

Abstract
From the first century AD, Europe has been interested by population movements, commonly known as Barbarian migrations. Among these processes, the one involving the Longobard culture interested a vast region, but its dynamics and demographic impact remains largely unknown. Here we report 87 new complete mitochondrial sequences coming from nine early-medieval cemeteries located along the area interested by the Longobard migration (Czech Republic, Hungary and Italy). From the same areas, we sampled necropoleis characterized by cultural markers associated with the Longobard culture (LC) and coeval burials where no such markers were found, or with a chronology slightly preceding the presumed arrival of the Longobards in that region (NLC). Population genetics analysis and demographic modeling highlighted a similarity between LC individuals, as reflected by the sharing of quite rare haplogroups and by the degree of genetic resemblance between Hungarian and Italian LC necropoleis estimated via a Bayesian approach, ABC. The demographic model receiving the strongest statistical support also postulates a contact between LC and NLC communities, thus indicating a complex dynamics of admixture in medieval Europe.
Subject terms: Population genetics, Evolutionary biology
Introduction
According to historical records, around the first century CE a Germanic population called “Longobard” was settled in the northern Elbe basin [1]. Around 500 CE the term “Longobard” reoccurs in the region north and west of the middle Danube, including Moravia and Hungary (Pannonia), and three generations later, in 568 CE, a people referred to as Longobards invaded and conquered much of Italy [2]. The geographical spread of the word “Longobard” might evoke a large migratory phenomenon; however, the effective impact of the Longobard migration is still highly debated. There is a clear connection between the funeral rituals and the material culture of Pannonia and Italy at the end of the 6th century, suggesting strong interaction and communication—possibly as a result of a migration recorded in written sources—between these two regions (see e.g. refs. [3, 4]). Archeological and written sources, however, are open to different interpretations, and are unable to tell us whether such similarities in the material culture result mainly from commercial exchanges or from actual displacement of people. If the latter is the case, a further question arises, namely the relative contribution of immigrants and previously settled people in the composition of the hybrid population. In particular, it is still highly debated whether and to what extent attributes such as grave goods and burial traditions are indicators of Longobard social identity, and whether the spread of these material markers across Europe is actually linked to population movements rather than to horizontal cultural transmission or trade. In this light, the analysis of ancient genetic data is fundamental to obtain a better understanding of past population dynamics and interactions.
The only ancient genetic data ever published from cemeteries associated with Longobard culture so far were sequences of the mtDNA control region from Piedmont, Italy [5, 6], and sequences of mtDNA control region and of informative positions in the coding region from the cemetery of Szólád in Hungary [7]. A broader analysis, based on larger assemblages of samples and genetic markers, is clearly necessary to provide a finer resolution of medieval populations’ movements and interactions in Europe. In this study, we sequenced complete mitochondrial genomes from nine early-medieval cemeteries located in the Czech Republic, Hungary and Italy, for a total of 87 individuals. In some of these cemeteries, a portion of the individuals are buried with cultural markers in these areas traditionally associated with the Longobard culture, such as wooden chambers, buried horses and weapons (hereby, we refer to these cemeteries as LC), as opposed to burial communities in which no artifacts or rituals associated by archeologists to Longobard culture have been found in any graves (for a more comprehensive description of the archeological context see ESM). These necropoleis, hereby referred as NLC, may represent local communities, as in the case of Italy, or other barbarian groups previously migrated to this region, as in the case of Hungary (see Materials and methods and ESM for details). This extended sampling strategy provides an excellent condition to investigate the degree of genetic affinity between coeval LC and NLC burials, and to shed light on early-medieval dynamics in Europe.
