

ABSTRACT
Small extracellular vesicles (EVs) are membrane enclosed structures that are usually released from cells upon exocytosis of multivesicular bodies (MVBs) as a collection of separate, free EVs. In this study, we analysed paraffin embedded sections of archived human colorectal cancer samples. We studied 3D reconstructions of confocal microscopic images complemented by HyVolution and STED imaging. Unexpectedly, we found evidence that large, MVB-like aggregates of ALIX/CD63 positive EV clusters were released en bloc by migrating tumour cells. These structures were often captured with partial or complete extra-cytoplasmic localization at the interface of the plasma membrane of the tumour cell and the stroma. Their diameter ranged between 0.62 and 1.94 μm (mean±S.D.: 1.17 ± 0.34 μm). High-resolution 3D reconstruction showed that these extracellular MVB-like EV clusters were composed of distinguishable internal particles of small EV size (mean±S.D.: 128.96 ± 16.73 nm). In vitro, HT29 colorectal cancer cells also showed the release of similar structures as confirmed by immunohistochemistry and immune electron microscopy. Our results provide evidence for an en bloc transmission of MVB-like EV clusters through the plasma membrane. Immunofluorescent-based detection of the MVB like small EV clusters in archived pathological samples may represent a novel and unique opportunity which enables analysis of EV release in situ in human tissues.
KEYWORDS: EV, migrating cancer cells, MVB, colorectal carcinoma
Introduction
Beside soluble mediators, cancer-stroma interactions also involve extracellular vesicles (EVs) that mediate complex biological information transfer via proteins, nucleic acids, lipids and metabolites. Small (< 200 nm diameter) EVs (sEVs) are typically released by the exocytosis of multivesicular bodies (MVBs) [1,2]. During the MVB exocytosis, separate, individual sEVs are considered to be released into the extracellular space, which cannot be resolved using conventional confocal microscopy limited by the light diffraction at ~250 nm [3]. Besides MVB exocytosis, cells can also release EVs (typically medium and large EVs) by budding of the plasma membrane [4,5]. Recent reports described the release of large, MVB like structures such as migrasomes [6], Nef protein-induced plasma membrane protrusions giving rise to released EV clusters [7], multivesicular spheres [8] and released multivesicular cargo [9]. These recent studies suggest that the examined cells can release sEV clusters (sEVCs).
Here we describe for the first time the release of ALG 2-interacting protein X (ALIX)/CD63 [1] positive MVB-like sEVC in formalin-fixed, paraffin-embedded (FFPE) tissue samples of colorectal carcinoma (CRC) patients as well as in in vitro cultures of HT29 CRC cells.
Materials and methods
Tissue samples
Surgically removed FFPE samples (fixed with 10% neutral buffered formalin) from metastatic CRC samples were embedded into tissue microarray (TMA) blocks (core diameter: 2 mm). We examined serial sections of two different tissue blocks from 31 CRC patients (Σ = 62 TMAs). The patients included 15 Dukes C patients (3 women (w)/12 men (m); mean age±S.D.: 58.20 ± 7.25 years; age range: 48–70 years) and 16 Dukes D ones (7w/9m; mean age±S.D.: 64.56 ± 8.37 years; age range: 51–80 years) (according to Astler-Coller-modified Dukes’ classification). Diagnoses were established on the basis of WHO criteria using H&E-stained serial sections by an expert pathologist [10]. This study was approved by the Semmelweis University Regional and Institutional Committee of Science and Research Ethics: ETT TUKEB 23,970/2011 and 8–23/2009–1018 EKU (ad.60/PI/09).
Cell culture
HT29 human colorectal cancer cells (ATTC_HTB-38) were grown in triplicates for 24 h on 8-well Nunc Lab-Tek (177,402, Nagle Nunc, Rochester, USA) for confocal microscopy and 8-well Ibitreat microscopy chamber (Ibidi GmbH, Martinsried, German) for STED microscopy (initial cell number: 5000 cell/300 µl/well). For electron microscopy (EM) and DAB immune EM, we cultured the cells on Lab-Tek chamber (initial cell number: 8000 cell/300 µl/well) for 72 h in RPMI 1640 medium (Biosera, Ringmer, UK) supplemented with 2 mM L-glutamine (Merck-Sigma-Aldrich, Darmstadt, Germany), 80 mg/2 ml gentamycin (Sandoz) and 10% sEV-depleted FBS. To preserve the MVB-like sEVCs, we decanted the culture medium carefully from the cultures, instead of aspirating it with a pipette. The sEV depletion was performed by ultracentrifugation of foetal bovine serum (Merck-Sigma-Aldrich, Darmstadt, Germany) in an Optima MAX-XP Benchtop Ultracentrifuge with an MLA-55 fixed-angle rotor (Beckman Coulter, Brea, USA) at 120,000 g for 16 h.
Isolation of MVB-like sEVCs from HT29 culture supernatant
Twenty-four hours conditioned medium samples of HT29 cells were decanted from the cell cultures and paraformaldehyde (PFA) was added to 4% final concentration. After 10 min at room temperature, 0.25% glutaraldehyde was added, and the samples were centrifug4ed at 2,000 g for 10 min. The pellet washed once with filtered 0.9% NaCl solution, and re-pelleted at 2,000 g for 10 min. Finally, the supernatant was removed carefully and 4% PFA in PBS was layered on top of the pellet.
Immunohistochemistry
FFPE TMA blocks and cultures of HT29 cells were immunostained using fluorochrome conjugated antibodies. Cell cultures were washed in TBS and fixed either with methanol and acetone (1:1), or in 4% neutral buffered formaldehyde (made from PFA), for 10 min. The latter samples were permeabilized TBS containing 0.2% Triton-X-100 (TBST) for 10 min. TMA sections mounted on adhesive glass slides (SuperFrost Plus, Thermo-Fisher) were dewaxed and treated for endogenous peroxidase blocking using 0.5% hydrogen peroxide in methanol for 20 min. Antigen retrieval was performed by heating dewaxed paraffin sections in 0.01M Tris-0.1M EDTA buffer pH 9.0 (TE) at ~100°C for 40 min in a JT 366 microwave oven (Whirlpool, Benton Harbor, MI). Nonspecific binding sites in cells and tissue sections were saturated using 5% BSA-TBST solution for 30 min. Antibodies specific for sEV markers ALIX (HPA011905, Sigma Aldrich, St Louis, USA; 1:400) and CD63 (NKI/C3, Leica-Novocastra, Wetzlar, Germany; 1:50), and LC3B (NB100-2220, Novus Biologicals, Centennial, Colorado, 1:200) proteins were used. Detection of RAB7 (R8779, Sigma Aldrich; St. Louis, MO, 1:200) protein. Pan-cytokeratin (AE1/AE3 from Dako, Glostrup, Denmark; 1:100) and cytokeratin 18 (Alexa Fluor 488-labelled, 18–0059 from Life Technologies, Grand Island, NY) antibodies were used to confirm the epithelial origin of cancer cells. Cell membrane E-cadherin was also immunostained using clones EP700Y and 36B5 (Thermo-LabVision, Fremont, CA; 1:100). Primary antibodies were detected using anti-rabbit or anti-mouse immunoglobulins conjugated with either Alexa Fluor 488, 546 or 514 (Life Technologies) in relevant combinations. The specificity of our EV markers was confirmed by detecting the co-localization of CD81 (SAB3500454, Sigma Aldrich; 1:200) and LAMP1 (FNab04684, Wuhan Fine Biotech, Wuhan, China; 1:100) proteins using polyclonal rabbit antibodies (Supplementary Figure 1). In these cases, the immunoperoxidase-based TSA (tyramide signal amplification) Plus system (Perkin-Elmer, Shelton, CT) was used, which allow double labelling combining antibodies from the same species, including FITC (green) and rhodamine (red) conjugated tyramide reagents for antigen demonstration.
The 2,000 g EV fractions were fixed with 4% PFA (20 min) and blocked with 10% FBS in PBS (60ʹ). Primary antibodies for RAB7 and LC3B were applied in 1:200 dilution using 10% FBS in PBS (4°C, overnight). After washing steps, donkey anti-mouse IgG conjugated with CF488A (Sigma Aldrich) and goat anti-rabbit IgG conjugated with Atto 647N (Sigma Aldrich) were used (1:400) in PBS containing 1% FBS (60ʹ). All incubations were done at room temperature (RT), where samples were washed using TBS (ph 7.4). