

Abstract
Missense variants located to the “molecular brake” in the tyrosine kinase hinge region of platelet-derived growth factor receptor-β, encoded by PFGFRB, can cause Penttinen-type (Val665Ala) and Penttinen-like (Asn666His) premature ageing syndromes, as well as infantile myofibromatosis (Asn666Lys and Pro660Thr). We have found the same de novo PDGFRB c.1997A>G p.(Asn666Ser) variants in two patients with lipodystrophy, acro-osteolysis and severely reduced vision due to corneal neovascularisation, reminiscent of a severe form of Penttinen syndrome with more pronounced connective tissue destruction. In line with this phenotype, patient skin fibroblasts were prone to apoptosis. Both in patient fibroblasts and stably transduced HeLa and HEK293 cells, autophosphorylation of PDGFRβ was observed, as well as increased phosphorylation of downstream signalling proteins such as STAT1, PLCγ1, PTPN11/SHP2-Tyr580 and AKT. Phosphorylation of MAPK3 (ERK1) and PTPN11/SHP2-Tyr542 appeared unaffected. This suggests that this missense change not only weakens tyrosine kinase autoinhibition, but also influences substrate binding, as both PTPN11 tyrosines (Tyr542 and Tyr580) usually are phosphorylated upon PDGFR activation. Imatinib was a strong inhibitor of phosphorylation of all these targets, suggesting an option for precision medicine based treatment.
Subject terms: Genetics research, Mutation, Protein-protein interaction networks
Introduction
Platelet-derived growth factor (PDGF) receptors are membrane-bound receptor tyrosine kinases (RTKs) important for connective tissue growth [1, 2]. After foetal development, signalling through PDGFRs is primarily turned on during connective tissue remodelling, as in wound healing. There are two PDGF receptors, α (PDGFRα) and β (PDGFRβ). Upon ligand binding to the receptors’ immunoglobulin-like extracellular domain, dimerisation that favours phosphorylation of tyrosine residues on the cytoplasmic domain occurs [3]. This, in turn, activates the kinase function of the receptor and exposes docking sites for proteins with Src-homology (SH) and phosphotyrosine-binding (PTB) domains, like protein-tyrosine phosphatase-nonreceptor-type 11 (PTPN11, also known as SHP2) and phospholipase-Cγ (PLCγ), signal transducers and activators of transcription (STATs), and several adaptor molecules [4, 5]. RTK activation is a complex process dependent on RTK type, density, endocytosis (often with continued signalling) and degradation (after ubiquitination). There are also positive and negative feedback loops that affect the likelihood of an RTK to be active or inactive, two receptor states that are in dynamic equilibrium with each other—sometimes described as receptor “breathing” [5].
So far, PDGFRB is the only of the two PDGF receptors where variants have been linked to congenital disease. Missense variants in PDGFRB have been associated with idiopathic basal ganglia calcifications (OMIM#615007; Leu658Pro and Arg987Trp) [6], infantile myofibromatosis (OMIM#228550; Arg561Cys, Pro660Thr and Asn666Lys) [7–9], Penttinen-type premature aging syndrome (OMIM#601812; Val665Ala) [10–12], Kosaki overgrowth syndrome (OMIM#616592; Pro584Arg and Trp566Arg) [13–15], and was recently found (Asn666His) in a single patient with Penttinen-overlapping features (hemangioma, craniosynostosis, intracranial cysts, gingival hypertrophy, acro-osteolysis, contractures and carpal tunnel syndrome) [16]. PDGFR targeted tyrosine kinase inhibitor treatment has been effective in two patients with germline activating PDGFRB variants; one a Penttinen-syndrome like patient with Asn666His [16], the other a child with infantile myofibromatosis and germline Arg681Cys [17].
Here, we describe two individuals with a severe form of Penttinen syndrome, both with a previously undescribed p.(Asn666Ser) amino acid substitution caused by the same de novo variant c.1997A>G in PDGFRB. Analysis of patient fibroblasts and transduced cell lines showed that the variant caused autophosphorylation of PDGFRβ and induced phosphorylation of several downstream signalling proteins: STAT1, PLCγ1, PTPN11-Y580 and AKT. In line with the lipodystrophic skin appearance, skin fibroblasts were prone to apoptosis. Imatinib was a strong inhibitor of constitutively activated Asn666Ser-substituted PDGFRβ, suggesting an option for patient treatment.
Materials and methods
The study was approved by the Regional Committee for Medical and Research Ethics, Western Norway (IRB # 00001872) (ref. no. 2010/3337 and 2014/59). The patients and their families were invited to participate in the study and were included after informed consent, and this included specific consent to publish facial photographs.