Materials and methods
Sample preparation and aDNA extraction
We collected teeth, postcranial elements and petrous bones for 135 individuals from nine early-medieval cemeteries located in the Czech Republic, Hungary and Italy (Fig. 1, Table 1 and Table S1). Archeologically, 5 of these cemeteries have been at least partly associated with the Longobard culture: Mušov in the Czech Republic (LCRMUS), Szólád (LHSZ) and Hegykő (LHHEG) in Hungary, Collegno (LICOL) and Fara Olivana (LIFAR) in Italy. The burial markers used to indicate this association were the presence of wooden chambers, buried horses and weapons, features collectively considered informative about the Longobard presence. We also evaluated the presence of various types of grave goods, such as jewelry, dress accessories, pottery and glass vessels. The other 4 sites—Fonyód (NLHFON), Hács-Béndekpuszta (NLHHACS) and Balantoszemes Szemesi-berek (NLHBAL) in Hungary, Torino-Giardini Reali (NLIGR) in Italy—while slightly antedating and geographically proximate to the other cemeteries, did not show evidence of cultural practices traditionally termed Longobard. In the Hungarian necropoleis, findings like artificially modified skulls and Gothic inscriptions of the Aryan Bible provide evidence supporting a Germanic origin of the buried individuals. In the Italian necropolis (Giardini Reali) the burial methods and the absence of grave goods suggest a local origin in the early Middle Ages. Additional information on the archeological context is available in the supplementary materials text. Bone samples were analyzed in the Molecular Anthropology Laboratory of the University of Florence, exclusively dedicated to ancient DNA analysis. Negative controls were used in all the experimental steps to monitor the absence of contaminants in reagent and environment. Long bones and tooth samples were cleaned by removing the surface layer using a dentist drill with disposable tips and exposed under UV light (λ = 254 nm) for 45 min on each side. Overall, 100 mg of powder were sampled from inside the compact portion of the bones and from the dentine of the tooth root and used in DNA extraction. Petrous bones were cleaned by brushing the surface and exposed under UV light (λ = 254 nm) for 45 min on each side. A disk saw was used to section the petrous bone and the inner surfaces were exposed under UV light (λ = 254 nm) before collecting the bone powder from the densest part of inner ear using a dentist drill with disposable tips as suggested in ref. [8]. DNA was extracted following the protocol proposed in ref. [9].
Fig. 1.

Geographical and genetic relationship between LC and NLC populations. Here and through the other figures LC cemeteries are represented by a circle while NLC ones are indicated by a square. Pie charts representing the frequencies of the major haplogroups in the populations. The size of the charts is proportional to the number of samples inside each necropolis
Table 1.
Cemetery information
| Cemetery | Country | Date/associated culture | Analyzed samples | Genotyped samples | |
|---|---|---|---|---|---|
| LCRMUS | Musov | Czech Republic | VI Century/Longobard | 10 | 7 |
| LHSZ | Szólád | Hungary | VI Century/Longobard | 46 | 39 |
| LHHEG | Hegykő | Hungary | V–VI Century/Longobard | 10 | 8 |
| LICOL | Collegno | Italy | VI–VII Century/Longobard | 36 | 23 |
| LIFAR | Fara Olivana | Italy | VI–VII Century/Longobard | 10 | 1 |
| NLHFON | Fonyód | Hungary | V Century/Non-Longobard | 5 | 3 |
| NLHHACS | Hács-Béndekpuszta | Hungary | V Century/Non-Longobard | 5 | 3 |
| NLHBAL | Balantoszemes-Szemesi Berek | Hungary | V Century/Non-Longobard | 10 | 2 |
| NLIGR | Torino-Giardini Reali | Italy | Early Medieval/Non-Longobard | 3 | 1 |
For each group, geographical and chronological distribution and culture are reported. The table also shows the number of analyzed samples for each cemetery and the number of mitochondrial genomes obtained
NGS library preparation and sequencing
NGS libraries were prepared starting from 20 µl of DNA extract for each specimen following a double-stranded DNA protocol [10] using a unique combination of two indexes per specimen. Libraries were enriched for mitochondrial DNA following a multiplexed capture protocol [11] and sequenced on an Illumina MiSeq run for 2 × 75 + 8 + 8 cycles.
Mitochondrial DNA sequence pre-processing and mapping
Sequences were demultiplexed and sorted according to the sample, then raw sequence data were analyzed using the pipeline described in ref. [12]. Merged reads were mapped on the revised Cambridge Reference Sequence, rCRS (GenBank Accession Number NC_012920.1). Reads with mapping quality below 30 were discarded. Consensus sequence for each sample was obtained considering positions covered at least threefold, and base calling was performed with at least 70% of concordance between reads. Misincorporation pattern was analyzed using MapDamage 2.0 [13] and contamination estimate was performed by contamMix [14]. Only samples with at least 92% of the mitochondrial genome covered at least threefold, with CtoT values higher than 17% and MAP authentic values higher than 92% according to contamMix were considered for population genetics analysis. This resulted in a total of 78 suitable sequences from cemeteries archeologically associated with the Longobard culture (7 from LCRMUS, 39 from LHSZ, 8 from LHHEG, 23 from LICOL, 1 from LIFAR) and 9 suitable sequences from cemeteries not associated to the Longobard culture (3 from NLHFON, 3 from NLHHACS, 2 from NLHBAL and 1 from NLIGR) (Table S2). All sequences have been deposited in GenBank under accession numbers MG182446–MG182470 and MG182472–MG182533 and polymorphisms are indicated in Table S3 with respect to the rCRS reference sequence.
