frequently, are also clinically relevant. This commonly occurs in patients with malabsorptive disorders, such as IBD or Celiac disease. Reduced intestinal iron absorption is also associated with gastric bypass surgery, use of proton-pump inhibitors for chronic gastric reflux, and in older individuals with gastritis and associated achlorhydria. Iron absorption is also impaired in those suffering from iron-refractory, iron-deficiency anemia (IRIDA), in which HEPC production is inappropriately high. These clinical disorders will be discussed in more detail in subsequent sections.
Iron Absorption Occurs Principally in the Proximal Small Bowel
Iron is absorbed predominantly in the duodenum and proximal jejunum (47, 57, 75, 140, 304, 306), but smaller, perhaps insignificant amounts, can also be absorbed from the stomach (62, 253), ileum (75, 137, 203), and colon (43, 203). The more efficient absorption in the proximal small bowel may relate to an intrinsic property of the gut mucosa in this region (71,101,186,243,255,261). Supporting this supposition is the fact that many iron transporters (e.g., DCYTB, DMT1, and FPN1) are most strongly expressed in this gut region, likely providing a mechanistic explanation for these observations (33,69,107,195,282). Iron absorption occurs mainly through mature, differentiated enterocytes of the mid-and upper villus (56), which express the proteins, which mediate iron flux (e.g., DCYTB, DMT1, FPN1, HEPH, etc.). Moreover, morphological adaptation of the intestinal mucosa occurs during ID, which increases the effective absorptive area (53), thus enhancing iron absorption. In hemolytic anemia, for example, enterocytes from lower portions of the villus can absorb iron (214), and during pregnancy, villus size increases (277). Also, during iron depletion, absorption occurs more distally in the small intestine (304), and villus width and length increase and more “mitotic figures” (indicating enhanced cell proliferation) are noted in intestinal crypts (53).
Influence of Dietary and Endogenous Factors on Iron Absorption
Iron in food is mainly heme-associated (heme iron) or inorganic (nonheme). Dietary heme iron comes from hemoglobin and myoglobin found predominantly in meat products. Heme iron is efficiently absorbed and mostly not significantly influenced by other dietary constituents. Nonheme iron, which constitutes the majority of dietary iron for most Americans, is found in plant foods and meat. It is highly insoluble since it exists mainly as Fe3+. Moreover, bioavailability of non-heme iron is affected by various dietary components. This is likely due to the fact that unlike with heme iron in which the iron atom is sequestered within the protoporphyrin ring, nonheme iron is mainly unbound, or loosely bound, to dietary components, and thus free to interact with other molecules. Dietary (e.g., ascorbic acid) and endogenous (e.g., gastric acid and citrate) factors help maintain inorganic iron in the more soluble ferrous (Fe2+) form, which is the substrate for the main intestinal iron transporter DMT1. Impaired gastric acid production may thus reduce the bioavailability of dietary nonheme iron (118, 260). The low pH environment resulting from gastric HCl production promotes iron absorption, as does an acidic microclimate that exists at the BB surface of enterocytes in the “unstirred” water layer (just beneath the mucus layer). This acidic microclimate is produced by the action of the BBM sodium-hydrogen exchanger (NHE3), which exchanges extracellular Na+ for intracellular H+. The resultant electrochemical H+ gradient from outside to inside cells provides the driving force for ferrous iron transport by DMT1 (182, 267), which is a ferrous iron/proton cotransporter. Some prebiotics (i.e., indigestible dietary fibers and starches that are fermented by gut bacteria) may also enhance iron absorption (16, 91, 165, 188).
In addition to promoting iron absorption in some cases (e.g., ascorbate), other dietary factors, mainly derived from plant foods, may impair iron absorption. Phytate and oxalate, polyphenols and tannins, which are abundant in some plant-based food, tightly bind nonheme iron in the gut lumen, thus decreasing bioavailability (130, 159, 275). Moreover, drugs (e.g., proton-pump inhibitors) or pathologic conditions (e.g., atrophic gastritis) that decrease gastric acid production likely decrease iron bioavailability and thus impair absorption (204, 265). Helicobacter pylori infection (74, 131), mucosal pathologies such as IBD and Celiac disease (73), and perturbations in intestinal motility also decrease iron absorption.
Intestinal Iron Absorption: General Properties
Differentiated epithelial cells (enterocytes) of the mid and upper villus in the duodenum and upper jejunum are the primary mediators of intestinal iron absorption (47, 56, 57, 75, 304, 306). Proteins involved in iron absorption and their functions are outlined in Table 1. The assimilation of dietary iron occurs in three distinct steps (Fig. 2): (i) iron import across the BBM; (ii) an intracellular phase where iron can be stored in ferritin, utilized for metabolic purposes or directed to the BLM for subsequent export; and (iii) iron export across the BLM. Several different forms of dietary iron can be utilized by humans (e.g., heme iron, nonheme iron, iron in ferritin from some plant foods, such as legumes, and lactoferrin from milk) and the enterocyte BBM has evolved to efficiently handle all of these different dietary forms. The absorption of inorganic, or nonheme, iron has been studied in the most detail over the past 20 years. These different sources of dietary iron are likely imported into enterocytes across the BBM by distinct mechanisms, but once absorbed they all contribute to a common cytosolic “labile” iron pool. All intracellular iron is then probably exported via a common FPN1-mediated pathway.
Table 1.
Iron Homeostasis-Related Proteins in Mammals
| Protein | Function |
|---|---|
| ABCG2* | Possible BBM heme exporter; protection against possible heme toxicity |
| DCYTB | Ferric iron reduction for absorption across BBM by DMT1 |
| DMT1 | Ferrous iron/ H+ cotransporter on enterocyte BBM; possible Cu transport during iron depletion |
| FLVCR | Heme export across BLM into circulation |
| FPN1 | Ferrous iron exporter on basolateral surface of enterocytes; hepcidin receptor |
| FTN | Iron storage within enterocytes; dietary form of iron found in legumes |
| HCP1 | Possible BBM heme importer; also a physiological proton-coupled folate transporter |
| HEPH | Ferroxidase; BLM of enterocytes; functionally coupled to iron export by FPN1 |
| HO-1/2 | Oxidation of heme molecule to release ferrous iron in endosomes |
| HEPC | Iron-regulatory, liver-derived, peptide hormone; binds to FPN1 and causes its internalization and degradation |
| LTF | Iron-binding protein in breast milk; LTF receptor expressed on apical surface of enterocytes |
ABCG2, breast cancer-resistance protein; DCYTB, duodenal cytochrome B; DMT1, divalent metal-ion transporter 1; FLVCR, feline leukemia virus, subgroup C, receptor; FPN1, ferroportin 1; FTN, ferritin; HCP1, heme carrier protein 1; HEPH, hephaestin; HO, heme oxygenase; HEPC, hepcidin; LTF, lactoferrin.
Iron absorption occurs very rapidly. For example, after the administration of a radioactive dose of iron into the lumen of the duodenum, radioactivity appears in the circulation within 15 s (306). Within several minutes, 60% to 80% of the total dose ultimately absorbed has entered the circulation (305, 306). A slower rate of transfer occurs for 12 to 48 h thereafter (278). This slower transfer phase could represent iron retained within ferritin that is slowly released (38, 86, 312). Not all ferritin iron is absorbed, however, as some is lost when mucosal cells are exfoliated into the gut lumen (38). Iron depletion increases the total amount of iron absorbed and also decreases the amount stored within ferritin in enterocytes (24, 234).
Iron absorption occurs in a biphasic manner, depending upon the luminal iron concentration (18, 38, 97, 290, 306). At the low end of the physiologic range of iron intakes, iron absorption increases linearly as iron concentration increases, but only up to a point. With higher luminal iron concentrations at the upper end of physiologic intakes, this direct, linear relationship is lost, demonstrating that the process is saturable. This phenomenon likely reflects the fact that iron absorption is carrier mediated. Absorption, however, never fully saturates, as with very high iron doses (e.g., with iron supplementation), a linear relationship between the luminal iron concentration and the amount absorbed is again observed. This then likely reflects non-specific, passive iron absorption via the paracellular pathway through tight junctions between enterocytes. This same basic phenomenon has also been frequently described in relation to the absorption of other essential minerals, notably calcium (3, 29). Very large doses of oral (i.e., supplemental) iron can thus override feedback mechanisms which normally limit iron absorption (86, 97), likely reflecting the nonspecific, paracellular component. The saturable, carrier-mediated component of iron transport probably represents the normal physiological DMT1/FPN1-mediated iron absorption pathway, which will be considered in detail below. Mechanisms that limit iron absorption with higher intakes (but still within the physiologic range) also are likely to relate to this pathway, since DMT1 has been shown to traffic-off of the BBM upon exposure to a high oral iron dose (181, 323). This phenomenon has been termed the “mucosal block” to iron absorption (90, 113, 215, 279). Early studies in humans utilized an iron tolerance test in iron-deficient adults (213). Sixty and thirty mg blocking doses inhibited absorption of a subsequent 10 mg iron test dose for up to 24 h. In addition, an oral iron dose caused a rapid decrease in the expression of DMT1 and DCYTB mRNAs, suggesting that decreased expression of the BBM iron transport machinery could contribute to the mucosal block (90).
The intestinal mucosa regulates dietary iron assimilation to prevent ID and toxic accumulations. Iron is imported across the BBM and into the enterocyte, but the ultimate fate of that absorbed iron depends on the intrinsic transport properties of the BLM. Iron not exported immediately can be stored within ferritin; prior to exfoliation of a particular enterocyte, this iron can be exported later into the portal circulation if needed to meet systemic iron demand. Previously, there was disagreement regarding whether BBM uptake or BLM efflux was the rate-limiting step in intestinal iron absorption [summarized in (7)]. Since the amount of iron crossing the BLM cannot exceed that crossing the BBM, logically, the efflux step would be rate-limiting. Early kinetic and physiological studies produced conflicting results on this topic, but the general consensus was that the basolateral transfer phase was indeed rate limiting. Recently, molecular and genetic studies have better clarified this issue, providing strong support for the regulatory role of BLM iron transfer in the regulation of whole-body iron homeostasis (5, 208). This topic will be discussed in greater detail in subsequent sections of this review.
Intestinal Iron Transport: Detailed Mechanistic Description
Iron import: Reduction of dietary nonheme (ferric) iron
Nonheme (or inorganic) iron is the most prevalent form of dietary iron, and almost all nonheme iron is in the oxidized, or ferric (Fe3+) form. Yet, ferrous iron (Fe2+) is more soluble and is the substrate for DMT1, the predominant BBM iron importer. Ferric iron must thus be reduced prior to transport by DMT1. As noted above, gastric secretions and dietary factors such as vitamin C promote iron reduction, but despite this, enzymatic iron reduction is probably required, particularly when metabolic demand for iron is elevated. Biochemical approaches led to the prediction that a BBM-associated ferric reductase (or “ferrireductase”) existed (244, 310). Subsequently, a transmembrane ferrireductase, called DCYTB, was identified (164, 194). DCYTB reduces iron in vitro and immunological techniques showed that inhibiting DCYTB activity impaired iron reduction in duodenal samples. Recent studies also showed that intracellular ascorbate provides the reducing equivalents (i.e., electrons) to DCYTB to allow conversion of Fe3+ to Fe2+ (164, 315). DCYTB is most robustly expressed in the proximal small intestine, and expression is enhanced by ID and hypoxia (168,194), known stimulators of iron absorption. Subsequent studies in DCYTB KO mice suggested that DCYTB is not essential for iron absorption (108) under physiological conditions. This may not be surprising, given that mice, unlike humans, synthesize and secrete ascorbate into the intestinal lumen. Later studies on DCYTB KO mice, however, demonstrated that lack of DCYTB slightly impaired iron absorption (193). More recent studies support the postulate that DCYTB is the only iron- / hypoxia-regulated ferrireductase in the mouse intestine (46, 169). Nonetheless, functional redundancy is likely provided by one or more additional (yet unidentified) BBM ferrireductases, and dietary and endogenous factors are clearly important as well.
Iron import: Divalent metal-ion transporter 1
Different forms of dietary iron can traverse the BBM of duodenal enterocytes, but the transport of nonheme iron seems to be quantitatively most significant (84, 102, 105, 107, 182). DMT1 facilitates the uptake of most nonheme iron from the intestinal lumen (182), but it is also expressed in many other cell types where it facilitates iron transport from endosomes into the cytosol after endocytosis of diferric-TF. Lack of DMT1 in the small intestine of mice (106), and mutations in SLC11A2 (the gene encoding DMT1) in rodents (83, 84), cause severe hypochromic, microcytic anemia, demonstrating that DMT1 is the predominant intestinal iron importer. DMT1 is an integral membrane protein with 12 predicted membrane-spanning domains (182). It transports ferrous iron (Fe2+) along with protons across the BBM; it thus functions as a secondary-active transporter, with the energy for iron transport being derived from the electrochemical H+ gradient across the apical membrane of enterocytes. DMT1 can also transport other divalent cations, including Cu (12, 139), Mn, Co, and Cd (107,133,268) in various model systems. Despite this, mutation or loss of DMT1 causes a severe iron-deficiency anemia in rodents (83, 266, 280) and humans (15, 198), with no perturbations related to these other metals being noted, expect for perhaps Mn (in mice) (289). Iron is thus likely the major physiological substrate of DMT1. Further supporting this postulate is the fact that intestine-specific DMT1 KO mice have an iron transport defect and are severely anemic; yet, copper and manganese absorption are unaltered (266). As mentioned above, DMT1 is a proton-coupled/ ferrous iron cotransporter; the movement of Fe2+ into enterocytes is thus coupled to the cotransport of protons (107). DMT1 is ideally suited for iron transport in the low pH environment of the duodenum. The proton gradient is likely provided by an apically expressed NHE3 (267), which is an antiporter that exchanges intracellular protons for extracellular Na+ ions. Moreover, functional investigations of DMT1 demonstrated that amino acid residues in several transmembrane domains are important for iron binding and uptake, and proton coupling (60, 209).
DMT1 functions in most body cells (where it is involved in iron export from endosomes as part of the TF cycle), but expression is particularly high in the duodenal epithelium, the main site of iron absorption (107). DMT1 is present on the BBM when large amounts of iron are being transporter, but the protein is predominantly found with intracellular membranes when body iron stores are adequate (33, 291, 323). Moreover, DMT1 was rapidly internalized from the BBM and degraded when rodents were given a bolus of iron (323). The iron-dependent trafficking of DMT1 likely plays a protective role to prevent toxic iron accumulations. Proteins which mediate DMT1 trafficking in hepatocytes have recently been described. For example, Nedd4 Family Interacting Proteins 1 and 2 (NDFIP1/2) act as adaptors to recruit a ubiquitin ligase to DMT1, thus facilitating proteasome-mediated degradation (85). Whether this occurs in the intestine is, however, unknown.
DMT1 expression is strongly induced by iron depletion, hypoxia, and other conditions, which stimulate iron absorption; conversely, expression is reduced when body iron stores are high and in response to infection and resultant inflammation (33, 51, 52, 88, 107, 173, 248). For example, DMT1 expression is upregulated after gastric bypass surgery for morbid obesity, which is frequently associated with iron depletion (187). Moreover, DMT1 expression is inhibited in individuals with IBD (e.g., Crohn’s disease, colitis) (313), perhaps contributing to the frequently described ID. A DMT1 transcript highly expressed in the duodenum contains an iron-responsive element (IRE) (a stem-loop structure) in the 3′ untranslated region. This structural element provides a mechanism for the iron-dependent regulation of DMT1 via the IRE/iron-regulatory proteins (IRP) system. During iron depletion, IRPs bind to the 3′ IRE and prevent degradation of the DMT1 mRNA molecule, thus allowing more DMT1 protein to be produced via enhanced translation. Conversely, when iron demand is low, IRPs do not interact with the DMT1 transcript IRE and it is destabilized, thus decreasing protein expression and reducing intestinal iron transport. Importantly, there are two 3′ DMT1 transcript splice variants, one containing an IRE (+IRE) and one without an IRE (-IRE); as mentioned earlier, the +IRE splice variant is the predominant variant expressed in the duodenal epithelium (284). Interestingly, the -IRE transcript variant is also regulated according to iron demand (i.e., higher in ID and lower during iron loading) (89), suggesting that other regulatory mechanisms exist (e.g., increased transcription of the SLC11A2 gene). In fact, the SLC11A2 gene is transactivated by a hypoxia-inducible transcription factor, HIF2α (190), which is stabilized during ID (with results in concurrent hypoxia). This topic is covered in more detail below. Furthermore, recent studies also provided evidence that HEPC signaling altered DMT1 expression/activity (48,197,318), including causing BBM DMT1 to be internalized and subsequently degraded in the proteasome (25), similar to HEPC regulation of FPN1 on the BLM.
Iron import: Heme-iron absorption
Dietary heme is derived principally from myoglobin, and to a lesser extent hemoglobin, in animal flesh, and is an important iron source for omnivorous humans (35). Heme is cleaved from myoglobin and hemoglobin by pancreatic proteases, and free heme is thus available for uptake across the BBM. Iron derived from heme has superior bioavailability as compared to nonheme iron. This may be due to the fact that iron is bound within the porphyrin ring structure in heme, so factors that may inhibit nonheme iron absorption (e.g., phytates, polyphenols, etc.) have less (if any) effect on heme absorption (303). In fact, only 10% of the iron in a typical Western diet is derived from heme, yet the “heme iron pool” contributes up to 50% of the total amount of dietary iron absorbed (20, 35).
Unlike for nonheme iron, the mechanism of heme absorption has remained enigmatic (303). A specific heme-binding protein on the BBM in several species was described, but it was never identified (100, 285, 311). Morphological studies demonstrated that heme was internalized into endocytic vesicles derived from the BBM, which subsequently fused with lysosomes in which the heme was degraded (224, 314), liberating the iron from the porphyrin ring. It is also possible that heme is transported directly across the BBM. Recent studies, in fact, described the identification of a low-affinity heme transporter, named heme-carrier protein 1 (HCP1) (269) (Fig. 2). Characteristics of HCP1 are consistent with a role in intestinal heme transport, including localization to the BBM and induction by hypoxia, which stimulates intestinal iron transport. Subsequent studies, however, demonstrated that HCP1 is also a high-affinity folate transporter, and it was renamed, proton-coupled folate transporter (PCFT) (239). Patients with mutations in the gene encoding HCP1/PCFT have folate deficiency, but iron metabolism does not seem to be affected (239). It thus seems unlikely that this protein is a physiologically-relevant heme transporter. In summary, the mechanism of intestinal heme absorption remains undefined.
After traversing the BBM, heme has two potential metabolic fates: catabolism within enterocytes or trafficking intact through the cell and across the BLM. The latter pathway is probably of lesser importance since radiotracer studies have shown that most of the radioactive iron found in the circulation following oral administration of heme with a radioactive iron center appears in the blood bound to TF (30, 55, 307). It thus seems that most heme-derived iron exits the cells by the same export pathway as absorbed nonheme iron. Nonetheless, two heme export proteins were identified in enterocytes, feline leukemia virus, subgroup C, receptor (FLVCR) and ABCG2 (or the breast cancer-resistance protein) (167). FLVCR reduces excess heme levels in developing erythrocytes, and may be involved in heme release from macrophages (143,241). ABCG2 facilitates heme export from developing erythroid cells (158). Both proteins are expressed in enterocytes and in cell culture models of the intestinal epithelium (with Abcg2 being apically expressed) (72, 241). Whether these proteins mediate intestinal heme iron absorption, or protect intestinal cells against heme-related toxicity, however, is unknown.