Patient recruitment
The two patients are adults of similar age, a woman from Norway (Fig. 1) and a man from Australia (Fig. 2). Both patients turned out to carry the same missense variant, NM_002609.3(PDGFRB) c.1997A>G p.(Asn666Ser), later verified to be de novo in both patients after patient and parental DNA testing by Sanger sequencing. Unique and identical phenotypes combined with identical de novo variant findings were considered as strong evidence for causality of the Asn666Ser missense change.
Fig. 1.

Top: facial photographs of the Norwegian patient from childhood until adult life. The maxillary hypoplasia was visible at age 1 year, giving a characteristic appearance. Joint contractures developed from age 4 years. She has had progressive scoliosis from age 14 years. Due to her fragile skin, operative correction has not been attempted. At age 18 years, she still had not lost her primary dentition, but the secondary incisors of the lower jaw were slowly emerging. Bottom: Corneal neovascularisation starting on her right eye at age 5–6 year causing severe visual impairment. She has fragile lipodystrophic skin with ulceration and osteolytic resorption of her feet and, to a lesser degree, her hands
Fig. 2.

Top: The patient from Australia has maxillary hypoplasia and eye protrusion due to shallow orbits and reduced visual acuity due to corneal neovascularisation. The skin is thin with clearly visible subcutaneous vessels. Bottom: He has contractures of large and small joints, scoliosis and skin ulceration, especially on the feet
Variant detection
After “false negative” results from in-house whole exome sequencing in 2013 due to low PFGDRB coverage after exome selection (only 6 reads corresponding to the variant position), whole genome sequencing of DNA from Patient 1/parents-trio and Patient 2 was performed by Macrogen (South Korea). In the Norwegian patient/parents-trio, SNP filtering was done by the following criteria: the variant should be de novo with a dbSNP frequency of less than 1%, it should be located in an exon or canonical splice site, and the sequencing depth should be at least 10 times. Three variants passed these filtering and quality criteria, but only one of these variant was also found in the Australian patient: chr5(GRCh37) g.149503839T>C, NM_002609.3(PDGFRB) c.1997A>G p.(Asn666Ser). This variant has been registered in LOVD (www.LOVD.nl) under patient ID#183370.
Patient skin fibroblast culture and lysis
Skin biopsy samples were obtained from the two patients (repeat biopsies were taken from the Norwegian patient to get enough cells) and healthy controls. The specimens were cut into small pieces for standard fibroblast culture at 37 °C with 5% CO2 in Amniochrome II medium with Amniochrome II Supplement for growth stimulation (Lonza, Verviers, Belgium). The cells were gradually transferred to Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (Lonza) supplemented with 10% foetal calf serum, penicillin, streptomycin and glutamine. When cells were 80% confluent, the medium was replaced with serum-free DMEM. After 16 h, cells were either treated with 0.1 μM imatinib (STI571, Selleckchem, Munich, Germany) or left untreated, and harvested after 6 h. Cells were lysed in 50 mM Tris-HCl pH 7.5 with 200 mM NaCl, 5 mM EDTA, 2% phosphatase inhibitor coctail-2 (from Sigma, St. Louis, MO), 1% Igepal, 1 mM phenylmethylsulfonylfluoride, 1× complete protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany), 0.5% Tween and 0.1% SDS, and stored at −20 °C before immunoblot analysis (−80 °C for long term storage).
Transgenic cell preparation and culture
A murine retroviral vector containing the PDGFRB open-reading frame was acquired from VectorBuilder (pMMLV[Exp]-hPDGFRB[NM_002609.3]:IRES:Puro, item #VB150910-10054 from Cyagen Biosciences, Santa Clara, CA). The PDGFRB c.1997A>G variant was inserted into the vector using the QuickChange Site-Directed Mutagenesis Kit (Catalog #200518, Agilent Technologies, Sydney, Australia) following the manufacturers recommendations. Mutant clones were verified by Sanger sequencing. Virus production was performed by transfecting Phoenix-AMPHO packaging cells (CRL-3213, ATCC, Manassas, VA) as described previously [18]. Two days after transfection, the medium was harvested and HEK293 cells (CRL-1573 from ATTC), HEK293T/17 cells (CRL-11268 from ATTC) and HeLa cells (CCL-2 from ATTC) were transduced following standard protocols [19]. All cells were grown in DMEM containing 10% foetal calf serum. Two days post-infection, stably transduced cells were selected by adding 10 ng/ml puromycin (cat# ant-pr-1, InvivoGen, San Diego, CA) to the culture medium, cells were thereafter kept in selection medium for additional 14 days. The cells were then treated with imatinib or left untreated, as described above.