Mitochondrial haplogroup definition and population genetics analyses
Mitochondrial haplogroups were assigned to the new mtDNA sequences according to PhyloTree Build 16 on Haplogrep [15, 16]. Phylogenetic networks were constructed using the Median Joining algorithm [17] implemented in Network 5.0 program (http://www.fluxus-technology.com). The ε value was set to 0 and the transversions were weighted 3× the weight of transitions. Networks were subjected to maximum parsimony post-analysis. Haplogroup frequencies for medieval populations were retrieved from previously published data in Csákyová et al.2016 [18] and references therein. As the vast majority of studies provided haplogroup frequencies inferred from HVRI, we reassigned the newly reported LC and NLC to haplogroups employing only this region. PCA based on haplogroup frequencies was conducted employing the function fviz_pca_biplot from the library factoextra [19] in R 3.4.0 [20]. The haplogroups frequencies of complete mitochondrial genomes from ancient populations (from Neolithic to medieval times) were taken from [21–32]. For a comparison with modern European variation, we used the data published by ref. [33], and we calculated FST values between pairs of populations with Arlequin 3.5 [34]. We compared LC and NLC complete mitochondrial genomes with a PCA using the function dudi.pca from the package adegenet [35]. We also computed pairwise differences between sequences with Arlequin v. 3.5 [34] and visualized them employing an MDS computed with the function cmdscale. To locate possible population structure inside our dataset, we assessed the best number of clusters inside it using the find.clusters function in adegenet [36], comparing the output of 10 independent runs using a custom-made R script. We then applied a DAPC analysis [37] on the dataset with 100,000 iterations.
Demographic simulations
To explicitly test the importance of NLC populations on the genetic makeup of migrating LC individuals, we conducted demographic simulations under an Approximate Bayesian Computation model framework. Accounting for the results of the exploratory analyses, we first hypothesized two models called admixture and continuity (Fig. 2a, b). The former recreate a scenario where a migrant population, LC from northern Europe, receive gene flow from local NLC individuals before moving to colonize other regions. The continuity model instead postulates no contact between LC and NLC populations, mimicking only the proposed Longobard migration from Czech Republic to Italy. As Hungarian NLC populations contained individuals found buried along with the artifacts are possibly associated with other Germanic cultures, we also developed two additional models named recent origin and recent origin + admixture (Fig. 2c, d). The recent origin model postulated a common ancestry between Hungarian NLC populations and LC ones sometimes around the first century CE, with a subsequent migration of the latter in Hungary. According to this model, the possible genetic similarities between Hungarian LC and NLC only derived from a recent common origin and not from local admixture events. The recent origin + admixture model presents, intuitively, the same general structure depicted in the recent origin scenario with the exception of an event of admixture in Hungary between LC populations and NLC ones. In all the tested models, we considered the Czech sample as the first source of migration. This does not mean that we considered the Czech Republic as the place of origin of Longobards (which, however, would have no consequences on the results of the analyses), but since inhumations rather than cremations only appear in that region, it is not possible to sample aDNA from other putative Longobard homelands. In order to provide an unbiased representation of genetic diversity inside our dataset, we first removed related samples based on the kinship analysis presented in ref. [38]. We also excluded from our demographic dataset 2 individuals from Szólád (LHSZ27A1/2) as the dating of their burial suggest a more recent origin with respect to surrounding graves. This process resulted in a reduced dataset of 78 unrelated sequences. We obtained 20,000 simulations for each of the proposed scenarios with the software package ABCtoolbox [39]. We performed the model selection procedure making use of the novel approach developed by Pudlo et al. [40], called ABC-rf, which rely on the “random-forest” machine learning approach [41]. Random forest uses the simulated datasets for each model in a reference table to predict the best suited model at each possible value of a set of covariates. After selecting it, another random forest obtained from regressing the probability of error of the same covariates determine the posterior probability. This procedure allows one to overcome the difficulties traditionally associated with the choice of summary statistics, while gaining a larger discriminative power among the competing models [41]. We built the reference table using the function abcrf from the package abcrf and employing a forest of 500 trees, as this number was suggested to provide the best trade-off between computational efficiency and statistical precision [41]. We carried out the actual model comparisons and obtained the posterior probabilities of the winning models using the function predict from the same package. To summarize the genetic information contained in our sequences, we considered the number of haplotypes, the number of private polymorphic sites, Tajima’s D, the mean number of pairwise differences for each population, the mean number of pairwise differences between populations and pairwise Fst. These summary statistics were obtained with arlsumstat [34]. We validated the model selection procedure calculating the classification error through the abcrf function of the abcrf R package. To do this, we used as pseudo-observed datasets each dataset of our reference table (Table S4). To verify whether the selected models were able to generate the observed data, we performed a linear discriminant analysis (LDA) using the function plot.abcrf. In order to estimate the parameters for the model chosen by the ABC-rf procedure (the admixture one), we ran further simulations reaching 1 million datasets. We applied a locally weighted multivariate regression [42] after a logtan transformation [43] of the 3000 best-fitting simulations to estimate the admixture model’s parameter using an R scripts from http://code.google.com/p/popabc/source/browse/#svn%2Ftrunk%2Fscripts, modified by SG. The complete list of models’ parameters and of the associated prior distributions are presented in Table S5.
