

Abstract
The landscape of head and neck squamous cell carcinoma (HNSCC) has been changing rapidly due to growing proportion of HPV-related disease and development of new therapeutic agents. At the same time, there has been a constant need for individually tailored treatment based on genetic biomarkers in order to optimize patient survival and alleviate treatment-related toxicities. In this regard, aberrations of PI3K pathway have important clinical implications in the treatment of HNSCC. They frequently constitute ‘gain of function’ mutations which trigger oncogenesis, and PI3K mutations can also lead to emergence of drug resistance after treatment with EGFR inhibitors. In this article, we review PI3K pathway as a target of treatment for HNSCC and summarize PI3K/mTOR inhibitors that are currently under clinical trials. In light of recent advancement of immune checkpoint inhibitors, consideration of PI3K inhibitors as potential immune modulators is also suggested.
Keywords: HNSCC, PI3K, mTOR, Akt, EGFR, PIK3CA, HPV, Drug resistance, Precision medicine
Background
Head and neck squamous cell carcinoma (HNSCC) arises from mucosal epithelium of oral cavity, pharynx and larynx. An estimate of 61,000 new cases of HNSCC were diagnosed in the US in 2016, with 13,190 deaths attributable to the disease [1]. Traditional risk factors include tobacco smoking, alcohol consumption, betel nut chewing and genetic predisposition such as Fanconi anemia [2–4]. Human papillomavirus (HPV) has recently emerged as a major and distinct risk factor for HNSCC. HPV-related HNSCC most commonly arises in oropharynx and has been associated with younger age of disease onset, less smoking history, better performance status and favorable prognosis [5]. The proportion of HPV-positive oropharyngeal squamous cell cancer has significantly increased for the past decade regardless of sex and race [6], raising the need for a separate therapeutic strategy.
Comprehensive genomic analysis of HNSCC revealed frequent alterations in genes encoding molecules in phosphoinositide 3-kinase (PI3K) pathway including PIK3CA, PTEN and PIK3R1 [7, 8]. In particular, HPV-related HNSCC frequently harbors mutations in the helical domain of PIK3CA, yet its biological significance has not been fully elucidated. In the era of precision medicine, it is becoming more important to understand key genomic alterations and their therapeutic implications [9]. This review will focus on the role of PI3K-Akt-mTOR pathway in relation to epidermal growth factor receptor (EGFR) and their clinical applications in HNSCC.
Phosphoinositide 3-kinase (PI3K) and PI3K-Akt-mTOR pathway
PI3K is a family of phospholipid kinase that is divided into three classes based on structure, function and substrate specificity. Class I PI3K is a heterodimer that consists of a regulatory and a catalytic subunit. It is further divided into class IA and IB. For class IA PI3K, there are three variants of catalytic subunit, p110α, p110β and p110δ (encoded by PIK3CA, PIK3CB and PIK3CD), and five variants of regulatory subunit, p85α, p55α, p50α (encoded by PIK3R1 and splice variants), p85β and p55δ (encoded by PIK3R2 and PIK3R3). p85 regulatory subunit contains Src homology 2 (SH2) domain which binds to phosphorylated Y-X-X-M motif in receptor tyrosine kinase [10]. It was found that five isoforms of regulatory subunit express different affinities to tyrosine kinases [11], and each p110 subunit is selectively recruited to receptor activation [12, 13]. These findings are consistent with selective mutation of p110 in various types of cancer and provides important prospect for targeted therapy. PIK3CA is one of the most commonly mutated and extensively studied oncogenes in various types of human cancer. An analysis of The Cancer Genome Atlas (TCGA) data showed that PIK3CA was the most frequently mutated gene in breast cancer samples, second most frequently mutated gene in uterine corpus endometrial cancer and third most commonly mutated gene in HNSCC [14]. PIK3CA is also heavily mutated in lung squamous cell carcinoma, urothelial carcinoma of bladder and colorectal adenocarcinoma [14]. Molecular composition of p110α, the product of PIK3CA, and p85α are illustrated in Fig. 1.
Fig. 1.
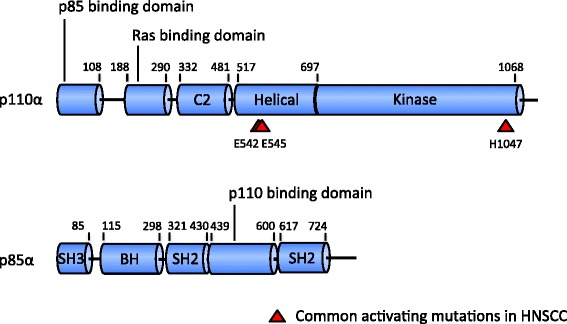
Linear composition of p110α and p85α molecules. Red arrowheads in p110α indicate ‘hotspot’ mutations. C2 in p110α is a putative membrane-binding domain. Breakpoint cluster region-homology (BH) domain in p85α has shown GTPase activating protein (GAP) activity toward Rab family. Rab GTPase induces degradation and deregulation of activated growth factor receptors, and mutated Rab GAP induces cell transformation [148]. However, it is unclear if this function is still active in complex with p110α [149]. BH domain in p85α is flanked by proline-rich domain, implying an auto-regulatory mechanism in interaction with its SH3 domain [150]
Class IB PI3K consists of p110γ catalytic subunit (encoded by PIK3CG) and p101 or p87 regulatory subunit (encoded by PIK3R5, PIK3R6). Class IA and IB PI3K phosphorylate 3-hydroxyl group of phosphatidylinositol (PI), phosphatidylinositol 4-phosphate (PIP) and phosphatidylinositol 4,5-bisphosphate (PIP2), producing phosphatidylinositol 3-phosphate (PI-3-P), phosphatidylinositol 3,4-bisphosphate (PI-3,4-P2) and phosphatidylinositol 3,4,5-triphosphate (PIP3), respectively [15]. Expressions of p110δ and p110γ are found exclusively in lymphocytic immune system whereas p110α and p110β are expressed ubiquitously [16]. Idelalisib, a drug used for treatment of lymphoma, is a selective inhibitor of p110δ which is abundantly expressed in malignant B cells [17].
Class II PI3K is a monomer of catalytic isoforms, C2α, C2β and C2γ (encoded by PIK3C2A, PIK3C2B and PIK3C2G), and lacks regulatory subunit. Class II lipid kinase produces PI-3,4-P2 from PIP and PI-3-P from PI. C2α isoform found in endosomes was suggested to play a role in angiogenesis and vascular barrier formation [18]. Class III PI3K is a heterodimer of a regulatory (Vps15, encoded by PIK3R4) subunit and a catalytic subunit (Vps34, encoded by PIK3C3), which converts PI to PI-3-P. Little is known about physiologic role of class III PI3K, but it was implicated in induction of autophagy in the state of nutrient deficiency [19].
The family of PI3K proteins mainly regulates cellular growth and cycle. Its activation is triggered by upstream receptor tyrosine kinase such as ErbB family receptor (including EGFR), platelet-derived growth factor receptor (PDGFR), insulin-like growth factor 1 receptor (IGF-1R) or G protein-coupled receptor (GPCR). PI3K attaches a phosphate group to the 3′ hydroxyl of the inositol head of PIP2, converting it to PIP3 [20]. Inositol phospholipids constitute a minor part of the cellular membrane and phosphorylation of inositol head has little effect on membrane structure. However, phosphorylated inositol head protruding from the membrane provides an anchoring site for secondary signaling molecules that are floating in the cytosol. Once PIP3 is formed by PI3K, cytosolic molecules such as Akt/Protein kinase B localize to plasma membrane and become tethered to the head of PIP3 via Pleckstrin homology (PH) domain in N terminal [21]. Activated Akt, in turn, phosphorylates a series of molecules including mechanistic target of rapamycin (mTOR) that promotes cell survival, proliferation and motility. The action of PI3K, conversion of PIP2 to PIP3, is negatively regulated by reverse phosphatases, such as phosphatase and tensin homolog (PTEN). Other cytoplasmic molecules that contain PH domain and interact with PIP3 include Rho-guanine nucleotide exchange factor (GEF). Rho family proteins, when activated by GEF, remodel cytoskeleton, decrease contact inhibition and increase cell motility, all of which elevate invasiveness in cancer cells [22].
Implications of PI3K pathway alteration for EGFR pathway in HNSCC
EGFR is a cell surface receptor tyrosine kinase in ErbB family and has been an attractive therapeutic target for various human cancers including HNSCC. The receptor becomes activated by ligand binding which transitions EGFR monomers to the allosteric homodimer. Receptor dimerization stimulates tyrosine kinase activity in C terminal domain and initiates downstream phosphorylation cascade through PI3K-Akt-mTOR, Raf-MEK-MAP kinase or JAK/STAT pathways (Fig. 2).
Fig. 2.
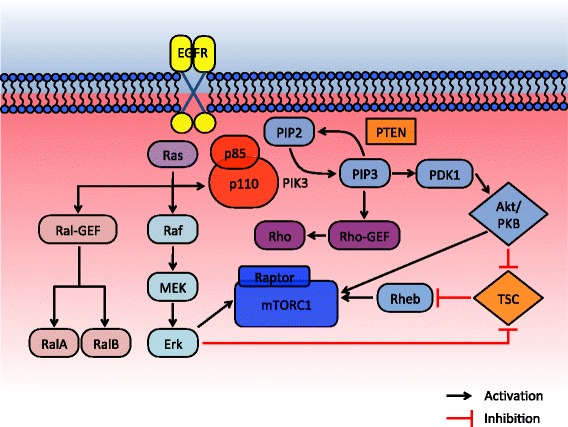
Interactive signaling pathway of EGFR-PI3K-mTOR. PI3K binds to cytoplasmic tail of receptor tyrosine kinase via SH domains within p85 regulatory subunit. Activation signal can also be transferred through Ras-binding domain in p110 catalytic subunit which tethers PI3K molecule to Ras protein in growth receptors. p110 activation by Ras binding is inhibited by p85 subunit which can be released by co-stimulation of SH domain by tyrosine kinase [151]
It has been well known that EGFR overexpression is involved in carcinogenesis of HNSCC [23, 24], and associated with poor prognosis [25, 26]. EGFR-targeting strategy with a monoclonal antibody, cetuximab, has prolonged survival of patients with locally advanced HNSCC in combination with radiotherapy [27]. Cetuximab is currently used with platinum-based chemotherapy as the first line treatment for HNSCC or for recurrent or metastatic (R/M) disease [28, 29]. However, efforts to develop a predictive biomarker for EGFR-targeting treatment have not been successful. In particular, overexpression of EGFR assessed by immunohistochemistry (IHC) could not be correlated with the level of treatment response to cetuximab [30–32]. Additionally, resistance to cetuximab has been widely observed in various types of cancer including HNSCC. Several evasive mechanisms may serve to restore original oncogene dependence, circumventing the initial targeting treatment. Receptors can potentially abrogate inhibitory action of therapeutic agents as they obtain second mutations that result in pharmacokinetic changes [33]. A well-known mutation of EGFR, T790M, enhances affinity of the kinase pocket for ATP, which competitively blocks binding of tyrosine kinase inhibitors [34]. Copy number gains of target genes also reactivate dependent pathway and counteract the treatment effect. For example, amplification of BRAF via copy number gains was found in 8% of the tumor samples from metastatic melanoma treated with BRAF inhibitors [35]. Studies with HNSCC demonstrated as well that copy number alteration by amplification of 7p11.2 accounts for a number of cases of EGFR activation [36–38]. It was also hypothesized that ligand overexpression or receptor cross phosphorylation triggers uncontrolled EGFR hyperactivity. A genetic profiling of HNSCC samples with EGFR activation revealed that EGFR ligands (including TGFα) were highly expressed in a subset, suggesting an establishment of an autocrine loop [39].
Alternatively, the function of target gene can be bypassed by activating downstream molecules of the signaling cascade or switching dependence to an alternative pathway for cell growth and proliferation [40]. As the tumor progresses and develops genomic heterogeneity, cells with genetic survival benefit outgrow through evolutionary selection pressure. In consistent with this theory, whole-exome sequencing of melanoma cells that are resistant to BRAF inhibitor revealed diverse genetic alterations in the downstream MAPK pathway [41]. Similarly, KRAS amplification or mutation was found in tumor samples from colorectal cancer patients who developed resistance to EGFR inhibitors [42]. Relevant to our review, compensatory activation of downstream pathway, mainly PI3K, has been proposed as one of the major resistance mechanisms to EGFR inhibitors in HNSCC. Gene expression of the molecules in PI3K pathway was elevated in cetuximab-resistant strains compared to cetuximab-susceptible cells [43], and addition of mTOR/PI3K inhibitor effectively achieved control of cell growth in HNSCC that acquired resistance to EGFR inhibitors [44, 45].
PI3K-mTOR alteration in HNSCC
66% of HNSCC harbor genomic alterations in one of the major components of PI3K pathway [46]. An analysis of whole-exome sequencing of 151 HNSCC tumors revealed that PI3K is the most commonly mutated mitogenic pathway among PI3K, JAK/STAT and MAPK and that presence of multiple mutations in PI3K signaling pathway is correlated with more advanced disease [8]. Physiologic data confirms that an aberrant PI3K-mTOR pathway is associated with cell motility, invasion and metastasis. PI3K-PTEN balance has a direct effect on chemotaxis and cell motility as it controls actin cytoskeleton via Rho family proteins, such as Rho, Rac and CDC42 [22, 47]. PIP3 and PIP2 determine epithelial polarity in individual cells, thus dysfunctional PI3K results in epithelial-mesenchymal transition, a critical event in tumor invasion [48].
PI3KCA is among the most frequently mutated genes in HNSCC, affected both in HPV-positive and negative diseases (56 and 34%, respectively) [7]. PIK3CA mutations in HPV-positive HNSCCs are concentrated in helical domain, whereas mutations are more spread out in HPV-negative diseases [9, 49]. TCGA data presents that 73% of PIK3CA mutations are located at E542, E545 in the helical domain and in H1047 in the kinase domain [7]. Frequency of these ‘hotspot’ mutations is also higher in HPV-positive oropharyngeal cancers [50].
Targeting PIK3CA alteration in human squamous cell xenografts has demonstrated susceptibility to treatment in vitro and in vivo, leading a path for its clinical implication. Inhibition of PI3K by competitive blockage of ATP binding site led to decreased phosphorylation of Akt in several studies [51–54]. In a number of the patient-derived xenografts harboring E545K and H1047R mutations, PI3K inhibitors were effective in achieving control of tumor growth [43, 55, 56]. Additionally, activation of PI3K/mTOR pathway from either mutation or gene amplification was positively correlated with tumor susceptibility to PI3K inhibitors in xenograft models [52, 57–59]. However, preclinical data also suggested that additional molecular change should interact with PIK3CA alteration for tumorigenesis. Cell lines engineered to harbor PIK3CA mutations in the ‘hotspots’ responded more favorably to PI3K/mTOR dual inhibition than PI3K inhibition only, indicating that tumor survival is not strictly dependent on the activated PI3K [60]. In a similar sense, PI3K inhibition demonstrated markedly synergistic effect when combined with EGFR or MEK inhibition [61]. Interestingly, PIK3CA activation in HPV-positive HNSCC did not necessarily lead to increased Akt target phosphorylation, but instead, led to increased mTOR activity and showed more sensitivity to PI3K/mTOR dual inhibition than Akt inhibition [62]. This finding can be extended to more favorable efficacy of PI3K/mTOR inhibitors over Akt inhibitors in clinical settings [63].
Locations of mutations affect PI3K structure and function, resulting in different responsiveness to inhibition and clinical outcome. Regulatory subunit p85 normally suppresses catalytic function of p110 at resting stage. Consequently, C terminal truncation or internal deletion of p85 releases p110 from negative regulation and constitutively activates the PI3K pathway [64, 65]. Additionally, as frequently mutated E542 and E545 in p110 are located at a distance from the kinase domain, it is plausible that mutations at these spots alter regulatory control of p85. Indeed, E545K mutation in the helical domain of p110 changes acid-base charge and disrupts inhibitory interaction between p85 and p110 [66]. H1047R mutation in the kinase domain, on the other hand, shifts orientation of the residue and changes conformation of the two loops of kinase that contact cell membrane. This allows for kinase access to phospholipid that is less regulated by p85 [67].
Independently from p110, p85 as a monomer also down-regulates PI3K activation: p85 is naturally more abundant than p110 and excess p85 monomers can sequestrate insulin receptor substrate 1 (IRS-1), an adaptor molecule that mediates signal transduction between IGF-1R and downstream PI3K [68]. Thus, in wild-type cells, the p85 monomer competes with the p85-p110 dimer for IRS binding and signal transduction. In heterozygous knock out cells, the amount of p85 monomers decreases more than p85-p110 dimers which up-regulates the PI3K pathway [69]. However, in null cells, complete absence of regulatory subunit to stabilize p110 leads to significantly decreased signal transduction causing cell apoptosis [69]. Although not as frequent as in PIK3CA, mutations in PIK3R1 (encoding p85α) can be found in 3% of HPV-positive HNSCC and 1% of HPV-negative HNSCC according to TCGA data [7].
Alteration of PTEN tumor suppressor gene is among the frequently found somatic mutations in human cancers as well as germline mutations causing hereditary cancer syndromes. PTEN dephosphorylates PIP3 to PIP2, inhibiting mitogenic signal transduction in the PI3K pathway. PTEN also interacts with PI3K, which plays a key role in chemotaxis and tumor metastasis [47, 48]. Clinical data has shown that loss of PTEN expression is a poor prognostic marker in oral squamous cell cancer [70]. However, PTEN loss was found in only a small number of HNSCC (8.16%), implying that it is a relatively minor component in PI3K pathway activation [8].
