

Abstract
The combination of VCl3(THF)3 and N,N-bis(trimethylsilyl)aniline (1a) is an efficient catalyst for the [2+2+1] coupling reaction of alkynes and azobenzenes, giving multisubstituted pyrroles. A plausible reaction mechanism involves the generation of a mono-(imido)vanadium(III) species as an initiation step, where 1a served as an imido source with concomitant release of 2 equiv of ClSiMe3, followed by a reaction with azobenzene to form a catalytically active bis(imido)-vanadium(V) species via N═N bond cleavage.
Multisubstituted pyrroles are synthetically valuable heteroaromatic compounds in terms of their versatility as building blocks of pharmaceuticals,1 natural products,2 functional materials,3 and dyes.4 Although cyclocondensation reactions such as the Paal–Knorr5 and Hantzsch6 reactions are well-established routes for synthesizing multisubstituted pyrroles, transition-metal-catalyzed multicomponent coupling reactions have been recently investigated as potent alternatives.7 Among them, the [2+2+1] cycloaddition reaction of alkynes and primary amines is attractive because many alkynes and primary amines are commercially available or easily synthesized.8 As shown in Scheme 1, AgBF4 catalyzed the oxidative coupling of activated alkynes and primary amines in the presence of stoichiometric amounts of iodobenzene diacetate (Scheme 1a),8g Cu(OAc)2 combined with Cs2CO3 under O2 catalyzed the coupling of 2-butynedioate and primary amines (Scheme 1b),8c and PdCl2 combined with super-stoichiometric amounts of CuCl and Na2CO3 catalyzed the coupling of diarylalkynes and primary amines (Scheme 1c).8d In these [2+2+1] cycloaddition reactions, activated alkynes bearing ester groups and stoichiometric amounts of oxidant and base were inevitably required.
Scheme 1.

Transition-Metal-Catalyzed [2+2+1] Pyrrole Formation Reactions
Recently, we reported a formal [2+2+1] coupling of alkynes and azobenzenes that was catalyzed by (imido)titanium complexes, either preformed or in situ generated from TiCl4(THF)2 and reductant (Scheme 1d).9 In an effort to expand the classes of catalysts and utility of this type of oxidative coupling, we herein report that vanadium complexes facilitate the [2+2+1] coupling of alkynes and azobenzenes, where an in situ generated catalyst system of VCl3(THF)3 combined with N,N-bis(trimethylsilyl)aniline (1a) showed the best catalytic activity (Scheme 1e). Furthermore, we confirmed that an in situ generated mono(imido)vanadium(III) intermediate cleaves the N═N double bond of azobenzene to give a bis(imido)vanadium(V) active species, which is responsible for the catalytic reaction, in contrast to the previously reported mono(imido)titanium(IV) active species for the Ti(II)/Ti(IV) catalytic cycle. These results highlight that diverse catalyst metals, architectures, and oxidation states can potentially play a role in complex, oxidative heterocycle-forming reactions.10
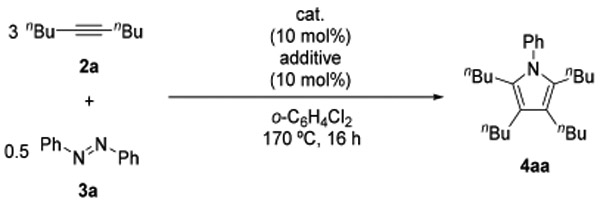
We have proposed that the previous Ti catalyst system for the [2+2+1] pyrrole formation reaction involved an (imido) titanium as the catalytically active species.9 Accordingly, we sought to find another effective catalyst system by varying transition metal chlorides upon activation by N,N-bis-(trimethylsilyl)aniline (1a), since some imido complexes of early transition metals have been synthesized by treating metal chlorides with 1a with concomitant loss of 2 equiv of ClSiMe3.9b,11. Thus, we searched for the best catalyst system for [2+2+1] pyrrole formation by reacting 5-decyne (2a) and azobenzene (3a) in the presence of various metal chlorides (10 mol %) and 1a (10 mol %) in o-dichlorobenzene at 170 °C for 16 h.