Most heme is degraded within the enterocyte, being mediated by heme oxygenases (HOs) (242,302). HO-1 is probably the main enzyme involved in catabolizing newly absorbed heme (167, 303), but definitive studies are currently lacking. In mice, lack of intestinal HO-1 did not impair heme absorption, although, as recognized by these authors, mice are not able to utilize heme iron very efficiently (80). Other recent studies suggested that HO-2 is the more likely of the two enzymes to be involved in releasing iron from intracellular heme (196). Both enzymes may contribute to heme degradation in enterocytes; but irrespective of which enzyme is predominant, iron released from heme probably enters the same intracellular transit pool as nonheme iron and is transported across the BLM via FPN1 (167, 242, 303).
As for nonheme iron, intestinal heme iron transport can be regulated by iron demand, being enhanced when metabolic iron requirements are elevated (257). Heme iron absorption, however, is not regulated over as wide a concentration range as for nonheme iron absorption (14,32,58,114). This is some-what paradoxical given that basolateral transfer is considered rate-limiting for absorption, and iron derived from heme and nonheme iron likely utilize the same export pathway. This apparent discrepancy may be explained by the fact that intracellular heme degradation is likely the rate-limiting step, not transit across the BBM. HO-2 shows limited regulation by iron status, but HO-1 expression is induced by ID, which may provide a mechanistic explanation for the more limited regulation of heme iron absorption (303).
Absorption of ferritin and lactoferrin
In addition to heme and nonheme iron, the human diet also contains protein-bound forms of iron that contribute to iron nutriture. Iron within plant cells is stored mainly in ferritin, which can contribute to dietary iron intake (287). Plant ferritins are currently being developed as a supplemental form of iron (327). Ferritin iron is bioavailable in laboratory rodents and humans (37, 63, 327). Whether ferritin is digested in the intestinal lumen releasing free iron or if it crosses the BBM intact is unknown. One recent study does, however, support the concept that dietary ferritin is hydrolyzed by the acidic gastric juices in the stomach, thus releasing iron for absorption (presumably by DMT1) (126). Ferritin can also be taken up intact via endocytosis in human Caco-2 cells, which are a commonly used in vitro model of the intestinal epithelium (141,259). A recent investigation supports the possibility that a similar phenomenon occurs in vivo (288). It was suggested that ferritin taken up by enterocytes can be retained and slowly release iron over time.
Lactoferrin is an iron-binding protein found in mammalian breast milk (180). It may act as a bacteriostatic agent, play a role in gut immunity and positively affect the growth and differentiation of the intestinal epithelium (178). Lactoferrin may also be a source of dietary iron for infants; however, lactoferrin KO mice survive the suckling period without developing any iron-related pathologies (300). The function of lactoferrin could be different in humans though. In fact, exogenously administered lactoferrin improved iron status in human supplementation trials (147, 154, 179, 220). A lactoferrin-binding protein/receptor was identified on the apical surface of enterocytes (135, 271, 281), but its exact function, nor its identity, have been definitively established. It was also suggested that lactoferrin participates in the acute-phase response to microbial infection by chelating bioavail-able iron (39). This report suggested that lactoferrin-iron complexes can be sequestered by secreted glyceraldehyde-3- phosphate dehydrogenase in the gut lumen, which then allows the lactoferrin-iron complex to be taken up into enterocytes (where the iron could subsequently be utilized for metabolic purposes or exported).
Iron absorption: The intracellular phase
How enterocytes handle newly absorbed dietary iron is incompletely understood. Iron likely exists as an exchangeable “pool”, but the nature of this iron pool is poorly defined. Iron in the cytoplasm probably binds to small organic acids such as citrate, or amino acids, or with low affinity to proteins (160). Membrane impermeable iron chelators, such as des-ferrioxamine, impair iron absorption, presumably by pulling iron away from these other molecules (given its extremely strong affinity for Fe2+) and preventing it from being utilized for metabolic purposes, stored in ferritin or exported by FPN1 (136,174,231). For another redox-active and potentially toxic transition metal, copper, several specific and high-affinity intracellular-binding proteins, or chaperones, have been identified (238). Surprisingly, for iron, only a single intracellular chaperone has been identified to date (see the succeeding text), but how and if it functions in the intestine is unclear. Intracellular iron has one of three fates: (i) it can be utilized for metabolic purposes in the cell (although most metabolic iron in mature enterocytes is acquired from diferric-TF prior to differentiation); (ii) it can be stored in ferritin. The iron within ferritin may be slowly released over time prior to exfoliation of the enterocyte into the intestinal lumen; or (iii) it can be exported across the BLM. The intracellular iron pool can also influence the expression of certain iron metabolism-related genes via a posttranscriptional regulatory loop (i.e., the IRE/IRP system), as discussed in the preceding test.
The best-characterized iron pool in enterocytes is contained within ferritin, an iron-storage protein (121,157). apo- Ferritin is comprised of 24 subunits which form a hollow sphere that can bind ~4500 iron atoms. Iron in excess of cellular metabolic needs, and not exported from cells to support systemic iron homeostasis, is incorporated into ferritin. Iron stored in ferritin can be released and utilized, should metabolic requirements increase (121). Otherwise, the iron in ferritin is lost in the feces after enterocyte apoptosis and subsequent exfoliation into the intestinal lumen. Excess iron storage in ferritin can cause ferritin complexes to fuse with lysosomes, leading to the degradation of the protein shell. The consequent amorphous mixture of peptides and iron oxide is referred to as hemosiderin (9,132,157,200). The ferritin transcript has a 5′ IRE, which leads to a translational block when bound by an IRP. The IRPs bind to ferritin when intracellular iron levels are low, but when iron is high, the IRPs do not bind and the translational block is removed. More ferritin protein is thus produced, and it sequesters excess iron and prevents it from accumulating to toxic levels (10, 121, 125). Ferritin may thus play a passive role in regulating intestinal iron absorption. Overexpression of ferritin, however, in cultured intestinal cells depletes cytosolic iron (227), and a similar effect may also occur in enterocytes in vivo (170). Enterocytes respond to iron depletion by increasing the expression of genes encoding the iron transport machinery (e.g., DCYTB, DMT1, FPN1, etc.). Ferritin may thus actively modulate the passage of iron across the enterocyte, and thus influence overall iron absorption. A recent study, in fact, supports this postulate by suggesting that ferritin functions in parallel with HEPC to prevent excessive iron absorption (294).
Recently, protein chaperones that might play a role in the intracellular trafficking of iron, were identified, poly (rC)-binding proteins 1/2 (PCBPP1/2). PCBP1 facilitated iron loading onto ferritin (270). More recently, PCBP2 was shown to interact with DMT1 and FPN1 in hepatocytes, thus possibly functioning in a similar manner to copper chaperones (319, 320). The in vivo significance of these observations is not clear, and whether these proteins function similarly in enterocytes has not yet been investigated.
Iron export: Ferroportin 1
Iron flux across the enterocyte BLM (i.e., the transfer phase) plays a critical, rate-limiting role in iron absorption. The BLM is the interface with the portal circulation, which transports nutrients from the gut to the liver, and as such, BLM iron efflux is controlled by systemic regulators which sense body iron needs. This in turn makes it the primary site for the regulation of dietary iron absorption. FPN1 is the only exporter of non-heme iron identified to date in mammals (1,69,194). FPN1 is widely expressed, as most cells must be able to export excess iron due to its potential toxicity. FPN1 is robustly expressed in cells that export large amounts of iron, including enterocytes (which absorb dietary iron), and macrophages of the RE system (including hepatic Kupffer cells) and hepatocytes (which store excess iron) (8, 152, 324).
FPN1 is an integral membrane protein with 12 predicted membrane-spanning domains (254). Like DMT1, it transports ferrous iron, but mechanisms of iron transport by FPN1 have not been elucidated in detail. Investigation of mutations in the SLC40A1 gene (encoding FPN1) in humans has led to the identification of a several residues involved in iron transport (228, 298); detailed structure-function studies, however, are lacking. Whether FPN1 functions as a monomer or a dimer is unclear, with some studies supporting function as a monomer (254), whereas others suggest that a dimer is more likely (66). FPN1 expression is strongest in the proximal small intestine, the major site of iron absorption (89, 195). Within the duodenal epithelium, expression is restricted to the mature absorptive enterocytes, and it has been localized to the BLM (1, 69, 195), consistent with a role in mediating iron export. Human FPN1 transports Fe, Zn and Co when heterologously expressed in Xenopus oocytes. Complementary in vivo studies by these investigators in mice, however, suggested that iron is the most important substrate (199). Moreover, Mn metabolism was perturbed in mice expressing a mutant form of FPN1 (i.e., flatiron mice), suggesting that FPN1 mediates some aspect of Mn metabolism (262, 263).
The essentiality of FPN1 in iron homeostasis was demonstrated by ablation of the gene in mice, and by studies in humans with mutations in the SLC40A1 gene (69, 70, 228). FPN1 KO in mice is embryonic lethal. Intestine-specific knockout of FPN1 causes severe iron-deficiency anemia, demonstrating that FPN1 is the main iron exporter (70). Mutations in SLC40A1 in humans occur infrequently, yet they represent an important class of human iron-loading disorders (64, 228, 298). Two basic clinical presentations are observed, depending on the specific mutation in SLC40A1. Mutations that alter localization and/or iron transport capacity impair intestinal iron absorption and increase iron accumulation in cells that store iron (i.e., RE macrophages and hepatocytes). Some patients instead have mutations that alter the interaction between circulating HEPC and FPN1, but do not directly impair the ability of FPN1 to transport iron. With these mutations, iron absorption is enhanced relative to iron needs since the usual feedback mechanism that limits iron uptake is perturbed (64).
FPN1 regulation is quite complex, occurring at transcriptional, posttranscriptional and posttranslational steps in the gene expression pathway. Duodenal FPN1 expression was shown to be inversely related to body iron load (1, 69, 195), which is predictable based upon the important role of the BLM iron transfer step. In the liver, however, FPN1 expression is low during ID and high when body iron levels are elevated (1), reflecting an opposite expression pattern to that in the gut. These opposing regulatory mechanisms may represent cellular responses to prevent ID or overload. Thus, when iron stores are depleted, high FPN1 expression in the gut promotes assimilation of dietary iron while low hepatic FPN1 expression promotes iron retention in the liver to meet the high metabolic demands of this organ. Conversely, when body iron stores are replete or high, low FPN1 expression in the gut prevents additional excess iron accumulation, while high expression in the liver, promotes iron flux thus preventing possible hepatic toxicity.
Differential regulation of FPN1 expression in these tissues relates to the fact that one FPN1 transcript variant contains a 5′ IRE. Similar to the ferritin transcript, when iron levels are elevated, the IRE will not be bound by an IRP and translation will proceed normally; conversely, when intracellular iron is depleted, an IRP will bind to the IRE and block translation (1). This does not, however, explain the opposite pattern of regulation in the intestinal epithelium. The fact that there are two FPN1 transcripts (i.e., splice variants), one that contains a 5′ IRE (+IRE) and one that does not (-IRE) (326), provides clarity on this issue. During iron depletion in the intestine, translation from the +IRE splice variant is diminished, but expression of the -IRE variant is increased, thereby promoting iron absorption. This increase in the expression of the -IRE FPN1 transcript variant in enterocytes probably reflects transcriptional induction by Hif2α in response to hypoxia, which results from ID (which impairs oxygen delivery to the gut) (190). Hypoxic regulation of FPN1 expression has been described (195). Dual transcriptional and translational regulation of FPN1 expression determines protein levels in enterocytes, but superimposed upon this is HEPC-mediated posttranslational regulation of FPN1 protein levels on the BLM. HEPC regulation of FPN1 is the critical factor, which allows iron absorption to be finely tuned according to alterations in iron demand, particularly during pathological tissue iron loading, and infection and inflammation.
Iron export: The multicopper ferroxidases hephaestin and ceruloplasmin
FPN1 exports ferrous iron but iron oxidation is required since ferric iron binds to TF, the main iron-transport protein in the blood. Iron efflux from duodenal enterocytes thus requires an iron exporter and a FOX. One such FOX in the intestine is HEPH (4, 297). Mice with mutations in the gene encoding HEPH (sex-linked anemia [sla] mice) have moderate iron-deficiency anemia in early life, which is probably caused mainly by impaired basolateral transfer of iron into the portal circulation (229, 230). Iron metabolism has also been investigated in HEPHKOmice and in mice lacking HEPH only in the intestine (92). HEPH KO mice have impaired intestinal iron transport, while adult, male HEPHint KO mice showed iron accumulation in duodenal enterocytes and mild ID. Another recent study showed that intestinal HEPH was required for optimal iron absorption in weanling, adult and pregnant mice under physiological conditions, but that it was not required to appropriately upregulate intestinal iron absorption during iron-deficiency or hemolytic anemia in adult mice of both sexes (https://doi.org/10.1182/bloodadvances.2017008359). Interestingly, in all these murine models of impaired HEPH function, growing, young mice are iron deficient and anemic, yet the anemia resolves as mice mature into adults. Other complementary FOXs may thus compensate for impaired HEPH activity. For example, FOX activity was documented in duodenal enterocytes of HEPH KO mice, perhaps revealing such alternative FOXs (245, 246).
HEPH expression is robust in mature enterocytes of the small intestine, and somewhat lower in other tissues (128). Surprisingly, it is abundantly expressed throughout the small intestine and into the colon (89). HEPH shares amino acid sequence homology with a circulating, liver-derived FOX, CP (297). Unlike CP, however, HEPH has a single C-terminal transmembrane domain, and it is thus membrane anchored. HEPH and CP both bind copper (which is incorporated co-translationally) and both can oxidize ferrous iron (40). Copper is required for enzymatic activity of both proteins, which likely explains why copper-deficient animals have defective iron absorption and consequent impaired erythropoiesis (59).
FPN1 and HEPH are functionally linked, so it is a logical postulate that they may physically interact. Although colocalization studies suggest that this is indeed a possibility, this has not been unequivocally demonstrated in vivo (116, 322). How the FPN1-HEPH functional couple occurs to mediate iron efflux is nonetheless poorly understood. In glioma cells and mouse bone marrow-derived macrophages, FOX activity of a membrane-anchored form of CP (GPI-CP) was required for iron export by FPN1 (65). In the absence of GPI-CP, ferrous iron remained bound to FPN1, triggering ubiquitination, internalization, and degradation of FPN1 in lysosomes. Iron oxidation thus stabilized FPN1 on the plasma membrane. The same basic interactive scheme may also relate to the FPN1- HEPH functional couple in the intestine, but this postulate has not been tested to date.
CP is secreted into the blood by the liver, where it facilitates iron oxidation and release from a variety of tissues (124). It could also promote iron absorption in the gut, but this possibility has not been definitively established. CP KO mice do not have an obvious defect in iron absorption (119), but subsequent studies have shown that CP may in part facilitate iron absorption when iron demands increase. For example, when erythropoiesis was enhanced in CP KO mice by phlebotomy, absorption did not increase to the same extent as in wild-type littermates (44). The possibility that CP contributes to intestinal iron absorption logically suggests redundancy in the oxidative mechanism and could explain why inhibition of HEPH activity does not lead to a very severe iron-deficiency anemia. Although circulating CP may promote iron absorption, it is likely not as effective as intestinal HEPH. This postulate is supported by the observation that HEPH is localized mainly in an unidentified intracellular compartment (162), although BLM localization has also been reported (116). Thus, if FPN1 and HEPH initially interact intracellularly to some extent within enterocytes, this could explain the relative greater efficiency of HEPH in promoting iron absorption since this location would be inaccessible to circulating CP.
Iron efflux: TF binding and distribution in the portal blood circulation
After export across the BLM, iron must enter the portal circulation, but exactly how this occurs is unknown. Newly absorbed iron binds to apo-TF, but the trans-capillary TF exchange rate cannot account for all absorbed iron (201,261). Moreover, if TF directly received iron from the BLM iron transport machinery, then apo-TF would bind to the BLM, but such a phenomenon has never been described. A recent study did not identify interactions between TF and Heph (129). Further, TF is not absolutely required for iron absorption as mice lacking TF (i.e., hypotransferrinemic mice), do not have impairments in iron absorption (19,61,272). Newly absorbed iron is thus likely to initially enter the interstitial fluids in the lamina propria as chelates with organic acids or amino acids, or bound with low affinity to plasma proteins (e.g., albumin), and subsequently be picked up by TF in the portal blood (which has a much higher affinity for ferric iron).
Copper: History and Background
It was established over two centuries ago that copper was present in lower marine invertebrates and plants, but it was not until the 1920s that copper was detected in animal tissues, when it was established that the human brain contains copper (23). Also around this time, a specific physiological role for copper was identified when it was found that experimental anemia in rats and in other mammals could be cured by administration of copper from a liver extract, along with iron salts (50). Evidence of copper being involved in a human disease was firmly established in the early twentieth century, with the initial description of Wilson’s Disease (WD). The fact that this disease was an inborn error of metabolism was not, however, understood until several decades thereafter (236). A relationship between copper depletion and anemia in humans was postulated in the 1930s, but conclusive experimental proof was not provided until sometime later. Overt copper deficiency in humans was first described in 1962 in patients with Menkes Disease (MD); however, the underlying genetic defect was not identified for another 10 years (236). It is now clearly established that copper is an essential nutrient for humans and other mammals. Copper is found in tissues and body fluids in parts per million (μg/g) to parts per billion (ng/g) amounts. Exquisite systems for regulating copper absorption, distribution, storage, utilization, and excretion have evolved in mammals, given that high or low copper levels cause severe homeostatic perturbations.
Copper: Biochemical and Physiological Properties and Regulation
The atomic mass of copper is ~63.5 Daltons. Two stable isotopes, 63Cu and 65Cu, and seven radioisotopes of copper exist, with 64Cu (~ 13 hr) and 67Cu (~70 hr) having the longest half-lives. These radioisotopes, along with the two stable isotopes, are most frequently used for studies related to copper metabolism and homeostasis. In biological systems, copper exists in two predominant oxidation states, Cu2+ (cupric) and Cu+ (cuprous). Like iron, copper is involved in redox reactions, during which it commonly shifts back and forth between the cupric and cuprous oxidation states. Cuprous copper (Cu+) is highly insoluble in aqueous solutions and is thus usually associated with other molecules within cells. Most copper involved in mammalian metabolism is bound to proteins, via specific interactions with amino acid side chains (i.e., R groups) that have the propensity to interact with positively charged ions. Copper enters the body from the diet, with the average intake being ~ 1.3 mg/d (Fig. 1). The amount extracted from the diet daily is ~0.8 mg/d, which is delivered to the liver. Excretion occurs predominantly via copper-transporting ATPase 2 (ATP7B) into the bile (~0.4 mg/d), with total fecal losses being ~1 mg/d. Copper is incorporated into CP and other cuproenzymes in the liver. CP is then secreted into the blood where it functions as a ferroxidase. Atomic copper, which also exits the liver, binds with serum proteins (e.g. albumin), and is by this mechanism transported to cells throughout the body. Homeostatic control of body copper levels includes modulation of copper absorption in the intestine and copper excretion in the liver.