ELISA and immunoblot analysis
For ELISA analysis, cells were harvested at 107 cells/ml in 1% NP-40, 20 mM Tris (pH 8.0), 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM activated sodium orthovanadate, 10 μg/ml Aprotinin, 10 μg/mL Leupeptin. To measure phosphorylated PDGFRβ, a DuoSet® IC PDGFRβ kit was used (# DYC1767-2, R&D systems) following the manufacturer’s recommendations. In this kit, an immobilised phospho-PDGFRβ antibody binds PDGFRβ (both phosphorylated and unphosphorylated) in the cell lysate. After washing, a HRP-conjugated antibody against phosphorylated tyrosine is used to detect receptor phosphorylation. This experiment was performed in duplicate.
For immunoblot analysis, proteins were separated on a high-resolution gel system (4–12% NuPAGE Novex Bis-Tris Gel; Life, Carlsbad, CA) according to the manufacturer’s instructions and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% non-fat dry milk (Bio-Rad), 1% glycine and 1% BSA in PBS-T buffer (standard PBS with 0.05% Tween 20), and incubated overnight at 4 °C with primary antibodies from Cell Signalling Technology (Danvers, MA) at recommended dilutions, catalogue numbers in the parentheses: phospho-Tyr70-STAT1 (#7649), STAT1 (#9172), phospho-Tyr783-PLCγ1 (#2821), PLCγ1 (#5690), phospho-Tyr542-PTPN11/SHP-2 (#3751), phospho-Tyr580-PTPN11/SHP-2 (#3703), PTPN11/SHP-2 (#3397), phospho-Ser473-AKT (#4060), AKT (#4691), phospho-Thr202/Tyr204-MAPK3/ERK1 (#4370) and MAPK3/ERK1 (#4695). Membranes were washed with PBS-T buffer and PBS and incubated with horseradish peroxidase-conjugated anti-rabbit IgG (#7074) for 1 h at room temperature. For phospho-Tyr580-PTPN11/SHP-2, we used a modified protocol and blocked with 5% non-fat dry milk in TBS-T, incubated overnight with primary antibody in 5% BSA in TBS-T followed by washing with TBS-T according to the manufacturer’s instructions. Proteins were visualised by the SuperSignal West Pico system alone or with added SuperSignal West Femto Maximum Sensitivity Substrate (ThermoFisher Scientific, Waltham, MA), using a ChemiDoc Touch Imaging System (BioRad). A protein standard (MagicMark, Life) was used as molecular weight marker. To control for equal loading, the membranes were washed in PBS and blocked, before they were incubated overnight with anti-GAPDH primary antibody (Sigma #G99545) and visualised as described above. Each experiment was performed in duplicates or more.
Measurement of apoptosis
Fibroblasts at 60–70% confluence were trypsinized, washed with chilled PBS and filtered through 70 micron filters. After centrifugation at 1000 rpm for 5 min at 4 °C, cells were resuspended in 200 μL of 1× annexin binding buffer (#V13246, ThermoFisher). Thereafter, 10 μL of annexin-V-FITC (#A13199, ThermoFisher) and 1 μL SYTOX Blue Dead Cell Stain (#S34857, Molecular Probes, Eugene, OR) were added, followed by dark incubation for 15 min at room temperature. The degree of apoptosis was determined by flow cytometry on a SONY SH800 Flow Cytometer. Normal skin fibroblasts treated for 20 h with the apoptosis inducing agent camptotechine (Sigma #C9911) at a final concentration of 10–6 M served as positive controls. For each sample of patient and control fibroblasts, a minimum 40,000 cells were used for each analysis using the FlowJo software. Each experiment was performed in triplicates.
Results
Two patients with strikingly similar progeria-like phenotypes, reminiscent of a severe form of Penttinen syndrome, are presented. Patient 1, a Norwegian woman now 24 years old, was born in pregnancy week 36 with birth weight 2450 g (˜10th centile), length 49 cm and head circumference 33 cm. She appeared normal, apart from large haemangiomas on both shoulders and close to the right eye, all three removed five days after birth. The histological diagnosis given at that time was juvenile haemangiopericytomas, later verification impossible. At age two weeks she was shunted for hydrocephalus and later re-operated twice. The first clinical suspicion of advanced biological age, i.e., of progeria, was at age 10 months. Unlike classical progeria of the Hutchinson-Gilford type (OMIM#186670), she has never had short stature with poor hair growth. Sanger sequencing of the LMNA gene did not reveal any disease-causing variants. Prominent features have been mid-face retraction due to maxillary hypoplasia, secondary exophthalmos due to shallow orbits, lipodystrophy, corneal neovascularisation and contractures of large (e.g., hips and knees) and small (metacarpophalangeal and interphalangeal) joints. Her length initially followed the 75th centile (mid parental height: 174.5 cm), later she became shorter due to progressive scoliosis from age 14 years. There is osteopenia in her vertebral column, but no degenerative changes. In her phalangeal and metacarpal/metatarsal bones, osteolytic changes were found. Head circumference has always been within upper normal range. Intelligence is normal. A wheelchair has since long been needed for mobility. The major health problems of adult life have been progressive scoliosis, progressive visual impairment due to conjunctival overgrowth of the cornea, osteolytic resorption of her feet and to a lesser degree her hands, and fragile lipodystrophic skin with ulceration, requiring frequent and painful wound revisions (Fig. 1). Internal organs are normal. She has good blood perfusion of her legs and feet, indicating that her skin ulcerations are not related to insufficient blood flow. Recently, after long standing ulceration refractive to therapy, she developed a squamous skin carcinoma on the sole of her right foot necessitating amputation below the knee. She has been offered experimental treatment with imatinib due to a favourable effect in vitro (see below). Imatinib is an established treatment of low-malignant dermatofibrosarcoma protuberans, associated with a COL12A1-PDGFRB fusion due to a 17;22 translocation causing PDGFRB overexpression [20]. So far, she has refused such treatment.
Patient 2 is a 24 years old Australian man. His birth weight was 3544 g (¨~50th centile), and he was born with haemangiomas on both flanks, the right ear, the right foot, the forehead and one on the inside of the skull. They all regressed upon steroid treatment. He was shunted for hydrocephalus as a new born and once again at age two years. His hydrocephalus was considered secondary to haemorrhage of the intracranial haemangioma. Seizures were present from the first day of life, and he was treated with phenobarbital until age 8 months. The seizures recurred at age 2 years and responded to carbamazepine. He has not had any seizures after 3 years of age. Early on he was tall (length along the 97th centile and head circumference on the 50th centile). Initial motor development was normal (e.g., he sat at age 6 months and walked at age 14 months). Similar to the Norwegian patient, he also had an aged appearance from an early age. The skin was thin with little or no subcutaneous fat, making subcutaneous vessels clearly visible. There were also areas of skin hypertrophy, especially on the extensor surfaces of major joints, and often co-occurring with skin depigmentation. In other areas, skin hyperpigmentation was a feature. Contractures due to scarring became increasingly prominent, of both large (e.g., the elbow on Fig. 2) and small joints, limiting the use of his hands and feet. Another prominent feature was maxillary hypoplasia and eye protrusion due to shallow orbits, resulting in a facial appearance that strikingly resembled Patient 1 (Figs 1 and 2). In childhood, scoliosis started to develop before age 10 years, and a wheelchair was needed for mobility. With time, the scoliosis and skin ulceration, especially on the feet and legs, became major health problems, as well as visual impairment (visual acuity 0.25 measured by Snellen chart) caused by pterygium formation and corneal drying due to problems closing the eyelids. He has not tried imatinib treatment, mainly because he has been lost to follow-up.
In these two patients with strikingly similar progeria-like phenotypes, whole genome sequencing revealed the same de novo missense change in PDGFRB in both patients: NM_002609.3(PDGFRB) c.1997A>G p.(Asn666Ser). In silico prediction programs (SIFT, Mutation taster, PolyPhen-2) all judged this Asn666Ser change of a highly conserved asparagin in the tyrosine kinase class III signature motif of PDGFRβ to be pathogenic. The tyrosine kinase class III signature motif corresponds to PDB entry #PS00240: G659-x-H-x-N-[LIVM]VN666LLGACT672, PDGFRβ amino acid positions indicated with superscripts. This sequence motif is located in the kinase hinge region between the autoinhibitory domain and the more C-terminal tyrosine kinase domain in the cytoplasmic part of the receptor tyrosine kinase (RTK). N666 in PDGFRβ corresponds to N549 in FGFR2, which constitutes the “molecular brake” of this RTK, i.e., an essential part of the autoinhibitory domain [3, 21].