The results of our initial catalyst screening are summarized in Table 1. The [2+2+1] coupling product, N-phenyl-2,3,4,5-tetra(n-butyl)pyrrole (4aa), was obtained in 23% yield with TiCl3(THF)3 and 1a, whereas the yield of 4aa was 4% for TiCl4(THF)2 and 1a (entries 1 and 2);12 both reactions produced a small amount of alkyne cyclotrimerization product, hexa(n-butyl)benzene. In contrast, VCl3(THF)3 quantitatively gave 4aa without any contamination of hexa(n-butyl)benzene (entry 3). CrCl3(THF)3 and MnCl2(THF)1.5 resulted in no reaction (entries 4 and 5). In the absence of 1a, the reactivity of VCl3(THF)3 was significantly suppressed, giving 4aa in 9% yield (entry 6). Among other early transition metal chlorides we screened in Table S1, WCl6 showed moderate catalytic activity to give 4aa in 65% yield.
Table 1.
Catalyst Screening a
 | |||
|---|---|---|---|
| entry | cat. | additive | yield [%]b |
| 1 | TiCl3(THF)3 | PhN(SiMe3)2 (1a) | 23 |
| 2 | TiCl4(THF)2 | PhN(SiMe3)2 (1a) | 4 |
| 3 | VCl3(THF)3 | PhN(SiMe3)2 (1a) | >99 (71)c |
| 4 | CrCl3(THF)3 | PhN(SiMe3)2 (1a) | trace |
| 5 | MnCl2(THF)1.5 | PhN(SiMe3)2 (1a) | n.d.d |
| 6 | VCl3(THF)3 | 9 | |
| 7 | V(═NPh)Cl3 | PhN(SiMe3)2 (1a) | >99 |
| 8 | V(═NPh)Cl3 | 6 | |
| 9 | V(═NtBu)Cl3 | PhN(SiMe3)2 (1a) | 98 |
| 10 | V(═N(2,6-iPr2C6H3))Cl3 | PhN(SiMe3)2 (1a) | 33 |
Reaction conditions: 2a, 3.00 mmol; 3a, 0.500 mmol; o-C6H4Cl2, 2.5 mL.
Determined by GC using pentadecane (internal standard).
Isolated yield.
Not detected.
Noteworthy was that the yield of 4aa by a preformed (imido)vanadium(V) complex, V(═NPh)Cl3, was sensitively affected by the addition of 1a: in the presence of 1a, V(═NPh)Cl3 produced 4aa in quantitative yield, whereas in the absence of 1a, V(═NPh)Cl3 resulted in poor yield of 4aa (entries 7 and 8). This result indicates that the formation of bis(imido)vanadium(V) species may be important for catalysis (vide infra). Bulkiness of the substituent on the imido group of the precatalyst was another factor to control the catalytic yield: V(═NtBu)Cl3/1a showed high yield, while bulkier V(═N(2,6-iPr2C6H3))Cl3/1a resulted in significantly lower yield (entries 9 and 10). We thus selected VCl3(THF)3/1a as the best catalytic system for further substrate scope.13
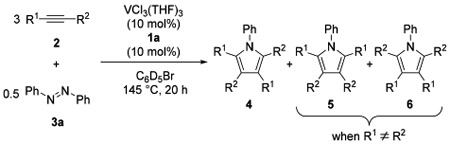
With the best catalytic system in hand, we explored the scope and limitations of alkynes for the vanadium-catalyzed pyrrole synthesis with 3a in C6D5Br, and the results of the NMR yields are shown in Table 2. Coupling reactions of symmetrical aliphatic alkynes such as 3-hexyne (2b) and 4-octyne (2c) afforded the corresponding pyrroles 4ba and 4ca in 74% and 92% yield, respectively (entries 1 and 2), whereas the yield of 4da from diphenylacetylene (2d) and 3a was low (entry 3). The unsymmetrical alkyne, 4-methyl-2-pentyne (2e), produced a mixture of 4ea, 5ea, and 6ea in 54% total yield with the ratio of 72:17:11 for the regioisomers (entry 4). When 1-phenyl-1-propyne (2f) was used as a substrate, the corresponding regioisomers of 4fa and 5fa were formed in the ratio of 53:47 in 34% total yield (entry 5). Regioselectivity of this reaction is assumed to be derived from the electronic effect of 2f.9c In contrast, good steric control of the regioselectivity was observed when 1-trimethylsilyl-1-hexyne (2g) was applied to the reaction, in which the single regioisomer 4ga was obtained in 42% yield (entry 6). Reaction with a bulky terminal alkyne, tert-butylacetylene (2h), also afforded the single regioisomer 4ha in 53% yield (entry 7), whereas all three regioisomers of 4ia, 5ia, and 6ia were detected by using n-butylacetylene (2i) (entry 8); both terminal alkynes 2h and 2i produced significant amounts of alkyne cyclotrimerization byproducts, even in the presence of excess amount of azobenzene in the case of 2i.14 We further tested the effect of various additives for synthesizing 4aa: ether, iodoarene, nitrile, amide, and triarylphosphine (1 equiv with respect to 3a) were tolerated in the reaction, an improved scope compared to the original Ti system (Table S4).15
Table 2.