Fig. 2.

Alternative models of relationship between LC and NLC populations. a Admixture, b continuity, c recent origin, d recent origin + admixture. Continuous lines represent populations that were included in the simulations while dotted lines characterize ghost populations
Results
Medieval Mitochondrial genomes from Czech Republic, Hungary and Italy
The 87 medieval mitogenomes were sequenced to an average coverage depth of 86.10× (from 6.66× to 201.89×, Table S2). Sequences were assigned to 71 distinct haplotypes, all falling within the expected overall mitochondrial diversity of western Eurasian mtDNA (Table S2). Indeed, most individuals belong to the H, T2 and J lineages (respectively, occurring in 33, 11 and 7 samples, Figs. 1 and 3), all of them commonly observed in Europe [44]. Mušov and Szólád LC cemeteries show similar frequencies of the H haplogroup (28% and 32%, respectively), while in Collegno this frequency is doubled (60%) (Fig. 1). The haplogroup distribution was rather different in Szólád and Hegykő (LC groups in Hungary). We found the U8 haplogroup uniquely present in Hegykő, with a frequency of 25%, while the H haplogroup is underrepresented with respect to the other samples. Phylogenetic links between haplotypes and their distribution among the archeological sites are shown in the Median Joining Network (Fig. 3). Haplotypes were broadly grouped into their respective lineages and no general structure associated with geography or culture seems to be present.
Fig. 3.

Median-joining network of the newly sequenced individuals. The size of the circles is proportional to the number of samples carrying a specific haplotype, and the background shading indicates the affiliation of the lineages to the major haplogroups
Relationship between LC/NLC groups and other European ancient and modern samples
To elucidate the affinities between our new LC/NLC samples and coeval individuals, we retrieved data on mtDNA haplogroup frequencies for 10 European medieval populations. A principal component analysis (PCA) on this expanded dataset highlighted the similarity between the LC graves of Szólád and medieval populations from Central Europe (Slovakia 800–1100 CE, Poland 1000–1400 CE) (Fig. 4). The Collegno individuals clustered midway between Slovakian and medieval samples from Southern Europe (Spain 500–600 CE, Italy 900–1400 CE). We extended the comparison to older samples, for which haplogroup frequencies calculated on complete mitochondrial genomes were available. The haplogroups composition of these ancient populations is shown in Fig S1. The most common haplogroup in our samples is H, which is also present at relatively high frequencies in almost all ancient populations. The rarer haplogroup N, found mostly in LCRMUS and LHSZ, appears to be present at low frequencies in ancient Hungarian samples (from Early Neolithic to medieval times), as well as in Britain, Central Europe and Balkan ancient samples (Fig S1). LICOL and LHSZ showed low frequencies of haplogroup I, which is documented in Central Europe and the Pontic Steppes since the Middle Bronze age, and then in the Hungarian Conquerors published by Neparaczki et al. [30].
Fig. 4.