To examine if N666S resulted in gain-of-function, both PDGFRβ-mediated autophosphorylation and downstream target phosphorylation were investigated. Phosporylation of PDGFRβ was markedly increased in PDGFRβ-N666S transduced HeLa cells, indicating that the N666S substitution did indeed diminish autoinhibition and stimulate RTK activation (Fig. 3, panel A). This was also associated with increased phosphorylation of the downstream signalling proteins STAT1, PLCγ1, PTPN11-Y580 and AKT, but not PTPN11-Y542 or MAPK3 in patient fibroblasts (Fig. 3, panel b). In PDGFRB mutant-transduced or wt-transduced HEK293 cells, similar findings were observed with the exception P-AKT since AKT appeared constitutively phosphorylated in these cells (Supplementary Fig. S1). When patient cells or transduced HEK293 cells were treated with the tyrosine kinase inhibitor imatinib, this apparent PDGFRβ-N666S-induced phosphorylation disappeared (Fig. 3 and S1), suggesting that imatinib could be a treatment option for these patients.
Fig. 3.

Panel a: ELISA studies of HeLa cells show 2–3 times more phosphorylated PDGFRβ in cells transduced with the PDGFRB-Asn666Ser vector than with the PDGFRB- wild type (wt) vector. Panel b: Western blots of cultured fibroblasts from a healthy control (Ctr) and patient-1 (N666S) with the Asn666Ser variant, untreated or treated with 0.1 μM imatinib (STI57). Specific antibodies were used to detect phosphorylated PLCγ1 (P-PLCγ1) and total PLCγ1 (PLCγ1), phosphorylated STAT1 (P-STAT1) and total STAT1 (STAT), phosphorylated-Tyr542-PTPN11, (P-PTPN11Y542) phosphorylated-Tyr580- PTPN11 (P-PTPN11Y580, and total PTPN11 (PTPN11), phosphorylated AKT (P-AKT) and total AKT (AKT), phosphorylated MAPK3 and total MAPK3. The amount of GAPDH (lines marked with asterisk (*)) was measured as a protein loading control. Molecular sizes are indicated in kDa. Similar results were seen using skin fibroblasts from Patient-2
We established three patient fibroblast cultures (two from Patient 1 and one from Patient 2). Normal cell morphology was seen in early passage patient fibroblast cultures. However, cell morphology started to change after 3–4 passages into more elongated cells that were multi-layered in some places. Later, cytoplasmic extensions appeared and apoptotic membrane blebbing became evident, and the cells ceased to grow (Supplementary Fig. S2). In contrast, control fibroblasts looked normal for >10 passages. Transduced HeLa and HEK293 cells did not have overt apoptosis, and no major differences regarding the morphology of these immortal cells could be observed. This apparent propensity of patient fibroblasts to enter apoptosis was confirmed by an apoptosis assay, followed by flow cytometric quantification. Much higher percentage of apoptotic cells (Fig. 4 and Supplementary Fig. S3), reflecting early (Q3 fraction) and late (Q2 fraction) apoptosis, was found in patient fibroblast cultures compared to control fibroblast cultures after a similar number of passages.
Fig. 4.
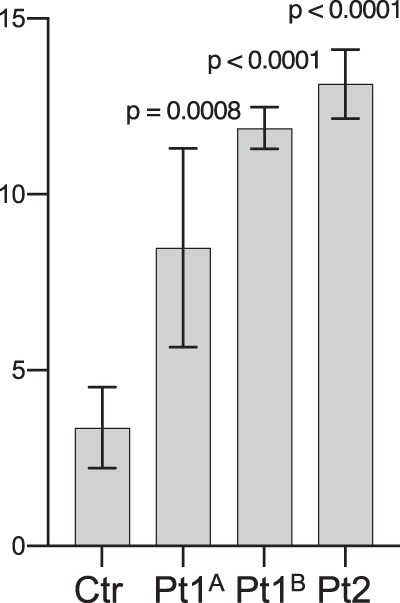
Apoptosis assay followed by flow cytometric quantification. Increased percentage of apoptotic cells (Q3 and Q2 fractions, reflecting early and late apoptosis, respectively) was found in two separate Norwegian (Pt1A and Pt1B) and a single Australian (Pt2) patient fibroblast cultures, and compared to three control fibroblast cultures (Ctr bar show merged results) after a similar number of passages. The values above the error bars represent p-values from an unpaired t-test
Discussion
In two patients with a striking progeria-like phenotype dominated by lipodystrophy, acro-osteolysis, corneal neovascularisation and scoliosis (Figs 1 and 2), we found the same de novo missense change in PDGFRB, predicting an Asn666Ser substitution. The patients’ phenotypes resemble a severe end of the Penttinen syndrome spectrum with more widespread destruction of connective tissue including bone [10–12], leading to extensive disfigurement or degradation of the hands and feet, and extensive skin ulcerations. Functional studies on patient fibroblasts, as well as transduced HeLa and cells, showed that Asn666Ser caused constitutive tyrosine kinase activation, like previously found for other Penttinen-syndrome associated variants (Fig. 3) [12, 16].
