

Abstract
Local, long-acting release fenretinide (4HPR) millicylindrical implants were prepared and evaluated for their release kinetics in vivo and their ability to suppress oral cancer tumor explant growth. Poly(lactic-co-glycolic acid)(PLGA) implants were prepared as a function of drug loading and the presence of various excipients (pore-formers, solubilizers, crystallization inhibitors) to enhance release of the insoluble 4HPR. Release kinetics and bioerosion of PLGA were monitored both in vitro in a PBS/Tween 80 buffer and in vivo by recovery of the drug remaining at the injection site. 4HPR was released from PLGA implants much slower in vivo than in the drug solubilizing media in vitro, with a 3-week lag phase and continuous release of > 2 months, but showed some release enhancement by addition of solubilizers. Water-soluble PVA/sucrose implants for release of 4HPR served to determine if drug dissolution provided suitable controlled release without the PLGA, and this formulation showed continuous drug release over 6 weeks in vivo. Placement of PLGA-4HPR implants adjacent to oral cancer tumor murine xenografts showed inhibition of tumor growth relative to sham implants, indicating the potential for the local 4HPR delivery approach to be useful for oral cancer chemoprevention.
Keywords: Fenretinide, Local delivery, In vivo release, PLGA, Controlled release, Oral cancer
1. Introduction
Oral squamous cell carcinoma (OSCC) is a worldwide health concern, and in 2016, ∼50,000 Americans were newly diagnosed with oral or pharyngeal cancer (http://oralcancerfoundation.org). The vast majority of oral cancers arise from malignant transformation of preneoplastic surface epithelial lesions (Reibel, 2003). Despite treatment advances that include intraoperative radiation, the 5-year survival rates of persons with OSCC is discouraging low (50%). Following initial therapy, HNSCC patients are managed by close clinical follow up often supplemented with imaging studies. Despite vigilant monitoring and well-recognized risk factors for recurrence, over one third of patients develop life-threatening and often untreatable recurrent OSCCs (Gleber-Netto et al. 2015). Replacement of the current “watchful waiting” strategy with well-tolerated and effective secondary OSCC chemoprevention could make a significant clinical impact for these individuals. As OSCC management requires extensive, often disfiguring surgery, treated OSCC patients often experience depression and reduced motivation. Development of an implantable delivery system to provide therapeutic drug levels without systemic drug-induced side effects and eliminate patient compliance issues could advance secondary OSCC chemoprevention. Previous studies from our labs have demonstrated that fenretinide (4HPR) inhibits focal adhesion kinase-extracellular matrix (FAK-ECM) interactions and significantly reduces invasion, which is the ultimate step in OSCC development (Han et al., 2015). To address OSCC’s redundant signaling cascades, secondary OSCC chemoprevention will ultimately require complementary agents (Mallery et al., 2017). Based on 4HPR’s multiple mechanisms of action including growth regulation and suppression of gratuitous signaling, (Han et al., 2015) it is our intention to include 4HPR in the secondary chemopreventive formulation.
Here, we chose to formulate 4HPR local controlled release (CR) implants using biodegradable poly(lactic-co-glycolic acid) (PLGA) to provide continuous release for > 30 d. Local CR drug delivery systems are presumed advantageous over daily oral dosage forms in that the once monthly dosing could lead to better treatment outcomes, elimination of the first-pass effect and patient compliance issues, lowered risk of systemic toxicity, and increased drug stability. PLGA is still the biodegradable controlled release polymer of choice in injectable depots owing to its safety, biodegradability, ease of processing, and tailorable release by adjusting its molecular weight, end-group capping, and ratio of lactide:glycolide monomers and ability to adjust the microclimate to stabilize compounds (Fredenberg et al., 2011; Hyon, 2000; Wischke and Schwendeman, 2008). To date, the FDA has approved several PLGA CR drug products including both systemic delivery products such as the Lupron Depot (leuprolide acetate microspheres for prostate cancer) and local delivery formulations including Ozurdex® (dexamethasone intravitreal implant for macular edema), and Atridox (doxycycline in situ forming implants [ISFIs] for periodontitis)(Wang et al., 2016). For our studies we chose to formulate 4HPR in millicylinders due to the ability to achieve high drug loading, and lower burst release compared to microspheres and ISFIs developed previously for systemic delivery (Wischke et al., 2010; Ying Zhang, in press). Additionally, the millicylinder formulations are desirable for future evaluation of in vivo tissue penetration and efficacy studies, and will allow for precise drug-tissue distribution measurements from the point of origin, and are expected to remain in place for better targeting of the local pre-cancerous region. Due to the small size of millicylinders (∼5 mg, 0.8 mm i.d. x 1 cm), they can be easily injected through a trocar syringe or surgically implanted.
Previous work has been done by our lab in formulating local 4HPR drug delivery systems including PLGA microspheres (Wischke et al., 2010), ISFIs (Wischke et al., 2010), and buccal mucoadhesive patches (Holpuch et al., 2012; Wu et al., 2012) as well as determining 4HPR solubility in various PLGA solubilizing solvents, release media compositions, and selected surfactants. In previous pharmacokinetic (PK) studies, we compared serum levels of 4HPR encapsulated in PLGA microspheres and ISFIs relative to a control drug suspension dosed subcutaneously (SC) in rats, and determined that PLGA CR formulations were successful at strongly reducing the burst release compared to the control suspension (Zhang et al., 2016). However, after 15 d, the amount of 4HPR released from the PLGA formulations coincided with those of the drug suspension and showed a steady decline for more than a month. Based on this data, after 2 weeks it was unclear whether the actual drug was exhibiting controlled release properties due to dissolution into surrounding interstitial fluid, or slow release from tissue, protein, and lipid “reservoirs” where 4HPR could have accumulated after fast dissolution.
These studies extended our previous work to include sustained duration (1–2 months) in vitro and in vivo of 4HPR encapsulated in PLGA millicylinders. Local delivery of hydrophobic 4HPR to aqueous interstitial fluid presents a significant challenge owing to its extreme water insolubility, with a logP of 6.31. We have selecting a continuously eroding PLGA polymer that will target the 1–2 month delivery period, although we considered the potential for the hydrophobic 4HPR to precipitate over time resulting in dissolution rate-controlled release instead of typical PLGA-erosion control. Initial parameters assessed included varying 4HPR loading, along with selected solubilizers and penetration enhancers in vitro and in vivo. To test if drug-dissolution was suitable to control release, 4HPR release from PLGA was also compared to water-soluble PVA/sucrose implants in vivo. Preliminary analysis of the efficacy of 4HPR-PLGA implants in an oral cancer xenograft mouse model was also examined.
2. Materials and methods
2.1. Materials
50:50 acid end-capped PLGA 503H (24–38 kDa), was purchased from Evonik, 4HPR was generously supplied by Merck Co, and acitretin was used as an internal standard (analytical grade, Sigma-Aldrich). Excipients used were sodium deoxycholate, (NaDC, 99% pure, Acros), polyvinylpyrrolidone (PVP K30, 40 kDa, BASF), hydroxypropyl methylcellulose K4M (HPMC K4M, Dow Chemical, Midland, MI), and β-CD (Sigma-Aldrich). Calcium deoxycholate (CaDC) was synthesized according to literature methods described in Supplemental information. Water-soluble implants were composed of polyvinyl alcohol (PVA, 9–10 kDa, 80% hydrolyzed, Sigma-Aldrich) and D-sucrose (Sigma-Aldrich). All other materials were reagent grade or better including: MgCO3, acetone, ethanol (EtOH), tetrahydrofuran (THF), and Tween 80. Solvents for UPLC-UV and MS analysis were HPLC or MS grade including acetonitrile (ACN), methanol (MeOH), double distilled water (ddH2O), phosphoric acid (H3PO4), ammonium formate (NH4COO) and formic acid (HCOOH). Silicon tubing (0.8 mm i.d.,) was purchased from BioRad Laboratories. Ethylenediaminetetraacetic acid (EDTA, analytical standard, LECO, St. Joseph, MI) and Bovine Serum Albumin (BSA, 99% pure, heat shock fraction, Sigma-Aldrich) were used as standards for nitrogen analysis.
2.2. Millicylinder preparation
To prepare PLGA millicylinders, 60% w/w 50:50 acid end-capped PLGA (24–38 kDa) was dissolved in acetone at room temperature by slowly vortexing in a capped 2 mL Eppendorf tube. 4HPR and excipients were added to the polymer mixture, and those excipients that were insoluble in acetone (including MgCO3 and NaDC) were cryomilled (Retsch swing mill cryomill, PN 20.749.001) and sieved to < 90 μm prior to their addition to the formulation. The solids were incorporated into polymer solution by stirring with a spatula. Additional acetone was added and recorded to obtain a solution with an extrudable viscosity. Note, that solutions with low viscosity result in hollow implants, and therefore the highest extrudable viscosity was sought to obtain solid, dense implants. Next, the resulting gel solution was loaded into a 3 mL syringe equipped with an 18G blunt end needle attached to silicone rubber tubing (0.8 mm i.d.), and slowly extruded. The implants were dried at room temperature for 2 d, and then transferred to vacuum oven and dried at 40 °C for an additional 2 d, after which the tubing was carefully removed and implants were cut to the desired length of 1 cm. Efforts were taken in all 4HPR experiments to minimize light exposure due to the drug’s known light instability.
Formulation efforts were necessary to accelerate 4HPR release rates. Initial studies optimized the 4HPR loading level (10, 20, 30% w/w). To improve release kinetics additional excipients were added including 20% NaDC or CaDC, along with investigating the effects of varying the concentration of the pore former MgCO3 (3, 10, 15% w/w). Other excipients were added to accelerate release, aid in drug solubilization, and prevent drug crystallization and included β-CD, HPMC K4M, and PVP K30. For the in vivo studies, a water soluble matrix 4HPR millicylinder was prepared with a PVA/ D-sucrose (40%, 30% w/v respectively in ddH20) matrix with and without excipients, followed by extrusion and drying in the same fashion as PLGA implants.
2.3. 4HPR solubility in the presence of selected excipients
Solubility studies were performed with selected excipients including NaDC, HPMC, β-CD, and PVP K30 at levels of 1, 2, 5, 10, 20% w/v in double distilled water (ddH2O). For all studies, 2 mg of 4HPR was added to 1 mL of solution and incubated at 37 °C while rotating, and protected from light with aluminum foil covered vials. The resulting suspension was centrifuged and the supernatant was analyzed by UPLC/UV as described in Section 2.7. Samples were taken on days 1 and 7, and day 7 solubility was reported. Previous work by our lab has determined 4HPR solubility parameters in selected organic solvents used in our assays and in presence of other solubilizing excipients (Wischke et al., 2010).
2.4. 4HPR implant loading
To determine the amount to 4HPR loaded into the implant, one millicylinder was weighed into a 15 mL centrifuge tube, PLGA and 4HPR were co-dissolved by addition of 0.5 mL THF, followed by precipitation of PLGA by addition of 9.5 mL EtOH. Next, the sample was centrifuged, and supernatant was assayed by UPLC/UV. Encapsulation efficiency was calculated based on actual loading divided by theoretical loading, which was invariable and ranged from 97 to 103%.
2.5. 4HPR in vitro release
4-HPR in vitro release from millicylinders was determined by placing one millicylinder (5–7 mg) in 5 mL Eppendorf tube containing 4 mL PBS pH 7.4 + 2% Tween 80, necessary to maintain sink conditions, and incubating at 37 °C on a shaking platform (200 rpm) while covering vials with aluminum foil to protect from light. The solutions were sampled by complete media replacement.
2.6. 4HPR in vivo release
To evaluate 4HPR release in vivo, millicylinders were weighed and implanted subcutaneously (SC) in the flanks of male Sprague Dawley rats using a 12 g trocar. Each rat could receive up to 6 implants. For each formulation, 3 implants were implanted in each rat, and 1 rat per time point (days 1, 7, 14, 28, 42, 60) was sacrificed. Millicylinders were carefully harvested to ensure complete removal and release from encompassing tissues. The wet millicylinders were weighed, dried by vacuum for 2 d, and weighed again to determine the mass loss and water uptake, and SEM images were acquired. The amount of 4HPR released was determined by assaying the amount of 4HPR remaining in recovered millicylinder by loading assay (Section 2.4).
2.7. 4HPR levels by UPLC-UV
4HPR levels in in vitro release media and millicylinders digests were determined by UPLC/UV. Because of 4HPR’s high extinction coefficient (47,900 L/(mol-cm) at 365 nm), excellent sensitivity was achieved via UPLC/UV with a LLOQ of 5 ng/mL. The reverse phase UPLC/UV analyses were carried out with a Waters Acquity UPLC system and Empower software under the following conditions: Acquity BEH C18 2.1 × 100 mm column, mobile phase 80:20 ACN: ddH2O + 0.1% H3PO4, isocratic flow rate of 0.65 mL/min, UV detection at 365 nm, and total analysis time of 2 min. 4HPR calibration standards were prepared in mobile phase (0.5–100 μg/mL) from a 0.5 mg/mL 4HPR stock solution in ACN, and reflects the large linear dynamic calibration range.
2.8. NaDC/CaDC levels by UPLC-MS
A UPLC-MS assay was developed for NaDC and CaDC in release media using Waters Acquity UPLC-MS (quadrupole mass analyzer, Acquity QDa) system and Empower software under the following conditions: Acquity BEH C18 2.1 × 100 mm column, mobile phase 37.5:37.5:25 MeOH: ACN: 0.02 M NH4COO pH adjusted to 4.3 with HCOOH, isocratic flow rate of 0.4 mL/min, and total analysis time of 3 min. MS was operated in positive ion mode and the molecular ion m/z 392.5 (deoxycholic acid) was monitored. NaDC calibration standards were prepared by dissolving a stock solution of 5 mg/mL NaDC in PBS, and working standards were prepared in the range of 10–500 μg/mL.
2.9. Millicylinder morphology via scanning electron microscopy (SEM)
The morphology of millicylinders was examined by scanning electron microscopy (SEM) using a Phillips XL FEG SEM. The millicylinders were dried, sputter coated with gold for 90 s, then imaged using a 3 kV electric beam. Cross section and surfaces of millicylinders were imaged and inspected qualitatively for discrete 4HPR crystals throughout the polymer matrix. When this happens, it is indicative that the drug has precipitated during millicylinder formation, and the drug solubility limit in the polymer has been exceeded.
2.10. Protein content in PLGA after in vivo release by LECO nitrogen analysis
The amount of protein infused into 4HPR PLGA millicylinders after in vivo release was determined by nitrogen analysis on LECO instrumentation (TruSpec CHN Micro, PN200–716, St. Joseph, MI). The LECO instrument was calibrated by weighing EDTA standards ranging from 0.2 to 3 mg, and percent nitrogen content was determined upon incineration of sample. To prepare samples for the protein analysis, the PLGA millicylinders (20% 4HPR + 20% NaDC + 15% MgCO3 + 1% PVP) were carefully removed from SC tissue after 28 and 42 d of in vivo release with minimal excess tissue encasing the implant. The drug was extracted from implant using the 4HPR loading assay (Section 2.4), supernatant was removed, and 4HPR levels were determined. The remaining precipitate (likely composed of PLGA and proteins, both of which are insoluble after the loading assay) was dried at 40 °C x 2 d in a vacuum oven, and dried mass was recorded. The percent nitrogen in the PLGA-protein pellet was measured, and percent protein was estimated by normalizing to the model protein BSA (16% w/w nitrogen), as albumin is the most abundant serum protein. To summarize this mass balance quantitation, the total composition of the millicylinder was determined by: 1) percent 4HPR by loading assay, 2) percent protein by nitrogen analysis, and 3) PLGA calculated from total mass of dried implant - (4HPR + protein).
2.11. Effects of 4HPR implants on OSCC tumor xenografts in nude mice
The efficacy of 4HPR-PLGA millicylinders designed for 30-day controlled release dosing were tested by their effect on OSCC tumor xenografts’ growth. These studies were conducted at Ohio State University animal facility in accordance to ULAR regulations. For tumor induction, 1 × 106 SCC2095sc human oral squamous cell carcinoma (OSCC) tumor cells (validated by STR analyses) were injected into right-back flank side athymic nude mice. Seven days after OSCC cell injection, mice had developed measurable OSCC tumor masses. Mice were randomly assigned to two groups (n = 2 control, n = 3 treated), including: 1) control sham- PLGA millicylinder and 2) two 4HPR-PLGA millicylinders implanted alongside of tumor, with treatment duration of 10 d. All mice were weighed and tumor volume was recorded each day. On day 10, mice were euthanized, 4HPR millicylinders were removed, wet mass recorded, then dried to obtain percent water uptake, then assayed for percent 4HPR release by loading assay.
4HPR levels in tumors were quantified via tumor homogenization followed by drug extraction. The wet mass of tumor was recorded, internal standard was spiked into tissue (50 μL of 5 μg/mL acitretin), and 2 mL ice cold RIPA lysis buffer (to lyse contents of cells) was added to the tumor and homogenized using mechanical homogenizer (10,000 rpms x 1 min). Next, 3 mL ice cold ACN was added, homogenates sonicated on ice for 10 min, followed by centrifugation at 10,000 rpm x 10 min at 4 °C. The supernatant was assayed by UPLC/UV and normalized by internal standard recovery. 4HPR levels in serum were determined by spiking 10 μL 5 μg/mL acitretin into 50 μL serum, adding 140 μL ice cold ACN, sonicating on ice x 10 min, followed by centrifugation, and assaying supernatant by UPLC.
3. Results and discussion
3.1. Effect of 4HPR loading on in vitro release
To determine the maximum drug loading that would allow for the most continuous and complete release, we selected PLGA 503H for controlled release of 4HPR based on our previous work with formulating 4HPR into microspheres and ISFIs (Wischke et al., 2010). The free-acid low molecular weight PLGA continuously erodes at physiological conditions to allow continuous release (Hirota et al., 2016). We have previously shown that the addition of a basic salt, magnesium carbonate (MgCO3), to the millicylinder can lead to more favorable release kinetics due to its slow pore forming capabilities (Desai et al., 2008), similar to what was originally patented for release of proteins (Bernstein et al., 1997). This poorly soluble base is a porosigen to facilitate drug release, which acts by reacting with PLGA monomers/oligomers to form water-soluble salts, which in turn create osmotic pressure and new pore diffusion pathways for the drug within the PLGA matrix (Desai et al., 2008; Kang and Schwendeman, 2002). Based on these considerations, 4HPR was encapsulated in PLGA 503H millicylinders containing 3% MgCO3. Loading levels of 4HPR were varied at 10, 20 and 30% and in vitro release was evaluated (as shown in Fig. 1). In the higher 4HPR loading levels (20, 30%) a 2-week lag phase was present, whereas 10% 4HPR had a more continuous release and released the greatest percentage after day 28 (53%). Therefore the 10% 4HPR-loaded implant formulation containing 3% MgCO3 was selected for in vivo release and characterization as described below.
Fig. 1.

Effect of 4HPR loading on in vitro release. The implants were composed of PLGA 503H + 3% MgCO3 + 10, 20 or 30% 4HPR. Values represent mean ± SE (n = 3).
While the mechanism of 4HPR release from PLGA is not clear based on available data, one possibility is that when drug loading is above 10% in the presence of only 3% MgCO3, the drug begins to precipitate the drug in the first 2 weeks. At 10% 4HPR loading and below, the drug is likewise expected to be dissolved in the polymer and may even be supersaturated, allowing it to diffuse through the polymer without a lag time. This important question of the drug loading effect was evaluated mechanistically in the Supplemental information (e.g., Table S1 and Fig. S3). Whereas no strong differences in polymer erosion were recorded after 28 d (Fig. S3), there was an inexplicable large water uptake by day 28 in the higher drug loaded implants. As the buffer solution contains 2% Tween 80, which strongly interacts with both water and the drug, this complex interaction in the PLGA polymer matrix could have been responsible for the water uptake, which also triggered drug release by the 28th day.
3.2. In vivo release of 4HPR-PLGA millicylinders with pore forming agent
The optimal releasing implant from Fig. 1 containing a pore forming agent was evaluated after SC implantation in rats. Kinetics of drug release, implant erosion, and water uptake were examined as shown in Fig. 2. Although the 10% 4HPR with 3% MgCO3 pore-forming solubilizing base formulation performed well in vitro, very little if any controlled release was observed in vivo, with much less drug released by day 28 (18% in vivo vs. 53% in vitro). This in vivo/in vitro discrepancy may reflect the presence of high levels of Tween 80 solubilizer present in the in vitro release media. The PLGA in vivo erosion kinetics is shown in Fig. 2b for the 4HPR formulation and the no-drug sham control (i.e. PLGA + 3% MgCO3). A substantial level of polymer erosion (60%) had occurred by day 28, however, 82% of drug still remained unreleased, indicating that the drug has much difficulty to diffuse owing to its poor water solubility once pores open during polymer erosion. Moreover, most of 4HPR release occurred on the first day. The erosion rates of drug and sham formulations were similar, with slightly faster erosion in the sham, which indicates that 4HPR does not significantly alter the PLGA 503H millicylinder standard erosion profile. As seen in Fig. 2b, there was a significant polymer mass loss that occurred between 14–28 days, which is consistent with expected PLGA 503H erosion kinetics from the dense implant. SEM images showing the erosion over 28 d for both the drug and sham implants can be found in Supplemental information, Fig. S1. The incomplete release from the implant along with the high PLGA erosion rate confirms our suspicion that the polymer has little effect on the late stage 4HPR release, and it is highly likely release is governed by the slow dissolution of the solid drug itself.
Fig. 2.
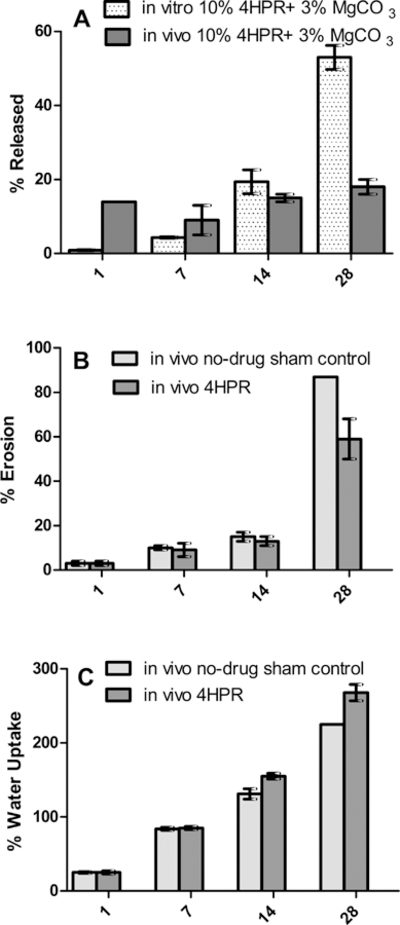
In vivo performance of 4HPR PLGA millicylinder formulation without solubilizers (10% 4HPR + 3% MgCO3). a) 4HPR release in vivo vs. in vitro (in a solubilizing media containing PBS+ 2%Tween), b) in vivo erosion of 4HPR PLGA implants compared to sham implants (PLGA + 3% MgCO3 only), and c) in vivo water uptake in 4HPR PLGA implants and sham PLGA implants. Values represent mean ± SE (n = 3).
The water uptake kinetics (Fig. 2c) of both the drug and sham implant were also similar, with 225% and 228% water uptake respectively. Water uptake is therefore not affected by the presence or absence of drug in the implant
3.3. Solubility and release enhancing excipients
Previous studies from our group indicated that 4HPR’s poor aqueous solubility adversely affected a 4HPR controlled release during the 1 month study (Zhang et al., 2016). To facilitate the release of the hydrophobic drug into aqueous interstitial fluid, we co-encapsulated 4HPR with PLGA and solubility enhancing excipients including NaDC via micelle formation and β-CD via inclusion complexes. Other excipients were also investigated based on their ability to inhibit drug crystallization and aide in implant disintegration including PVP K30 (Parchment et al., 2014; Srinarong et al., 2010; SURMAN et al., 2013), and HPMC K4M (Do et al., 2015; Siepmann and Peppas, 2001). HPMC has also been utilized to facilitate drug release due to its swelling behavior that promotes pore formation. All excipients studied have been categorized as generally regarded as safe (GRAS) by the US FDA, and many are currently used in FDA approved drug products (Strickley, 2004). NaDC selection was based on numerous studies on the solubilization and permeation enhancing behavior of bile salts (D’Archivio, et al., 1997; Gu et al., 1992; Lichtenberg et al., 1988; Reis et al., 2004) inclusive of retinoid solubilization (Li et al., 1996; Mohapatra and Mishra, 2011). The aqueous solubility of 4HPR at 37 ˚C in the presence of excipients is shown in Fig. 3. NaDC provided the best solubilization with 20% NaDC providing 1030 μg/mL 4HPR solubility, i.e., a 200:1 mass ratio of NaDC: 4HPR. β-CD resulted in a 40 μg/ml 4HPR solubility at 20% excipient level. The crystallization inhibitor/pore forming agent HPMC did not solubilize 4HPR, while the disintegrant/crystallization inhibitor PVP K30 provided only minimal solubility enhancement of 0.66 μg/mL at 20% PVP compared to 0.2 μg/ml 4HPR aqueous solubility without excipients. Despite little solubility enhancement achieved with HPMC and PVP, these agents were still pursued as they could improve release kinetics by alternative aforementioned mechanisms.