Targeting PI3K-Akt-mTOR pathway in clinic
PI3K inhibitor
Buparlisib (BKM120)
Buparlisib is an orally bioavailable pan-PI3K inhibitor, targeting the ATP binding site of p110 kinase domain. Its inhibitory potency is equitable on class IA isoforms of p110α, β and δ, but slightly less against class IB p110γ [51]. An in vitro study demonstrated IC50 values for Akt inhibition of 104 ± 18, 234 ± 47 and 463 ± 87 nmol/L for PI3Kα, β and δ, respectively [51]. Buparlisib is rapidly absorbed orally and its serum concentration increases proportionately to dosage [71]. The molecule also penetrates blood brain barrier and administration of buparlisib by gavage effectively controlled metastatic growth of human breast cancer in mouse brain [72]. Based on preclinical data, its antitumor activity was also attributed to suppression of microtubular dynamics [73], and antiangiongenic effect [51]. A combination of buparlisib, cetuximab and radiation exerted a synergistic antiproliferative effect on human head and neck cancer cell lines [74, 75]. In vivo, buparlisib inhibited PI3K activity in cell lines with wild-type PIK3CA as well as mutant form harboring any hotspot mutation of E542K, E545K or H1047R [76]. In a phase I dose-escalation study for advanced solid tumors, most common side effects included rash, abnormal hepatic function, alteration in glucose metabolism and fatigue [71]. In a recent randomized phase II trial with R/M HNSCC, adding buparlisib to paclitaxel improved progression-free survival (PFS) to 4–6 months compared to 3–5 months in the placebo plus paclitaxel group (p = 0.011) [77]. In this trial, comparable proportions of the patients had a mutation in PIK3CA, 11% and 13% in the buparlisib and control arm, respectively. Patients taking buparlisib also maintained stable quality of life and demonstrated good tolerance to the treatment compared to the placebo group, as similar proportions of patients discontinued the treatment due to adverse effects [77]. However, this study failed to demonstrate significant improvement in overall survival (OS) with buparlisib partly because of insufficient power. There are several ongoing clinical trials to evaluate the efficacy and safety of buparlisib with or without additional therapy (Table 1).
Table 1.
Clinical trials evaluating PI3K or mTOR inhibitor in patients with HNSCC
| Agent | Clinical Trial Identifier | Other Targeted Agent | Additional Therapy | Conditions | Phase | Status |
|---|---|---|---|---|---|---|
| PI3K inhibitor | ||||||
| Alpelisib (BYL719) | NCT02145312 | – | – | R/M HNSCC, failed to respond to platinum-based therapy | II | Not yet recruiting |
| NCT02537223 | – | Cisplatin, radiation | Locoregionally advanced HNSCC, not previously treated | I | Active, recruiting | |
| NCT01602315 | Cetuximab | – | R/M HNSCC | I/II | Terminated (sponsor withdrawal) | |
| NCT02298595 | Cetuximab | Cisplatin | HPV-associated oropharyngeal SCC | I/II | Not yet recruiting | |
| Buparlisib (BKM120) | NCT01816984 | Cetuximab | – | R/M HNC | I/II | Active, not recruiting |
| NCT01737450 | – | – | Recurrent or progressive HNC | II | Active, recruiting | |
| NCT02113878 | – | Cisplatin, radiation | Locally advanced HNSCC | I | Active, recruiting | |
| PX-866 | NCT01252628 | Cetuximab | R/M HNSCC | II | Completed | |
| NCT01204099 | Docetaxel | Locally advanced or R/M HNSCC | II | Completed | ||
| Copanlisib | NCT02822482 | Cetuximab | – | HNSCC with PI3KCA mutation/amplification or PTEN loss | I/II | Active, recruiting |
| INCB050465 | NCT02646748 | Itacitinib | Pembrolizumab | Advanced solid tumors | I | Active, recruiting |
| mTOR inhibitor | ||||||
| Sirolimus | NCT01195922 | – | – | Advanced HNSCC, not previously treated | I/II | Completed |
| Temsirolimus | NCT01172769 | – | – | R/M HNSCC | II | Completed |
| NCT01009203 | Erlotinib | – | Advanced HNSCC, refractory to platinum | II | Terminated (high patient withdrawal rate) | |
| NCT01016769 | – | Paclitaxel, carboplatin | R/M HNSCC | I/II | Active, not recruiting | |
| NCT02215720 | Cetuximab | – | Advanced or metastatic solid tumors | I | Active, recruiting | |
| NCT00703625 | – | Docetaxel | Resistant solid malignancies | I | Completed | |
| Everolimus (RAD001) | NCT01332279 | Erlotinib | Radiation | Recurrent HNC, previously treated with radiation | I | Withdrawn (sponsor withdrawal) |
| NCT01313390 | – | Docetaxel | R/M HNSCC | I/II | Terminated (lack of recruitment) | |
| NCT01009346 | Cetuximab | Cisplatin, carboplatin | R/M HNSCC | I/II | Terminated (toxicity) | |
| NCT01051791 | – | – | R/M HNSCC | II | Active, not recruiting | |
| PI3K/mTOR dual inhibitor | ||||||
| SF1126 | NCT02644122 | – | – | R/M HNSCC | II | Active, recruiting |
| Gedatolisib | NCT03065062 | Palbociclib | – | Advanced HNSCC | I | Active, recruiting |
| Dactolisib (BEZ235) | NCT00620594 | – | – | Advanced solid tumors | I | Completed |
| PI3K/HDAC dual inhibitor | ||||||
| CUDC-907 | NCT02307240 | – | – | Advanced or relapsed solid tumors | I | Active, recruiting |
PX-866
PX-866 is an analog of wortmannin that irreversibly inhibits class I PI3K by binding to Lys in ATP catalytic site [78]. Potent and irreversible binding of PX-866 enables sub-nanomolar IC50 values of 0.1, 1.0 and 2.9 nmol/L for PI3Kα PI3Kγ and PI3Kδ, respectively, in contrast to much higher IC50 of > 300 nmol/L for PI3Kβ [79]. In vivo studies revealed antitumor activities of PX-866 against human colon cancer, ovarian cancer and lung cancer xenografts [80]. It enhanced antitumor activities of cisplatin and radiation treatment in colon cancer and ovarian cancer cells, respectively [80]. PX-866 also effectively overcame resistance to EGFR inhibitor in human lung cancer cells lacking expression of ErbB-3 [79]. PX-866 induced cessation of tumor growth in xenograft models of human HNSCC which included one case of PIK3CA gene amplification and another case of E545K [43]. However, clinical trials of PX-866 failed to show promising results. In phase II clinical trials, combined use of PX-866 with either cetuximab or docetaxel failed to achieve improved PFS or OS compared to each treatment alone [81, 82].
Alpelisib (BYL719)
Theoretically, a selective inhibitor of PI3Kα can achieve antitumor activity without affecting other isoforms of PI3K, allowing for a more favorable side effect profile. Alpelisib was designed as a specific inhibitor of PI3Kα, the product of frequently mutated PIK3CA [83]. The molecule inhibits wild-type PI3Kα (IC50 = 4.6 nmol/L) as well as PI3Kα with common PI3KCA mutations, such as E545K or H1047R (IC50 = 4 nmol/L), more potently than PI3Kδ (IC50 = 290 nmol/L) or PI3Kγ (IC50 = 250 nmol/L) [52]. Preclinical data also suggested that PIK3CA mutation makes cancer cells more vulnerable to PI3K inhibition by alpelisib. In vitro pharmacologic sensitivity screen among a broad panel of cancer cell lines revealed that sensitivity to alpelisib was positively associated with the presence of PIK3CA mutation, amplification or copy number gain [84], which was confirmed by an in vivo study using mouse models [52]. In a HNSCC cell line (Cal-33) and a patient-derived xenograft model, both harboring H1047R mutation in PIK3CA, administration of alpelisib using nanoparticles induced inhibition of tumor growth and sensitization to radiation [55]. Compared to HNSCC cell lines with wild-type PIK3CA, cell lines with PIK3CA H1047R mutation were more susceptible to antiproliferative effect of alpelisib [56]. In another in vivo study, PIK3CA mutation, regardless of its location, was the strongest predictive feature that correlated with favorable response to alpelisib [52]. Compensatory hyperactivation of PIK3CA is one of the major mechanisms of treatment resistance, thus PI3K inhibitors are being tested with other targeted therapies, such as EGFR inhibitors. Inhibition of PI3K with alpelisib enhanced tumor sensitivity to cetuximab in HNSCC xenograft models [85]. A phase I trial of alpelisib combined with cetuximab in R/M HNSCC resulted in one partial response (PR), three unconfirmed PRs and five stable diseases (SDs) among 32 cases with relatively good patient tolerance [86]. PI3K activation status was unknown in this trial. In a more recent phase I trial of alpelisib, any of complete response (CR), PR or SD was achieved in 13 out of 19 study participants with PIK3CA-mutant HNSCC (NCT01219699) [87].
Copanlisib
Copanlisib is a potent inhibitor of class I PI3K with sub-nanomolar IC50. The molecule exhibits preferential activity against PI3Kα and PI3Kδ over PI3Kβ and PI3Kγ (IC50 values of 0.5 and 0.7 nmol/L over 3.7 and 6.4 nmol/L, respectively) [57, 88]. It demonstrated superior inhibitory effect in cells with PIK3CA activating mutations over wild-type in breast cancer and non-small cell lung cancer xenografts [57]. Phase I trials in patients with advanced or refractory solid tumors presented good patient tolerance and evidence of disease control [89, 90]. Efficacy and safety of combined copanlisib and cetuximab for HNSCC is under study (NCT02822482).
mTOR inhibitor
Sirolimus (rapamycin)
Sirolimus was initially developed as an antifungal metabolite, extracted from the bacterium Streptomyces hygroscopicus [91]. However, since its immunosuppressive and antiproliferative properties were revealed, this macrolide molecule has been more widely used for oncologic treatment and for prevention of graft rejection or coronary stent blockage. Sirolimus binds with FKBP12 (12 kDa FK506-binding protein) to form a gain-of-function complex that function as an inhibitor of mTOR complex 1 (mTORC1) [92]. This compound, as a result, inhibits metabolic alteration and cell proliferation which is triggered by upstream gain-of-function mutations, such as PI3K and Akt. Sirolimus demonstrated antiproliferative activity in HNSCC cell lines inducing synergistic effect with chemotherapeutic agents or radiation [93, 94]. In HNSCC xenograft models with activated PI3K-Akt pathway, administration of sirolimus induced marked inhibition of tumor growth and cell apoptosis [58, 59]. It also suppressed lymphangiogenesis in HNSCC xenograft models and prevented spread of the cancer cells to adjacent lymph nodes [95]. In a phase I trial of sirolimus and bevacizumab for patients with advanced malignancies, no objective response was observed among the participants with HNSCC [96]. However, among the patients with stage II-IVA, untreated HNSCC, neoadjuvant trial of sirolimus followed by definitive therapy (surgery or chemoradiation) demonstrated significant clinical responses (one CR, one PR and 14 SDs among 16 patients) with good patient tolerance [97]. Sirolimus is known for poor bioavailability and low predictability of serum concentration after intestinal absorption, thus its narrow therapeutic window and a long half-life require regular drug concentration monitoring [98]. Based on these concerns, analogs of sirolimus have been developed to improve pharmacokinetic properties.
Temsirolimus
Temsirolimus is a water-soluble analog of sirolimus and can be administered parenterally [99]. It undergoes hydrolysis after administration to form sirolimus, but the medication itself is also capable of inhibiting mTOR. Temsirolimus is currently FDA approved for the treatment of advanced renal cell carcinoma [100]. Several preclinical studies proved that a combination of temsirolimus and cetuximab induces synergistic antitumor effect, as it mitigates or prevents compensatory downstream mTOR over-activation induced by EGFR inhibitor [101–105]. There have been a number of phase I/II trials using temsirolimus in patients with HNSCC. In a phase I study of temsirolimus used with carboplatin and paclitaxel in R/M HNSCC, 22% of the patients exhibited objective PRs [106]. The information regarding PI3K activation status was lacking in this study. In TEMHEAD trial, a phase II study of temsirolimus in R/M HNSCC refractory to platinum and cetuximab, tumor shrinkage occurred in 39.4% of the patients mostly within the first six weeks of the treatment. However, no objective response was achieved, nor did PI3KCA mutational status (H1048Y and G1050S) predict treatment success [107]. In another trial including a broad range of advanced malignancies, the combination of bevacizumab, cetuximab and temsirolimus was effective in achieving PRs in 25% of the patients with HNSCC, but a few patients were withdrawn from the trial because of toxicities [108]. In this study, treatment-responders did not carry PIK3CA mutation in HNSCC cells. A trial combining temsirolimus with erlotinib for R/M HNSCC was closed early due to toxicity and patient death [109]. In a phase I pharmacokinetic study of temsirolimus, dose-limiting toxicities occurred such as thrombocytopenia, stomatitis or mucositis, asthenia, manic-depressive syndrome and rash [110]. Thus, the treatment effect of temsirolimus should be evaluated against potential toxicities and more clinical trials are ongoing.
Everolimus (RAD001)
Everolimus is a hydroxyethyl derivative of rapamycin, offering improved oral bioavailability. The medication has a short half-life, allowing for quick establishment of stable status and improved drug safety [111]. After intestinal absorption, everolimus is not converted to rapamycin, instead forms a complex with FKBP12 and inhibits mTOR [112]. It is currently approved by the FDA for treatment of multiple malignancies including advanced breast cancer, kidney cancer, neuroendocrine tumor (NET) of pancreas, progressive NET of GI and lung, tuberous sclerosis-associated renal angiomyolipoma and subependymal giant cell astrocytoma [113]. Although everolimus was effective in arresting tumor growth in HNSCC xenograft models [114, 115], clinical data was not as encouraging. Several phase I studies demonstrated PRs among patients with HNSCC [116–119], but the doses of everolimus used were different depending on other treatments combined, such as platinum, docetaxel, cetuximab or radiation. Phase II trials with everolimus also failed to demonstrate clinical benefit for HNSCC. Either as monotherapy or combination with erlotinib, treatment with everolimus was not successful in achieving objective response in patients with previously treated R/M HNSCC [120, 121]. There is a currently active clinical trial testing everolimus monotherapy in patients with R/M HNSCC (NCT01051791).
PI3K/mTOR dual inhibitor
SF1126
SF1126 is a peptide-conjugated prodrug of LY294002, with improved water solubility and pharmacokinetics. RGDS conjugation enables the molecule to bind to specific integrins within the tumor, enhancing drug permeability [53]. LY294002 is a pan-PI3K inhibitor, with IC50 values of 720 nmol/L, 306 nmol/L, 1.33 μmol/L and 1.6 μmol/L for PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ respectively, and similar IC50 for mTOR (1.5 μmol/L) [53, 122]. In a phase I trial, SF1126 as a single agent was effective in maintaining stable diseases in patients with GIST and clear cell renal cancer, and in combination with rituximab decreased absolute lymphocyte count and lymph node/spleen size in CLL [123]. SF1126 monotherapy is now being evaluated for treatment of R/M HNSCC (NCT02644122).
Gedatolisib
Gedatolisib is a potent and reversible inhibitor of class I PI3K and mTOR. IC50 values for PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ and mTOR are 0.4 nmol/L, 6 nmol/L, 8 nmol/L, 6 nmol/L and 10 nmol/L, respectively [124]. The inhibitory activity against PI3Kα with hotspot mutations, such as E545K and H1047R, are comparatively low (0.6 nmol/L and 0.8 nmol/L) [124]. Its antitumor activity was demonstrated in in vitro studies using mutant cells harboring E545K or H1047R in PIK3CA as well as wild-type [124, 125]. Gedatolisib also inhibited cell proliferation and increased radiosensitivity of human nasopharyngeal cancer cells with PI3K/mTOR hyperactivation [126]. Additionally, use of gedatolisib in EGFR inhibitor-resistant HNSCC suppressed cell survival and induced apoptosis [45]. Phase I trials with gedatolisib for patients with advanced cancer demonstrated potential antitumor activities with PRs and acceptable tolerance [127, 128]. However, no apparent relationship between PIK3CA alteration and treatment response was observed in these trials. There is an ongoing phase I trial of gedatolisib combined with palbociclib (CDK4/CDK6 inhibitor) for advanced solid tumors including HNSCC (NCT03065062).
Dactolisib (BEZ235)
Dactolisib is an ATP-competitive dual inhibitor of PI3K and mTOR, exerts more potency on PI3Kα, PI3Kδ, PI3Kγ and mTOR (IC50 values of 4, 7, 5 and 21 nmol/L, respectively) than PI3Kβ (IC50 = 75 nmol/L) [54, 129]. Dactolisib exhibited potent antiproliferative activity, halting cell cycles at G1 [54] and attenuating VEGF expression [129]. HNSCC cell lines with H1047R mutation were more susceptible to inhibition with lower IC50, whereas E545K conferred only slightly increased sensitivity [60]. In clinical settings, however, there has been little evidence to support drug efficacy and safety. When dactolisib was used for patients with castration-resistant prostate cancer or everolimus-resistant pancreatic NET, the trials were discontinued due to dose-limiting toxicities, such as stomatitis, vomiting, diarrhea or hyperglycemia [130, 131]. Combination of dactolisib and everolimus tested in patients with various advanced solid tumors, including one case of HNSCC, failed to demonstrate objective response [132]. Another phase I trial of dactolisib treatment for various, advanced solid tumors is now complete and the result is being awaited (NCT00620594).
PI3K/HDAC dual inhibitor
CUDC-907
CUDC-907 is an orally administered inhibitor of class I PI3K isoforms and histone deacetylase (HDAC). IC50 values for PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ are 19, 54, 38 and 311 nmol/L, respectively [133]. Simultaneous inhibition of PI3K and HDAC has demonstrated synergistic effect compared to the combined level of growth suppression achieved by single compound of HDAC inhibitor, vorinostat, and PI3K inhibitor, GDC-0941 [133]. CUDC907 has proved to be therapeutic against B cell lymphoma by decreasing MYC protein levels [134]. The effect of dual inhibition synergistically induced apoptosis of MYC-altered cells in diffuse large B-cell lymphoma (DLBCL) [135]. For cancer cells that developed resistance to PI3K inhibition through alternative pathway activation, concurrent inhibition of HDAC can down-regulate other signaling proteins and circumvent treatment resistance. This potential benefit of dual inhibition was supported by an in vitro finding which demonstrated that administration of HDAC inhibitor successfully overcame resistance to mTOR inhibitor in lymphoma cells [136]. An in vivo study has also revealed that dual inhibition of PI3K and HDAC can defeat cancer resistance to platinum-based treatment by suppressing multidrug resistance transporters and DNA repairs [137]. The first phase I trial of CUDC-907 for the treatment of relapsed/refractory lymphoma achieved two CRs and three PRs in patients with DLBCL [138]. There is an actively ongoing phase I trial of CUDC-907 for the patients with advanced or relapsed solid tumors (NCT02307240), and another phase I trial for the patients with metastatic or locally advanced thyroid cancer (NCT03002623).