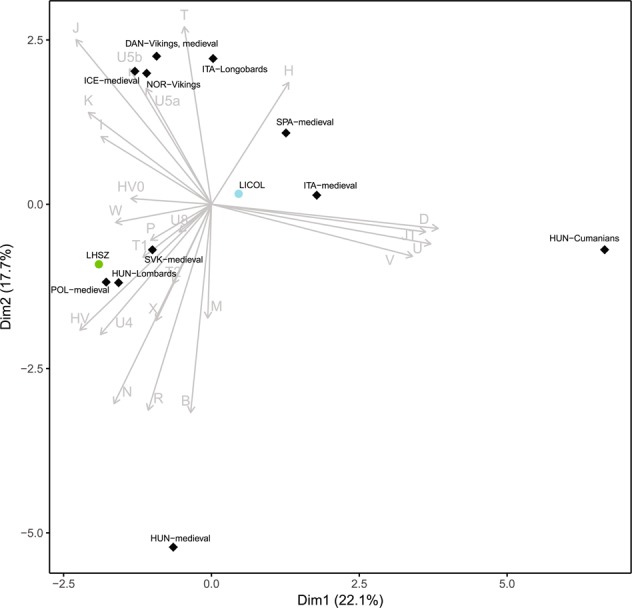
PCA based on haplogroup frequencies of LC and medieval populations. Circles represent LC necropoleis, whereas diamonds characterize the 11 medieval European populations
Next, we compared both LC and NLC necropoleis with 14 modern-day Eurasian populations (Fig. S2). In general, NLC samples showed shorter genetic distances with modern European populations; among the LC samples, the one from Szólád is the less differentiated from the modern genetic variation. Noticeably, The Collegno LC sample shows the lowest genetic distance with the modern Hungarian sample, and then, to a lesser extent, with the Italian one.
LC/NLC mitochondrial genetic variation and archeological context
We explored the relationships between LC and NLC individuals through a principal component analysis (PCA, Fig. S3A). The first two axes of the PCA suggest a certain similarity between groups, as NLC individuals are found across all of the range of genetic variation shown by the LC samples. However, the NLC sample is much smaller than the LC sample, and so this finding does not suggest that the NLC individuals might represent just a subset of the genetic variation in the populations they belong to. There is also no clear geographical structure between samples in our dataset, with individuals from Italy, Hungary and Czech Republic clustering together. However, the first PC clearly separates a group of 12 LC individuals found at Szólád, Collegno and Mušov from a group composed by both LC and NLC individuals. The same pattern is also found when pairwise differences among individuals are plotted by multidimensional scaling (MDS, Fig. S4). To further investigate this peculiar genetic structure, we performed a k-means analysis and a discriminant analysis of principal components (DAPC) on the whole dataset. At K = 4 the 12 LC individuals located by the first PC form a cluster together, and stay together even at K = 7, the most supported number of clusters (Fig. S3B-C-D). It is worth noting that the partition of LC and NLC individuals in different clusters actually follows their haplogroup differentiation, with these 12 LC individuals belonging to haplogroups that are generally rare in modern and ancient European populations (I, N, W and X). The macrohaplogroups I and W (that were present in ancient populations from Britain, Central Europe and Steppe, see Fig. S1) now show their highest frequency in northern Europe (e.g. Finland [45]). The peculiarity of this group is strengthened by archeological information from the Szólád cemetery, where 8 of the 12 individuals in this group originated, indicating that all these samples were found buried with typical Longobard artifacts and grave assemblages. We do not find the same tight association for the 3 samples from Collegno, where the 3 graves are indeed devoid of evident Germanic cultural markers; however, they are not placed in a separate and marginal location—as for the tombs without grave goods found in Szólád—but among graves with wooden chambers and weapons. In this light, the individuals buried in this manner may have been members of the same community as well, but belonging to the lowest social level. Finally, this group also includes an individual from the Mušov graveyard. This finding is particularly interesting in light of the fact that the Mušov necropolis has been only tentatively associated with Longobard occupation (see ESM for details), based on the presence of few archeological markers. This Mušov individual belongs to haplogroup N, and actually represents the only member of the Mušov sample not showing a common haplogroup (everybody else belongs to the H or T haplogroup).
Testing models of Longobard migrations
To shed light on these early-Medieval populations’ dynamics, we used an Approximate Bayesian Computation approach. We compared the possibility of admixture between LC and local NLC populations with a scenario postulating a simpler migration of LC individuals with no additional contact (Fig. 2a, b). We also tested two additional scenarios to explicitly consider that the similarities between LC and NLC individuals in Hungary may derive only from a recent common origin rather than local admixture (Fig. 2c, d). The admixture model received strong support, with probability of 86% (Table S4). The four models were well recognized by the ABC procedure we followed, as indicated by both the classification error (Table S6) and by the LDA plot (Fig. S5 and S6). This result can be interpreted as reflecting gene flow from NLC inhabitants of the region into a migrating LC group. Indeed, when we estimated the extent of these admixture events, we observed that more than 80% of the genetic makeup of the Hungarian LC population could be traced to NLC people already inhabiting the region, while the Czech LC contributed around 18% (Table S7 and Fig. S7). This could either indicate a reduced contribution of Czech LC to the genetic makeup of the Hungarian LC, or that the Mušov cemetery, while showing archeological signs of Longobard occupation, is not a good proxy of the LC population that moved from the Czech Republic to Hungary. The Collegno individuals can, instead, trace more than 70% of their genetic makeup to LC populations migrating from Hungary, confirming the high degree of similarity shown by the exploratory analyses and supporting the migration hypothesis based on archeological data.
Discussion
In this work, we extracted and analyzed complete mitochondrial genomes from 87 early-Medieval individuals sampled in 9 necropoleis. On the basis of archeological information, we have classified these cemeteries as putatively occupied by Longobards or by different early-Medieval communities. These genetic data have been used to explore, for the first time, the genetic variation and structure of these groups, so as to understand whether, and to what extent, different communities found along the route of the Longobard migration may resemble each other biologically. We also explicitly tested migration models, in which genetic exchange between neighboring communities was possible. Our exploratory analyses highlighted a degree of genetic similarity between the LC and NLC communities, as shown by the level of haplogroups sharing (Fig. 1) and by the short genetic distance between LC and NLC groups (Fig. S2). This result is not surprising, given the well-known overall genetic similarity among all European populations. The most represented macrohaplogroup was H, found at high frequency in both LC and NLC groups. Other haplogroups shared between LC and NLC were T, J, U5 and K. However, a peculiar set of samples, including only LC individuals from different European regions, appeared to be well distinct from the rest of the samples, consistent with the results of migrational exchanges between Pannonia and Italy. In most cases, these individuals were also associated with burials with typical Germanic grave goods, a term that here defines an assemblage of objects including Longobard artifacts, but not only them. This particular association, together with the presence in this cluster of haplogroups that reach high frequency in Northern European populations, suggests a possible link between this core group of individuals and the proposed homeland of different ancient barbarian Germanic groups, which will be further investigated. Therefore, at present we do not have evidence that the populations associated with northern Europe were not already in the region decades prior to the time that the Longobards entered the area. However, these groups were likely related with the Gothic confederation, hence having different culture and provenience than Longobards. As both archeological and bioarchaeometric analysis of the Szólád LC cemetery suggested that it was in use only for a short period of time, we suggest a recent arrival of LC communities in Hungary [7].
To better understand whether the observed genetic similarities across Europe could indeed result from migration, and to quantify the degree of resemblance among LC and NLC neighbor groups, we compared evolutionary/demographic models accounting for the degree of genetic variation here observed in the LC and NLC samples. The strongest support was observed for a model including admixture between LC and NLC populations with respect to models postulating recent common origin of the Hungarian samples, or assuming genetic isolation during the putative Longobard migration from Pannonia to Italy. This result highlights the importance of the genetic exchanges during the migration period between individuals belonging to different communities. The other interesting result was the high degree of genetic resemblance between the LC cemeteries in Hungary and Collegno, in Italy. We indeed estimated that about 70% of the lineages found in Collegno actually derived from the Hungarian LC groups, in agreement with previous archeological and historical hypotheses. This supports the view that the spread of Longobards into Italy actually involved movements of people, who gave a substantial contribution to the gene pool of the resulting populations. As a consequence of these genetic similarities, the Collegno LC sample shows higher genetic affinities with modern Hungarians than with Italians (Fig. S2). This is even more remarkable thinking that, in many studied cases, military invasions are movements of males, and hence do not have consequences at the mtDNA level. Here, instead, we have evidence of maternally linked genetic similarities between LC in Hungary and Italy, supporting the view that immigration from Central Europe involved females as well as males.
Analysis of nuclear data, as well as more genetic information from necropoleis not associated with the Longobard culture, would help elucidate in more detail these past population dynamics. However, the resolution provided by the mitochondrial genomes presented in this study, combined with detailed archeological information, has allowed us to explicitly test demographic hypotheses, thus significantly improving our knowledge about different aspects of the complex pattern of migrations involving Longobard populations.
Supplementary information
Acknowledgements
We would like to thank Angela Livia Casadei for her help in the analysis of genetic data. This study has been supported by the following institutions and foundations: Riksbankens Jubileumsfond, Alexander von Humboldt Stiftung, Bundesministerium für Bildung und Forschung, Gerard B. Lambert Foundation, Università degli Studi di Padova, Institute for Advanced Study. KV and PG were supported by NSF #1450606. GB, AB and FT were supported by the ERC Advanced Grant Agreement No. 295733, ‘LanGeLin’ project.
Data and software availability
All newly generated data have been deposited in GenBank: MG182446–MG182470 and MG182472–MG182533.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Stefania Vai, Andrea Brunelli, Alessandra Modi
Contributor Information
David Caramelli, Phone: +390552757744, Email: david.caramelli@unifi.it.
Silvia Ghirotto, Phone: +39 0532 455951, Email: ghrslv@unife.it.
Supplementary information
The online version of this article (10.1038/s41431-018-0319-8) contains supplementary material, which is available to authorized users.
References
- 1.Bemmann J, Schmauder M, editors. Die Langobarden in den Schriftquellen bis zu den Markomannenkriegen. Proceedings of the Mitteleuropa Langobarden – Awaren – Slawen. p. 43–50; Frankfurt a.M., Bonn: Habelt, 2008.
- 2.Geary P. The myth of nations. The medieval origins of Europe. Princeton: Princeton University Press; 2002. [Google Scholar]
- 3.Koncz I. A historical date and its archaeological consequences. Acta Archaeol Acad Sci Hung 2015;66:315–40.
- 4.Giostra C. Goths and Lombards in Italy: the potential of archaeology with respect to ethnocultural identification. Eur J Post-Class Archaeol. 2011;1:7–36. [Google Scholar]
- 5.Vai S, Ghirotto S, Pilli E, et al. Genealogical relationships between early medieval and modern inhabitants of Piedmont. PLoS ONE. 2015;10:e0116801. doi: 10.1371/journal.pone.0116801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vai S, Lari M, Ghirotto S, Rizzi E, Achilli A, Olivieri A, et al. Diachronic and synchronic genetic analysis of ancient Piedmont population. J Biol Res. 2012;1:LXXXV.
- 7.Alt KW, Knipper C, Peters D, et al. Lombards on the move-an integrative study of the migration period cemetery at Szolad, Hungary. PLoS ONE. 2014;9:e110793. doi: 10.1371/journal.pone.0110793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinhasi R, Fernandes D, Sirak K, et al. Optimal ancient DNA yields from the inner ear part of the human petrous bone. PLoS ONE. 2015;10:e0129102. doi: 10.1371/journal.pone.0129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dabney J, Knapp M, Glocke I, et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci USA. 2013;110:15758–63. doi: 10.1073/pnas.1314445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010:prot5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 11.Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS ONE. 2010;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Modi A, Tassi F, Susca RR, et al. Complete mitochondrial sequences from Mesolithic Sardinia. Sci Rep. 2017;7:42869. doi: 10.1038/srep42869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jónsson H, Ginolhac A, Schubert M, Johnson PLF, Orlando L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29:1682–4. doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Q, Mittnik A, Johnson Philip LF, et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr Biol. 2013;23:553–9. doi: 10.1016/j.cub.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kloss-Brandstatter A, Pacher D, Schonherr S, et al. HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum Mutat. 2011;32:25–32. doi: 10.1002/humu.21382. [DOI] [PubMed] [Google Scholar]
- 16.van Oven M. PhyloTree Build 17: Growing the human mitochondrial DNA tree. Forensic Sci Int. 2015;5:e392–e394. [Google Scholar]
- 17.Bandelt HJ, Forster P, Sykes BC, Richards MB. Mitochondrial portraits of human populations using median networks. Genetics. 1995;141:743–53. doi: 10.1093/genetics/141.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Csakyova V, Szecsenyi-Nagy A, Csosz A, et al. Maternal genetic composition of a medieval population from a Hungarian-Slavic contact zone in Central Europe. PLoS ONE. 2016;11:e0151206. doi: 10.1371/journal.pone.0151206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kassambara A, Mundt F. Factoextra: extract and visualize the results of multivariate data analyses. R package version. J Stat Softw. 2016;25:1–18.