Several copper-dependent enzymes and copper-binding proteins have been identified in mammals (listed in Table 2). In mammalian biology, copper serves a predominant role as an enzymatic cofactor for a several copper-containing enzymes (or cuproenzymes). These enzymes are mainly oxidases, and collectively, they are involved in single electron transfer reactions between a substrate and molecular oxygen using either reduced (Cu+) or oxidized (Cu2+) copper atoms. Descriptions of these proteins and their physiochemical properties and functions have been published elsewhere (77, 192), and are beyond the scope of this review article. Non-enzymatic functions of copper have also been described, where it plays a role in diverse physiological processes, including angiogenesis, oxygen transport in the blood, neurotransmitter homeostasis, and regulation of gene expression. Moreover, mRNA levels for many proteins involved in copper homeostasis in mammals (e.g., CTR1, ATP7A, and ATP7B) do not change in response to dietary copper intake levels, demonstrating a lack of control at the level of gene transcription or transcript stability. Regulation of copper intake and efflux may instead be controlled at a posttranscriptional level, predominantly by protein trafficking, as exemplified by the copper-transporting ATPases moving from the TGN to either the enterocyte BLM (ATP7A) or to the canalicular membrane of hepatocytes (ATP7B) when copper is in excess. One exception is that expression of ATP7A in the duodenal epithelium has recently been shown to be induced at the transcriptional level (by HIF2α) during iron deprivation (316, 317).
Table 2.
Copper-Dependent Enzymes and Selected Copper-Binding Proteins in Mammals
| Copper-containing enzymes* | Function | Effect of copper deficiency |
|---|---|---|
| Amine oxidases | Deamination of mono- and diamines | Variable |
| Lysyl oxidase | Processing of elastin and collagen | Bone and cartilage stability impaired |
| Ceruloplasmin (CP) | Ferroxidase; iron release from stores | Iron accumulation in liver, brain, pancreas |
| Hephaestin (HEPH) | Ferroxidase; intestinal iron transport | Iron accumulation in enterocytes; iron deficiency |
| Dopamine β-monoxygenase | Catecholamine metabolism | Neuropathologies, hypomyelination of nerve fibers |
| Tyrosinase | Pigmentation; melanin biosynthesis | Hypopigmentation; abnormal keratinization of hair |
| Peptidylglycine α-amidating monoyxgenase | Activation of peptide hormones | Altered CNS hormone production; CVS dysfunction |
| Cytochrome C oxidase (CCO) | Electron transport; ATP production | Decreased capacity for oxidative phosphorylation |
| Superoxide dismutase 1 (SOD1) | Antioxidant defense | Increased susceptibility to oxygen free radicals |
| Superoxide dismutase (extracellular) (SOD3) | Antioxidant defense | Increased susceptibility to oxygen free radicals |
| Zyklopen | Ferroxidase; placental iron efflux | Unknown |
| Monoamine oxidase A and B | Degrades amine neurotransmitters | Unknown |
| Copper-binding proteins** | Copper-dependent function | |
| a2-Macroglobulin | Transport of copper from site of absorption in intestine to liver | |
| Albumin | Transport of copper from site of absorption in intestine to liver; copper transport in blood | |
| ATOX1 | Copper chaperone for copper-transporting ATPases; copper-dependent transcription factor | |
| ATP7A | Transports copper into TGN and out of cells; Menkes disease gene | |
| ATP7B | Transports copper into TGN; copper excretion in bile; Wilson’s disease gene | |
| COMMD1 | Interacts with ATP7B in liver, copper excretion; Bedlington terrier copper toxicosis disease gene | |
| CCS | Copper chaperone for SOD1 in cytosol | |
| CTR1 | Plasma membrane copper transporter; necessary for copper uptake in intestine, liver, heart | |
| Metallothionein I/II (MTI/II) | Intracellular copper storage proteins (also bind zinc and cadmium) | |
| XIAP | Ubiquitination of COMMD1 and CCS; mediates proteasomal degradation | |
Many of these proteins are not mentioned elsewhere in this review.
Only those proteins mentioned in this review are listed here.
Copper: Bioavailability and Nutrient Interactions
Total copper amounts in adults range from 50 to 120 mg. Copper is not stored in the human body to any extent; tissue copper levels thus likely reflect copper associated with various enzymes (i.e., cuproenzymes) and copper-binding proteins. The typical diet of an adult in the U.S. supplies slightly more copper than is recommended (RDA = 0.9 mg/d). The best dietary sources of copper are shellfish, seeds, nuts, organ meats, wheat bran cereal, whole grain products, and chocolate-containing foods. Vegan diets contain adequate copper, but absorption is lower from plant foods, which is predictable given the array of dietary factors known to interact with positively charged metal ions (discussed earlier for iron). Copper is also found in vitamin and mineral supplements. In these products, however, copper is often in the cupric oxide form which has lower solubility and thus bioavailability.
The relative amount of dietary copper is a major predictor of the efficiency of intestinal absorption. So, when dietary copper levels are high, percent absorption decreases, and conversely, when intake is low, percent absorption increases. Several dietary factors decrease copper bioavailability, including carbohydrates, ascorbic acid, some amino acids, zinc, iron, and molybdenum (192), particularly when intake of these dietary constituents is high. Large doses of supplemental zinc can induce severe systemic copper deficiency. This same phenomenon has also recently been reported in several elderly patients that have utilized excessive amounts of zinccontaining denture creams (68, 123, 205, 328). Furthermore, the impact of these and potentially other dietary constituents on intestinal copper absorption may be more significant in neonates, since digestive function and homeostatic regulation of biliary copper excretion are not well developed at this developmental stage.
Of the nutrients discussed above, Cu metabolism is best known to be influenced by vitamin C, iron, and zinc. Alterations in dietary copper levels are also thought to potentially affect the metabolism of other nutrients, but except for iron, this topic will not be considered further here. Supplemental doses of vitamin C can cause copper deficiency in laboratory rodents, and the same phenomenon may be applicable to some humans as well. For example, plasma vitamin C levels inversely correlated with serum CP and antioxidant activity in premature infants (235). Consistent with this observation in infants, other investigations in humans have also demonstrated that high vitamin C intake may decrease serum FOX (i.e., CP) activity. Depression of CP activity is suggestive of copper depletion, since CP is a recognized biomarker of moderate to severe copper deficiency. Additionally, iron is known to influence copper metabolism and vice versa (54). Physiologically relevant iron-copper interplay in the mammalian small intestine includes iron regulating expression of an intestinal copper exporter, ATP7A and copper regulating expression and activity of the multicopper FOX, HEPH (42,251,252). Moreover, hepatic copper levels are influenced by body iron status, with low iron inducing hepatic copper accumulation (87). In addition, hemoglobin production is impaired by copper depletion in the setting of normal serum iron levels, implicating copper in some aspect of hemoglobin synthesis (87). Exactly how copper influences this process is, however, unknown, but it may relate to iron import into or utilization in mitochondria. Furthermore, in regards to zinc and its influence on copper homeostasis, high zinc intake inhibits intestinal copper absorption. Induction of MT, an intracellular copper/zinc-binding protein, could provide a mechanistic explanation for this observation, especially since MT has a higher affinity for copper than zinc (144). Mice lacking both isoforms of MT, however, also became copper depleted upon high zinc exposure, so other mechanisms are likely to be involved (249). A similar phenomenon has been reported in humans who became copper deficient upon consumption of supplements containing 50 mg of zinc daily for several weeks to months (82). This latter observation is what informed the establishment of the UL for zinc intake in adult humans of 40 mg/d.
Copper: Absorption, Transport, Storage, and Excretion
Like for iron, efficient adaptive mechanisms have evolved in humans to prevent copper deficiency and protect against copper toxicity. This is important since copper depletion impairs the activity of several cuproenzymes, which mediate important physiologic functions, including connective tissue formation, pigmentation, iron homeostasis, neurotransmitter synthesis, and others. One such mechanism involves adaptation of intestinal copper absorption according to dietary copper intake levels. So, when intakes are low, percent absorption increases and vice versa. When copper intake is sustained at very low levels (<0.7 mg/d), however, the intestine can no longer compensate, and copper depletion ensues. This intake level is significantly below the average intake level in the United States (~1.2 mg/d), so this scenario is less likely for most healthy adults. Conversely, when copper intake is high, percent absorption decreases, copper may be sequestered within MT in enterocytes, and biliary copper excretion increases. Unlike for iron, humans have the ability to excrete excess copper in the bile. With normal dietary intakes, ~10% of copper is retained, which reflects intestinal absorption as well as biliary excretion of recently absorbed copper.
Dietary copper and copper secreted into the intestine both contribute to the intestinal copper pool. Copper is present in hepatic and pancreatic secretions, and in other GI secretions (e.g., electrolyte and fluid secreted by intestinal crypts); however, copper in bile is probably complexed with bile salts and no longer bioavailable. Intestinal copper absorption has been investigated by several investigators over the past several years. Most dietary copper is in the cupric (Cu2+) state, but intestinal copper importers transport cuprous (Cu+) copper. Dietary copper must thus be reduced prior to absorption (Fig. 2). At least three cupric reductases have been identified (cytochrome b [558] ferric/cupric reductase (151), STEAP2, and DCYTB), but the precise roles of each in intestinal copper absorption has not been clearly defined. DCYTB can reduce ferric iron, but it may also be able to reduce cupric copper, as recently reported (315). Other candidate cupric reductases include the Six Transmembrane Epithelial Antigen Of The Prostate (STEAP) family proteins (217), which have been defined as metalloreductases. After reduction, cuprous copper can be transported into enterocytes by copper transporter 1 (CTR1) (211). A critical role for CTR1 was established in mice, since animals with significantly reduced CTR1 expression in the intestine (CTR1 intestine-specific KO mice) become severely copper depleted. Interestingly, the mutant mice accumulate copper within duodenal enterocytes, yet the copper cannot be utilized. This probably reflects an endocytic process whereby CTR1 and dietary copper are endocytosed, and CTR1 then pumps the copper from the endosome into the cytosol for utilization (211) (Fig. 2). DMT1 can also transport copper, as has been established by several groups (11,12,175), although DMT1 may only transport copper under certain circumstances but not necessarily during physiological conditions (133, 266). Copper transport by DMT1 seems particularly plausible during iron depletion when DMT1protein levels in the gut are dramatically increased and in the setting of no (or few) competing iron atoms (139, 248, 264, 329).
Since free copper is highly reactive, it is almost always bound to cellular proteins and other molecules, which reduces its reactivity. In fact, some have estimated that there may be a single free (unbound) copper atom in a typical cell. Mammalian cells have thus evolved specific regulatory mechanisms to handle absorbed copper and to get it to its final intracellular location. This is accomplished by a host of intracellular copper-binding proteins, referred to as copper chaperones. For example, one such protein, Cytochrome C Oxidase Copper Chaperone (COX17), delivers copper to the mitochondria to support biosynthesis of cytochrome C oxidase (221), which functions in the electron transport chain. Another copper-binding chaperone, antioxidant protein 1 (ATOX1) delivers copper to ATP7A for transport into the trans-Golgi network, which promotes the biosynthesis of cuproenzymes (166). Intracellular copper must also be distributed in the cytosol to Cu, Zn-superoxide dismutase, and this is accomplished by copper chaperone for SOD1 (CCS) (36). Furthermore, when copper is in excess, it can be bound to MT in the cytosol, which prevents the accumulation of unbound (i.e., reactive and toxic) copper and subsequently leads to copper loss upon exfoliation of mature enterocytes into the gut lumen. Copper export from enterocytes is mediated by ATP7A, which traffics to the BLM (from the TGN) to promote copper efflux (226). Copper remains in the cuprous state as it traverse and exits enterocytes. Once copper exits enterocytes, the oxidizing environment of the interstitial fluids presumably converts cuprous copper (Cu+) to cupric copper (Cu2+), which then binds to albumin or α2-macroglobulin for delivery in the portal blood to the liver.
Once newly absorbed copper reaches the liver, it is reduced by an unidentified reductase (possibly a STEAP protein) and then imported into hepatocytes by CTR1 (Fig. 3) (153). Similar to what occurs in enterocytes, once inside liver cells, copper interacts with chaperone proteins which distribute it to various cuproenzymes or copper transporters. An ATP7A homolog, ATP7B transports copper into the TGN, supporting the biosynthesis of CP and other cuproproteins. When copper accumulates in excess of metabolic needs, in a similar fashion to ATP7A trafficking in enterocytes, ATP7B shifts from the TGN to the canalicular membrane of the hepatocyte, which permits copper excretion into the biliary tree. This then represents the primary excretory route for endogenous copper. Biliary copper, which is complexed with bile salts and thus not bioavailable, and unabsorbed dietary copper, is lost in the stools. Copper excretion is immature (and less efficient) during fetal and neonatal life, perhaps explaining the higher hepatic copper levels noted at these pre- and postnatal developmental stages. Cholestasis, which is most common in the elderly, may also lead to hepatic copper accumulation.
Figure 3.
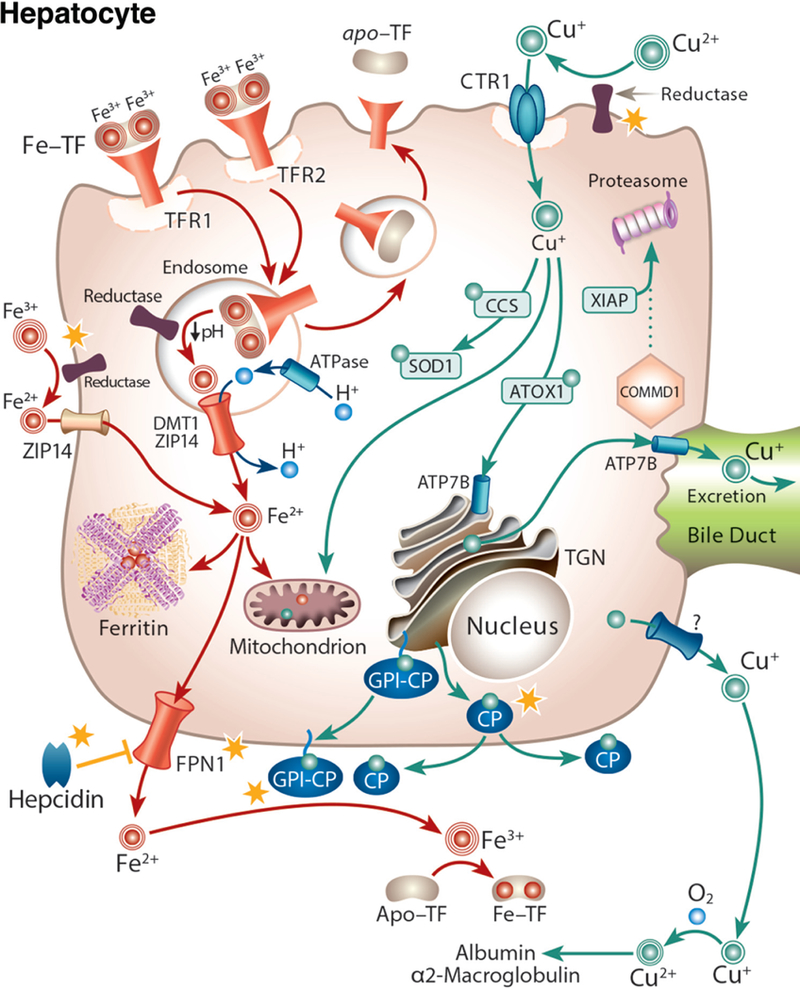
Iron-copper metabolism in a hepatocyte, highlighting points of intersection between these two essential trace minerals. Iron-copper interactions within hepatocytes are indicated by yellow stars. Hepatocytes produce and secrete the iron-regulatory, peptide hormone hepcidin (not shown), which alters intestinal iron absorption (thus justifying the consideration of liver iron homeostasis in this review). Hepcidin also acts in an autocrine fashion to block iron release from hepatocytes (bottom left). Copper may stabilize hepcidin, and thus influence its activity. Hepatocytes also play a principal role in copper metabolism by mediating the excretion of excess copper in bile. These cells assimilate iron via receptor-mediated endocytosis of diferric-TF via transferrin receptors (TFR1/2). Iron is subsequently released from TF by the action of an H+-ATPase in endosomes, reduced (perhaps by STEAP3), and is then transported into the cytosol by DMT1 (or ZIP14). Under pathological conditions of iron overload, nontransferrin bound (ferric) iron in the blood may be reduced and taken up into hepatocytes by ZIP14. This reductase may also reduce copper. Iron is used in cells for metabolic purposes, stored in ferritin, or exported by FPN1 (which may be influenced by copper levels). After reduction, cuprous copper is taken up into hepatocytes via CTR1 and distributed by chaperones. ATOX1 delivers copper to ATP7B, which transports copper into the TGN for incorporation into cuproenzymes, including the FOXs CP and GPI-CP. These FOXs mediate the oxidation of ferrous iron (in an autocrine manner) after release by hepatocytes or other cells (by paracrine of endocrine actions) to permit ferric iron binding to TF in the interstitial fluids. ATP7B also transports excess copper across the canalicular membrane into bile for excretion. ATP7B activity is modulated by COMMD1, and XIAP, a ubiquitin ligase which mediates proteasomal degradation of COMMD1.
Intersection of Iron and Copper Metabolism in the Intestine
Interactions at the enterocyte BBM
The influence of copper on intestinal iron homeostasis has been reported, appreciated, and investigated for several decades or more (87). Mechanistic insight into how this occurs is, however, only now emerging. Recent investigations by many groups around the globe have reported significant iron-copper interactions in duodenal enterocytes, which is important since iron absorption ultimately determines overall body iron levels (since excess iron cannot be excreted) and iron is absorbed in the proximal small intestine. The DCYTB ferrireductase expressed on the apical surface of enterocytes can reduce iron and copper (315), demonstrating a potential point of intersection. Furthermore, another recent report demonstrated that DCYTB was upregulated in the duodenum of copper-deficient mice (191). This possibly related to copper-deficiency anemia and the ensuing intestinal hypoxia, which stabilized a hypoxia-inducible transcription factor, HIF2α, which then transactivated DCYTB expression. Additional studies done in intestinal cell lines showed that disrupting intracellular copper homeostasis, by silencing ATP7A expression, greatly reduced DCYTB expression, again supporting the concept that DCYTB may be influenced by copper levels (111). The BBM iron importer DMT1 is another probable point of intersection between iron and copper in the intestinal epithelium. DMT1 was reported to transport multiple divalent cations initially upon its discovery (107), but subsequent investigations have called into question the biological significance of some of the originally reported DMT1 substrates. More recent work supports the concept that DMT1 transports a much smaller number of divalent cations, principally iron, cobalt, and manganese (133). During physiological conditions, at least in mice, it could be that iron is the most important (or only relevant) dietary substrate of intestinal DMT1 (266). If the same is true in other mammalian species, including humans, has not been experimentally determined. Furthermore, expression of human DMT1 in Xenopus oocytes did not increase copper transport, whether copper was in the cupric or cuprous state (133). It is, however, possible that mechanisms of transport differ between amphibian and mammalian cells (e.g., different membrane lipid compositions, a different assortment of integral membrane proteins, etc., could lead to distinct differences in transport protein function when comparing the two cell types). Several groups have, however, shown that DMT1 can transport copper (11, 12, 175), but in most cases, the physiological significance of this was not established. Moreover, it is possible that DMT1 transports copper only under certain physiologic circumstances, such as during iron deprivation when DMT1 protein expression is hugely increased (248). Indeed, it was recently reported that overexpressed DMT1 in a HEK293 cell model (human embryonic kidney cells) transported copper (139), but only when cells were deprived of iron (using a specific chelator). These authors also showed, in duodenal loop experiments, that lack of DMT1 impaired copper absorption in iron-deficient (i.e., Belgrade) rats (as compared to normal littermate controls). DMT1 could thus, at least in part, mediate increased copper transport into duodenal enterocytes during iron deprivation, which would support the observation that copper content increased in the duodenal epithelium during ID (78, 248). Given these disparate results, further experimentation and documentation is required to establish a possible role of intestinal DMT1 in copper homeostasis during ID, and during physiological (and possibly pathological) conditions in other mammalian species (besides mice).