Fig. 3.

Effect of excipient concentration on 4HPR solubility in deionized water at 37 °C.
We have proceeded to develop 4HPR-PLGA millicylinders that were capable of accelerating in vitro release (Supporting Information, Fig. S2) by 1) increasing 4HPR solubility by addition of 20% NaDC, and 2) increasing PLGA porosity to aide in drug diffusion by increasing (MgCO3). The addition of the 20% NaDC allowed us to achieve a 20% drug loading, while decreasing the 3-week lag phase. In this solubilized formulation (20% NaDC), increasing MgCO3 from 3% to 15% further enhanced the extent of 4HPR release after 28 d from 50%–71%. Next the effects of specialized excipients (β-CD, HPMC, and PVP) added to PLGA + 20% 4HPR + 20% NaDC + 15% MgCO3 millicylinders were examined in vitro and in vivo as shown in the following section.
3.4. In vivo releases from 4HPR- PLGA millicylinders with solubilizers and crystallization inhibitors
The next set of PLGA millicylinders formulations aimed to increase 4HPR release in vivo by co-incorporating a high level of solubilizers, pore forming agents, and specialized excipients. The baseline formulation tested was 20% 4HPR + 20% NaDC + 15% MgCO3. The specialized excipients included: 5% β-CD to enhance solubility by formation of an inclusion complex with 4HPR, 1% HPMC K4M to promote swelling, aide in pore formation, and inhibit crystallization, and 1% PVP K30 to enhance solubility (Parchment et al., 2014), inhibit crystallization (SURMAN et al., 2013), and aid in disintegration (Srinarong et al., 2010). The in vivo/in vitro release with these specialized excipients is shown in Fig. 4a. Much slower release was observed in vivo for all compared to in vitro (day 28 release was ∼25% vs. ∼80%), and release in vivo in these cases was only slightly improved compared to the initial formulation containing 10% 4HPR + 3% MgCO3 (Fig. 2a) without solubilizers (25% vs 18% by day 28). Inclusion of the crystallization inhibitor PVP to the baseline formulation was effective at providing controlled release of 4HPR, whereas the other excipients additives had an initial burst release with no further drug release until after 4 weeks.
Fig. 4.

a)In vivo /in vitro release for PLGA 503H + 20% 4HPR + 15% MgCO3 + 20% NaDC (baseline) and specialized excipients including 5% B-CD, 1% HPMC K4M, and 1% PVP K30. b) Implant erosion, and c) implant water uptake in vivo. (mean ± SE, n = 3).
The PLGA erosion was evaluated, and all these formulations showed similar erosion profiles, which were unaffected by HPMC, β-CD, or PVP (Fig. 4b). The erosion profiles were as follows: 0–15% of eroded on day 1, 15–25% eroded by day 7, and 20–30% eroded by day 28. Greater variability on day 28 likely reflects the extent of surrounding connective tissue attachment to the implant. Higher levels of integrated connective tissue could result in an overestimate of mass (and under-estimate of PLGA erosion). On day 28, implants that contained high levels of MgCO3 and NaDC demonstrated significantly less PLGA erosion compared to the non-solubilized implant (30% vs. 60%), but nonetheless released more drug. These findings are consistent with the in vitro mechanistic data provided in the Supplemental information (Fig. S3), which showed that NaDC and MgCO3 not only inhibit PLGA erosion, but also promote implant water uptake due to their osmotic properties. These results indicate that 4HPR release from PLGA implants in vivo is not an erosion-controlled process over the 28 d period.
Implant water uptake was evaluated as shown in Fig. 4c, and was similar for these 4 formulations, where all excipients (B-CD, HPMC, and PVP) promoted the same level of water uptake as the baseline formulation containing 20% NaDC and 15% MgCO3, yielding ∼ 75%, 150%, 250% water uptake on days 1, 7, 28, respectively. When compared to the initial formulation without solubilizers (Fig. 2c), we see that the presence of these osmotic agents only significantly increased water uptake on day 1, while after 7 and 28 d, they behaved similarly (25%, 155%, 225%), but was much less compared to in vitro (> 500% after 28 d). This trends indicate that the tissue environment decreases implant water uptake, and again confirms that drug release in vivo from PLGA implants is not driven by implant water uptake.
Thus far we have shown that addition of solubilizing, pore forming agents and specialized excipients to the PLGA implants, while effective in vitro at accelerating 4HPR release, had little effect in vivo on accelerating 4HPR release after 4 weeks (∼25% drug release). Furthermore, we have concluded from these two in vivo experiments (with and without solubilizers) that neither PLGA erosion rate nor implant water uptake can explain the slow in vivo drug release. Therefore, the likely cause is very slow dissolution of the drug. To test this, 4HPR in vivo release from PLGA implants with solubilizers was compared to a water-soluble matrix implant (PVA/sucrose) over 2 months. In addition, a second PVA/sucrose formulation incorporated a solubilizer and crystallization inhibitor (NaDC, PVP). Two salt forms of deoxycholic acid (sodium and calcium) were utilized in the PLGA formulations, with the intent that the less water soluble CaDC would reduce initial burst release and thereby facilitate 4HPR release from implant at later time points. A water-soluble matrix implant was prepared in a PVA/sucrose vehicle with and without solubilizers (20% 4HPR + 20% NaDC + 1% PVP), and compared to PLGA formulation previously shown to have the most desirable release (Fig. 4, 20% 4HPR + 15% MgCO3 + 1% PVPK30 + 20% NaDC or CaDC). The in vivo evaluation (Fig. 5a), which depicts controlled 4HPR release over 2 months for the PLGA formulations, demonstrated that bile salt composition (sodium vs. less soluble calcium salt) had little effect on the rate of 4HPR release, both exhibiting a 3-week lag period. Notably, both bile salts showed nearly identical 4HPR release profiles, despite achieving a slower release of the deoxycholic acid (DC−) solubilizer over 14 d with the calcium salt, compared to an 80% burst release with NaDC (Fig. 5c). These results indicate that 4HPR release from PLGA millicylinders may not be controlled by solubilization, at least not by NaDC.
Fig. 5.

a) In vivo release of 4HPR from CR PLGA millicylinders vs. that from water soluble matrix PVA/sucrose implants with and without solubilizers. The PLGA implants contained 20% 4HPR + 15% MgCO3 + 1% PVP + 20% NaDC or CaDC, while the PVA/sucrose implants contained either 20% 4HPR or additional solubilizer and crystallization inhibitor (20% NaDC + 1% PVP). b) Implant images prior to harvesting from rat SC tissue on day 28 (yellow represents 4HPR). C) In vivo release of sodium and calcium deoxycholate (DC−) from PLGA millicylinders. (mean ± SE, n = 3).
When examining the drug release from the water soluble PVA/sucrose implants, we observed that 4HPR was released steadily during the first 3 weeks, then a large surge in drug release, followed by a controlled release pattern similar to the PLGA implants in weeks 4–6 (Fig. 5a). The additional solubilizing excipients in this PVA/sucrose implant led to an accelerated 4HPR release after 28 d, whereby 78% was released vs. 59% for those without the extra excipients. In the PLGA formulations, only 32% 4HPR was released after 1 month, and the presence of the 3-week lag phase was likely due to the rate of PLGA erosion, as we have previously showed (Fig. 4c) that PLGA did not exhibit appreciable erosion in vivo until after 2 weeks. After 1 month, the water-soluble excipients would have dissipated from the implantation site, leading us to the logical conclusion that the controlled release is due to dissolution of the hydrophobic drug solid into the surrounding aqueous tissues. After 6 weeks, both water soluble PVA/ sucrose implants formulations (with and without solubilizers) had performed similarly, and released ∼90% 4HPR, whereas the PLGA only released ∼50% 4HPR. Similarities in the release slopes of PLGA and PVA/sucrose implants between weeks 4–6 suggests that late stage release of 4HPR from PLGA is controlled by dissolution of the drug.
The images of the implants after a month in vivo release in rat SC tissue are shown in Fig. 5b, and we see that the PLGA implants maintained their cylindrical shapes, with no apparent characteristic yellow colored 4HPR diffusion into the surrounding tissue. The PVA/sucrose implants were no longer in their millicylindrical form, and the addition of the NaDC and PVP solubilizers enabled greater 4HPR tissue dispersion. We further note that the two implants (PVA/sucrose, PLGA) could potentially be used together for further extended release as the water soluble PVA/sucrose formulation can be used to overcome the initial lag phase, while PLGA can provide a 2 + month long acting release of 4HPR.
These data from 4HPR in vivo release from millicylinders align with results from our previous PK study, where a slow and continuous release of 4HPR from a SC injected drug suspension occurred after 2 weeks, following a large burst release, confirming that the controlled release properties are due to the dissolution of the hydrophobic drug. The fact that 4HPR release from PVA/sucrose water-soluble matrix formulation was faster than the PLGA and different from the solid control used in PK study, suggests that the PVA/sucrose formulation may release faster than pure 4HPR crystals. We were unable to test 4HPR crystals here because we could not collect them to determine the fraction drug remaining, but future PK analyses could be used to answer this question.
These PLGA-4HPR millicylinders were further examined to determine what other physiological factors besides polymer properties were hindering drug release and dispersion into tissue. These tests included SEM images of tissue-millicylinder interactions and nitrogen analysis to estimate protein levels within the millicylinders. The SEM images of a PLGA millicylinder (20% 4HPR + 15% MgCO3 + 1% PVP + 20% NaDC) after 28 d in the SC environment are displayed in Fig. 6. In these micrographs, the tissue binding to the millicylinders is quite apparent, where the millicylinder has become completely encased within a fibrous tissue capsule. The resistance exerted by this tissue encasing could be partially responsible for the inhibition of drug release from the millicylinders. Upon harvesting of the millicylinder, the tissue encasement was removed and the extracted millicylinder (Fig. 6c,d) was further examined. Observation of the implant cross sections showed mostly crystalline species, perhaps drug or protein, along with remnants of the porous PLGA matrix. The low local pH within the PLGA implant could be contributing to plasma protein precipitation. The composition of this drug-polymer-protein mass was investigated after removing from the tissue encasement and the amount of protein was estimated via LECO nitrogen analysis. After 28 d, the extracted millicylinder contained 23 ± 1% w/w 4HPR of the extracted mass, similar to the initial drug loading of 20%. From the LECO analysis, the estimated percent protein (normalized to BSA) bound in this extracted mass was 25 ± 4%, and PLGA was calculated to be 52 ± 5%, which aligns well with the in vitro erosion data for this formulation of 60%. After 42 d, this extracted PLGA-4HPR-protein was composed of 16% 4HPR, ∼55% protein, and ∼29% PLGA. From this analysis, we observe high levels of protein within the PLGA-4HPR implant, which have played a role in the drug release.
Fig. 6.

SEM images depicting 4HPR-PLGA millicylinders interactions with the tissue environment after 28 d in vivo. PLGA millicylinder shown was composed of 20% 4HPR + 15% MgCO3 + 1% PVP + 20% NaDC. a) Cross section of millicylinder encased with tissue, b) zoomed in to show tissue growth into the millicylinder, c) 4HPR-PLGA millicylinder removed from tissue encasing, and d) cross section (zoomed in to show precipitated 4HPR, permeated tissue components, and remaining PLGA matrix).
3.5. Evaluation of in vivo efficacy
The optimized and characterized formulation of 503H + 20% 4HPR + 20% NaDC + 15% MgCO3 + 1% PVP was tested in nude mice bearing OSCC xenografts. Effects on tumor growth relative to blank PLGA + PBS OSCC tumor explants were assessed via daily bidirectional caliper tumor measurements. As shown in Fig. 7, the 4HPR-PLGA millicylinders were effective at reducing the rate of tumor growth over 10 d (1.3 ± 0.3 fold increase in volume from initial, n = 3) compared to the control PBS/PLGA millicylinder (2.0 ± 0.3 fold increase, n = 2) (Note: smaller sample sizes were used than desired due to lack of tumor formation in some mice. Due to low animal numbers statistical analyses were not suitable). The efficacy of 4HPR-PLGA millicylinder in these studies served as proof of concept for more comprehensive studies which showed that 4HPR was effective in combination with other chemotherapeutic agents such as tocilizumab and 2-methoxyestradial at reducing rate of tumor growth (Mallery et al., 2017). Shown in Fig. 8 are Hematoxylin and eosin (H&E) stained sections from the OSCC tumor cell xenograft from both the 4HPR dosed group and control group, which demonstrate tumor necrosis in close proximity to the 4HPR implant, while tumor tissue proliferated a few millimeters from the implants. Therapeutic efficacious 4HPR levels (> 10 μM)(Wu et al., 2012) of 90 ± 76 μM were achieved in tumors after CR delivery with 4HPR-PLGA millicylinders, although levels were highly variable as reflected in the large standard error of the mean. 4HPR was not detected in serum, which is a favorable aspect that local drug delivery is capable of alleviating potentially toxic systemic side effects. Additionally, the UPLC chromatograms of the 4HPR dosed tumor extract showed no active metabolite 4-oxo-4-HPR when compared to the calibration standard (Representative chromatogram in Supplemental information, Fig. S5).