Inhibition of PI3K pathway and immune system
It has been well known that inhibitors of mTOR, such as sirolimus, modulate immune system. Clinically, they have been used as immune suppressive agents to prevent rejection for patients who had undergone organ transplant. In fact, PI3K family controls many aspects of cell development, differentiation and function in both innate and adaptive immune system [139]. Especially, PI3Kγ and PI3Kδ are highly expressed in all subtypes of leukocyte, and inhibition of PI3Kγ suppressed progression of breast cancer in an animal model by inhibiting tumor inflammation and myeloid cell-mediated angiogenesis [140]. Furthermore, it has been revealed that PI3Kγ in macrophage has a critical role in the interplay between immune stimulation and suppression during inflammation or cancer development [141]. Class I PI3K signaling becomes activated by antigen receptors expressed by T and B cells, altering adaptive immune system. Therefore, inhibition of PI3Kδ dampens regulatory T cells, enhances activity of cytotoxic T cells and induces tumor regression as shown in animal models of melanoma, lung cancer, thymoma and breast cancer [142]. Various mutations in genes encoding PI3Kδ may as well lead to immunodeficiency syndromes [143].
Immune checkpoint inhibitors such as anti-programmed death 1 (anti-PD1) antibodies have demonstrated remarkable activities in HNSCC [144, 145]. Interestingly, the level of immune checkpoint ligands such as programmed death ligand 1 (PD-L1) appears to be regulated by the PI3K-Akt-mTOR pathway: inhibition of PI3K, Akt or mTOR decreased expression of PD-L1 in a non-small cell lung cancer model in vitro and in vivo [146]. Furthermore, combination of PI3Kγ blockade and immune checkpoint blockade with anti-PD1 therapy induced a synergistic growth inhibitory effect in animal models of both HPV-positive and negative HNSCC [141]. In this study, the authors showed that PI3Kγ in macrophages plays a key role in inducing immune suppression by inhibiting NFκB pathway. Inhibition of PI3Kγ in macrophages, therefore, stimulated NFκB activation and promoted an immunostimulatory transcriptional program, restoring T cell activation. Another report suggests that PI3K-Akt pathway activation may mediate Tim-3 expression in HNSCC, which is associated with more exhausted phenotype of tumor infiltrating lymphocytes, and cause resistance to immune checkpoint blockade [147]. However, the role of PI3K pathway in cancer immunology needs to be clinically investigated further. There are phase I trials of combining PI3Kδ inhibitor (INCB050465) with pembrolizumab in advanced solid tumors (NCT02646748), and combining PI3Kβ inhibitor (GSK2636771) with pembrolizumab in advanced melanoma (NCT03131908). With recent approvals of immune checkpoint inhibitors for the treatment of R/M HNSCC, effects of adding PI3K inhibitors to immune checkpoint inhibitors will be further explored.
Conclusions
PI3K plays a key role in the progression of HNSCC and development of resistance against cetuximab. Genomic alterations affecting PI3K are common among both HPV-positive and HPV-negative diseases and serve as an attractive target for the treatment of HNSCC. Early clinical trials evaluating PI3K inhibitors have shown disappointing results, but further evaluation with more potent agents and careful patient selection might lead to development of effective PI3K inhibitors in HNSCC. In light of recent success of immune checkpoint inhibitors, potential impacts of PI3K inhibition on immune system should be considered in the future development of PI3K-targeted therapy.
Funding
There was no financial support or funding for this review article.
Abbreviations
- anti-PD1
anti-programmed death 1
- BH
Breakpoint cluster region-homology
- CR
Complete response
- DLBCL
Diffuse large B-cell lymphoma
- EGFR
Epidermal growth factor receptor
- Erk
Extracellular signal-regulated kinase
- FKBP12
12 kDa FK506-binding protein
- GAP
GTPase activating protein
- GEF
Guanine nucleotide exchange factor
- GPCR
G protein-coupled receptor
- HDAC
Histone deacetylase
- HNSCC
Head and neck squamous cell carcinoma
- HPV
Human papillomavirus
- IGF-1R
Insulin-like growth factor 1 receptor
- IHC
Immunohistochemistry
- IRS-1
Insulin receptor substrate 1
- MEK
MAPK(mitogen-activated protein kinase)/Erk kinase
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex1
- NET
Neuroendocrine tumor
- OS
Overall survival
- PDGFR
Platelet-derived growth factor receptor
- PDK1
Phosphoinositide-dependent kinase 1
- PD-L1
Programmed death-ligand 1
- PFS
Progression-free survival
- PH
Pleckstrin homology
- PI
Phosphatidylinositol
- PI-3,4-P2
Phosphatidylinositol 3,4-bisphosphate
- PI3K
Phosphoinositide 3-kinase
- PI-3-P
Phosphatidylinositol 3-phosphate
- PIP
Phosphatidylinositol 4-phosphate
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- PIP3
Phosphatidylinositol 3,4,5-triphosphate
- PKB
Protein kinase B
- PR
Partial response
- PTEN
Phosphatase and tensin homolog
- Ral
Ras-like protein
- Rheb
Ras homolog enriched in brain
- SD
Stable disease
- SH
Src homology
- TCGA
The cancer genome atlas
- TSC
Tuberous sclerosis complex
Authors’ contributions
KJ searched literature and RM compiled current clinical trials. KJ and HK wrote the initial manuscript. RM and HK provided revisions. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Kyungsuk Jung, Email: kyungsuk.jung@fccc.edu.
Hyunseok Kang, Email: hkang30@jhmi.edu.
Ranee Mehra, Email: rmehra1@jhmi.edu.
References
- 1.American Cancer Society . Cancer Facts & Figures. Atlanta: American Cancer Society; 2016. [Google Scholar]
- 2.Maier H, Dietz A, Gewelke U, Heller WD, Tobacco WH. Alcohol and the risk of head and neck cancer. Clin Investig. 1992;70(3–4):320–327. doi: 10.1007/BF00184668. [DOI] [PubMed] [Google Scholar]
- 3.Goldenberg D, Lee J, Koch WM, Kim MM, Trink B, Sidransky D, et al. Habitual risk factors for head and neck cancer. Otolaryngol Head Neck Surg. 2004;131(6):986–993. doi: 10.1016/j.otohns.2004.02.035. [DOI] [PubMed] [Google Scholar]
- 4.Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, Huvos AG, et al. High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg. 2003;129(1):106–112. doi: 10.1001/archotol.129.1.106. [DOI] [PubMed] [Google Scholar]
- 5.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D'Souza G, Westra WH, Wang SJ, van Zante A, Wentz A, Kluz N, et al. Differences in the prevalence of human papillomavirus (HPV) in head and neck squamous cell cancers by sex, race, anatomic tumor site, and HPV detection method. JAMA Oncol. 2017;3(2):169-77. [DOI] [PMC free article] [PubMed]
- 7.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3(7):761–769. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang H, Kiess A, Chung CH. Emerging biomarkers in head and neck cancer in the era of genomics. Nat Rev Clin Oncol. 2015;12(1):11–26. doi: 10.1038/nrclinonc.2014.192. [DOI] [PubMed] [Google Scholar]
- 10.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72(5):767–778. doi: 10.1016/0092-8674(93)90404-E. [DOI] [PubMed] [Google Scholar]
- 11.Inukai K, Funaki M, Anai M, Ogihara T, Katagiri H, Fukushima Y, et al. Five isoforms of the phosphatidylinositol 3-kinase regulatory subunit exhibit different associations with receptor tyrosine kinases and their tyrosine phosphorylations. FEBS Lett. 2001;490(1–2):32–38. doi: 10.1016/S0014-5793(01)02132-9. [DOI] [PubMed] [Google Scholar]
- 12.Foukas LC, Claret M, Pearce W, Okkenhaug K, Meek S, Peskett E, et al. Critical role for the p110alpha phosphoinositide-3-OH kinase in growth and metabolic regulation. Nature. 2006;441(7091):366–370. doi: 10.1038/nature04694. [DOI] [PubMed] [Google Scholar]
- 13.Papakonstanti EA, Zwaenepoel O, Bilancio A, Burns E, Nock GE, Houseman B, et al. Distinct roles of class IA PI3K isoforms in primary and immortalised macrophages. J Cell Sci. 2008;121(Pt 24):4124–4133. doi: 10.1242/jcs.032763. [DOI] [PubMed] [Google Scholar]
- 14.Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leevers SJ, Vanhaesebroeck B, Waterfield MD. Signalling through phosphoinositide 3-kinases: the lipids take Centre stage. Curr Opin Cell Biol. 1999;11(2):219–225. doi: 10.1016/S0955-0674(99)80029-5. [DOI] [PubMed] [Google Scholar]
- 16.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3(4):317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 17.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997–1007. doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoshioka K, Yoshida K, Cui H, Wakayama T, Takuwa N, Okamoto Y, et al. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med. 2012;18(10):1560–1569. doi: 10.1038/nm.2928. [DOI] [PubMed] [Google Scholar]
- 19.Backer JM. The regulation and function of class III PI3Ks: novel roles for Vps 34. Biochem J. 2008;410(1):1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 20.Lee JY, Engelman JA, Cantley LC. Biochemistry. PI3K charges ahead. Science. 2007;317(5835):206–207. doi: 10.1126/science.1146073. [DOI] [PubMed] [Google Scholar]
- 21.Scheid MP, Woodgett JR. PKB/AKT: functional insights from genetic models. Nat Rev Mol Cell Biol. 2001;2(10):760–768. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 22.Komiya Y, Onodera Y, Kuroiwa M, Nomimura S, Kubo Y, Nam JM, et al. The rho guanine nucleotide exchange factor ARHGEF5 promotes tumor malignancy via epithelial-mesenchymal transition. Oncogene. 2016;5(9):e258. doi: 10.1038/oncsis.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozanne B, Richards CS, Hendler F, Burns D, Gusterson B. Over-expression of the EGF receptor is a hallmark of squamous cell carcinomas. J Pathol. 1986;149(1):9–14. doi: 10.1002/path.1711490104. [DOI] [PubMed] [Google Scholar]
- 24.Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53(15):3579–3584. [PubMed] [Google Scholar]
- 25.Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, et al. impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62(24):7350–7356. [PubMed] [Google Scholar]
- 26.Keren S, Shoude Z, Lu Z, Beibei Y. Role of EGFR as a prognostic factor for survival in head and neck cancer: a meta-analysis. Tumour Biol. 2014;35(3):2285–2295. doi: 10.1007/s13277-013-1303-0. [DOI] [PubMed] [Google Scholar]
- 27.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 28.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–1127. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 29.Erbitux. http://pi.lilly.com/us/erbitux-uspi.pdf. Accessed 5 May 2017.