Interactions at the enterocyte BLM
The iron efflux, or transfer, step may also be influenced by copper. Iron export from duodenal enterocytes involves functional coupling between an iron exporter (FPN1) and an iron oxidase (HEPH, CP, and possibly other unidentified proteins). FPN1 is the sole iron exporter identified thus far, and recent investigations have considered whether its expression or activity is modified by copper. Copper depletion of mice increased FPN1 mRNA levels, perhaps involving HIF2α-mediated transactivation of the SLC40A1 gene (encoding FPN1) (191). Another recent investigation, however, provided opposing results (237). In this study, copper depletion of mice and rats did not influence FPN1 expression or activity. Another investigation showed that copper concentrations were reduced in some tissues of FPN1 mutant (i.e., flatiron) mice, suggesting that FPN1 transports copper or otherwise somehow influences copper homeostasis (262). Additionally, in vitro experimentation in rat intestinal epithelial (IEC-6) cells, demonstrated that SLC40A1 gene transcription was stimulated when ATP7A was silenced by siRNAs, which presumably perturbed intracellular copper homeostasis (104). Transactivation of the SLC40A1 gene correlated with enhanced iron efflux from cells with diminished ATP7A expression, again exemplifying potential regulation of FPN1 expression by copper. In sum, these studies are suggestive that FPN1 expression or activity is influenced by copper, but additional investigation is required to provide definitive evidence of such copper-dependent regulation.
Another clear link between iron and copper metabolism in the mammalian intestine is the multi-copper FOX HEPH, a copper-dependent protein that functions in iron metabolism. Since HEPH requires copper for catalysis, it is a logical postulate that its expression or activity is influenced by copper levels. Recently, it was shown that intestinal FOX activity, which was attributed to HEPH, was diminished in copper-depleted mice, and the authors suggested that this contributed to the noted systemic ID (42). Another investigation provided evidence that intestinal iron absorption was downregulated in copper-deprived rats (250), which correlated with depression of intestinal HEPH activity (252). Furthermore, when copper-depleted rats were refed copper, intestinal iron absorption was normalized, which correlated with increases in HEPH protein expression on immunoblots (251). These more recent investigations in laboratory rodents support a much earlier study, which showed that intestinal iron absorption was impaired in copper-depleted pigs (172). Furthermore, other recent reports showed that HEPH directly interacted with FPN1 in rat enterocytes (322), and that this interaction was reduced upon iron feeding (321). Copper restriction, by influencing HEPH biosynthesis, could thus indirectly alter FPN1 protein levels on the enterocyte BLM by changing the dynamic interactions between HEPH and FPN1 (and thus possibly altering iron flux). These studies, in sum, provide evidence that copper depletion impairs intestinal iron absorption and that this is associated with inhibition of HEPH expression and activity. Other mechanisms could, however, also be involved since collectively, these investigations did not provide direct proof that decreased HEPH activity was the sole mediator of impaired iron absorption.
Copper metabolism is altered during iron deficiency
Copper homeostasis in the mammalian intestine is likely altered during ID. The seminal event causing this may be the induction of DMT1 expression by the IRE/IRP system in response to decreases in intracellular iron levels and also transactivation of SLC11A2 gene expression by HIF2α in response to hypoxia. Huge increases in BBM DMT1 protein levels, and the absence of competing iron atoms, may enhance DMT1-mediated copper transport into duodenal enterocytes (139). Supporting this supposition are the observations that enterocyte copper levels increase during ID (248) and also that expression of MTI and MTII is strongly induced (>20-fold) (51, 52, 103). MTs interact with multiple metals, but their affinity for copper is particularly high (144). Copper has been shown to transactivate MT gene expression by influencing DNA-binding activity of an intracellular metal-sensing, trans-acting factor, MTF-1 (325). Induction of MT expression may thus be a direct result of increasing intracellular copper levels in duodenal enterocytes. Moreover, expression of the copper transporter ATP7A is strongly induced during iron depletion (52,103,248), which could influence intracellular copper homeostasis and/or alter the biosynthesis of various cuproenzymes. How induction of ATP7A alters basic cellular physiology would depend upon its relative distribution between the plasma membrane and the TGN, but this has not been experimentally addressed to date so the impact of ATP7A induction is not currently understood in detail. Importantly, these coordinated molecular events related to copper homeostasis occur when iron transport is enhanced, suggesting that copper and/or ATP7A (in relation to copper metabolism or possibly independent of its role in copper metabolism) may positively influence intestinal iron metabolism. The possibility that ATP7A is important for intestinal iron flux was indeed recently tested in cell culture models of the mammalian intestinal epithelium, rat IEC-6, and human Caco-2 cells (111). This investigation provided evidence that depression of ATP7A expression impaired iron flux, supporting the postulate that ATP7A (and/or copper) positively influences intestinal iron absorption.
In the setting of induction of DMT1 expression, increased intracellular copper levels and induction of MT expression, and upregulation of intestinal ATP7A expression, hepatic copper concentrations also increase dramatically (52, 248). Hepatic copper loading, has in fact, been frequently observed during iron depletion in many mammalian species (87). The mechanism that causes liver copper concentrations to increase during ID, and the physiologic purpose of such, are not completely understood. Given upregulation of DMT1, which may transport copper, and ATP7A, which is a copper exporter, it is a logical postulate that enhanced intestinal copper flux during ID increases copper flow to the liver, which may then lead to hepatic copper accumulation. This also implies that biliary copper excretion is not enhanced, since this would presumably prevent hepatic copper accumulation. The physiologic purpose of copper accumulation could relate to enhancing biosynthesis of the circulating FOX CP, which mediates iron release from stores. This has been shown in iron-depleted rats (247). Hepatic iron-copper interactions will be considered in more detail in a subsequent section of this review.
Iron-copper interactions in the intestine are further exemplified by coordinate mechanisms that mediate the compensatory physiological response of the intestine to iron depletion. Low iron decreases hemoglobin production, thus leading to tissue hypoxia. Low iron/hypoxia in the gut stabilizes a hypoxia-responsive transcription factor, HIF2α, which transactivates several genes in duodenal enterocytes related to iron absorption (190,264). These include DCYTB, DMT1, FPN1 (283), and ATP7A (316). This molecular mechanism of upregulating key iron homeostasis-related genes is particularly intriguing in the context of this article, since the HIF transcriptional complex (i.e., a hypoxia-responsive HIFα subunit and a constitutively expressed HIFβ subunit) is stabilized by copper (79, 189). Increased copper in enterocytes (248) and the liver, and in blood during ID (53,76) may thus potentiate HIF activity, leading to enhanced transcription of genes that mediate whole-body and intestinal iron metabolism. It was also recently reported that HIFα subunits are stabilized by iron deprivation (in the absence of hypoxia) in in vivo and in vitro experimental settings (222). This may be caused by depressed activity of the HIF prolyl hydroxylases (since they are iron-dependent proteins), which mediate oxygen-dependent degradation of the HIFα subunits during normoxia. A recent study in human Caco-2 cells further supported these observations by demonstrating that iron deprivation (using an iron chelator) preferentially increased expression of known HIF-responsive genes (127). Regulation of gene expression by the HIFs is thus probably influenced by iron and copper levels, which is relevant here since genes regulated by HIF signaling relate to iron and copper homeostasis.
As alluded in the preceding text, it was recently demonstrated that the ATP7A copper transporter is a HIF2α target in the rat intestine (316, 317). A subsequent study provided further experimental support for the co-regulation of iron and copper transport-related genes (316) in the duodenal epithelium. These authors demonstrated that HIF2α induced ATP7A expression during hypoxia and that this induction also required the SP1 trans-acting factor. It was hypothesized that this same mechanism could also be involved in the upregulation of DCYTB and DMT1 during ID/hypoxia. This mechanism may also have implications for copper import by the CTR1 copper transporter, since the CTR1 gene was transactivated by HIF2α under basal conditions (233). These intriguing observations support the concept that expression of iron and copper homeostasis-related proteins may be regulated by a conserved mechanism, providing further evidence of the potential relevance of iron-copper interactions in the duodenal epithelium.
Additional investigations utilizing the human Caco-2 cell model of human enterocytes also revealed potentially important intersections between iron and copper metabolism. For example, one study demonstrated that iron or copper depletion increased influx of both metals (176), and further that iron or copper chelation increased iron transport. Copper supplemented Caco-2 cells had increased expression of genes encoding iron transporters, again demonstrating iron-copper interplay (117). Based upon these latter findings, it was hypothesized that copper exposure decreased intracellular iron concentrations, which then secondarily enhanced iron transport. The molecular mechanisms, which explain these observations are currently undefined, but nonetheless, these studies collectively further exemplify potentially important iron-copper interactions in IECs, thus providing the impetus for further investigation.
Iron overload and copper metabolism
It has been suggested that iron can interfere with copper utilization (148). An experiment in rats showed that higher dietary iron can increase the dietary requirement for copper (149). Moreover, high iron consumption has been reported to interfere with copper absorption in infants and adults (148). It has been further suggested that iron overload can perturb copper utilization (295, 296). For example, immunoreactive CP was reported to be decreased in hereditary hemochromatosis (HH), a genetic iron-loading disorder (67). Additional recent investigations in rats (112) and mice (110) demonstrated that high-iron intake can lead to copper depletion. In these studies, high-iron fed rodents developed severe copper deficiency-related pathologies, including growth retardation, cardiac hypertrophy, anemia, and impaired production of the liver-derived circulating FOX CP, which is a biomarker of moderate to severe copper deficiency (110, 112). Increasing dietary Cu prevented the development of these pathologies in rats, proving that copper deficiency was the underlying cause (112). In mice, high iron intake was suggested to interfere with copper absorption and tissue distribution (110). Collectively, these observations may be of particular physiological relevance since: (i) many Americans may have marginal copper intakes (148, 150); (ii) refined grain products are fortified with iron in the United States, increasing dietary iron consumption; and (iii) many individuals also consume iron supplements. This then leads to concern that higher iron consumption may disrupt copper homeostasis in some individuals, with possible pathological consequences. Copper depletion associated with high-iron intake could be most detrimental during pregnancy as copper deficiency has severe effects on the developing fetus (93, 293), and in children during periods of rapid growth. Most importantly, iron supplementation is widely recommended for pregnant women (34, 96, 256, 274). It has in fact, been suggested that iron supplements should contain extra copper (148), which is a reasonable recommendation since the upper tolerable intake limit (UL) for copper is > 10-fold above the RDA for copper, so a slight increase in copper intake should be harmless in the majority of the U.S. population. Overall, this is an important area of scientific pursuit that has been largely unexplored in recent years, and additional future experimentation is clearly warranted.
Intersection of Iron and Copper Metabolism in the Liver
Delivery of diet-derived iron and copper to the liver
Dietary iron is absorbed predominantly in the duodenum and upper jejunum, as described earlier. Subsequent to traversing the intestinal epithelium, iron enters the portal blood circulation as diferric-TF and travels to the liver. Diferric-TF enters hepatocytes by receptor-mediated endocytosis via TFR1 expressed on the cell surface (Fig. 3). Another TFR isoform, TFR2, may mediate uptake of diferric-TF in the liver as well (142). In human liver, TFR2 may be the predominant isoform (45). TFR2 may also function as an “iron sensor,” and thus be involved in the regulation of Hamp gene transcription in hepatocytes. After entry into hepatocytes, diferric- TF-containing endosomes are acidified by the action of an H+-ATPase, which results in iron release from TF into the endosomal lumen. Ferric iron is then reduced within the endosome by an unknown ferrireductase (possibly a STEAP family protein). Ferrous iron is then transported into the cytosol of the cell to enter the labile iron pool. This intracellular transfer step may be mediated by DMT1, as was originally proposed; however, mice lacking DMT1 (i.e., DMT1 KO mice) can take up iron into the liver normally (99). The same holds true for mice lacking DMT1 only in hepatocytes (i.e., hepatocyte-specific DMT1 KO mice) (299). Recent studies have provided evidence that endosomal iron transfer in hepatocytes may be mediated by ZIP14, which can transport iron and other metals (e.g., zinc, manganese) (94,177). Once in the cytosol, iron in the liver is utilized for metabolic purposes (e.g., synthesis of iron-containing proteins), stored in ferritin (if in excess), or exported (presumably by FPN1). Iron exported by FPN1 is in the ferrous state, so released iron must be oxidized by CP and/or GPI-CP to allow binding to TF and distribution in the blood.
After traversing the intestinal mucosa, diet-derived copper is bound in the portal blood by albumin or α2-macroglobulin. Recall that cuprous copper exported by ATP7A is probably spontaneously oxidized to cupric copper (Cu2+) by dissolved oxygen in the interstitial fluids. Further, since copper enters hepatocytes via CTR1, which transports Cu+ (212), cupric copper must be first reduced (Fig. 3). Like for iron, this may be accomplished by a STEAP family member protein (153). The physiologic role of hepatic CTR1 was probed by generating hepatocyte-specific CTR1 KO mice (146). Mice lacking CTR1 only in the liver showed a ~50% reduction in liver copper content, which correlated with similar decreases in the activity of some cuproenzymes. Other unidentified mechanisms must thus exist for copper to enter the liver. Once in hepatocytes, copper is bound to intracellular chaperones (as in enterocytes), which distribute copper to sites of the biosynthesis of copper-containing proteins. ATP7B, an ATP7A homolog, pumps copper into the TGN to support cuproen-zyme synthesis (including the biosynthesis of CP). When copper levels are elevated, ATP7B traffics to the canalicular membrane where it facilitates copper efflux into the bile (258). Copper excreted in bile is probably not available for reabsorption since it is complexed with bile salts. In addition to ATP7B, biliary copper secretion requires copper metabolism MURR1 domain 1 (COMMD1) (183). COMMD1 protein expression is controlled by X-linked inhibitor of apoptosis (XIAP), which ubiquitinates COMMD1 and targets it for degradation in the proteasome (184).
The interplay of iron and copper in the mammalian liver
The multi-copper FOX CP represents one well-known point of intersection between iron and copper in the liver since it is a copper-dependent protein that has a principal function in iron metabolism (i.e., release of iron from stores, including the liver, and some other tissues, e.g., the brain). Oxidation of ferrous iron released from tissues requires CP to permit binding to TF and distribution in the blood. The physiological role of CP in humans is well established since patients with inactivating CP mutations have been identified. CP mutation results in a very rare genetic disease called aceruloplasminemia (120). Patients devoid of CP load iron in parenchymal tissues, which causes oxidative damage to the liver (and to a lesser extent to other tissues) (122, 286), progressive neurological and retinal degeneration, and diabetes (155). These symptoms are very similar to what is seen in the genetic iron-loading disorder HH, which is caused by dysregulation of hepatic hepcidin expression. The membrane-anchored form of CP (GPI-CP) is also expressed in liver, so it presumably complements circulating CP function in this (and other) organs (202). Mice lacking CP have a similar phenotype to patients with aceruloplasminemia (119). Moreover, copper-depleted rodents accumulate liver iron, probably since CP activity is diminished in copper deficiency (28). Additionally, circulating CP levels increase during iron depletion (134, 247), perhaps due to enhanced biosynthesis of the holo (i.e., active, copper-containing) form of the enzyme, which could be potentiated by copper loading in hepatocytes. Furthermore, CP may be linked to alterations in hepatic iron metabolism in those suffering from an increasingly common liver disorder, nonalcoholic fatty liver disease (NAFLD) (2).
The STEAP family of metalloreductases represent another possible link between iron and copper metabolism in the liver (153). Some STEAP proteins, STEAP2/3/4 in particular, can reduce both iron and copper, which enhances transport of both metals into cells (95, 217). Mice lacking STEAP3 (i.e., STEAP3 knockout mice) have impaired utilization of TF-bound iron in erythroid cells (216), which results in the development of iron-deficiency anemia. This may be caused by defective ferric iron reduction in endosomes, which would impair iron efflux into the cytosol (by DMT1 or ZIP14) and subsequent utilization for erythropoiesis. Interestingly, STEAP3 is robustly expressed in hepatocytes, which acquire iron from diferric-TF. STEAP3 could thus play an important role in hepatic iron metabolism. Surprisingly though, STEAP3 KO mice have higher hepatic iron and copper concentrations than control littermates (163). Although these observations do not necessarily clarify the role of STEAP3 in hepatic iron/copper homeostasis, they provide impetus for further analysis of STEAP3 in liver mineral metabolism.
An inverse relationship has been noted between iron and copper status and liver accumulation of both metals. For example, iron-depleted rats load hepatic copper in the liver (53, 219, 248, 276), and copper-deprived rats (and mice) load hepatic iron (49, 202, 273, 309). Hepatic iron loading in copper deficiency is probably explained by depression of CP activity, which would impair iron release from the liver (and other tissues) (218). Why hepatic copper levels increase during ID, and the physiologic/pathologic consequences of such are not known. Hepatic copper loading during ID may be associated with increased copper absorption and/or decreased biliary copper excretion. The first possibility is supported by the following molecular events which occur during ID: (i) DCYTB expression increases (46) and it can reduce copper (in addition to iron) (315); (ii) DMT1 expression increases dramatically, and it was shown to transport copper under these conditions (139); and (iii) ATP7A is strongly induced in parallel with DCYTB and DMT1 and one of its functions is to export excess copper into the portal circulation (51, 52). Earlier studies in iron-deprived rats, however, found no increase in copper absorption (232), but the experimental approach only considered total body copper accumulation as a surrogate for absorption (i.e., possible copper redistribution was not accounted for). Copper accumulated in the intestinal mucosa during ID (78, 248) could have positive influences on enterocyte iron homeostasis, and some of this excess copper could be transferred to the liver (potentially increasing copper content there). Hepatic copper accumulation could also be caused by lack of mobilization of copper into bile for excretion, impaired cuproenzyme synthesis (e.g., CP, which would lead to hepatic copper accumulation since most serum copper is in CP), or impaired non-CP copper export from the liver (note that some copper exits the liver via other mechanisms that do not involve CP secretion). Thus, copper redistribution and accumulation in some tissues could positively influence iron metabolism, while whole-body copper levels are not altered.