Fig. 7.

Pilot studies to assess the effects of 4HPR-PLGA millicylinders (503H + 20% 4HPR + 20% NaDC + 15% MgCO3 + 1% PVP) on established xenograft tumors that were developed by implantation of human OSCC cells (2095sc, 106 in Matrigel) injected into the flanks of nude mice. Relative to the control (drug free) PLGA millicylinder, tumors that were injected with the 4-HPR implants showed reduced growth. (n = 2 control, n = 3 4-HPR PLGA implants).
Fig. 8.
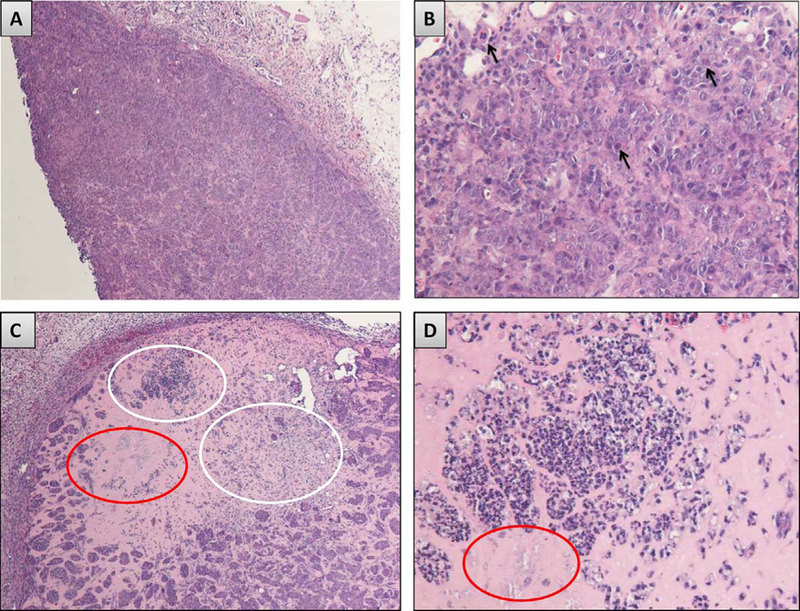
Hematoxylin and eosin (H&E) stained sections from the OSCC tumor cell xenograft studies. A) of OSCC tumor tissue harvested from the control (nontreated) nude mice shows a large aggregation of intact tumor cell nests and cords in a well vascularized tumor stroma. B) Numerous mitotic figures along with scattered apoptotic and dyskeratotic cells are observed in the control group tumor nests. In contrast, C) the 4HPR treated group shows foci of necrosis (white elipse) and residual ghosts of cell nests (red elipse) in residual Matrigel. Appreciably smaller OSCC tumor islands are also visible. D) Higher magnification representation of the necrotic tumor cells and cell island ghosts. Image scale: A, C 40X, B, D 200X.
The release performance of the PLGA-4HPR after implantation alongside the perimeter of the tumor is listed in Table 1. The PLGA-4HPR millicylinders released 16.0 ± 2.0% of their load after 10 d equating to ∼160 μg 4HPR (x 2 millicylinders = 320 μg dosed), which was approximately double of the released amount of the same formulation implanted in flanks of rats (7.5 ± 2.8% released on day 7). This difference could be due to the different properties of tissue environments, where the tissue surrounding the tumor may have greater blood flow and acidic pH compared to the SC tissue. The 4HPR-PLGA implants had greater water uptake compared to the sham PLGA implants (367% vs. 197%, Table 1), which aligns well with our mechanistic characterization studies (Supplemental information).
Table 1.
Tumor efficacy study with 4HPR-PLGA millicylinders. Mean ± SE, n = 3.
| Group | Water uptake | 4HPR release (day 10) | 4HPR level in tumor (μM) |
|---|---|---|---|
| Sham PLGA | 367 ± 21 % | - | - |
| 4HPR-PLGA | 197 ± 22 % | 16 ± 2% | 90 ± 76 |
4. Conclusion
Here we describe our approach to control the release of 4HPR in SC tissues and tumor environments. We found that 4HPR releases from PLGA millicylinders much slower in vivo compared to in vitro, likely due to difference in solubilization in the in vitro release media compared to in vivo. After exhibiting a significant lag phase in vivo, PLGA implants that included various excipients provide release to proceed continuously for at least 2 months. The lag phase can be obviated by formulating a millicylinder without PLGA but with soluble components (PVA/sucrose/PVP/NaDC) to promote dissolution and crystallization inhibition. Indeed, the shorter and longer acting formulations could be useful in the future as a combination, as the overlapping release may be desired. Initial evaluations of the efficacy of the PLGA implants demonstrate inhibition of OSCC tumor xenographs with local controlled release. Therefore, these data motivate further development of the local controlled release approach for chemoprevention with 4HPR.
Supplementary Material
Acknowledgement
This work was supported by National Institutes of Health R01CA171329 and R01CA211611.