- 30.Licitra L, Storkel S, Kerr KM, Van Cutsem E, Pirker R, Hirsch FR, et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer. 2013;49(6):1161–1168. doi: 10.1016/j.ejca.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 31.Tinhofer I, Klinghammer K, Weichert W, Knodler M, Stenzinger A, Gauler T, et al. Expression of amphiregulin and EGFRvIII affect outcome of patients with squamous cell carcinoma of the head and neck receiving cetuximab-docetaxel treatment. Clin Cancer Res. 2011;17(15):5197–5204. doi: 10.1158/1078-0432.CCR-10-3338. [DOI] [PubMed] [Google Scholar]
- 32.Ang KK, Zhang Q, Rosenthal DI, Nguyen-Tan PF, Sherman EJ, Weber RS, et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J Clin Oncol. 2014;32(27):2940–2950. doi: 10.1200/JCO.2013.53.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 35.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20(7):1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- 36.Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker J, Parker N, et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol. 2006;24(25):4170–4176. doi: 10.1200/JCO.2006.07.2587. [DOI] [PubMed] [Google Scholar]
- 37.Temam S, Kawaguchi H, El-Naggar AK, Jelinek J, Tang H, Liu DD, et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25(16):2164–2170. doi: 10.1200/JCO.2006.06.6605. [DOI] [PubMed] [Google Scholar]
- 38.Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC, et al. Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma. Cancer Res. 2009;69(6):2568–2576. doi: 10.1158/0008-5472.CAN-08-3199. [DOI] [PubMed] [Google Scholar]
- 39.Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5(5):489–500. doi: 10.1016/S1535-6108(04)00112-6. [DOI] [PubMed] [Google Scholar]
- 40.Alifrangis CC, McDermott U. Reading between the lines; understanding drug response in the post genomic era. Mol Oncol. 2014;8(6):1112–1119. doi: 10.1016/j.molonc.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4(1):94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keysar SB, Astling DP, Anderson RT, Vogler BW, Bowles DW, Morton JJ, et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol Oncol. 2013;7(4):776–790. doi: 10.1016/j.molonc.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Degese MS, et al. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst. 2014;106(9):dju215. [DOI] [PMC free article] [PubMed]
- 45.D'Amato V, Rosa R, D'Amato C, Formisano L, Marciano R, Nappi L, et al. The dual PI3K/mTOR inhibitor PKI-587 enhances sensitivity to cetuximab in EGFR-resistant human head and neck cancer models. Br J Cancer. 2014;110(12):2887–2895. doi: 10.1038/bjc.2014.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vander Broek R, Mohan S, Eytan DF, Chen Z, Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis. 2015;21(7):815–825. doi: 10.1111/odi.12206. [DOI] [PubMed] [Google Scholar]
- 47.Kolsch V, Charest PG, Firtel RA. The regulation of cell motility and chemotaxis by phospholipid signaling. J Cell Sci. 2008;121(Pt 5):551–559. doi: 10.1242/jcs.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin-Belmonte F, Mostov K. Regulation of cell polarity during epithelial morphogenesis. Curr Opin Cell Biol. 2008;20(2):227–234. doi: 10.1016/j.ceb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 49.Koncar RF, Feldman R, Bahassi EM, Hashemi Sadraei N. Comparative molecular profiling of HPV-induced squamous cell carcinomas. Cancer Med. 2017;6(7):1673–1685. doi: 10.1002/cam4.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nichols AC, Palma DA, Chow W, Tan S, Rajakumar C, Rizzo G, et al. High frequency of activating PIK3CA mutations in human papillomavirus-positive oropharyngeal cancer. JAMA Otolaryngol Head Neck Surg. 2013;139(6):617–622. doi: 10.1001/jamaoto.2013.3210. [DOI] [PubMed] [Google Scholar]
- 51.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 52.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 53.Garlich JR, De P, Dey N, Su JD, Peng X, Miller A, et al. A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res. 2008;68(1):206–215. doi: 10.1158/0008-5472.CAN-07-0669. [DOI] [PubMed] [Google Scholar]
- 54.Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 55.Mizrachi A, Shamay Y, Shah J, Brook S, Soong J, Rajasekhar VK, et al. Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma. Nat Commun. 2017;8:14292. doi: 10.1038/ncomms14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keam B, Kim S, Ahn YO, Kim TM, Lee SH, Kim DW, et al. In vitro anticancer activity of PI3K alpha selective inhibitor BYL719 in head and neck cancer. Anticancer Res. 2015;35(1):175–182. [PubMed] [Google Scholar]
- 57.Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12(11):2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 58.Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res. 2005;65(21):9953–9961. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- 59.Liu D, Hou P, Liu Z, Wu G, Xing M. Genetic alterations in the phosphoinositide 3-kinase/Akt signaling pathway confer sensitivity of thyroid cancer cells to therapeutic targeting of Akt and mammalian target of rapamycin. Cancer Res. 2009;69(18):7311–7319. doi: 10.1158/0008-5472.CAN-09-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wirtz ED, Hoshino D, Maldonado AT, Tyson DR, Weaver AM. Response of head and neck squamous cell carcinoma cells carrying PIK3CA mutations to selected targeted therapies. JAMA Otolaryngol Head Neck Surg. 2015;141(6):543–549. doi: 10.1001/jamaoto.2015.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mazumdar T, Byers LA, Ng PK, Mills GB, Peng S, Diao L, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther. 2014;13(11):2738–2750. doi: 10.1158/1535-7163.MCT-13-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sewell A, Brown B, Biktasova A, Mills GB, Lu Y, Tyson DR, et al. Reverse-phase protein array profiling of oropharyngeal cancer and significance of PIK3CA mutations in HPV-associated head and neck cancer. Clin Cancer Res. 2014;20(9):2300–2311. doi: 10.1158/1078-0432.CCR-13-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Argiris A, Cohen E, Karrison T, Esparaz B, Mauer A, Ansari R, et al. A phase II trial of perifosine, an oral alkylphospholipid, in recurrent or metastatic head and neck cancer. Cancer Biol Ther. 2006;5(7):766–770. doi: 10.4161/cbt.5.7.2874. [DOI] [PubMed] [Google Scholar]
- 64.Luo J, Cantley LC. The negative regulation of phosphoinositide 3-kinase signaling by p85 and it's implication in cancer. Cell Cycle. 2005;4(10):1309–1312. doi: 10.4161/cc.4.10.2062. [DOI] [PubMed] [Google Scholar]
- 65.Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, et al. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61(20):7426–7429. [PubMed] [Google Scholar]
- 66.Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, et al. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317(5835):239–242. doi: 10.1126/science.1135394. [DOI] [PubMed] [Google Scholar]
- 67.Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106(40):16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luo J, Field SJ, Lee JY, Engelman JA, Cantley LC. The p85 regulatory subunit of phosphoinositide 3-kinase down-regulates IRS-1 signaling via the formation of a sequestration complex. J Cell Biol. 2005;170(3):455–464. doi: 10.1083/jcb.200503088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22(3):965–977. doi: 10.1128/MCB.22.3.965-977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee JI, Soria JC, Hassan KA, El-Naggar AK, Tang X, Liu DD, et al. Loss of PTEN expression as a prognostic marker for tongue cancer. Arch Otolaryngol Head Neck Surg. 2001;127(12):1441–1445. doi: 10.1001/archotol.127.12.1441. [DOI] [PubMed] [Google Scholar]
- 71.Ando Y, Inada-Inoue M, Mitsuma A, Yoshino T, Ohtsu A, Suenaga N, et al. Phase I dose-escalation study of buparlisib (BKM120), an oral pan-class I PI3K inhibitor, in Japanese patients with advanced solid tumors. Cancer Sci. 2014;105(3):347–353. doi: 10.1111/cas.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nanni P, Nicoletti G, Palladini A, Croci S, Murgo A, Ianzano ML, et al. Multiorgan metastasis of human HER-2+ breast cancer in Rag2−/−;Il2rg−/− mice and treatment with PI3K inhibitor. PLoS One. 2012;7(6):e39626. doi: 10.1371/journal.pone.0039626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11(8):1747–1757. doi: 10.1158/1535-7163.MCT-11-1021. [DOI] [PubMed] [Google Scholar]
- 74.Lattanzio L, Tonissi F, Monteverde M, Vivenza D, Russi E, Milano G, et al. Treatment effect of buparlisib, cetuximab and irradiation in wild-type or PI3KCA-mutated head and neck cancer cell lines. Investig New Drugs. 2015;33(2):310–320. doi: 10.1007/s10637-015-0210-1. [DOI] [PubMed] [Google Scholar]
- 75.Bozec A, Ebran N, Radosevic-Robin N, Chamorey E, Yahia HB, Marcie S, et al. Combination of phosphotidylinositol-3-kinase targeting with cetuximab and irradiation: a preclinical study on an orthotopic xenograft model of head and neck cancer. Head Neck. 2017;39(1):151–159. doi: 10.1002/hed.24560. [DOI] [PubMed] [Google Scholar]