Lastly, possible copper-dependent regulation of the iron-regulatory hormone hepcidin has been recently experimentally assessed. For example, one recent study showed that copper treatment induced hepcidin expression in hepatoma cells via transactivation of the Hamp gene promoter by the metal-responsive transcription factor MTF1 (13). In addition, hepcidin expression is diminished in rats (27, 138) and mice (42) in response to copper depletion. Moreover, the biologically active from of the hepcidin peptide (hepcidin-25), binds copper with high affinity (292). It was suggested that copper was required for hepcidin activity, suggesting that perturbations in copper status could potentially alter iron metabolism via changes in hepcidin function. Another recent study, however, concluded that the level of interaction between HEPC and copper was likely insufficient to influence the ability of HEPC to regulate iron homeostasis (161). Moreover, the copper-binding properties of hepcidin-25 were recently utilized in the development of a novel detection assay for human hepcidin in serum (156). In this assay, hepcidin present in serum is labeled with copper, and then copper is quantified by HPLC and ICP-MS, supposedly then reflecting the amount of hepcidin in the original sample. Furthermore, copper may enhance the antimicrobial activity and bactericidal properties of hepcidin-25 (185).
Intersection of Iron-Copper Metabolism: Fruitful Areas for Future Research
Iron-copper interactions have been recognized since the 1800s, but the molecular basis for many of these interactions is just now being elucidated. Recent investigations from research groups worldwide have begun to provide mechanistic insight into the intersection of iron and copper metabolism, and the physiologic/pathological significance of these interactions. This review pointed out areas of recent progress and also highlighted others where follow-up investigative work has been lacking. Additional experimentation in these areas should provide novel insight into how copper influences iron metabolism, which is of clinical significance given the morbidity and mortality associated with iron-deficiency and iron-overload-related pathologies in humans. Recent investigation of copper and copper transporters in relation to neurodegenerative disease, immune function, and cancer (109, 301) provides rationale for further consideration of how iron influences copper homeostasis. Many potentially important, unanswered questions remain regarding the intersection of these two essential trace minerals, including: (i) Does DMT1 potentiate copper absorption during ID? Does it transport copper during other physiologic or pathologic conditions in humans? Do alterations in dietary copper intake impact iron transport by DMT1?; (ii Is DCYTB a “cuprireductase” in the duodenal epithelium, thus potentiating copper absorption? Are other metalloreductases present on the enterocyte BBM, and if so, are they ferri- and cuprireductases?; (iii) Does circulating CP complement intestinal HEPH function and thus positively influence intestinal iron transport? Are changes in CP activity associated with the compensatory response of the intestinal epithelium to iron deprivation? Does decreased CP activity during copper deprivation negatively impact intestinal iron transport in humans?; (iv) Are impairments in iron absorption during copper deficiency only related to inhibition of HEPH expression and function? Does copper depletion alter the FPN1-HEPH (or other FOXs) functional couple and thus modulate the rate-limiting step of iron absorption (i.e., the transfer step)?; (v) How does enhanced copper import into enterocytes impact iron absorption during iron depletion? Does copper redistribution in duodenal enterocytes during ID impact iron metabolism? Does ATP7A function in ente-rocytes positively influence iron absorption? Its coregulation with iron transporters suggests that it indeed does.; (vi) Does copper impact HIF activity in cells that play a principle role in regulating iron homeostasis (enterocytes, hepatocytes, and RE macrophages)? This is an important question since copper is required for HIF-dependent transactivation of gene expression in some model systems (79, 240).; (vii) What causes hepatic copper loading during iron depletion, and how might this impact whole-body iron metabolism? Does hepatic copper accumulation potentiate the biosynthesis of CP in hepatocytes?; (viii) What is the mechanism by which copper depletion causes anemia (i.e., what is the specific copper-dependent step in iron utilization by developing erythrocytes)? (ix) And lastly, does iron supplementation increase the risk for developing copper deficiency-related pathologies in humans? Emerging evidence suggests that this may indeed be the case. If so then, should iron supplements contain copper so as to avoid any untoward effects of high-iron supplementation on copper metabolism?
Concluding Remarks
Recent investigations into the intersection of iron and copper metabolism in rats and mice have revealed novel nuances of mineral homeostasis in mammals and have increased our understanding of pathological mechanisms of disease. Importantly, many of these studies have clear relevance to human physiology and pathophysiology. One important outcome of the efforts of numerous investigators over the past several decades relates to the influence of copper on iron metabolism. This is particularly critical given that dysregulation of iron homeostasis directly relates to the appearance of numerous clinically-relevant pathologies in humans. Strikingly, copper accumulates in tissues, which regulate overall body iron homeostasis during ID, including the duodenal epithelium, the liver, and blood. Copper redistribution to these tissues likely represents physiological compensation to enhance iron mobilization from the diet and stores to support erythropoiesis. Extra copper in enterocytes may enhance iron transport and delivery to the liver. Hepatic copper loading may potentiate CP production, which would increase iron mobilization from stores to the bone marrow. The multi-copper FOXs clearly mediate important functions related to iron metabolism, and logically, their activity is altered by copper loading or depletion, but additional copper-dependent mechanisms, which influence iron homeostasis, are likely to be discovered in the future. Moreover, investigations of copper depletion have also revealed that copper and cuproenzymes can influence iron homeostasis, independent of influences on HEPH or CP expression or activity. Given the established physiologic relationships between iron and copper, it is important to determine if copper may also influence iron homeostasis during states of pathologic iron-overload (e.g., HH, and iron-loading anemias such as β-thalassemia). Emerging evidence provides us with important clues that iron overload disrupts copper homeostasis, but the converse situation (i.e., copper influencing the iron overload phenotype) is just now being experimentally considered.
Didactic Synopsis.
Major teaching points
Iron and copper are essential nutrients for humans since they mediate numerous important physiologic functions; deficiency of either is associated with significant pathophysiologic outcomes.
Iron and copper exist in two oxidation states in biological systems, and high redox potentials lead to toxicity in cells and tissues when in excess.
Iron and copper atoms have similar physiochemical properties, and as such, interactions between them are predictable.
Both minerals are absorbed by duodenal enterocytes, after first being reduced in the gut lumen from their predominant dietary forms.
Intestinal and hepatic ferrireductases, such as duodenal cytochrome B or STEAP proteins, may promote iron and copper reduction, which is required for absorption into enterocytes and subsequent uptake into hepatocytes.
Two multicopper ferroxidases perhaps best exemplify iron-copper interactions: hephaestin (HEPH) in duodenal enterocytes and ceruloplasmin (CP) circulating in the plasma. HEPH is required for optimal intestinal iron absorption during physiologic conditions and during pregnancy, while CP is required for iron release from stores and other tissues (e.g., brain).
During iron deficiency, copper transport into duodenal enterocytes increases, possibly promoting iron absorption. This may be mediated by the principal intestinal iron transporter, divalent metal-ion transporter 1 (DMT1), which can also transport copper.
Hepatic copper accumulation during iron deficiency may enhance biosynthesis of CP.
Intestinal genes encoding iron transporters are regulated by a hypoxia-inducible transcription factor, HIF2α. Copper enhances the DNA-binding activity of the HIFs, exemplifying another way in which copper may influence iron homeostasis.
Iron overload, as seen in the genetic disease hereditary hemochromatosis, may impair copper utilization. Moreover, high-dose iron supplementation may increase risk for copper depletion in humans, and as such, it has been suggested that iron supplements should also contain copper.
Acknowledgements
The authors of this manuscript are supported by grants R01 DK074867 from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and R01 DK109717 from NIDDK and the Office of Dietary Supplements (J. F. Collins, PI).
References
- 1.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 275: 19906–19912, 2000. [DOI] [PubMed] [Google Scholar]
- 2.Aigner E, Theurl I, Haufe H, Seifert M, Hohla F, Scharinger L, Stickel F, Mourlane F, Weiss G, Datz C. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology 135: 680–688, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Alexander RT, Rievaj J, Dimke H. Paracellular calcium transport across renal and intestinal epithelia. Biochem Cell Biol 92: 467–480, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Anderson GJ, Frazer DM, McKie AT, Vulpe CD. The ceruloplasmin homolog hephaestin and the control of intestinal iron absorption. Blood Cells Mol Dis 29: 367–375, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Anderson GJ, Frazer DM, McLaren GD. Iron absorption and metabolism. Curr Opin Gastroenterol 25: 129–135, 2009. [DOI] [PubMed] [Google Scholar]
- 6.Anderson GJ, Powell LW, Halliday JW. The endocytosis of transferrin by rat intestinal epithelial cells. Gastroenterology 106: 414–422, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Anderson GJ, Vulpe CD. Regulation of intestinal iron transport In: Templeton DM, editor. Molecular and Cellular Iron Transport. New York: Marcel Dekker, 2002, pp.559–598. [Google Scholar]
- 8.Anderson GJ, Vulpe CD. Mammalian iron transport. Cell Mol Life Sci 66: 3241–3261, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andrews SC, Treffry A, Harrison PM. Siderosomal ferritin. The missing link between ferritin and haemosiderin? Biochem J 245:439–446, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arosio P, Levi S. Ferritins: Structure and functional aspects In: Templeton DM, editor. Molecular and Cellular Iron Transport. New York: Marcel Dekker, 2002, pp. 125–154. [Google Scholar]
- 11.Arredondo M, Mendiburo MJ, Flores S, Singleton ST, Garrick MD. Mouse divalent metal transporter 1 is a copper transporter in HEK293 cells. Biometals 27: 115–123, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Arredondo M, Munoz P, Mura CV, Nunez MT. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. Am J Physiol Cell Physiol 284: C1525–C1530, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Balesaria S, Ramesh B, McArdle H, Bayele HK, Srai SK. Divalent metal-dependent regulation of hepcidin expression by MTF-1. FEBS Lett 584:719–725, 2010. [DOI] [PubMed] [Google Scholar]
- 14.Bannerman RM. Quantitative aspects of hemoglobin-iron absorption. J Lab Clin Med 65: 944–950, 1965. [PubMed] [Google Scholar]
- 15.Bardou-Jacquet E, Island ML, Jouanolle AM, Detivaud L, Fatih N, Ropert M, Brissot E, Mosser A, Maisonneuve H, Brissot P, Loreal O. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol Dis 47: 243–248, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Baye K, Guyot JP, Mouquet-Rivier C. The unresolved role of dietary fibers on mineral absorption. Crit Rev Food Sci Nutr 57:949–957, 2017. [DOI] [PubMed] [Google Scholar]
- 17.Iron Beard J. In: Bowman BA, editor. Present Knowledge in Nutrition. Washington, D.C.: ILSI Press, 2006, pp. 430–444. [Google Scholar]
- 18.Becker G, Korpilla-Schafer S, Osterloh K, Forth W. Capacity of the mucosal transfer system and absorption of iron after oral administration in rats. Blut 38: 127–134, 1979. [DOI] [PubMed] [Google Scholar]
- 19.Bernstein SE. Hereditary hypotransferrinemia with hemosiderosis, a murine disorder resembling human atransferrinemia. J Lab Clin Med 110: 690–705, 1987. [PubMed] [Google Scholar]
- 20.Bezwoda WR, Bothwell TH, Charlton RW, Torrance JD, MacPhail AP, Derman DP, Mayet F. The relative dietary importance of haem and non-haem iron. S Afr Med J 64: 552–556, 1983. [PubMed] [Google Scholar]
- 21.Black MM, Quigg AM, Hurley KM, Pepper MR. Iron deficiency and iron-deficiency anemia in the first two years of life: Strategies to prevent loss of developmental potential. Nutr Rev 69(Suppl 1): S64–S70, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Blanck HM, Cogswell ME, Gillespie C, Reyes M. Iron supplement use and iron status among US adults: Results from the third National Health and Nutrition Examination Survey. Am J Clin Nutr 82: 1024–1031, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Bodansky M The zinc and copper content of the human brain. J Biol Chem 48:361–364, 1921. [Google Scholar]
- 24.Boender CA, Verloop MC. Iron absorption, iron loss and iron retention in man: Studies after oral administration of a tracer dose of 59FeSO4 and 131-BaSO4. Br J Haematol 17: 45–58, 1969. [DOI] [PubMed] [Google Scholar]
- 25.Brasse-Lagnel C, Karim Z, Letteron P, Bekri S, Bado A, Beaumont C. Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology 140: 1261–1271 e1261, 2011. [DOI] [PubMed] [Google Scholar]
- 26.Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 361:669–673, 2003. [DOI] [PubMed] [Google Scholar]
- 27.Broderius M, Mostad E, Prohaska JR. Suppressed hepcidin expression correlates with hypotransferrinemia in copper-deficient rat pups but not dams. Genes Nutr 7: 405–414, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broderius M, Mostad E, Wendroth K, Prohaska JR. Levels of plasma ceruloplasmin protein are markedly lower following dietary copper deficiency in rodents. Comp Biochem Physiol C Toxicol Pharmacol 151: 473–479, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bronner F Calcium absorption—a paradigm for mineral absorption. J Nutr 128:917–920, 1998. [DOI] [PubMed] [Google Scholar]
- 30.Brown EB, Hwang YF, Nicol S, Ternberg J. Absorption of radiationlabeled hemoglobin by dogs. J Lab Clin Med 72: 58–64, 1968. [PubMed] [Google Scholar]
- 31.Brubaker C, Sturgeon P. Copper deficiency in infants; a syndrome characterized by hypocupremia, iron deficiency anemia, and hypoproteinemia. AMA 92: 254–265, 1956. [PubMed] [Google Scholar]
- 32.Callender ST, Mallett BJ, Smith MD. Absorption of haemoglobin iron. Br J Haematol 3: 186–192, 1957. [DOI] [PubMed] [Google Scholar]
- 33.Canonne-Hergaux F, Gruenheid S, Ponka P, Gros P. Cellular and subcellular localization of the Nramp2 iron transporter in the intestinal brush border and regulation by dietary iron. Blood 93: 4406–4417, 1999. [PubMed] [Google Scholar]
- 34.Cao C, O’Brien KO. Pregnancy and iron homeostasis: An update. Nutr Rev 71: 35–51, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Carpenter CE, Mahoney AW. Contributions of heme and nonheme iron to human nutrition. Crit Rev Food Sci Nutr 31: 333–367, 1992. [DOI] [PubMed] [Google Scholar]
- 36.Casareno RL, Waggoner D, Gitlin JD. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J Biol Chem 273: 23625–23628, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Chang YJ, Jo MY, Hwang EH, Park CU, Kim KS. Recovery from iron deficiency in rats by the intake of recombinant yeast producing human H-ferritin. Nutrition 21: 520–524, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Charlton RW, Jacobs P, Torrance JD, Bothwell TH. The role of the intestinal mucosa in iron absorption. J Clin Invest 44: 543–554, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chauhan AS, Rawat P, Malhotra H, Sheokand N, Kumar M, Patidar A, Chaudhary S, Jakhar P, Raje CI, Raje M. Secreted multifunctional Glyceraldehyde-3-phosphate dehydrogenase sequesters lactoferrin and iron into cells via a non-canonical pathway. Sci Rep 5: 18465, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H, Attieh ZK, Su T, Syed BA, Gao H, Alaeddine RM, Fox TC, Usta J, Naylor CE, Evans RW, McKie AT, Anderson GJ, Vulpe CD. Hephaestin is a ferroxidase that maintains partial activity in sex-linked anemia mice. Blood 103: 3933–3939, 2004. [DOI] [PubMed] [Google Scholar]
- 41.Chen H, Attieh ZK, Syed BA, Kuo YM, Stevens V, Fuqua BK, Andersen HS, Naylor CE, Evans RW, Gambling L, Danzeisen R, Bacouri- Haidar M, Usta J, Vulpe CD, McArdle HJ. Identification of zyklopen, a new member of the vertebrate multicopper ferroxidase family, and characterization in rodents and human cells. J Nutr 140: 1728–1735, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen H, Huang G, Su T, Gao H, Attieh ZK, McKie AT, Anderson GJ, Vulpe CD. Decreased hephaestin activity in the intestine of copper-deficient mice causes systemic iron deficiency. J Nutr 136: 1236–1241, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Chernelch M, Fawwaz R, Sargent T, Winchell HS.Effect of phlebotomy and pH on iron absorption from the colon. J Nucl Med 11: 25–27, 1970. [PubMed] [Google Scholar]
- 44.Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab 2: 309–319, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Chloupkova M, Zhang AS, Enns CA. Stoichiometries of transferrin receptors 1 and 2 in human liver. Blood Cells Mol Dis 44: 28–33, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi J, Masaratana P, Latunde-Dada GO, Arno M, Simpson RJ, McKie AT. Duodenal reductase activity and spleen iron stores are reduced and erythropoiesis is abnormal in Dcytb knockout mice exposed to hypoxic conditions. J Nutr 142: 1929–1934, 2012. [DOI] [PubMed] [Google Scholar]
- 47.Chowrimootoo G, Debnam ES, Srai SK, Epstein O. Regional characteristics of intestinal iron absorption in the guinea-pig. Exp Physiol 77: 177–183, 1992. [DOI] [PubMed] [Google Scholar]