Scope and Limitations of Alkynes a
 | ||
|---|---|---|
| entry | alkyne | yield [%](4:5:6)b |
| 1 | R1 = R2 = Et (2b) | 74 |
| 2 | R1 = R2 = nPr (2c) | 92 |
| 3 | R1 = R2 = Ph (2d) | 16c |
| 4 | R1 = Me, R2 = iPr (2e) | 54 (72:17:11) |
| 5 | R1 = Me, R2 = Ph (2f) | 34 (53:47:0) |
| 6d | R1 = SiMe3, R2 = nBu (2g) | 42 (100:0:0) |
| 7 | R1 = H, R2 = tBu (2h) | 53 (100:0:0) |
| 8e | R1 = H, R2 = nBu (2i) | 17 (53:35:12) |
Reaction conditions: 2, 0.988 mmol; 3a, 0.165 mmol; C6D5Br, 0.5 mL.
NMR yield based on 3a using 1,3,5-trimethoxybenzene (internal standard).
Isolated yield.
For 40 h.
The reaction was performed using 1 equiv of 2i and 6 equiv of 3a.
We next examined the effect of the para-substituents of azobenzenes. The coupling reaction with 4,4′-dimethoxyazo-benzene (3b) or 4,4′-dimethylazobenzene (3c) afforded the corresponding pyrroles 4ab and 4ac in 66% and 69% yield, whereas reaction with 4,4′-dibromoazobenzene (3d) or 4,4′-bis(trifluoromethoxy)azobenzene (3e) resulted in the formation of 4ad or 4ae in 91% and 94% yield, respectively (Table S5). Hammett analysis of the catalytic reaction using 3a–e revealed that the reaction rate was accelerated by the electron-withdrawing substituents (ρ = 1.97, Figure S38), being in good accordance with the tendency observed for the Ti-catalyzed system.9d,15 In addition, a good positive correlation was observed between the initial reaction rate and reduction potentials of azobenzenes, indicating that the reduction potential of azobenzenes is significant to determine the reaction rate (Figure S40).15
To gain insight into a reaction mechanism, we carried out a kinetic study for pyrrole formation under the optimized reaction conditions where the catalyst was in situ generated by treating VCl3(THF)3 with 1a and analyzed the resulting data by variable time normalization analysis.16 The obtained rate law is provided in eq 1. The reaction obeyed a second-order with respect to the concentration of VCl3(THF)3, which was in sharp contrast to the half-order rate dependence on the catalyst concentration for the Ti-catalyzed system,9d suggesting the involvement of a dimeric vanadium species for azobenzene N═N double-bond cleavage. For the substrates, we found a first-order rate dependence for the concentration of 5-decyne, while the dependency of the concentration of azobenzene was slightly deviated from a first-order rate, being 0.8.
| (1) |
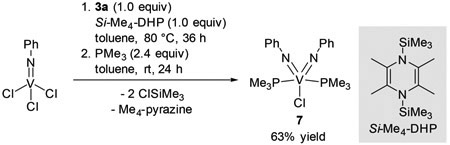
During the catalytic reaction, an imidovanadium(III) species is assumed to be responsible for the N═N bond cleavage, giving a bis(imido)vanadium(V) species. We thus examined an in situ generated “V(═NPh)Cl” species for the catalytic reaction. V(═NPh)Cl3 was treated with an organosilicon reducing reagent, Si-Me4-DHP,17 in the presence of 2a and 3a in C6D5Br, giving “V(═NPh)Cl”. The stoichiometry of this reaction was clarified by the observation of 2 equiv of ClSiMe3 and 1 equiv of 2,3,5,6-tetramethylpyrazine (Me4-pyrazine) by 1H NMR analysis (Figure S44).15 Subsequent treatment of the reaction mixture at 145 °C for 20 h resulted in the formation of 4aa in 91% yield, indicating that a similar imidovanadium(III) species may be generated from VCl3(THF)3 and 1a in the reaction mixture.
We further examined the reaction of the in situ generated imidovanadium(III) species “V(═NPh)Cl” with 3a in toluene at 80 °C, followed by subsequent addition of PMe3 to trap a bis(imido)vanadium complex, V(═NPh)2Cl(PMe3)2 (7), in 63% yield (eq 2). In this reaction, half an equivalent of 3a with
 |
(2) |
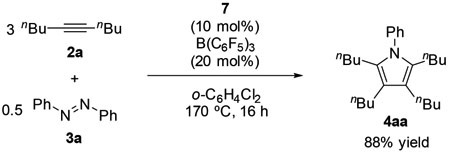
respect to V(═NPh)Cl3 was consumed. The molecular structure of 7 was confirmed by NMR and single-crystal X-ray diffraction analysis (Figure 1).18 Complex 7 catalyzed the pyrrole formation in the presence of B(C6F5)3 as a phosphine scavenger in 88% yield (eq 3), clearly indicating that the
 |
(3) |
bis(imido)vanadium(V) species is a catalytically relevant species. We also found that complex 7 was generated by treating VCl3(THF)3 with 1a in the presence of 3a in C6D5Br at 145 °C followed by addition of PMe3.15
Figure 1.

Molecular structure of complex 7 with 30% thermal ellipsoids. All hydrogen atoms are omitted for clarity.
On the basis of the kinetic study and the control experiments, we propose a plausible catalytic cycle for the vanadium-catalyzed [2+2+1] coupling of 5-decyne (2a) and azobenzene (3a) (Scheme 2). Reaction of VCl3(THF)3 with 1a generates an on-cycle mono(imido)vanadium(III) species A together with a release of 2 equiv of ClSiMe3 under the reaction conditions. The V(III) species A reductively cleaves the N═N double bond of 3a upon reaction with half an equivalent of 3a, giving a bis(imido)vanadium species B,19 presumably through a dimerization process similar to that proposed for the Ti system.9d,e This is consistent with the second-order rate dependence on the concentration of the catalyst and the requirement of the imido ligand for showing high catalytic activity. In addition, formation of B is consistent with the generation of 7 after exposure of VCl3(THF)3 to 1a and 3a under the reaction conditions and trapping with PMe3. One of two V═N moieties in B reacts with alkyne via [2+2] addition to form an azavanadacyclobutene C.20 Next, subsequent insertion of the second alkyne into the V–C bond affords an azavanadacyclohexadiene intermediate D. The formation of azametallacyclohexadienes from imidometal complexes and alkynes was already noted for molybdenum,21b tungsten,21b titanium,21a and zirconium21c complexes. After the second insertion, reductive elimination from D forms the corresponding pyrrole and mono(imido)vanadium(III) species E, in which the low-valent vanadium center might be stabilized by the coordination of generated pyrrole.22 Odom and co-workers observed the similar reductive elimination from azamolybda- or azatungstacyclohexadiene, giving pyrrole derivatives 21b. Finally, the pyrrole is dissociated to regenerate a mono(imido)vanadium(III) species A. In fact, no reaction was observed for complex 7 and 3-hexyne (2b) at room temperature in the presence of B(C6F5)3, while we only found the formation of N-phenyl-2,3,4,5-tetraethylpyrrole (4ba) without any intermediates such as C, D, and E upon heating. Based on the kinetic study, the mono(imido)vanadium(III) A is the resting state for generating catalytically active bis(imido) vanadium B via a bimetallic mechanism, and further reaction of B with 1 equiv of alkyne is the rate-determining step in this catalytic reaction. The key differences between this V-catalyzed reaction and the previous Ti-catalyzed reaction is low-valent species as a resting state during this catalytic cycle, which is consistent with the relative reducing ability of Ti(II) and V(III).
Scheme 2.