- 76.Kong D, Yamori T, Yamazaki K, Dan S. In vitro multifaceted activities of a specific group of novel phosphatidylinositol 3-kinase inhibitors on hotspot mutant PIK3CA. Investig New Drugs. 2014;32(6):1134–1143. doi: 10.1007/s10637-014-0152-z. [DOI] [PubMed] [Google Scholar]
- 77.Soulieres D, Faivre S, Mesia R, Remenar E, Li SH, Karpenko A, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017;18(3):323–335. doi: 10.1016/S1470-2045(17)30064-5. [DOI] [PubMed] [Google Scholar]
- 78.Zask A, Kaplan J, Toral-Barza L, Hollander I, Young M, Tischler M, et al. Synthesis and structure-activity relationships of ring-opened 17-hydroxywortmannins: potent phosphoinositide 3-kinase inhibitors with improved properties and anticancer efficacy. J Med Chem. 2008;51(5):1319–1323. doi: 10.1021/jm7012858. [DOI] [PubMed] [Google Scholar]
- 79.Ihle NT, Paine-Murrieta G, Berggren MI, Baker A, Tate WR, Wipf P, et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther. 2005;4(9):1349–1357. doi: 10.1158/1535-7163.MCT-05-0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3(7):763–772. [PubMed] [Google Scholar]
- 81.Jimeno A, Bauman JE, Weissman C, Adkins D, Schnadig I, Beauregard P, et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015;51(4):383–388. doi: 10.1016/j.oraloncology.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jimeno A, Shirai K, Choi M, Laskin J, Kochenderfer M, Spira A, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol. 2015;26(3):556–561. doi: 10.1093/annonc/mdu574. [DOI] [PubMed] [Google Scholar]
- 83.Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 84.Huang A, Fritsch C, Wilson C, Reddy A, Liu M, Lehar J, et al. Abstract 3749: single agent activity of PIK3CA inhibitor BYL719 in a broad cancer cell line panel. Cancer Res. 2012;72(8 Supplement):3749. doi: 10.1158/1538-7445.AM2012-3749. [DOI] [Google Scholar]
- 85.Sheng Q, Wang H, Das R, Chen Y, Liang J, Cao A, et al. Abstract 4261: targeting HER3 and PI3K in head and neck squamous cancer cells. Cancer Res. 2013;73(8 Supplement):4261. doi: 10.1158/1538-7445.AM2013-4261. [DOI] [Google Scholar]
- 86.Razak ARA, Ahn MJ, Yen CJ, Solomon BJ, Lee SH, Wang HM, et al. Phase Ib/II study of the PI3K alpha inhibitor BYL719 in combination with cetuximab in recurrent/metastatic squamous cell cancer of the head and neck (SCCHN). J Clin Oncol. 2014;32(15)
- 87.Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J Clin Oncol. 2018;36(13):1291-9. [DOI] [PMC free article] [PubMed]
- 88.Scott WJ, Hentemann MF, Rowley RB, Bull CO, Jenkins S, Bullion AM, et al. Discovery and SAR of novel 2,3-Dihydroimidazo[1,2-c]quinazoline PI3K inhibitors: identification of Copanlisib (BAY 80-6946) ChemMedChem. 2016;11(14):1517–1530. doi: 10.1002/cmdc.201600148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Doi T, Fuse N, Yoshino T, Kojima T, Bando H, Miyamoto H, et al. A phase I study of intravenous PI3K inhibitor copanlisib in Japanese patients with advanced or refractory solid tumors. Cancer Chemother Pharmacol. 2017;79(1):89–98. doi: 10.1007/s00280-016-3198-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin's lymphomas. Ann Oncol. 2016;27(10):1928–1940. doi: 10.1093/annonc/mdw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med. 2012;1(1):29. doi: 10.1186/2001-1326-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–379. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aissat N, Le Tourneau C, Ghoul A, Serova M, Bieche I, Lokiec F, et al. Antiproliferative effects of rapamycin as a single agent and in combination with carboplatin and paclitaxel in head and neck cancer cell lines. Cancer Chemother Pharmacol. 2008;62(2):305–313. doi: 10.1007/s00280-007-0609-2. [DOI] [PubMed] [Google Scholar]
- 94.Shinohara ET, Maity A, Jha N, Lustig RA. Sirolimus as a potential radiosensitizer in squamous cell cancer of the head and neck. Head Neck. 2009;31(3):406–411. doi: 10.1002/hed.20898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel V, Marsh CA, Dorsam RT, Mikelis CM, Masedunskas A, Amornphimoltham P, et al. Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res. 2011;71(22):7103–7112. doi: 10.1158/0008-5472.CAN-10-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cohen EE, Sharma MR, Janisch L, Llobrera M, House L, Wu K, et al. A phase I study of sirolimus and bevacizumab in patients with advanced malignancies. Eur J Cancer. 2011;47(10):1484–1489. doi: 10.1016/j.ejca.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shirai K, Day TA, Szabo E, Van Waes C, O'Brien PE, Matheus MG, et al. A pilot, single arm, prospective trial using neoadjuvant rapamycin prior to definitive therapy in head and neck squamous cell carcinoma. J Clin Oncol. 2015;33(15)
- 98.Mukherjee S, Mukherjee U. A comprehensive review of immunosuppression used for liver transplantation. J Transp Secur 2009; 2009:701464. [DOI] [PMC free article] [PubMed]
- 99.Dancey J, Sausville EA. Issues and progress with protein kinase inhibitors for cancer treatment. Nat Rev Drug Discov. 2003;2(4):296–313. doi: 10.1038/nrd1066. [DOI] [PubMed] [Google Scholar]
- 100.Torisel. http://labeling.pfizer.com/showlabeling.aspx?id=490. Accessed 24 Apr 2017.
- 101.Bozec A, Etienne-Grimaldi MC, Fischel JL, Sudaka A, Toussan N, Formento P, et al. The mTOR-targeting drug temsirolimus enhances the growth-inhibiting effects of the cetuximab-bevacizumab-irradiation combination on head and neck cancer xenografts. Oral Oncol. 2011;47(5):340–344. doi: 10.1016/j.oraloncology.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 102.Jimeno A, Kulesza P, Wheelhouse J, Chan A, Zhang X, Kincaid E, et al. Dual EGFR and mTOR targeting in squamous cell carcinoma models, and development of early markers of efficacy. Br J Cancer. 2007;96(6):952–959. doi: 10.1038/sj.bjc.6603656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lattanzio L, Milano G, Monteverde M, Tonissi F, Vivenza D, Merlano M, et al. Schedule-dependent interaction between temsirolimus and cetuximab in head and neck cancer: a preclinical study. Anti-Cancer Drugs. 2016;27(6):533–539. doi: 10.1097/CAD.0000000000000360. [DOI] [PubMed] [Google Scholar]
- 104.Bozec A, Ebran N, Radosevic-Robin N, Sudaka A, Monteverde M, Toussan N, et al. Combination of mTOR and EGFR targeting in an orthotopic xenograft model of head and neck cancer. Laryngoscope. 2016;126(4):E156–E163. doi: 10.1002/lary.25754. [DOI] [PubMed] [Google Scholar]
- 105.Nathan CO, Amirghahari N, Rong X, Giordano T, Sibley D, Nordberg M, et al. Mammalian target of rapamycin inhibitors as possible adjuvant therapy for microscopic residual disease in head and neck squamous cell cancer. Cancer Res. 2007;67(5):2160–2168. doi: 10.1158/0008-5472.CAN-06-2449. [DOI] [PubMed] [Google Scholar]
- 106.Fury MG, Sherman E, Ho A, Katabi N, Sima C, Kelly KW, et al. A phase I study of temsirolimus plus carboplatin plus paclitaxel for patients with recurrent or metastatic (R/M) head and neck squamous cell cancer (HNSCC) Cancer Chemother Pharmacol. 2012;70(1):121–128. doi: 10.1007/s00280-012-1894-y. [DOI] [PubMed] [Google Scholar]
- 107.Grunwald V, Keilholz U, Boehm A, Guntinas-Lichius O, Hennemann B, Schmoll HJ, et al. TEMHEAD: a single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN group (AIO) Ann Oncol. 2015;26(3):561–567. doi: 10.1093/annonc/mdu571. [DOI] [PubMed] [Google Scholar]
- 108.Liu X, Kambrick S, Fu S, Naing A, Subbiah V, Blumenschein GR, et al. advanced malignancies treated with a combination of the VEGF inhibitor bevacizumab, anti-EGFR antibody cetuximab, and the mTOR inhibitor temsirolimus. Oncotarget. 2016;7(17):23227–23238. doi: 10.18632/oncotarget.7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bauman JE, Arias-Pulido H, Lee SJ, Fekrazad MH, Ozawa H, Fertig E, et al. A phase II study of temsirolimus and erlotinib in patients with recurrent and/or metastatic, platinum-refractory head and neck squamous cell carcinoma. Oral Oncol. 2013;49(5):461–467. doi: 10.1016/j.oraloncology.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raymond E, Alexandre J, Faivre S, Vera K, Materman E, Boni J, et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22(12):2336–2347. doi: 10.1200/JCO.2004.08.116. [DOI] [PubMed] [Google Scholar]
- 111.Tanaka C, O'Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, et al. Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol. 2008;26(10):1596–1602. doi: 10.1200/JCO.2007.14.1127. [DOI] [PubMed] [Google Scholar]
- 112.Nguyen SA, Walker D, Gillespie MB, Gutkind JS, Day TA. mTOR inhibitors and its role in the treatment of head and neck squamous cell carcinoma. Curr Treat Options in Oncol. 2012;13(1):71–81. doi: 10.1007/s11864-011-0180-2. [DOI] [PubMed] [Google Scholar]
- 113.Afinitor. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/afinitor.pdf. Accessed 24 Apr 2017.