- 48.Chung B, Chaston T, Marks J, Srai SK, Sharp PA. Hepcidin decreases iron transporter expression in vivo in mouse duodenum and spleen and in vitro in THP-1 macrophages and intestinal Caco-2 cells. J Nutr 139: 1457–1462, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Chung J, Prohaska JR, Wessling-Resnick M. Ferroportin-1 is not upregulated in copper-deficient mice. J Nutr 134: 517–521, 2004. [DOI] [PubMed] [Google Scholar]
- 50.Cohn EJ, Minot GR, Fulton JF, Ulrichs HF, Sargent FC, Weare JH, Murphy WP. The nature of the material in liver effective in pernicious anemia: I. J Biol Chem 1xxiv:1xix, 1927. [Google Scholar]
- 51.Collins JF. Gene chip analyses reveal differential genetic responses to iron deficiency in rat duodenum and jejunum. Biol Res 39: 25–37, 2006. [PMC free article] [PubMed] [Google Scholar]
- 52.Collins JF, Franck CA, Kowdley KV, Ghishan FK. Identification of differentially expressed genes in response to dietary iron deprivation in rat duodenum. Am J Physiol Gastrointest Liver Physiol 288: G964–G971, 2005. [DOI] [PubMed] [Google Scholar]
- 53.Collins JF, Hu Z, Ranganathan PN, Feng D, Garrick LM, Garrick MD, Browne RW. Induction of arachidonate 12-lipoxygenase (Alox15) in intestine of iron-deficient rats correlates with the production of biologically active lipid mediators. Am J Physiol Gastrointest Liver Physiol 294: G948–G962, 2008. [DOI] [PubMed] [Google Scholar]
- 54.Collins JF, Prohaska JR, Knutson MD. Metabolic crossroads of iron and copper. Nutr Rev 68: 133–147, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Conrad ME, Benjamin BI, Williams HL, Foy AL. Human absorption of hemoglobin-iron. Gastroenterology 53: 5–10, 1967. [PubMed] [Google Scholar]
- 56.Conrad ME Jr., Crosby WH Intestinal mucosal mechanisms controlling Iron absorption. Blood 22: 406–415, 1963. [PubMed] [Google Scholar]
- 57.Conrad ME, Parmley RT, Osterloh K. Small intestinal regulation of iron absorption in the rat. J Lab Clin Med 110: 418–426, 1987. [PubMed] [Google Scholar]
- 58.Conrad ME, Weintraub LR, Sears DA, Crosby WH. Absorption of hemoglobin iron. Am J Physiol 211:1123–1130, 1966. [DOI] [PubMed] [Google Scholar]
- 59.Coppen DE, Davies NT. Studies on the roles of apotransferrin and caeruloplasmin (EC 1.16.3.1)on iron absorption in copper-deficient rats using an isolated vascularly- and luminally-perfused intestinal preparation. Br J Nutr 60: 361–373, 1988. [DOI] [PubMed] [Google Scholar]
- 60.Courville P, Chaloupka R, Cellier MF. Recent progress in structurefunction analyses of Nramp proton-dependent metal-ion transporters. Biochem Cell Biol 84: 960–978, 2006. [DOI] [PubMed] [Google Scholar]
- 61.Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: A rodent model for hemochromatosis Proc Natl Acad Sci U S A 84: 3457–3461, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dagg JH, Kuhn IN, Templeton FE, Finch CA. Gastric absorption of iron Gastroenterology 53: 918–922, 1967. [PubMed] [Google Scholar]
- 63.Davila-Hicks P, Theil EC, Lonnerdal B. Iron in ferritin or in salts (ferrous sulfate) is equally bioavailable in nonanemic women. Am J Clin Nutr 80: 936–940, 2004. [DOI] [PubMed] [Google Scholar]
- 64.De Domenico I, McVey Ward D, Nemeth E, Ganz T, Corradini E, Ferrara F, Musci G, Pietrangelo A, Kaplan J. Molecular and clinical correlates in iron overload associated with mutations in ferroportin Haematologica 91: 1092–1095, 2006. [PMC free article] [PubMed] [Google Scholar]
- 65.De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, Musci G, Kaplan J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J 26: 2823–2831, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Domenico I, Ward DM, Musci G, Kaplan J. Evidence for the mul¬timeric structure of ferroportin. Blood 109: 2205–2209, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dib N, Valsesia E, Malinge MC, Mauras Y, Misrahi M, Cales P. Late onset of Wilson’s disease in a family with genetic haemochromatosis. Eur J Gastroenterol Hepatol 18: 43–47, 2006. [DOI] [PubMed] [Google Scholar]
- 68.Doherty K, Connor M, Cruickshank R. Zinc-containing denture adhe¬sive: A potential source of excess zinc resulting in copper deficiency myelopathy. Br Dent J 210: 523–525, 2011. [DOI] [PubMed] [Google Scholar]
- 69.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 403: 776–781, 2000. [DOI] [PubMed] [Google Scholar]
- 70.Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, Andrews NC. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell metabolism 1: 191–200, 2005. [DOI] [PubMed] [Google Scholar]
- 71.Dowdle EB, Schachter D, Schenker H. Active transport of Fe59 by everted segments of rat duodenum. Am J Physiol 198: 609–613, 1960. [DOI] [PubMed] [Google Scholar]
- 72.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22: 7340–7358, 2003. [DOI] [PubMed] [Google Scholar]
- 73.Dube C, Rostom A, Sy R, Cranney A, Saloojee N, Garritty C, Sampson M, Zhang L, Yazdi F, Mamaladze V, Pan I, Macneil J, Mack D, Patel D, Moher D. The prevalence of celiac disease in average-risk and at-risk Western European populations: A systematic review. Gastroenterology 128: S57–S67, 2005. [DOI] [PubMed] [Google Scholar]
- 74.DuBois S, Kearney DJ. Iron-deficiency anemia and Helicobacter pylori infection: A review of the evidence. Am J Gastroenterol 100: 453–459, 2005. [DOI] [PubMed] [Google Scholar]
- 75.Duthie HL. The relative importance of the duodenum in the intestinal absorption of iron. Br J Haematol 10: 59–68, 1964. [DOI] [PubMed] [Google Scholar]
- 76.Ece A, Uyanik BS, Iscan A, Ertan P, Yigitoglu MR. Increased serum copper and decreased serum zinc levels in children with iron deficiency anemia. Biol Trace Elem Res 59: 31–39, 1997. [DOI] [PubMed] [Google Scholar]
- 77.Copper EDH In:O’Dell BLS RA, editors. Clinical Nutrition in Health and Disease: Handbook of Nutritionally Essential Mineral Elements. New York: Marcel Dekker, 1997, pp. 231–373. [Google Scholar]
- 78.El-Shobaki FA, Rummel W. Binding of copper to mucosal transferrin and inhibition of intestinal iron absorption in rats. Res Exp Med (Berl) 174: 187–195, 1979. [DOI] [PubMed] [Google Scholar]
- 79.Feng W, Ye F, Xue W, Zhou Z, Kang YJ. Copperregulationofhypoxia- inducible factor-1 activity. Mol Pharmacol 75: 174–182, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fillebeen C, Gkouvatsos K, Fragoso G, Calve A, Garcia-Santos D, Buffler M, Becker C, Schumann K, Ponka P, Santos MM, Pantopoulos K. Mice are poor heme absorbers and do not require intestinal Hmox1 for dietary heme iron assimilation. Haematologica 100: e334–e337, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Finch CA, Ragan HA, Dyer IA, Cook JD. Body iron loss in animals. Proc Soc Exp Biol Med 159: 335–338, 1978. [DOI] [PubMed] [Google Scholar]
- 82.Fischer PW, Giroux A, L’Abbe MR. Effect of zinc supplementation on copper status in adult man. Am J Clin Nutr 40: 743–746, 1984. [DOI] [PubMed] [Google Scholar]
- 83.Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: Evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A 95: 1148–1153, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fleming MD, Trenor CC 3rd, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet 16: 383–386, 1997. [DOI] [PubMed] [Google Scholar]
- 85.Foot NJ, Dalton HE, Shearwin-Whyatt LM, Dorstyn L, Tan SS, Yang b, Kumar S. Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2. Blood 112: 4268–4275, 2008. [DOI] [PubMed] [Google Scholar]
- 86.Forth W, Rummel W. Iron absorption. Physiol Rev 53: 724–792, 1973. [DOI] [PubMed] [Google Scholar]
- 87.Fox PL. The copper-iron chronicles: The story of an intimate relationship. Biometals 16: 9–40, 2003. [DOI] [PubMed] [Google Scholar]
- 88.Frazer DM, Inglis HR, Wilkins SJ, Millard KN, Steele TM, McLaren GD, McKie AT, Vulpe CD, Anderson GJ. Delayed hepcidin response explains the lag period in iron absorption following a stimulus to increase erythropoiesis. Gut 53: 1509–1515, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Frazer DM, Vulpe CD, McKie AT, Wilkins Sj, Trinder D, Cleghorn GJ, Anderson GJ. Cloning and gastrointestinal expression of rat hephaestin: Relationship to other iron transport proteins. Am J Physiol Gastrointest LiverPhysiol 281: G931–G939, 2001. [DOI] [PubMed] [Google Scholar]
- 90.Frazer DM, Wilkins SJ, Becker EM, Murphy TL, Vulpe CD, McKie AT, Anderson GJ. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut 52: 340–346, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Freitas Kde C, Amancio OM, de Morais MB. High-performance inulin and oligofructose prebiotics increase the intestinal absorption of iron in rats with iron deficiency anaemia during the growth phase. Br J Nutr 108: 1008–1016, 2012. [DOI] [PubMed] [Google Scholar]
- 92.Fuqua BK, Lu Y, Darshan D, Frazer DM, Wilkins SJ, Wolkow N, Bell AG, Hsu J, Yu CC, Chen H, Dunaief JL, Anderson GJ, Vulpe CD. The multicopper ferroxidase hephaestin enhances intestinal iron absorption in mice. PLoS One 9: e98792, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gambling L, Kennedy C, McArdle HJ. Iron and copper in fetal development. Semin Cell Dev Biol 22: 637–644, 2011. [DOI] [PubMed] [Google Scholar]
- 94.Gao J, Zhao N, Knutson MD, Enns CA. The hereditary hemochromatosis protein, HFE, inhibits iron uptake via down-regulation of Zip14 in HepG2 cells. JBiol Chem 283: 21462–21468, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gauss GH, Kleven MD, Sendamarai AK, Fleming MD, Lawrence CM. The crystal structure of six-transmembrane epithelial antigen of the prostate 4 (Steap4), aferri/cuprireductase, suggests a novel interdomain flavin-binding site. J Biol Chem 288: 20668–20682, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gautam CS, Saha L, Sekhri K, Saha PK. Iron deficiency in pregnancy and the rationality of iron supplements prescribed during pregnancy. Medscape J Med 10: 283, 2008. [PMC free article] [PubMed] [Google Scholar]
- 97.Gitlin D, Cruchaud A. On the kinetics of iron absorption in mice. J Clin Invest 41:344–350, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gitlin JD, Schroeder JJ, Lee-Ambrose LM, Cousins RJ. Mechanisms of caeruloplasmin biosynthesis in normal and copper-deficient rats. Biochem J 282(Pt3): 835–839, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Graham RM, Chua AC, Herbison CE, Olynyk JK, Trinder D. Liver iron transport. World J Gastroenterol 13: 4725–4736, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grasbeck R, Kouvonen I, Lundberg M, Tenhunen R. An intestinal receptor for heme. Scand J Haematol 23: 5–9, 1979. [DOI] [PubMed] [Google Scholar]
- 101.Greenberger NJ, Balcerzak SP, Ackerman GA. Iron uptake by isolated intestinal brush borders: Changes induced by alterations in iron stores. J Lab Clin Med 73:711–721, 1969. [PubMed] [Google Scholar]
- 102.Gulec S, Anderson GJ, Collins JF. Mechanistic and regulatory aspects of intestinal iron absorption. Am J Physiol Gastrointest Liver Physiol 307: G397–G409, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gulec S, Collins JF. Investigation of ironmetabolismin mice expressing a mutant Menke’s copper transporting ATPase (Atp7a) protein with diminished activity (Brindled; Mo (Br) (/y)). PLoS One 8: e66010, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gulec S, Collins JF. Silencing the Menkes copper-transporting ATPase (Atp7a) gene in rat intestinal epithelial (IEC-6) cells increases iron flux via transcriptional induction of ferroportin 1 (Fpn1). J Nutr 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gulec S, Collins JF. Molecular mediators governing iron-copper interactions. Annu Rev Nutr 34: 95–116, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Robine S, Andrews NC.Slc11a2is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J Clin Invest 115: 1258–1266, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: 482–488, 1997. [DOI] [PubMed] [Google Scholar]
- 108.Gunshin H, Starr CN, Direnzo C, Fleming MD, Jin J, Greer EL, Sellers VM, Galica SM, Andrews NC. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood 106: 2879–2883, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gupta A, Lutsenko S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future medicinal chemistry 1: 1125–1142, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ha JH, Doguer C, Collins JF. Consumption of a high-iron diet disrupts homeostatic regulation of intestinal copper absorption in adolescent mice. Am J Physiol Gastrointest Liver Physiol 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ha JH, Doguer C, Collins JF. Knockdown of copper-transporting ATPase 1 (Atp7a) impairs iron flux in fully-differentiated rat (IEC-6) and human (Caco-2) intestinal epithelial cells. Metallomics 8: 963–972, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ha JH, Doguer C, Wang X, Flores SR, Collins JF. High-iron consumption impairs growth and causes copper-deficiency anemia in weanling Sprague-Dawley rats. PLoS One 11: e0161033, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hahn PF, Bale WF, Ross JF, Balfour WM, Whipple GH. Radioactive iron absorption by gastro-intestinal tract: Influence of anemia, anoxia, and antecedent feeding distribution in growing dogs. J Exp Med 78: 169–188, 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hallberg L, Solvell L. Absorption of hemoglobin iron in man. Acta MedScand 181: 335–354, 1967. [DOI] [PubMed] [Google Scholar]
- 115.Halterman JS, Kaczorowski JM, Aligne CA, Auinger P, Szilagyi PG. Iron deficiency and cognitive achievement among school-aged children and adolescents in the United States. Pediatrics 107: 1381–1386, 2001. [DOI] [PubMed] [Google Scholar]
- 116.Han O, Kim EY. Colocalization of ferroportin-1 with hephaestin on the basolateral membrane of human intestinal absorptive cells. J Cell Biochem 101: 1000–1010, 2007. [DOI] [PubMed] [Google Scholar]
- 117.Han O, Wessling-Resnick M. Copper repletion enhances apical iron uptake and transepithelial iron transport by Caco-2 cells. Am J Physiol Gastrointest Liver Physiol 282: G527–G533, 2002. [DOI] [PubMed] [Google Scholar]
- 118.Harris PR, Serrano CA, Villagran A, Walker MM, Thomson M, Duarte I, Windle HJ, Crabtree JE. Helicobacter pylori-associated hypochlorhy- dria in children, and development of iron deficiency. J Clin Pathol 66: 343–347, 2013. [DOI] [PubMed] [Google Scholar]
- 119.Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc NatlAcadSciUSA 96: 10812–10817, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivray RT, Gitlin JD. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci U S A 92: 2539–2543, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Harrison PM, Arosio P. The ferritins: Molecular properties, iron storage function and cellularregulation. Biochim Biophys Acta 1275: 161–203, 1996. [DOI] [PubMed] [Google Scholar]
- 122.Hayashi H, Hattori A, Tatsumi Y, Hayashi K, Katano Y, Ueyama J, Wakusawa S, Yano M, Goto H. Various copper and iron overload patterns in the livers of patients with Wilson disease and idiopathic copper toxicosis. Med Mol Morphol 46: 133–140, 2013. [DOI] [PubMed] [Google Scholar]
- 123.Hedera P, Peltier A, Fink JK, Wilcock S, London Z, Brewer GJ. Myelopolyneuropathy and pancytopenia due to copper deficiency and high zinc levels of unknown origin II. The denture cream is a primary source of excessive zinc. Neurotoxicology 30: 996–999, 2009. [DOI] [PubMed] [Google Scholar]
- 124.Hellman NE, Gitlin JD. Ceruloplasmin metabolism and function. Annu Rev Nutr 22: 439–458, 2002. [DOI] [PubMed] [Google Scholar]
- 125.Hintze KJ, Theil EC. Cellular regulation and molecular interactions of the ferritins. Cell Mol Life Sci 63: 591–600, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hoppler M, Schonbachler A, Meile L, Hurrell RF, Walczyk T. Ferritin- iron is released during boiling and in vitro gastric digestion. JNutr 138: 878–884, 2008. [DOI] [PubMed] [Google Scholar]
- 127.Hu Z, Gulec S, Collins JF. Cross-species comparison of genomewide gene expression profiles reveals induction of hypoxia-inducible factor- responsive genes in iron-deprived intestinal epithelial cells. Am J Physiol Cell Physiol 299: C930–C938, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hudson DM, Curtis SB, Smith VC, Griffiths TA, Wong AY, Scudamore CH, Buchan AM, MacGillivray RT. Human hephaestin expression is not limited to enterocytes of the gastrointestinal tract but is also found in the antrum, the enteric nervous system, and pancreatic {beta}-cells. Am J Physiol Gastrointest Liver Physiol 298: G425–G432, 2010. [DOI] [PubMed] [Google Scholar]
- 129.Hudson DM, Krisinger MJ, Griffiths TA, Macgillivray RT. Neither human hephaestin nor ceruloplasmin forms a stable complex with transferrin. J Cell Biochem 103: 1849–1855, 2008. [DOI] [PubMed] [Google Scholar]
- 130.Hurrell R, Egli I. Iron bioavailability and dietary reference values. Am J Clin Nutr 91: 1461S–1467S,2010. [DOI] [PubMed] [Google Scholar]
- 131.Hutchinson C, Geissler CA, Powell JJ, Bomford A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut 56: 1291–1295, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Iancu TC. Biological and ultrastructural aspects of iron overload: An overview. Pediatr Pathol 10: 281–296, 1990. [DOI] [PubMed] [Google Scholar]
- 133.Illing AC, Shawki A, Cunningham CL, Mackenzie B. Substrate profile and metal-ion selectivity of human divalent metal-ion transporter-1. JBiol Chem 287: 30485–30496, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Iwanska S, Strusinska D. Copper metabolism in different states of erythropoiesis activity. Acta Physiol Pol 29: 465–474, 1978. [PubMed] [Google Scholar]
- 135.Iyer S, Lonnerdal B. Lactoferrin, lactoferrin receptors and iron metabolism. Eur J Clin Nutr 47: 232–241, 1993. [PubMed] [Google Scholar]
- 136.Jacobs A, Kaye MD, Trevett D. The chelation of iron during intestinal absorption. J Lab Clin Med 74: 212–217, 1969. [PubMed] [Google Scholar]
- 137.Jacobs P, Bothwell TH, Charlton RW. Intestinal iron transport: Studies using a loop of gut with an artificial circulation. Am J Physiol 210: 694–700, 1966. [DOI] [PubMed] [Google Scholar]
- 138.Jenkitkasemwong S, Broderius M, Nam H, Prohaska JR, Knutson MD. Anemic copper-deficient rats, but not mice, display low hepcidin expression and high ferroportin levels. J Nutr 140: 723–730, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiang L, Garrick MD, Garrick LM, Zhao L, Collins JF. Divalent metal transporter 1 (Dmt1) mediates copper transport in the duodenum of iron-deficient rats and when overexpressed in iron-deprived HEK-293 cells. JNutr 143: 1927–1933, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johnson G, Jacobs P, Purves LR. Iron binding proteins ofiron-absorbing rat intestinal mucosa. J Clin Invest 71: 1467–1476, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kalgaonkar S, Lonnerdal B. Receptor-mediated uptake of ferritin- bound iron by human intestinal Caco-2 cells. J Nutr Biochem 20: 304–311, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP. Molecular cloning oftransferrin receptor 2. Anew member of the transferrin receptor-like family. J Biol Chem 274: 20826–20832, 1999. [DOI] [PubMed] [Google Scholar]