Plausible Reaction Mechanism for Vanadium-Catalyzed [2+2+1] Coupling of Alkynes and Azobenzenes
In summary, we have demonstrated catalytic [2+2+1] pyrrole formation from alkynes and azobenzenes by VCl3(THF)3 upon activation with 1a as an imido source. Control experiments and kinetic studies provided a plausible reaction mechanism, in which bis(imido)vanadium(V) species is assumed to be a catalytic active species. Bimetallic activation of azobenzene by a mono(imido)vanadium(III) species is a key step in the catalytic cycle, as evidenced by the observation of the second-order rate dependence on the concentration of the catalyst. Further development of this vanadium-catalyzed pyrrole formation reaction is ongoing in our laboratories.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mr. Hibiki Ochi for the initial experiment results. K.K. thanks the financial support by the JSPS Research Fellowships for Young Scientist. H.T. acknowledges the financial support by JSPS KAKENHI Grant No. 15KK0185, a Fund for the Promotion of Joint International Research (Fostering Joint International Research), and Multidisciplinary Research Laboratory System of Graduate School of Engineering Science, Osaka University. K.M. acknowledges financial support by JSPS KAKENHI Grant Nos. 15H05808 and 15K21707 in Precisely Designed Catalysts with Customized Scaffolding (No. 2702). Financial support was provided by the National Institutes of Health (1R35GM119457) and the Alfred P. Sloan Foundation (I.A.T. is a 2017 Sloan Fellow). Equipment for the UMN Chemistry Department NMR facility was supported through a grant from the National Institutes of Health (S10OD011952) with matching funds from the University of Minnesota.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b13390.
NMR characterization of complex 7 and the coupling products; full experimental details regarding the kinetic study (PDF)
X-ray crystallographic data for complex 7 (CIF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Bhardwaj V; Gumber D; Abbot V; Dhiman S; Sharma P Pyrrole: A Resourceful Small Molecule in Key Medicinal Heteroaromatics. RSC Adv. 2015, 5, 15233. [Google Scholar]; (b) Baumann M; Baxendale IR; Ley SV; Nikbin N An Overview of the Key Routes to the Best Selling 5-Membered Ring Heterocyclic Pharmaceuticals. Beilstein J. Org. Chem 2011, 7, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Walsh CT; Garneau-Tsodikova S; Howard-Jones AR Biological Formation of Pyrroles: Nature’s Logic and Enzymatic Machinery. Nat. Prod. Rep 2006, 23, 517. [DOI] [PubMed] [Google Scholar]
- (3).Novák P; Müller K; Santhanam KSV; Haas O Electrochemically Active Polymers for Rechargeable Batteries. Chem. Rev 1997, 97, 207. [DOI] [PubMed] [Google Scholar]
- (4).Loudelt A; Burgess K BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev 2007, 107, 4891. [DOI] [PubMed] [Google Scholar]
- (5) (a).Knorr L Synthese von Furfuranderivaten aus dem Diacetbernsteinsäureester. Ber. Dtsch. Chem. Ges 1884, 17, 2863. [Google Scholar]; (b) Paal C Ueber die Derivate des Acetophenonacetessigesters und des Acetonylacetessigesters. Ber. Dtsch. Chem. Ges 1884, 17, 2756. [Google Scholar]
- (6).Hantzsch A Neue Bildungsweise von Pyrrolderivaten. Ber. Dtsch. Chem. Ges 1890, 23, 1474. [Google Scholar]