- 114.Li SH, Lin WC, Huang TL, Chen CH, Chiu TJ, Fang FM, et al. Significance of mammalian target of rapamycin in patients with locally advanced stage IV head and neck squamous cell carcinoma receiving induction chemotherapy with docetaxel, cisplatin, and fluorouracil. Head Neck. 2016;38(Suppl 1):E844–E852. doi: 10.1002/hed.24111. [DOI] [PubMed] [Google Scholar]
- 115.Klinghammer K, Raguse JD, Plath T, Albers AE, Joehrens K, Zakarneh A, et al. A comprehensively characterized large panel of head and neck cancer patient-derived xenografts identifies the mTOR inhibitor everolimus as potential new treatment option. Int J Cancer. 2015;136(12):2940–2948. doi: 10.1002/ijc.29344. [DOI] [PubMed] [Google Scholar]
- 116.Fury MG, Lee NY, Sherman E, Ho AL, Rao S, Heguy A, et al. A phase 1 study of everolimus + weekly cisplatin + intensity modulated radiation therapy in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2013;87(3):479–486. doi: 10.1016/j.ijrobp.2013.06.2043. [DOI] [PubMed] [Google Scholar]
- 117.Fury MG, Sherman E, Haque S, Korte S, Lisa D, Shen R, et al. A phase I study of daily everolimus plus low-dose weekly cisplatin for patients with advanced solid tumors. Cancer Chemother Pharmacol. 2012;69(3):591–598. doi: 10.1007/s00280-011-1734-5. [DOI] [PubMed] [Google Scholar]
- 118.Fury MG, Sherman E, Ho AL, Xiao H, Tsai F, Nwankwo O, et al. A phase 1 study of everolimus plus docetaxel plus cisplatin as induction chemotherapy for patients with locally and/or regionally advanced head and neck cancer. Cancer. 2013;119(10):1823–1831. doi: 10.1002/cncr.27986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saba NF, Hurwitz SJ, Magliocca K, Kim S, Owonikoko TK, Harvey D, et al. Phase 1 and pharmacokinetic study of everolimus in combination with cetuximab and carboplatin for recurrent/metastatic squamous cell carcinoma of the head and neck. Cancer. 2014;120(24):3940–3951. doi: 10.1002/cncr.28965. [DOI] [PubMed] [Google Scholar]
- 120.Geiger JL, Bauman JE, Gibson MK, Gooding WE, Varadarajan P, Kotsakis A, et al. Phase II trial of everolimus in patients with previously treated recurrent or metastatic head and neck squamous cell carcinoma. Head Neck. 2016;38(12):1759–1764. doi: 10.1002/hed.24501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Massarelli E, Lin H, Ginsberg LE, Tran HT, Lee JJ, Canales JR, et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2015;26(7):1476–1480. doi: 10.1093/annonc/mdv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cleary JM, Shapiro GI. Development of phosphoinositide-3 kinase pathway inhibitors for advanced cancer. Curr Oncol Rep. 2010;12(2):87–94. doi: 10.1007/s11912-010-0091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mahadevan D, Chiorean EG, Harris WB, Von Hoff DD, Stejskal-Barnett A, Qi W, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer. 2012;48(18):3319–3327. doi: 10.1016/j.ejca.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, et al. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011;17(10):3193–3203. doi: 10.1158/1078-0432.CCR-10-1694. [DOI] [PubMed] [Google Scholar]
- 125.Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, et al. Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J Med Chem. 2010;53(6):2636–2645. doi: 10.1021/jm901830p. [DOI] [PubMed] [Google Scholar]
- 126.Liu T, Sun Q, Li Q, Yang H, Zhang Y, Wang R, et al. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol Cancer Ther. 2015;14(2):429–439. doi: 10.1158/1535-7163.MCT-14-0548. [DOI] [PubMed] [Google Scholar]
- 127.Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, et al. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced Cancer. Clin Cancer Res. 2015;21(8):1888–1895. doi: 10.1158/1078-0432.CCR-14-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wainberg ZA, Alsina M, Soares HP, Brana I, Britten CD, Del Conte G, et al. A multi-arm phase I study of the PI3K/mTOR inhibitors PF-04691502 and Gedatolisib (PF-05212384) plus irinotecan or the MEK inhibitor PD-0325901 in advanced Cancer. Target Oncol. 2017;12(6):775–785. doi: 10.1007/s11523-017-0530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8(8):2204–2210. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Massard C, Chi KN, Castellano D, de Bono J, Gravis G, Dirix L, et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur J Cancer. 2017;76:36–44. doi: 10.1016/j.ejca.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 131.Fazio N, Buzzoni R, Baudin E, Antonuzzo L, Hubner RA, Lahner H, et al. A phase II study of BEZ235 in patients with Everolimus-resistant, advanced pancreatic neuroendocrine Tumours. Anticancer Res. 2016;36(2):713–719. [PMC free article] [PubMed] [Google Scholar]
- 132.Wise-Draper TM, Moorthy G, Salkeni MA, Karim NA, Thomas HE, Mercer CA, et al. A phase Ib study of the dual PI3K/mTOR inhibitor Dactolisib (BEZ235) combined with Everolimus in patients with advanced solid malignancies. Target Oncol 2017. [DOI] [PMC free article] [PubMed]
- 133.Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012;18(15):4104–4113. doi: 10.1158/1078-0432.CCR-12-0055. [DOI] [PubMed] [Google Scholar]
- 134.Mondello P, Derenzini E, Asgari Z, Philip J, Brea EJ, Seshan V, et al. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget. 2017;8(8):14017–14028. doi: 10.18632/oncotarget.14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sun K, Atoyan R, Borek MA, Dellarocca S, Samson ME, Ma AW, et al. Dual HDAC and PI3K inhibitor CUDC-907 downregulates MYC and suppresses growth of MYC-dependent cancers. Mol Cancer Ther. 2017;16(2):285–299. doi: 10.1158/1535-7163.MCT-16-0390. [DOI] [PubMed] [Google Scholar]
- 136.Gupta M, Ansell SM, Novak AJ, Kumar S, Kaufmann SH, Witzig TE. Inhibition of histone deacetylase overcomes rapamycin-mediated resistance in diffuse large B-cell lymphoma by inhibiting Akt signaling through mTORC2. Blood. 2009;114(14):2926–2935. doi: 10.1182/blood-2009-05-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.To KKW, Fu LW. CUDC-907, a dual HDAC and PI3K inhibitor, reverses platinum drug resistance. Investig New Drugs 2017. [DOI] [PubMed]
- 138.Younes A, Berdeja JG, Patel MR, Flinn I, Gerecitano JF, Neelapu SS, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016;17(5):622–631. doi: 10.1016/S1470-2045(15)00584-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. 2013;31:675–704. doi: 10.1146/annurev-immunol-032712-095946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011;19(6):715–727. doi: 10.1016/j.ccr.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510(7505):407–411. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol. 2016;16(11):702–714. doi: 10.1038/nri.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ferris RL, Blumenschein G, Jr., Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 2016;375(19):1856–1867. [DOI] [PMC free article] [PubMed]
- 145.Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for platinum- and Cetuximab-refractory head and neck Cancer: results from a single-arm, phase II study. J Clin Oncol. 2017;35(14):1542–1549. doi: 10.1200/JCO.2016.70.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lastwika KJ, Wilson W, 3rd, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung Cancer. Cancer Res 2016;76(2):227–238. [DOI] [PubMed]
- 147.Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology. 2017;6(1):e1261779. doi: 10.1080/2162402X.2016.1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chamberlain MD, Chan T, Oberg JC, Hawrysh AD, James KM, Saxena A, et al. Disrupted RabGAP function of the p85 subunit of phosphatidylinositol 3-kinase results in cell transformation. J Biol Chem. 2008;283(23):15861–15868. doi: 10.1074/jbc.M800941200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 150.Kapeller R, Prasad KV, Janssen O, Hou W, Schaffhausen BS, Rudd CE, et al. Identification of two SH3-binding motifs in the regulatory subunit of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269(3):1927–1933. [PubMed] [Google Scholar]
- 151.Jimenez C, Hernandez C, Pimentel B, Carrera AC. The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras. J Biol Chem. 2002;277(44):41556–41562. doi: 10.1074/jbc.M205893200. [DOI] [PubMed] [Google Scholar]