- 143.Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319: 825–828, 2008. [DOI] [PubMed] [Google Scholar]
- 144.Kelly EJ, Palmiter RD. A murine model of Menkes disease reveals a physiological function of metallothionein. Nat Genet 13: 219–222, 1996. [DOI] [PubMed] [Google Scholar]
- 145.Kim BE, Turski ML, Nose Y, Casad M, Rockman HA, Thiele DJ. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell metab 11: 353–363, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kim H, Son HY, Bailey SM, Lee J. Deletion of hepatic Ctr1 reveals its function in copper acquisition and compensatory mechanisms for copper homeostasis. Am J Physiol Gastrointest Liver Physiol 296: G356–G364, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.King JC Jr., Cummings GE, Guo N, Trivedi L, Readmond BX, Keane V, Feigelman S, de Waard R. A double-blind, placebo-controlled, pilot study of bovine lactoferrin supplementation in bottle-fed infants. J Pedi- atr Gastroenterol Nutr 44: 245–251, 2007. [DOI] [PubMed] [Google Scholar]
- 148.Klevay LM. IHD from copper deficiency: A unified theory. Nutr Res Rev 29: 172–179, 2016. [DOI] [PubMed] [Google Scholar]
- 149.Klevay LM. Iron overload can induce mild copper deficiency. J Trace Elem Med Biol 14: 237–240, 2001. [DOI] [PubMed] [Google Scholar]
- 150.Klevay LM. Is the Western diet adequate in copper? J Trace Elem Med Biol 25:204–212, 2011. [DOI] [PubMed] [Google Scholar]
- 151.Knopfel M, Solioz M. Characterization of a cytochrome b(558) fer- ric/cupric reductase from rabbit duodenal brush border membranes. Biochem Biophys Res Commun 291: 220–225, 2002. [DOI] [PubMed] [Google Scholar]
- 152.Knutson M, Wessling-Resnick M. Iron metabolism in the reticuloendothelial system. CritRev Biochem Mol Biol 38: 61–88, 2003. [DOI] [PubMed] [Google Scholar]
- 153.Knutson MD. Steap proteins: Implications for iron and copper metabolism. Nutr Rev 65: 335–340, 2007. [DOI] [PubMed] [Google Scholar]
- 154.Koikawa N, Nagaoka I, Yamaguchi M, Hamano H, Yamauchi K, Sawaki K. Preventive effect of lactoferrin intake on anemia in female long distance runners. Biosci Biotechnol Biochem 72: 931–935, 2008. [DOI] [PubMed] [Google Scholar]
- 155.Kono S Aceruloplasminemia: An update. Int Rev Neurobiol 110: 125–151, 2013. [DOI] [PubMed] [Google Scholar]
- 156.Konz T, Alonso-Garcia J, Montes-Bayon M, Sanz-Medel A. Comparison of copper labeling followed by liquid chromatography-inductively coupled plasma mass spectrometry and immunochemical assays for serum hepcidin-25 determination. Anal Chim Acta 799: 1–7, 2013. [DOI] [PubMed] [Google Scholar]
- 157.Koorts AM, Viljoen M. Ferritin and ferritin isoforms I: Structure- function relationships, synthesis, degradation and secretion. Arch Physiol Biochem 113: 30–54, 2007. [DOI] [PubMed] [Google Scholar]
- 158.Krishnamurthy P, Schuetz JD. Role of ABCG2/BCRP in biology and medicine. Annu Rev Pharmacol Toxicol 46: 381–410, 2006. [DOI] [PubMed] [Google Scholar]
- 159.Kruger J, Taylor JR, Du X, De Moura FF, Lonnerdal B, Oelofse A. Effect of phytate reduction of sorghum, through genetic modification, on iron and zinc availability as assessed by an in vitro dialysability bioaccessibility assay, Caco-2 cell uptake assay, and suckling rat pup absorptionmodel. Food Chem 141: 1019–1025, 2013. [DOI] [PubMed] [Google Scholar]
- 160.Kruszewski M Labile iron pool: The main determinant of cellular response to oxidative stress. MutatRes 531: 81–92, 2003. [DOI] [PubMed] [Google Scholar]
- 161.Kulprachakarn K, Chen YL, Kong X, Arno MC, Hider RC, Srichairatanakool S, Bansal SS. Copper(II) binding properties of hep- cidin. J Biol Inorg Chem 21: 329–338, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kuo YM, Su T, Chen H, Attieh Z, Syed BA, McKie AT, Anderson GJ, Gitschier J, Vulpe CD. Mislocalisation of hephaestin, a multicopper ferroxidase involved in basolateral intestinal iron transport, in the sex linked anaemia mouse. Gut 53: 201–206, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lambe T, Simpson RJ, Dawson S, Bouriez-Jones T, Crockford TL, Lepherd M, Latunde-Dada GO, Robinson H, Raja KB, Campagna DR, Villarreal G Jr., Ellory JC, Goodnow CC, Fleming MD, McKie AT, Cornall RJ. Identification of a Steap3 endosomal targeting motif essential for normal iron metabolism. Blood 113: 1805–1808, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lane DJ, Bae DH, Merlot AM, Sahni S, Richardson DR. Duodenal cytochrome b (DCYTB) in iron metabolism: An update on function and regulation. Nutrients 7: 2274–2296, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Laparra JM, Diez-Municio M, Herrero M, Moreno FJ. Structural differences of prebiotic oligosaccharides influence their capability to enhance iron absorption in deficient rats. Food Funct 5: 2430–2437, 2014. [DOI] [PubMed] [Google Scholar]
- 166.Larin D, Mekios C, Das K, Ross B, Yang AS, Gilliam TC. Characterization of the interaction between the Wilson and Menkes disease proteins and the cytoplasmic copper chaperone, HAH1p. J Biol Chem 274: 28497–28504, 1999. [DOI] [PubMed] [Google Scholar]
- 167.Latunde-Dada GO, Simpson RJ, McKie AT. Recent advances in mammalian haem transport. Trends Biochem Sci 31: 182–188, 2006. [DOI] [PubMed] [Google Scholar]
- 168.Latunde-Dada GO, Van der Westhuizen J, Vulpe CD, Anderson GJ, Simpson RJ, McKie AT. Molecular and functional roles of duodenal cytochrome B (Dcytb) in iron metabolism. Blood Cells Mol Dis 29: 356–360, 2002. [DOI] [PubMed] [Google Scholar]
- 169.Latunde-Dada GO, Xiang L, Simpson RJ, McKie AT. Duodenal cytochrome b (Cybrd 1) and HIF-2alpha expression during acute hypoxic exposure in mice. Eur J Nutr 50: 699–704, 2011. [DOI] [PubMed] [Google Scholar]
- 170.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R III, Grinberg A, Love P, Tresser N, Rouault TA. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 27: 209–214, 2001. [DOI] [PubMed] [Google Scholar]
- 171.Le CH. The Prevalence of Anemia and Moderate-Severe Anemia in the US Population (NHANES 2003–2012). PloS one 11: e0166635, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee GR, Nacht S, Lukens JN, Cartwright GE. Iron metabolism in copper-deficient swine. J Clin Invest 47: 2058–2069, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee PL, Gelbart T, West C, Halloran C, Beutler E. The human Nramp2 gene: Characterization of the gene structure, alternative splicing, promoter region and polymorphisms. Blood Cells Mol Dis 24: 199–215, 1998. [DOI] [PubMed] [Google Scholar]
- 174.Levine DS, Huebers HA, Rubin CE, Finch CA. Blocking action of parenteral desferrioxamine on iron absorption in rodents and men. Gastroenterology 95: 1242–1248, 1988. [DOI] [PubMed] [Google Scholar]
- 175.Lin C, Zhang Z, Wang T, Chen C, James Kang Y. Copper uptake by DMT1: A compensatory mechanism for CTR1 deficiency in human umbilical vein endothelial cells. Metallomics 7: 1285–1289, 2015. [DOI] [PubMed] [Google Scholar]
- 176.Linder MC, Zerounian NR, Moriya M, Malpe R. Iron and copper homeostasis and intestinal absorption using the Caco2 cell model. Biometals 16: 145–160, 2003. [DOI] [PubMed] [Google Scholar]
- 177.Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc NatlAcadSci USA 103: 13612–13617, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lonnerdal B Nutritional roles of lactoferrin. Curr Opin Clin Nutr Metab Care 12: 293–297, 2009. [DOI] [PubMed] [Google Scholar]
- 179.Lonnerdal B, Bryant A. Absorption of iron from recombinant human lactoferrin in young US women. Am J Clin Nutr 83: 305–309, 2006. [DOI] [PubMed] [Google Scholar]
- 180.Lonnerdal B, Iyer S. Lactoferrin: Molecular structure and biological function. Annu Rev Nutr 15: 93–110, 1995. [DOI] [PubMed] [Google Scholar]
- 181.Ma Y, Specian RD, Yeh KY, Yeh M, Rodriguez-Paris J, Glass J. The transcytosis of divalent metal transporter 1 and apo-transferrin during iron uptake in intestinal epithelium. Am J Physiol Gastrointest Liver Physiol 283: G965–G974, 2002. [DOI] [PubMed] [Google Scholar]
- 182.Mackenzie B, Garrick MD. Iron Imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol 289: G981–G986, 2005. [DOI] [PubMed] [Google Scholar]
- 183.Maine GN, Burstein E. COMMD proteins: COMMing to the scene. Cell Mol Life Sci 64: 1997–2005, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Maine GN, Mao X, Muller PA, Komarck CM, Klomp LW, Burstein E. COMMD1 expression is controlled by critical residues that determine XIAP binding. Biochem J 417: 601–609, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Maisetta G, Petruzzelli R, Brancatisano FL, Esin S, Vitali A, Campa M, Batoni G. Antimicrobial activity of human hepcidin 20 and 25 against clinically relevant bacterial strains: Effect of copper and acidic pH. Peptides 31: 1995–2002, 2010. [DOI] [PubMed] [Google Scholar]
- 186.Manis JG, Schachter D. Active transport of iron by intestine: Effects of oral iron and pregnancy. Am J Physiol 203: 81–86, 1962. [DOI] [PubMed] [Google Scholar]
- 187.Marambio A, Watkins G, Castro F, Riffo A, Zuniga R, Jans J, Villanueva ME, Diaz G. Changes in iron transporter divalent metal transporter 1 in proximal jejunum after gastric bypass. World J Gastroenterol 20: 6534–6540, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Marciano R, Santamarina AB, de Santana AA, Silva Mde L, Amancio OM, do Nascimento CM, Oyama LM, de Morais MB. Effects of pre- biotic supplementation on the expression of proteins regulating iron absorption in anaemic growing rats. Br J Nutr 113: 901–908, 2015. [DOI] [PubMed] [Google Scholar]
- 189.Martin F, Linden T, Katschinski DM, Oehme F, Flamme I, Mukhopadhyay CK, Eckhardt K, Troger J, Barth S, Camenisch G, Wenger RH. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: Implications for ceruloplasmin regulation. Blood 105: 4613–4619, 2005. [DOI] [PubMed] [Google Scholar]
- 190.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest 119: 1159–1166, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Matak P, Zumerle S, Mastrogiannaki M, El Balkhi S, Delga S, Mathieu JR, Canonne-Hergaux F, Poupon J, Sharp PA, Vaulont S, Peyssonnaux C Copper deficiency leads to anemia, duodenal hypoxia, upregulation of HIF-2alpha and altered expression of iron absorption genes in mice. PLoS One 8: e59538, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.MC L. Biochemistry and molecular biology of copper in mammals In: EJ M, editor. Handbook of Copper Pharmacology and Toxicology. Totowa, NJ: Humana Press, 2003, pp. 3–32. [Google Scholar]
- 193.McKie AT. The role of Dcytb in iron metabolism: An update. Biochem Soc Trans 36: 1239–1241, 2008. [DOI] [PubMed] [Google Scholar]
- 194.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, Peters TJ, Raja KB, Shirali S, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 291: 1755–1759, 2001. [DOI] [PubMed] [Google Scholar]
- 195.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell 5: 299–309, 2000. [DOI] [PubMed] [Google Scholar]
- 196.Melis MA, Cau M, Congiu R, Sole G, Barella S, Cao A, Westerman M, Cazzola M, Galanello R. A mutation in the TMPRSS6 gene, encoding a transmembrane serine protease that suppresses hepcidin production, in familial iron deficiency anemia refractory to oral iron. Haematologica 93: 1473–1479,2008. [DOI] [PubMed] [Google Scholar]
- 197.Mena NP, Esparza A, Tapia V, Valdes P, Nunez MT. Hepcidin inhibits apical iron uptake in intestinal cells. Am J Physiol Gastrointest Liver Physiol 294: G192–G198, 2008. [DOI] [PubMed] [Google Scholar]
- 198.Mims MP, Guan Y, Pospisilova D, Priwitzerova M, Indrak K, Ponka P, Divoky V, Prchal JT. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood 105: 1337–1342, 2005. [DOI] [PubMed] [Google Scholar]
- 199.Mitchell CJ, Shawki A, Ganz T, Nemeth E, Mackenzie B. Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. AmJPhysiol Cell Physiol 306: C450–C459,2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Miyazaki E, Kato J, Kobune M, Okumura K, Sasaki K, Shintani N, Arosio P, Niitsu Y. Denatured H-ferritin subunit is a major constituent of haemosiderin in the liver of patients with iron overload. Gut 50: 413–419, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Morgan EH. The role of plasma transferrin in iron absorption in the rat. Q J Exp Physiol Cogn Med Sci 65: 239–252, 1980. [DOI] [PubMed] [Google Scholar]
- 202.Mostad EJ, Prohaska JR. Glycosylphosphatidylinositol-linked cerulo- plasmin is expressed in multiple rodent organs and is lower following dietary copper deficiency. Exp Biol Med (Maywood) 236: 298–308, 2011. [DOI] [PubMed] [Google Scholar]
- 203.Murray MJ, Delaney JP, Stein N.Useof isolated subcutaneous intestinal loops for direct study of intestinal absorption of radioisotopes in dogs. Am J Dig Dis 9: 684–689, 1964. [DOI] [PubMed] [Google Scholar]
- 204.Nand S, Tanvetyanon T. Proton pump inhibitors and iron deficiency: Is the connection real? South Med J 97:799, 2004. [DOI] [PubMed] [Google Scholar]
- 205.Nations SP, Boyer PJ, Love LA, Burritt MF, Butz JA, Wolfe GI, Hynan LS, Reisch J, Trivedi JR. Denture cream: An unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology 71: 639–643, 2008. [DOI] [PubMed] [Google Scholar]
- 206.Nemeth E, Ganz T. The role of hepcidin in iron metabolism. Acta Haematol 122: 78–86, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood 105: 1803–1806, 2005. [DOI] [PubMed] [Google Scholar]
- 208.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090–2093, 2004. [DOI] [PubMed] [Google Scholar]
- 209.Nevo Y Site-directed mutagenesis investigation of coupling properties of metal ion transport by DCT1. Biochim Biophys Acta 1778: 334–341, 2008. [DOI] [PubMed] [Google Scholar]
- 210.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc NatlAcadSci USA 98: 8780–8785, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. CellMetab 4: 235–244, 2006. [DOI] [PubMed] [Google Scholar]
- 212.Nose Y, Rees EM, Thiele DJ. Structure of the Ctr1 copper trans’PORE’ter reveals novel architecture. Trends Biochem Sci 31:604–607, 2006. [DOI] [PubMed] [Google Scholar]
- 213.O’Neil-Cutting MA, Crosby WH. Blocking of iron absorption by a preliminary oral dose of iron. Arch Intern Med 147: 489–491, 1987. [PubMed] [Google Scholar]
- 214.O’Riordan DK, Sharp P, Sykes RM, Srai SK, Epstein O, Debnam ES. Cellular mechanisms underlying the increased duodenal iron absorption in rats in response to phenylhydrazine-induced haemolytic anaemia. Eur J Clin Invest 25: 722–727, 1995. [DOI] [PubMed] [Google Scholar]
- 215.Oates PS, Trinder D, Morgan EH. Gastrointestinal function, divalent metal transporter-1 expression and intestinal iron absorption. Pflugers Arch 440: 496–502, 2000. [DOI] [PubMed] [Google Scholar]
- 216.Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37: 1264–1269, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood 108: 1388–1394, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Osaki S, Johnson DA, Frieden E. The mobilization of iron from the perfused mammalian liver by a serum copper enzyme, ferroxidase I. JBiol Chem 246: 3018–3023, 1971. [PubMed] [Google Scholar]
- 219.Owen CA Jr. Effects of iron on copper metabolism and copper on iron metabolism in rats. Am J Physiol 224: 514–518, 1973. [DOI] [PubMed] [Google Scholar]
- 220.Paesano R, Torcia F, Berlutti F, Pacifici E, Ebano V, Moscarini M, Valenti P. Oral administration of lactoferrin increases hemoglobin and total serum iron in pregnant women. Biochem Cell Biol 84: 377–380, 2006. [DOI] [PubMed] [Google Scholar]
- 221.Palumaa P, Kangur L, Voronova A, Sillard R Metal-binding mechanism of Cox17, a copper chaperone for cytochrome c oxidase. Biochem J 382: 307–314,2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, Simon MC. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol 27: 912–925, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, Nemeth E, Thompson J, Risler JK, Zaborowska C, Babakaiff R, Radomski CC, Pape TD, Davidas O, Christakis J, Brissot P, Lockitch G, Ganz T, Hayden MR, Goldberg YP. Mutations in HFE2 cause iron overload in chromosome lq-linked juvenile hemochromatosis. Nat Genet 36: 77–82, 2004. [DOI] [PubMed] [Google Scholar]
- 224.Parmley RT, Barton JC, Conrad ME, Austin RL, Holland RM. Ultra- structural cytochemistry and radioautography of hemoglobin-iron absorption. Exp Mol Pathol 34: 131–144, 1981. [DOI] [PubMed] [Google Scholar]
- 225.Patel BN, David S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem 272: 20185–20190, 1997. [DOI] [PubMed] [Google Scholar]
- 226.Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: A novel mechanism of regulated trafficking. EMBO J 15: 6084–6095, 1996. [PMC free article] [PubMed] [Google Scholar]
- 227.Picard V, Epsztejn S, Santambrogio P, Cabantchik ZI, Beaumont C. Role of ferritin in the control of the labile iron pool in murine ery-throleukemia cells. J Biol Chem 273: 15382–15386, 1998. [DOI] [PubMed] [Google Scholar]
- 228.Pietrangelo A Non-HFE hemochromatosis. Hepatology 39: 21–29, 2004. [DOI] [PubMed] [Google Scholar]
- 229.Pinkerton PH. Histological evidence of disordered iron transport in the x-linked hypochromic anaemia of mice. J Pathol Bacteriol 95:155–165, 1968. [DOI] [PubMed] [Google Scholar]
- 230.Pinkerton PH, Bannerman RM. Hereditary defect in iron absorption in mice. Nature 216: 482–483, 1967. [DOI] [PubMed] [Google Scholar]
- 231.Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Iron absorption in iron-loading anaemias: Effect of subcutaneous desferrioxamine infusions. Lancet 2: 737–739, 1977. [DOI] [PubMed] [Google Scholar]
- 232.Pollack S, George JN, Reba RC, Kaufman RM, Crosby WH. The absorption of nonferrous metals in iron deficiency. J Clin Invest 44: 1470–1473, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Pourvali K, Matak P, Latunde-Dada GO, Solomou S, Mastrogiannaki M, Peyssonnaux C, Sharp PA. Basal expression of copper transporter 1 in intestinal epithelial cells is regulated by hypoxia-inducible factor 2alpha. FEBS letters 586: 2423–2427, 2012. [DOI] [PubMed] [Google Scholar]
- 234.Powell LW, Campbell CB, Wilson E. Intestinal mucosal uptake of iron and iron retention in idiopathic haemochromatosis as evidence for a mucosal abnormality. Gut 11: 727–731, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Powers HJ, Loban A, Silvers K, Gibson AT. Vitamin C at concentrations observed in premature babies inhibits the ferroxidase activity of caeruloplasmin. Free Radic Res 22: 57–65, 1995. [DOI] [PubMed] [Google Scholar]
- 236.Prohaska JR. Genetic diseases of copper metabolism. Clin Physiol Biochem 4: 87–93, 1986. [PubMed] [Google Scholar]
- 237.Prohaska JR, Broderius M. Copper deficiency has minimal impact on ferroportin expression or function. Biometals 25: 633–642, 2012. [DOI] [PubMed] [Google Scholar]
- 238.Prohaska JR, Gybina AA. Intracellular copper transport in mammals. JNutr 134: 1003–1006, 2004. [DOI] [PubMed] [Google Scholar]
- 239.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127: 917–928, 2006. [DOI] [PubMed] [Google Scholar]
- 240.Qiu L, Ding X, Zhang Z, Kang YJ. Copper is required for cobalt- induced transcriptional activity of hypoxia-inducible factor-1. J Pharmacol Exp Ther 342: 561–567, 2012. [DOI] [PubMed] [Google Scholar]
- 241.Quigley JG, Yang Z, Worthington MT, Phillips JD, Sabo KM, Sabath DE, Berg CL, Sassa S, Wood BL, Abkowitz JL. Identification of a human heme exporter that is essential for erythropoiesis. Cell 118: 757–766, 2004. [DOI] [PubMed] [Google Scholar]
- 242.Raffin SB, Woo CH, Roost KT, Price DC, Schmid R. Intestinal absorption of hemoglobin iron-heme cleavage by mucosal heme oxygenase. J Clin Invest 54: 1344–1352, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Raja KB, Bjarnason I, Simpson RJ, Peters TJ. In vitro measurement and adaptive response of Fe3+ uptake by mouse intestine. Cell Biochem Funct 5:69–76, 1987. [DOI] [PubMed] [Google Scholar]
- 244.Raja KB, Simpson RJ, Peters TJ. Investigation of a role for reduction in ferric iron uptake by mouse duodenum. Biochim Biophys Acta 1135: 141–146, 1992. [DOI] [PubMed] [Google Scholar]
- 245.Ranganathan PN, Lu Y, Fuqua BK, Collins JF. Discovery of a cytoso¬lic/soluble ferroxidase in rodent enterocytes. Proc Natl Acad Sci USA 109: 3564–3569, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Ranganathan PN, Lu Y, Fuqua BK, Collins JF. Immunoreactive hep-haestin and ferroxidase activity are present in the cytosolic fraction of rat enterocytes. Biometals 25: 687–695, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ranganathan PN, Lu Y, Jiang L, Kim C, Collins JF. Serum cerulo- plasmin protein expression and activity increases in iron-deficient rats and is further enhanced by higher dietary copper intake. Blood 118: 3146–3153, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Ravia JJ, Stephen RM, Ghishan FK, Collins JF. Menkes Copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brnsh-border and basolateral membrane domains. J Biol Chem 280: 36221–36227, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Reeves PG. Copper metabolism in metallothionein-null mice fed a high zinc diet. JNutrBiochem 9: 598–601, 1998. [Google Scholar]
- 250.Reeves PG, DeMars LC. Copper deficiency reduces iron absorption and biological half-life in male rats. J Nutr 134: 1953–1957, 2004. [DOI] [PubMed] [Google Scholar]
- 251.Reeves PG, Demars LC. Repletion of copper-deficient rats with dietary copper restores duodenal hephaestin protein and iron absorption. Exp Biol Med (Maywood) 230: 320–325, 2005. [DOI] [PubMed] [Google Scholar]
- 252.Reeves PG, Demars LC, Johnson WT, Lukaski HC. Dietary copper deficiency reduces iron absorption and duodenal enterocyte hephaestin protein in male and female rats. JNutr 135: 92–98, 2005. [DOI] [PubMed] [Google Scholar]
- 253.Rhodes J, Beton D, Brown DA. Absorption of iron instilled into the stomach, duodenum, and jejunum. Gut 9: 323–324, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Rice AE, Mendez MJ, Hokanson CA, Rees DC, Bjorkman PJ. Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J Mol Biol 386: 717–732, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Richter GW, Lee YH. Absorption of iron from gut into blood: Sex- and time-related studies in rats. Experientia 38: 583–585, 1982. [DOI] [PubMed] [Google Scholar]
- 256.Rioux FM, LeBlanc CP. Iron supplementation during pregnancy: What are the risks and benefits of current practices? Appl Physiol Nutr Metab 32: 282–288, 2007. [DOI] [PubMed] [Google Scholar]
- 257.Roberts SK, Henderson RW, Young GP. Modulation of uptake of heme by rat small intestinal mucosa in iron deficiency. Am J Physiol 265: G712–G718, 1993. [DOI] [PubMed] [Google Scholar]
- 258.Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology 119:782–793,2000. [DOI] [PubMed] [Google Scholar]
- 259.San Martin CD, Garri C, Pizarro F, Walter T, Theil EC, Nunez MT. Caco-2 intestinal epithelial cells absorb soybean ferritin by mu2 (AP2)- dependent endocytosis. J Nutr 138: 659–666, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sato Y, Yoneyama O, Azumaya M, Takeuchi M, Sasaki SY, Yokoyama J, Shioji K, Kawauchi Y, Hashimoto S, Nishigaki Y, Kobayashi M, Sugimura K, Honma T, Narisawa R, Aoyagi Y. The relationship between iron deficiency in patients with Helicobacter pylori-infected nodular gastritis and the serum prohepcidin level. Helicobacter 20: 11–18, 2015. [DOI] [PubMed] [Google Scholar]
- 261.Schumann K, Osterloh K, Forth W. Independence of in vitro iron absorption from mucosal transferrin content in rat jejunal and ileal segments. Blut 53: 391–400, 1986. [DOI] [PubMed] [Google Scholar]
- 262.Seo YA, Elkhader JA, Wessling-Resnick M. Distribution of manganese and other biometals in flatiron mice. Biometals 29: 147–155, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Seo YA, Wessling-Resnick M. Ferroportin deficiency impairs manganese metabolism in flatiron mice. FASEB J 29: 2726–2733, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia- inducible transcription factors are essential for iron absorption following iron deficiency. Cell metab 9: 152–164, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Sharma VR, Brannon MA, Carloss EA. Effect of omeprazole on oral iron replacement in patients with iron deficiency anemia. South Med J 97: 887–889, 2004. [DOI] [PubMed] [Google Scholar]
- 266.Shawki A, Anthony SR, Nose Y, Engevik MA, Niespodzany EJ, Barrientos T, Ohrvik H, Worrell RT, Thiele DJ, Mackenzie B Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese. Am J Physiol Gastrointest LiverPhysiol 309: G635–G647, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Shawki A, Engevik MA, Kim RS, Knight PB, Baik RA, Anthony SR, Worrell RT, Shull GE, Mackenzie B. Intestinal brush-border Na+/H+ exchanger-3 drives H+-coupled iron absorption in the mouse. Am J Physiol Gastrointest Liver Physiol 311: G423–G430, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Shawki A, Knight PB, Maliken BD, Niespodzany EJ, Mackenzie B. H(+)-coupled divalent metal-ion transporter-1: Functional properties, physiological roles and therapeutics. Curr Top Membr 70: 169–214, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Identification of an intestinal heme transporter. Cell 122: 789–801, 2005. [DOI] [PubMed] [Google Scholar]
- 270.Shi H, Bencze KZ, Stemmler TL, Philpott CC. A cytosolic iron chaperone that delivers iron to ferritin. Science 320: 1207–1210, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Shin K, Wakabayashi H, Yamauchi K, Yaeshima T, Iwatsuki K. Recombinant human intelectin binds bovine lactoferrin and its peptides. Biol Pharm Bull 31: 1605–1608, 2008. [DOI] [PubMed] [Google Scholar]
- 272.Simpson RJ, Lombard M, Raja KB, Thatcher R, Peters TJ. Iron absorption by hypotransferrinaemic mice. Br J Haematol 78: 565–570, 1991. [DOI] [PubMed] [Google Scholar]
- 273.Singh NP, Medeiros DM. Effect of copper deficiency and Sodium intake upon liver lipid and mineral composition in the rat. Biol Trace Elem Res 6: 423–429, 1984. [DOI] [PubMed] [Google Scholar]
- 274.Siu AL, U.S. Preventive Services Tasks Force. Screening for iron deficiency anemia and iron supplementation in pregnant women to improve maternal health and birth outcomes: Recommendation statement. Am Fam Physician 93: 133–136,2016. [PubMed] [Google Scholar]
- 275.Sotelo A, Gonzalez-Osnaya L, Sanchez-Chinchillas A, Trejo A. Role of oxate, phytate, tannins and cooking on iron bioavailability from foods commonly consumed in Mexico. Int J Food Sci Nutr 61: 29–39, 2010. [DOI] [PubMed] [Google Scholar]
- 276.Sourkes TL, Lloyd K, Birnbaum H Inverse relationship ofheptic copper and iron concentrations in rats fed deficient diets. Can J Biochem 46: 267–271, 1968. [DOI] [PubMed] [Google Scholar]
- 277.Southon S, Wright AJ, Fairweather-Tait SJ. The effect of differences in dietary iron intake on 59Fe absorption and duodenal morphology in pregnant rats. Br J Nutr 62: 707–717, 1989. [DOI] [PubMed] [Google Scholar]
- 278.Stewart WB, Gambino SR. Kinetics of iron absorption in normal dogs. Am J Physiol 201: 67–70, 1961. [Google Scholar]
- 279.Stewart WB, Yuile CL, Claiborne HA, Snowman RT, Whipple GH. Radioiron absorption in anemic dogs; fluctuations in the mucosal block and evidence for a gradient of absorption in the gastrointestinal tract. J Exp Med 92: 375–382, 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Su MA, Trenor CC, Fleming JC, Fleming MD, Andrews NC. The G185R mutation disrupts function of the iron transporter Nramp2. Blood 92:2157–2163, 1998. [PubMed] [Google Scholar]
- 281.Suzuki YA, Shin K, Lonnerdal B. Molecular cloning and functional expression of a human intestinal lactoferrin receptor. Biochemistry 40: 15771–15779,2001. [DOI] [PubMed] [Google Scholar]
- 282.Takeuchi K, Bjarnason I, Laftah AH, Latunde-Dada GO, Simpson RJ, McKie AT. Expression of iron absorption genes in mouse large intestine. Scand J Gastroenterol 40: 169–177, 2005. [DOI] [PubMed] [Google Scholar]
- 283.Taylor M, Qu A, Anderson ER, Matsubara T, Martin A, Gonzalez FJ, Shah YM. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 140: 2044–2055, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Tchernitchko D, Bourgeois M, Martin ME, Beaumont C. Expression of the two mRNA isoforms of the iron transporter Nramp2/DMTI in mice and function of the iron responsive element. Biochem J 363: 449–455, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Tenhunen R, Grasbeck R, Kouvonen I, Lundberg M. An intestinal receptor for heme: Its partial characterization. Int J Biochem 12: 713–716, 1980. [DOI] [PubMed] [Google Scholar]
- 286.Thackeray EW, Sanderson SO, Fox JC, Kumar N. Hepatic iron overload or cirrhosis may occur in acquired copper deficiency and is likely mediated by hypoceruloplasminemia. J Clin Gastroenterol 45: 153–158, 2011. [DOI] [PubMed] [Google Scholar]
- 287.Theil EC. Iron, ferritin, and nutrition. Annu Rev Nutr 24:327–343,2004. [DOI] [PubMed] [Google Scholar]
- 288.Theil EC, Chen H, Miranda C, Janser H, Elsenhans B, Nunez MT, Pizarro F, Schumann K. Absorption of iron from ferritin is independent of heme iron and ferrous salts in women and rat intestinal segments. JNutr 142:478–483,2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Thompson K, Molina RM, Donaghey T, Schwob JE, Brain JD, Wessling-Resnick M. Olfactory uptake of manganese requires DMT1 and is enhanced by anemia. FASEB J 21: 223–230, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Thomson AB, Valberg LS. Kinetics of intestinal iron absorption in the rat: Effect of cobalt. AmJPhysiol 220: 1080–1085, 1971. [DOI] [PubMed] [Google Scholar]
- 291.Trinder D, Oates PS, Thomas C, Sadleir J, Morgan EH. Localisation of divalent metal transporter 1 (DMT1) to the microvillus membrane of rat duodenal enterocytes in iron deficiency, but to hepatocytes in iron overload. Gut 46: 270–276, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Tselepis C, Ford SJ, McKie AT, Vogel W, Zoller H, Simpson RJ, Diaz Castro J, Iqbal TH, Ward DG. Characterization of the transition-metal- binding properties of hepcidin. Biochem J 427: 289–296, 2010. [DOI] [PubMed] [Google Scholar]
- 293.Uriu-Adams JY, Scherr RE, Lanoue L, Keen CL. Influence of copper on early development: Prenatal and postnatal considerations. Biofactors 36: 136–152, 2010. [DOI] [PubMed] [Google Scholar]
- 294.Vanoaica L, Darshan D, Richman L, Schumann K, Kuhn LC. Intestinal ferritin H is required for an accurate control of iron absorption. Cell Metab 12: 273–282, 2010. [DOI] [PubMed] [Google Scholar]
- 295.Videt-Gibou D, Belliard S, Bardou-Jacquet E, Troadec MB, Le Lan C, Jouanolle AM, Loreal O, Rivalan J, Brissot P. Iron excess treatable by copper supplementation in acquired aceruloplasminemia: A new form of secondary human iron overload? Blood 114: 2360–2361, 2009. [DOI] [PubMed] [Google Scholar]
- 296.Videt-Gibou D, Belliard S, Rivalan J, Menard D, Edan G. [Acquired copper deficiency myelopathy]. RevNeurol (Paris) 166:639–643,2010. [DOI] [PubMed] [Google Scholar]
- 297.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier J, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. NatGenet 21: 195–199, 1999. [DOI] [PubMed] [Google Scholar]
- 298.Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol 13: 4690–4698, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Wang CY, Knutson MD. Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice. Hepatology 58: 788–798, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Ward PP, Mendoza-Meneses M, Cunningham GA, Conneely OM. Iron status in mice carrying a targeted disruption of lactoferrin. Mol Cell Biol 23: 178–185,2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Wee NK, Weinstein DC, Fraser ST, Assinder SJ. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int J Biochem Cell Biol 45: 960–963, 2013. [DOI] [PubMed] [Google Scholar]
- 302.Weintraub LR, Weinstein MB, Huser HJ, Rafal S. Absorption of hemoglobin iron: The role of a heme-splitting substance in the intestinal mucosa. J Clin Invest 47: 531–539, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.West AR, Oates PS. Mechanisms of heme iron absorption: Current questions and controversies. World J Gastroenterol l4: 4101–4110, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Wheby MS. Site of iron absorption in man. Scand J Haematol 7:56–62, 1970. [DOI] [PubMed] [Google Scholar]
- 305.Wheby MS, Crosby WH. The gastrointestinal tract and iron absorption. Blood 22: 416–428, 1963. [PubMed] [Google Scholar]
- 306.Wheby MS, Jones LG, Crosby WH. Studies on iron absorption. Intestinal regulatory mechanisms. J Clin Invest 43: 1433–1442, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wheby MS, Suttle GE, Ford KT III. Intestinal absorption of hemoglobin iron. Gastroenterology 58: 647–654, 1970. [PubMed] [Google Scholar]
- 308.Widdowson EM, McCance RA. The absorption and excretion of iron before, during and after a period of very high intake. Biochem J 31: 2029–2034, 1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Williams DM, Kennedy FS, Green BG. Hepatic iron accumulation in copper-deficient rats. Br J Nutr 50: 653–660, 1983. [DOI] [PubMed] [Google Scholar]
- 310.Wollenberg P, Rummel W. Dependence of intestinal iron absorption on the valency state of iron. Naunyn Schmiedebergs Arch Pharmacol 336: 578–582, 1987. [DOI] [PubMed] [Google Scholar]
- 311.Worthington MT, Cohn SM, Miller SK, Luo RQ, Berg CL. Charac¬terization of a human plasma membrane heme transporter in intestinal and hepatocyte cell lines. Am J Physiol Gastrointest Liver Physiol 280: G1172–G1177, 2001. [DOI] [PubMed] [Google Scholar]
- 312.Worwood M, Jacobs A. The subcellular distribution of orally adminis¬tered 59Fe in rat small intestinal mucosa. Br J Haematol 20: 587–597, 1971. [DOI] [PubMed] [Google Scholar]
- 313.Wu W, Song Y, He C, Liu C, Wu R, Fang L, Cong Y, Miao Y, Liu Z. Divalent metal-ion transporter 1 is decreased in intestinal epithelial cells and contributes to the anemia in inflammatory bowel disease. Sci Rep5: 16344, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Wyllie JC, Kaufman N. An electron microscopic study ofheme uptake by rat duodenum. Lab Invest 47: 471–476, 1982. [PubMed] [Google Scholar]
- 315.Wyman S, Simpson RJ, McKie AT, Sharp PA. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett 582: 1901–1906, 2008. [DOI] [PubMed] [Google Scholar]
- 316.Xie L, Collins JF. Transcription factors Sp1 and Hif2alpha mediate induction of the copper-transporting ATPase (Atp7a) gene in intestinal epithelial cells during hypoxia. J Biol Chem 288: 23943–23952, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Xie L, Collins JF. Transcriptional regulation of the Menkes copper ATPase (Atp7a) gene by hypoxia-inducible factor (HIF2{alpha}) in intestinal epithelial cells. AmJ Physiol Cell Physiol 300:C1298–C1305, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Yamaji S, Sharp P, Ramesh B, Srai SK. Inhibition of iron transport across human intestinal epithelial cells by hepcidin. Blood 104: 2178–2180, 2004. [DOI] [PubMed] [Google Scholar]
- 319.Yanatori I, Richardson DR, Imada K, Kishi F. Iron export through the transporter ferroportin 1 is modulated by the iron chaperone PCBP2. J Biol Chem 291: 17303–17318, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Yanatori I, Yasui Y, Tabuchi M, Kishi F. Chaperone protein involved in transmembrane transport of iron. Biochem J 462: 25–37, 2014. [DOI] [PubMed] [Google Scholar]
- 321.Yeh KY, Yeh M, Glass J. Interactions between ferroportin and hep- haestin in rat enterocytes are reduced after iron ingestion. Gastroenterology 141: 292–299, 299.e1, 2011. [DOI] [PubMed] [Google Scholar]
- 322.Yeh KY, Yeh M, Mims L, Glass J. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am J Physiol Gastrointest Liver Physiol 296: G55–G65, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Yeh KY, Yeh M, Watkins JA, Rodriguez-Paris J, Glass J. Dietary iron induces rapid changes in rat intestinal divalent metal transporter expression. Am J Physiol Gastrointest Liver Physiol 279: G1070–G1079, 2000. [DOI] [PubMed] [Google Scholar]
- 324.Zhang AS, Xiong S, Tsukamoto H, Enns CA. Localization of iron metabolism-related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes. Blood 103: 1509–1514, 2004. [DOI] [PubMed] [Google Scholar]
- 325.Zhang B, Georgiev O, Hagmann M, Gunes C, Cramer M, Faller P, Vasak M, Schaffner W. Activity of metal-responsive transcription factor 1 by toxic heavy metals and H2O2 in vitro is modulated by metallothionein. Mol Cell Biol 23: 8471–8485, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab 9: 461–473, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Zhao G Phytoferritin and its implications for human health and nutrition. Biochim Biophys Acta 1800: 815–823, 2010. [DOI] [PubMed] [Google Scholar]
- 328.Zittel S, Ufer F, Gerloff C, Munchau A, Rosenkranz M. Severe myelopathy after denture cream use—is copper deficiency or excess zinc the cause? Clin Neurol Neurosurg 121: 17–18, 2014. [DOI] [PubMed] [Google Scholar]
- 329.Zoller H, Koch RO, Theurl I, Obrist P, Pietrangelo A, Montosi G, Haile DJ, Vogel W, Weiss G. Expression of the duodenal iron transporters divalent-metal transporter 1 and ferroportin 1 in iron deficiency and iron overload. Gastroenterology 120: 1412–1419, 2001. [DOI] [PubMed] [Google Scholar]