The phenotypic spectrum seen in patients with different missense variants in PDGFRB range from basal ganglia calcifications, infantile myofibromatosis and premature aging to somatic overgrowth with neurologic deterioration [6–14, 16]. The basal ganglia calcification-related Leu658Pro and Arg987Trp substitutions were found to abolish kinase activity (Leu658Pro) or destabilise PDGFRβ (Arg987Trp) [22]. Leu658Pro is also in the hinge region of PDGFRβ, very close to the “molecular brake region” where our patients’ Asn666Ser substitution is located. Possibly, a proline at this location stabilises the RTK in an inactive conformation and explains the defective signalling. Other hinge-region missense variants are likely or proven to be gain-of-function variants with increased tyrosine kinase activity [12, 16, 23]. These distinct conditions have overlapping phenotypic features. Replacement of Asn666 with other amino acids seems to diminish the degree of kinase autoinhibition by disturbing the brake either directly or indirectly and promote PDGFRβ phosphorylation [21]. Intriguingly, PDGFRβ-Asn666Ser native (i.e., patient fibroblast, Fig. 3b) or transduced (HEK293, Fig. S1) cells displayed constitutively increased phosphorylation of PTPN11-Y580, but not PTPN11-Y542, which usually is co-phosphorylated with Y580 upon conventional ligand-induced PDGFRβ activation [24]. It remains to be seen if these hinge-region (and other) PDGFRβ substitutions also influence substrate binding, i.e., not only affect the equilibrium between inactive and active RTK conformations.
We found evidence that PDGFRB-Asn666Ser caused phosphoinositide 3-kinase (PI3K)/AKT signalling (phosphorylation of AKT and PLCγ) and increased STAT-mediated regulation of transcription (phosphorylation of STAT1) even in the absence of receptor ligand (PDGF-BB). Phosphorylation of MAPK3 was not induced (Fig. 3 and S1), despite reports indicating that PTPN11-Y580 is essential for sustained ERK phosphorylation [24, 25]. It has been shown that ligand binding to either phosphorylated Y542 or Y580 play regulatory roles for PTPN11 activity [26], and thus for growth factor-mediated signal transduction through PTPN11 [5, 27]. The results were similar but not identical to a cell line transfected with the PDGFRB-Val665Ala variant found in patients with classical Penttinen syndrome, where constitutive PLCγ and STAT3 but not AKT phosphorylation was found [12]. We did not investigate STAT3 phosphorylation, but STAT1 phosphorylation was constitutively increased (Fig. 3 and S1). STAT1 has recently been found necessary for the severe autoinflammation and wasting seen in mice with an activating PDGFRB variant (Asp849Val) [28]. PDGFRB activation on a STAT1 negative KO-mouse background was associated with overgrowth and prolonged life span, while the presence of STAT1 caused autoinflammation and wasting. Our patients’ phenotypes are certainly reminiscent of the latter.
It appears that increased tissue destruction may contribute to acro-osteolysis, scoliosis, lipodystrophy and chronic skin ulceration. Extensive apoptosis was observed in short-term patient-derived primary fibroblast cultures (Fig. 4). Of note, this apoptotic tendency coexisted with findings suggesting stimulation of tissue growth, like the patients’ upper normal or tall statures (in childhood on the 75th and 97th centiles), the congenital haemangiomas and the corneal neovascularisation. It is well known that increased connective tissue growth can be caused by increased PDGFRβ signalling [4, 28], and there are also previous indications that PDGFRβ overstimulation may cause apoptosis [29, 30].
The hot spots for PDGRB-activating missense mutations involved in cancer are those affecting the juxtamembrane domain and the activation loop [22]. In PDGFRα, substitutions of amino acid Asn659, corresponding to Asn666 in PDGFRβ, was a common finding in cancer cells (see the COSMIC database; http://cancer.sanger.ac.uk/cosmic), especially Asn659Tyr and Asn659Lys. The Asn659Lys change corresponds to the PDGFRβ Asn666Lys variant associated with infantile myofibromatosis. Unlike PDGFRα, PDGFRβ has few changes reported in COSMIC. Val665Ala (the Penttinen syndrome variant) and Asn666Ser (our patients’ variant) are reported only once. It is not known if there is an increased cancer risk due to these PDGFRβ variants. Of note, the squamous cell skin carcinoma of the Norwegian patient developed in an area of long-standing hyperkeratoses and ulcerations. Thus, the underlying cause might be chronic wound healing and not necessarily constitutive PDGFRβ activation.
Tyrosine kinase-activating PDGFRB variants can be sensitive to tyrosine kinase inhibitors like imatinib, sunitinib, nilotinib and ponatinib [9, 16, 17, 23]. Since imatinib not only inhibits ABL tyrosine kinase but also PDGFRβ, the drug has been approved for treatment of dermatofibrosarcoma protuberans caused by PDGFRβ overexpression [2, 20]. Drug tolerance is generally good with few side effects, but an effect on growth can be seen in children [31]. These tyrosine kinase inhibitors were found to be effective in a patient with myofibromatosis and a germ-line PDGFRB variant (Arg681Cys) [17], and a Penttinen-syndrome like patient with the Asn666His variant that was treated with sunitinib for >1 year with good effect on joint contractures and quality of life parameters [16]. We found imatinib to be an efficient inhibitor also of Asn666Ser-activated PDGFRB, preventing in vitro phosphorylation of PLCγ, STAT1 and AKT in both patient fibroblasts and transduced HEK293 cells (Fig. 3 and S1). Taken together, it is likely that imatinib represents a treatment option also for our progeria patients. Such treatment may improve skin atrophy, stop acro-osteolysis and prevent visual loss. One of our patients has been offered treatment, but declined, and the other has been lost to follow-up.
In summary, we describe two individuals with a striking progeria-like condition that resemble a severe end of the Penttinene syndrome spectrum with more widespread destruction of connective tissue. Both patients have the same de novo Asn666Ser missense variant in PDGFRB that cause constitutive RTK autophosphorylation and activation of downstream interaction partners. Imatinib was an excellent in vitro inhibitor of Asn666Ser substituted PDGFRβ, suggesting an option for treatment.
Supplementary information
Acknowledgements
We would like to thank Unni Larsen for technical assistance and Raoul CM Hennekam for professional assistance. The work was supported by grants from the Western Norway Regional Health Authority (911466, 911977, and 912161 to C.B.) and Dr. Jon S. Larsens Foundation (to C.B.).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41431-018-0323-z) contains supplementary material, which is available to authorised users.
References
- 1.Chen PH, Chen X, He X. Platelet-derived growth factors and their receptors: structural and functional perspectives. Biochim Biophys Acta. 2013;1834:2176–86. doi: 10.1016/j.bbapap.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen H, Marsiglia WM, Cho MK, et al. Elucidation of a four-site allosteric network in fibroblast growth factor receptor tyrosine kinases. Elife. 2017;6:e21137. [DOI] [PMC free article] [PubMed]
- 4.Heldin CH. Targeting the PDGF signaling pathway in the treatment of non-malignant diseases. J Neuroimmune Pharmacol. 2014;9:69–79. doi: 10.1007/s11481-013-9484-2. [DOI] [PubMed] [Google Scholar]
- 5.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolas G, Pottier C, Maltete D, et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology. 2013;80:181–7. doi: 10.1212/WNL.0b013e31827ccf34. [DOI] [PubMed] [Google Scholar]
- 7.Cheung YH, Gayden T, Campeau PM, et al. A recurrent PDGFRB mutation causes familial infantile myofibromatosis. Am J Hum Genet. 2013;92:996–1000. doi: 10.1016/j.ajhg.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martignetti JA, Tian L, Li D, et al. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. Am J Hum Genet. 2013;92:1001–7. doi: 10.1016/j.ajhg.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arts FA, Sciot R, Brichard B, et al. PDGFRB gain-of-function mutations in sporadic infantile myofibromatosis. Hum Mol Genet. 2017;26:1801–10. doi: 10.1093/hmg/ddx081. [DOI] [PubMed] [Google Scholar]
- 10.Penttinen M, Niemi KM, Vinkka-Puhakka H, Johansson R, Aula P. New progeroid disorder. Am J Med Genet. 1997;69:182–7. doi: 10.1002/(SICI)1096-8628(19970317)69:2<182::AID-AJMG13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 11.Zufferey F, Hadj-Rabia S, De Sandre-Giovannoli A, et al. Acro-osteolysis, keloid like-lesions, distinctive facial features, and overgrowth: two newly recognized patients with premature aging syndrome, Penttinen type. Am J Med Genet A. 2013;161A:1786–91. doi: 10.1002/ajmg.a.35984. [DOI] [PubMed] [Google Scholar]