References
- Bernstein Howard, Zhang Yan, Khan M. Amin, Tracy A. Mark, 1997. Modulated release from biocompatible polymers, US 5656297 [Google Scholar]
- D’Archivio AA, Galantini L, Gavuzzo E, Giglio E, Mazza F, 1997. Calcium ion binding to bile salts. Langmuir 13, 3090–3095. [Google Scholar]
- Desai KGH, Mallery SR, Schwendeman SP, 2008. Effect of formulation parameters on 2-methoxyestradiol release from injectable cylindrical poly(dl-lactide-co-glycolide) implants. Eur. J. Pharm. Biopharm 70, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MP, Neut C, Metz H, Delcourt E, Siepmann J, Mäder K, Siepmann F, 2015. Mechanistic analysis of PLGA/HPMC-based in-situ forming implants for periodontitis treatment. Eur. J. Pharm. Biopharm 94, 273–283. [DOI] [PubMed] [Google Scholar]
- Fredenberg S, Wahlgren M, Reslow M, Axelsson A, 2011. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm 415, 34–52. [DOI] [PubMed] [Google Scholar]
- Gleber-Netto FO, Braakhuis BJM, Triantafyllou A, Takes RP, Kelner N, Rodrigo JP, Strojan P, Vander Poorten V, Rapidis AD, Rinaldo A, Brakenhoff RH, Ferlito A, Kowalski LP, 2015. Molecular events in relapsed oral squamous cell carcinoma: recurrence vs secondary primary tumor. Oral Oncol 51 (8), 738–744. [DOI] [PubMed] [Google Scholar]
- Gu JJ, Hofmann AF, Ton-Nu HT, Schteingart CD, Mysels KJ, 1992. Solubility of calcium salts of unconjugated and conjugated natural bile acids. J. Lipid Res 33, 635–646. [PubMed] [Google Scholar]
- Han BB, Li S, Tong M, Holpuch AS, Spinney R, Wang D, Border MB, Liu Z, Sarode S, Pei P, Schwendeman SP, Mallery SR, 2015. Fenretinide perturbs focal adhesion kinase in premalignant and malignant human oral keratinocytes. Fenretinide’s chemopreventive mechanisms include ECM interactions. Cancer Prev. Res 8, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Doty AC, Ackermann R, Zhou J, Olsen KF, Feng MR, Wang Y, Choi S, Qu W, Schwendeman AS, Schwendeman SP, 2016. Characterizing release mechanisms of leuprolide acetate-loaded PLGA microspheres for IVIVC development I: in vitro evaluation. J. Control Release 244, 302–313. [DOI] [PubMed] [Google Scholar]
- Holpuch AS, Phelps MP, Desai KG, Chen W, Koutras GM, Han BB, Warner BM, Pei P, Seghi GA, Tong M, Border MB, Fields HW, Stoner GD, Larsen PE, Liu Z, Schwendeman SP, Mallery SR, 2012. Evaluation of a mucoadhesive fenretinide patch for local intraoral delivery: a strategy to reintroduce fenretinide for oral cancer chemoprevention. Carcinogenesis 33, 1098–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyon SH, 2000. Biodegradable poly (lactic acid) microspheres for drug delivery systems. Yonsei Med. J 41, 720–734. [DOI] [PubMed] [Google Scholar]
- Kang J, Schwendeman SP, 2002. Comparison of the effects of Mg(OH)2 and sucrose on the stability of bovine serum albumin encapsulated in injectable poly(d,l-lactide-coglycolide) implants. Biomaterials 23, 239–245. [DOI] [PubMed] [Google Scholar]
- Li C-Y, Zimmerman C, Wiedmann T, 1996. Solubilization of retinoids by bile salt/phospholipid aggregates. Pharm. Res 13, 907–913. [DOI] [PubMed] [Google Scholar]
- Lichtenberg D, Younis N, Bor A, Kushnir T, Shefi M, Almog S, Nir S, 1988. On the solubility of calcium deoxycholate: kinetics of precipitation and the effect of conjugated bile salts and lecithin. Chem. Phys. Lipid 46, 279–291. [DOI] [PubMed] [Google Scholar]
- Mallery SR, Wang D, Santiago B, Pei P, Schwendeman S, Nieto K, Spinney R, Tong M, Koutras G, Han BB, Holpuch AS, Lang JC, 2017. Benefits of multifaceted chemopreventives in the suppression of the oral squamous cell carcinoma (OSCC) tumorigenic phenotype. Cancer Prev. Res 10, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra M, Mishra AK, 2011. Effect of Submicellar Concentrations of Conjugated and Unconjugated Bile Salts on the Lipid Bilayer Membrane. Langmuir 27, 13461–13467. [DOI] [PubMed] [Google Scholar]
- Parchment RE, Jasti BR, Boinpally RR, Rose SE, Holsapple ET, 2014. Liposomal Nanoparticles and Other Formulations of Fenretinide for use in Therapy and Drug Delivery. US 8709379. [Google Scholar]
- Reibel J, 2003. Prognosis of oral pre-malignant lesions: significance of clinical, histopathological, and molecular biological characteristics. Crit. Rev. Oral Biol. Med 14, 47–62. [DOI] [PubMed] [Google Scholar]
- Reis S, Moutinho CG, Matos C, de Castro B, Gameiro P, Lima JLFC, 2004. Noninvasive methods to determine the critical micelle concentration of some bile acid salts. Anal. Biochem 334, 117–126. [DOI] [PubMed] [Google Scholar]
- Siepmann J, Peppas NA, 2001. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv. Drug Deliv. Rev 48, 139–157. [DOI] [PubMed] [Google Scholar]
- Srinarong P, Kouwen S, Visser MR, Hinrichs WLJ, Frijlink HW, 2010. Effect of drug-carrier interaction on the dissolution behavior of solid dispersion tablets. Pharm. Dev. Technol 15, 460–468. [DOI] [PubMed] [Google Scholar]
- Strickley R, 2004. Solubilizing excipients in oral and injectable formulations. Pharm. Res 21, 201–230. [DOI] [PubMed] [Google Scholar]
- Surman P, Binnie FC, VOS MG, 2013. Pharmaceutical methods and topical compositions containing acitretin. Patent application, WO2013050874 A1. [Google Scholar]
- Wang Y, Wen Q, Choi S, 2016. FDA’s regulatory science program for generic PLA/PLGA-based drug products. Am. Pharm. Rev [Google Scholar]
- Wischke C, Schwendeman SP, 2008. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm 364, 298–327. [DOI] [PubMed] [Google Scholar]
- Wischke C, Zhang Y, Mittal S, Schwendeman SP, 2010. Development of PLGA-based injectabledelivery systems for hydrophobic fenretinide. Pharm. Res 27, 2063–2074. [DOI] [PubMed] [Google Scholar]
- Wu X, Desai KG, Mallery SR, Holpuch AS, Phelps MP, Schwendeman SP, 2012. Mucoadhesive fenretinide patches for site-specific chemoprevention of oral cancer: enhancement of oral mucosal permeation of fenretinide by coincorporation of propylene glycol and menthol. Mol. Pharm 9, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wischke C, Mittal S, Mitra A, Schwendeman SP, 2016. Design of controlled release PLGA microspheres for hydrophobic fenretinide. Mol. Pharm 13, 2622–2630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.