- 20.R Develompment Core Team. R: a language and environment for statistical computing. Vienna, Austria: the R Foundation for Statistical Computing. http://www.R-project.org/; 2011.
- 21.Tassi F, Vai S, Ghirotto S, Lari M, Modi A, Pilli E, et al. Genome diversity in the Neolithic Globular Amphorae culture and the spread of Indo-European languages. Proc Biol Sci. 2017;284:20171540. [DOI] [PMC free article] [PubMed]
- 22.Schiffels S, Haak W, Paajanen P, et al. Iron Age and Anglo-Saxon genomes from East England reveal British migration history. Nat Commun. 2016;7:10408. doi: 10.1038/ncomms10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allentoft ME, Sikora M, Sjogren KG, et al. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–72. doi: 10.1038/nature14507. [DOI] [PubMed] [Google Scholar]
- 24.Gunther T, Valdiosera C, Malmstrom H, et al. Ancient genomes link early farmers from Atapuerca in Spain to modern-day Basques. Proc Natl Acad Sci USA. 2015;112:11917–22. doi: 10.1073/pnas.1509851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haak W, Lazaridis I, Patterson N, et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522:207–11. doi: 10.1038/nature14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipson M, Szecsenyi-Nagy A, Mallick S, et al. Parallel palaeogenomic transects reveal complex genetic history of early European farmers. Nature. 2017;551:368–72. doi: 10.1038/nature24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martiniano R, Caffell A, Holst M, et al. Genomic signals of migration and continuity in Britain before the Anglo-Saxons. Nat Commun. 2016;7:10326. doi: 10.1038/ncomms10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathieson I, Lazaridis I, Rohland N, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mittnik A, Wang CC, Pfrengle S, et al. The genetic prehistory of the Baltic Sea region. Nat Commun. 2018;9:442. doi: 10.1038/s41467-018-02825-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neparaczki E, Kocsy K, Toth GE, et al. Revising mtDNA haplotypes of the ancient Hungarian conquerors with next generation sequencing. PLoS ONE. 2017;12:e0174886. doi: 10.1371/journal.pone.0174886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olalde I, Brace S, Allentoft ME, et al. The Beaker phenomenon and the genomic transformation of northwest Europe. Nature. 2018;555:190–6. doi: 10.1038/nature25738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rusu I, Modi A, Vai S, et al. Maternal DNA lineages at the gate of Europe in the 10th century AD. PLoS ONE. 2018;13:e0193578. doi: 10.1371/journal.pone.0193578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batini C, Hallast P, Vagene AJ, et al. Population resequencing of European mitochondrial genomes highlights sex-bias in Bronze Age demographic expansions. Sci Rep. 2017;7:12086. doi: 10.1038/s41598-017-11307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 35.Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–5. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 36.Jombart T, Ahmed I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics. 2011;27:3070–1. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amorim CEG, Vai S, Posth C, Modi A, Koncz I, Hakenbeck S, et al. Understanding 6th-century barbarian social organization and migration through paleogenomics. Nat Commun. 2018;9:3547. [DOI] [PMC free article] [PubMed]
- 39.Wegmann D, Leuenberger C, Neuenschwander S, Excoffier L. ABCtoolbox: a versatile toolkit for approximate Bayesian computations. BMC Bioinformatics. 2010;11:116. doi: 10.1186/1471-2105-11-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pudlo P, Marin JM, Estoup A, Cornuet JM, Gautier M, Robert CP. Reliable ABC model choice via random forests. Bioinformatics. 2016;32:859–66. doi: 10.1093/bioinformatics/btv684. [DOI] [PubMed] [Google Scholar]
- 41.Breiman L. Random forests. Mach Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 42.Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–35. doi: 10.1093/genetics/162.4.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamilton G, Stoneking M, Excoffier L. Molecular analysis reveals tighter social regulation of immigration in patrilocal populations than in matrilocal populations. Proc Natl Acad Sci USA. 2005;102:7476–80. doi: 10.1073/pnas.0409253102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kivisild T. Maternal ancestry and population history from whole mitochondrial genomes. Investig Genet. 2015;6:3. doi: 10.1186/s13323-015-0022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hedman M, Brandstatter A, Pimenoff V, et al. Finnish mitochondrial DNA HVS-I and HVS-II population data. Forensic Sci Int. 2007;172:171–8. doi: 10.1016/j.forsciint.2006.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All newly generated data have been deposited in GenBank: MG182446–MG182470 and MG182472–MG182533.