- 12.Johnston JJ, Sanchez-Contreras MY, Keppler-Noreuil KM, et al. A point mutation in PDGFRB causes autosomal-dominant penttinen syndrome. Am J Hum Genet. 2015;97:465–74. doi: 10.1016/j.ajhg.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takenouchi T, Yamaguchi Y, Tanikawa A, Kosaki R, Okano H, Kosaki K. Novel overgrowth syndrome phenotype due to recurrent de novo PDGFRB mutation. J Pediatr. 2015;166:483–6. doi: 10.1016/j.jpeds.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 14.Minatogawa M, Takenouchi T, Tsuyusaki Y, et al. Expansion of the phenotype of Kosaki overgrowth syndrome. Am J Med Genet A. 2017;173:2422–7. doi: 10.1002/ajmg.a.38310. [DOI] [PubMed] [Google Scholar]
- 15.Gawlinski P, Pelc M, Ciara E, et al. Phenotype expansion and development in Kosaki overgrowth syndrome. Clin Genet. 2018;93:919–24. doi: 10.1111/cge.13192. [DOI] [PubMed] [Google Scholar]
- 16.Pond D, Arts FA, Mendelsohn NJ, Demoulin JB, Scharer G, Messinger Y. A patient with germ-line gain-of-function PDGFRB p.N666H mutation and marked clinical response to imatinib. Genet Med. 2018;20:142–50. doi: 10.1038/gim.2017.104. [DOI] [PubMed] [Google Scholar]
- 17.Mudry P, Slaby O, Neradil J, et al. Case report: rapid and durable response to PDGFR targeted therapy in a child with refractory multiple infantile myofibromatosis and a heterozygous germline mutation of the PDGFRB gene. BMC Cancer. 2017;17:119. doi: 10.1186/s12885-017-3115-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swift S, Lorens J, Achacoso P, Nolan GP. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol. 2001;Chapter 10:Unit 10 17C. doi: 10.1002/0471142735.im1017cs31. [DOI] [PubMed] [Google Scholar]
- 20.Ugurel S, Mentzel T, Utikal J, et al. Neoadjuvant imatinib in advanced primary or locally recurrent dermatofibrosarcoma protuberans: a multicenter phase II DeCOG trial with long-term follow-up. Clin Cancer Res. 2014;20:499–510. doi: 10.1158/1078-0432.CCR-13-1411. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Ma J, Li W, et al. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–30. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arts FA, Velghe AI, Stevens M, Renauld JC, Essaghir A, Demoulin JB. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J Cell Mol Med. 2015;19:239–48. doi: 10.1111/jcmm.12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arts FA, Chand D, Pecquet C, et al. PDGFRB mutants found in patients with familial infantile myofibromatosis or overgrowth syndrome are oncogenic and sensitive to imatinib. Oncogene. 2016;35:3239–48. doi: 10.1038/onc.2015.383. [DOI] [PubMed] [Google Scholar]
- 24.Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem. 2003;278:41677–84. doi: 10.1074/jbc.M306461200. [DOI] [PubMed] [Google Scholar]
- 25.Tajan M, de Rocca Serra A, Valet P, Edouard T, Yart A. SHP2 sails from physiology to pathology. Eur J Med Genet. 2015;58:509–25. doi: 10.1016/j.ejmg.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Sun J, Lu S, Ouyang M, et al. Antagonism between binding site affinity and conformational dynamics tunes alternative cis-interactions within Shp2. Nat Commun. 2013;4:2037. doi: 10.1038/ncomms3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang SQ, Tsiaras WG, Araki T, et al. Receptor-specific regulation of phosphatidylinositol 3’-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol. 2002;22:4062–72. doi: 10.1128/MCB.22.12.4062-4072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He C, Medley SC, Kim J, et al. STAT1 modulates tissue wasting or overgrowth downstream from PDGFRbeta. Genes Dev. 2017;31:1666–78. doi: 10.1101/gad.300384.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petti LM, Ray FA. Transformation of mortal human fibroblasts and activation of a growth inhibitory pathway by the bovine papillomavirus E5 oncoprotein. Cell Growth Differ. 2000;11:395–408. [PubMed] [Google Scholar]
- 30.Zhang Y, Lehman JM, Petti LM. Apoptosis of mortal human fibroblasts transformed by the bovine papillomavirus E5 oncoprotein. Mol Cancer Res. 2002;1:122–36. [PubMed] [Google Scholar]
- 31.Millot F, Guilhot J, Baruchel A, et al. Growth deceleration in children treated with imatinib for chronic myeloid leukaemia. Eur J Cancer. 2014;50:3206–11. doi: 10.1016/j.ejca.2014.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.