- (7) Representative reviews for multicomponent pyrrole formation: (a).Estévez V, Villacampa M, Menéndez JC Recent Advances in the Synthesis of Pyrroles by Multicomponent Reactions. Chem. Soc. Rev 2014, 43, 4633. [DOI] [PubMed] [Google Scholar]; (b) Gulevich AV; Dudnik AS; Chernyak N; Gevorgyan V Transition Metal-Mediated Synthesis of Monocyclic Aromatic Heterocycles. Chem. Rev 2013, 113, 3084. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Estévez V; Villacampa M; Menéndez JC Multicomponent Reactions for the Synthesis of Pyrroles. Chem. Soc. Rev 2010, 39, 4402. [DOI] [PubMed] [Google Scholar]
- (8) Selected examples of [2+2+1] pyrrole formation: (a).Matsui K, Shibuya M, Yamamoto Y Synthesis of Pyrroles via Ruthenium-catalyzed Nitrogen-transfer [2+2+1] Cycloaddition of α,ω-Diynes Using Sulfoximines as Nitrene Surrogates. Commun. Chem 2018, 1, 21. [Google Scholar]; (b) Chong Q; Xin X; Wang C; Wu F; Wan B Synthesis of Polysubstituted Pyrroles via Ag(I)-mediated Conjugate Addition and Cyclization Reaction of Terminal Alkynes with Amines. Tetrahedron 2014, 70, 490. [Google Scholar]; (c) Zhang L; Wang X; Li S; Wu J Synthesis of Pyrrole-2,3,4,5-tetracarboxylates via a Copper-catalyzed Reaction of Amine with But-2-ynedioate. Tetrahedron 2013, 69, 3805. [Google Scholar]; (d) Chen X; Li X; Wang N; Jin J; Lu P; Wang Y Palladium-Catalyzed Reaction of Arylamine and Diarylacetylene: Solvent-Controlled Construction of 2,3-Diarylindoles and Pentaarylpyrroles. Eur. J. Org. Chem 2012, 2012, 4380. [Google Scholar]; (e) Chen X; Jin J; Wang Y; Lu P Palladium-Catalyzed Synthesis of 7,9-Diaryl-8 H-acenaphtho[1,2-c]-pyrroles and Their Application in Explosives Detection. Chem. - Eur. J 2011, 17, 9920. [DOI] [PubMed] [Google Scholar]; (f) Wu T-C; Tai C-C; Tiao H-C; Kuo M-Y; Wu Y-T Nickel-Catalyzed, Cascade Cycloadditions of 1-Ethynyl-8-halonaphthalenes with Nitriles: Synthesis, Structure, and Physical Properties of New Pyrroloarenes. Chem. - Eur. J 2011, 17, 1930. [DOI] [PubMed] [Google Scholar]; (g) Liu W; Jiang H; Huang L One-Pot Silver-Catalyzed and PIDA-Mediated Sequential Reactions: Synthesis of Polysubstituted Pyrroles Directly from Alkynoates and Amines. Org. Lett 2010, 12, 312. [DOI] [PubMed] [Google Scholar]; (h) Yamamoto Y; Kinpara K; Saigoku T; Takagishi H; Okuda S; Nishiyama H; Itoh K Cp*RuCl-Catalyzed [2+2+2] Cycloadditions of α,ω-Diynes with Electron-Deficient Carbon–Heteroatom Multiple Bonds Leading to Heterocycles. J. Am. Chem. Soc 2005, 127, 605. [DOI] [PubMed] [Google Scholar]
- (9) (a).Chiu H-C; See XY; Tonks IA Dative Directing Group Effects in Ti-Catalyzed [2+2+1] Pyrrole Synthesis: Chemo- and Regioselective Alkyne Heterocoupling. ACS Catal 2019, 9, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davis-Gilbert ZW; Kawakita K; Blechschmidt DR; Tsurugi H; Mashima K; Tonks IA In Situ Catalyst Generation and Benchtop-Compatible Entry Points for TiII/TiIV Redox Catalytic Reactions. Organometallics 2018, 37, 4439. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chiu H-C; Tonks IA Trimethylsilyl-Protected Alkynes as Selective Cross-Coupling Partners in Titanium-Catalyzed [2+2+1] Pyrrole Synthesis. Angew. Chem., Int. Ed 2018, 57, 6090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Davis-Gilbert ZW; Wen X; Goodpaster JD; Tonks IA Mechanism of Ti-Catalyzed Oxidative Nitrene Transfer in [2+2+1] Pyrrole Synthesis from Alkynes and Azobenzene. J. Am. Chem. Soc 2018, 140, 7267. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Guo J; Deng X; Song C; Lu Y; Qu S; Dang Y; Wang Z-X Differences between the Elimination of Early and Late Transition Metals: DFT Mechanistic Insights into the Titanium-catalyzed Synthesis of Pyrroles from Alkynes and Diazenes. Chem. Sci 2017, 8, 2413. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Davis-Gilbert ZW; Hue RJ; Tonks IA Catalytic Formal [2+2+1] Synthesis of Pyrroles from Alkynes and Diazenes via TiII/TiIV Redox Catalysis. Nat. Chem 2016, 8, 63. [DOI] [PubMed] [Google Scholar]
- (10).Beaumier EP; Pearce AJ; See XY; Tonks IA Modern Applications of Low-valent Early Transition Metals in Synthesis and Catalysis. Nat. Rev. Chem 2019, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11) Selected examples of using N,N-bis(trimethylsilyl)aniline derivatives as an imido source: (a).Benito JM, de Jesús E, de la Mata FJ, Flores JC, Gómez R, Gómez-Sal P Carbosilane Dendrimers Containing Peripheral Cyclopentadienyl Niobium- and Tantalum-imido Complexes. J. Organomet. Chem 2006, 691, 3602. [Google Scholar]; (b) Antiñolo A; Dorado I; Fajardo M; Garcés A; Kubicki MM; López-Mardomingo C; Otero A; Prashar S Synthesis and Reactivity of New Mono- and Dinuclear Niobium and Tantalum Imido Complexes: X-ray Crystal Structure of [Ta(η5-C5H4SiMe3)-Cl2{=NC6Me4-4-(N(SiMe3)2)}]. J. Organomet. Chem 2006, 691, 1361. [Google Scholar]; (c) Pennington DA; Horton PN; Hursthouse MB; Bochmann M; Lancaster SJ Synthesis and Catalytic Activity of Dinuclear Imido Titanium Complexes: the Molecular Structure of [Ti(NPh)Cl(μ-Cl)(THF)2]2. Polyhedron 2005, 24, 151. [Google Scholar]; (d) Antiñolo A; Dorado I; Fajardo M; Garcés A; López-Solera I; López-Mardomingo C; Kubicki MM; Otero A; Prashar S Synthesis, Structural Characterisation and Reactivity of New Dinuclear Monocyclopentadienyl Imidoniobium and -tantalum Complexes – X-ray Crystal Structures of [{Nb(η5-C5H4SiMe3)Cl2}2(μ-1,4-NC6H4N)], [{Ta(η5-C5Me5)Cl2}2(μ-1,4-NC6H4N)] and [{Ta(η5-C5Me5)(CH2SiMe3)2}2(μ-1,4-NC6H4N)]. Eur. J. Inorg. Chem 2004, 2004, 1299. [Google Scholar]; (e) Antiñolo A; Dorado I; Fajardo M; Garcés A; Kubicki MM; López-Mardomingo C; Otero A Synthesis and Structural Characterisation of New Organo-diimido Tantalum and Niobium Complexes. Dalton Trans 2003, 910. [Google Scholar]; (f) Benito JM; de Jesús E; de la Mata FJ; Flores JC; Gómez R; Gómez-Sal P Arylimido Niobium(V) Complexes: Mononuclear and Dendritic Derivatives. J. Organomet. Chem 2002, 664, 258. [Google Scholar]; (g) Dorado I; Garcés A; López-Mardomingo C; Fajardo M; Rodríguez A; Antiñolo A; Otero A Synthesis and Structural Characterization of New Organo-diimido and Organo-imido Niobium and Titanium Complexes. J. Chem. Soc., Dalton Trans 2000, 2375. [Google Scholar]
- (12).Titanium chloride/1a catalyzed the pyrrole formation in over 70% yield under higher concentration of the substrates ([2a] = 2.4 M, [3a] = 0.4 M).
- (13).Effects of additional reaction parameters such as solvent, temperature, other metal chlorides, and other additives were included in the Supporting Information.
- (14).The result of the three-component coupling reaction is shown in the Supporting Information.
- (15).See the Supporting Information.
- (16) (a).Burés JA Simple Graphical Method to Determine the Order in Catalyst. Angew. Chem., Int. Ed 2016, 55, 2028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burés J Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles. Angew. Chem., Int. Ed 2016, 55, 16084. [DOI] [PubMed] [Google Scholar]
- (17) (a).Tsurugi H; Mashima K A New Protocol to Generate Catalytically Active Species of Group 4–6 Metals by Organosilicon-Based Salt-Free Reductants. Chem. - Eur. J 2019, 25, 913. [DOI] [PubMed] [Google Scholar]; (b) Saito T; Nishiyama H; Kawakita K; Nechayev M; Kriegel B; Tsurugi H; Arnold J; Mashima K Reduction of (tBuN=)NbCl3(py)2 in a Salt-Free Manner for Generating Nb(IV) Dinuclear Complexes and Their Reactivity toward Benzo[c]cinnoline. Inorg. Chem 2015, 54, 6004. [DOI] [PubMed] [Google Scholar]; (c) Saito T; Nishiyama H; Tanahashi H; Kawakita K; Tsurugi H; Mashima K 1,4-Bis(trimethylsilyl)-1,4-diaza-2,5-cyclo-hexadienes as Strong Salt-Free Reductants for Generating Low-Valent Early Transition Metals with Electron-Donating Ligands. J. Am. Chem. Soc 2014, 136, 5161. [DOI] [PubMed] [Google Scholar]
- (18) (a).La Pierre HS; Arnold J; Toste FD Z-Selective Semihydrogenation of Alkynes Catalyzed by a Cationic Vanadium Bisimido Complex. Angew. Chem., Int. Ed 2011, 50, 3900. [DOI] [PubMed] [Google Scholar]; (b) La Pierre HS; Minasian SG; Abubekerov M; Kozimor SA; Shuh DK; Tyliszczak T; Arnold J; Bergman RG; Toste FD Vanadium Bisimide Bonding Investigated by X-ray Crystallography, 51V and 13C Nuclear Magnetic Resonance Spectroscopy, and V L3,2-Edge X-ray Absorption Near-Edge Structure Spectroscopy. Inorg. Chem 2013, 52, 11650. [DOI] [PubMed] [Google Scholar]
- (19) (a).Milsmann C; Turner ZR; Semproni SP; Chirik PJ Azo N=N Bond Cleavage with a Redox-Active Vanadium Compound Involving Metal–Ligand Cooperativity. Angew. Chem., Int. Ed 2012, 51, 5386. [DOI] [PubMed] [Google Scholar]; (b) Komuro T; Matsuo T; Kawaguchi H; Tatsumi K Synthesis of a Vanadium(III) Tris(arylthiolato) Complex and Its Reactions with Azide and Azo Compounds: Formation of a Sulfenamide Complex via Cleavage of an Azo N=N Bond. Inorg. Chem 2005, 44, 175. [DOI] [PubMed] [Google Scholar]
- (20).DeWith J; Horton AD; Orpen AG Unusual [2+2] Cycloaddition Adducts of an Imidovanadium Complex with Alkynes and Ethene: Conversion to η3-1-Azaallyl and Ethenyl Complexes. Organometallics 1993, 12, 1493. [Google Scholar]
- (21) (a).Vujkovic N; Ward BD; Maisse-François A; Wadepohl H; Mountford P; Gade LH Imido-Alkyne Coupling in Titanium Complexes: New Insights into the Alkyne Hydroamination Reaction. Organometallics 2007, 26, 5522. [Google Scholar]; (b) Lokare KS; Ciszewski JT; Odom AL Group-6 Imido Activation by a Ring-Strained Alkyne. Organometallics 2004, 23, 5386. [Google Scholar]; (c) Walsh PJ The Synthesis and Reactivity of Zirconium-Nitrogen Double and Single Bonds. Ph.D. Thesis, University of California, Berkeley, 1990. [Google Scholar]
- (22) (a).Glueck DS; Wu J; Hollander FJ; Bergman RG Monomeric (Pentamethylcyclopentadienyl)iridium Imido Compounds: Synthesis, Structure, and Reactivity. J. Am. Chem. Soc 1991, 113, 2041. [Google Scholar]; (b) Kuhn N; Horn E-M; Zauder E; Bläser D; Boese R Stable Sandwich Complexes with Pentamethylpyrrole Ligands. Angew. Chem., Int. Ed. Engl 1988, 27, 579. [Google Scholar]; (c) Huttner G; Mills OS Übergangsmetallkomplexe cyclischer π-Liganden, III. Kristall- und Molekülstruktur von Tricarbonyl(N-methyl-pyrrol)-chrom(0). Chem. Ber 1972, 105, 301. [Google Scholar]; (d) Öfele K; Dotzauer E Cr(CO)3-π-komplexe von Pyrrol und Pyrrolderivaten. J. Organomet. Chem 1971, 30, 211. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.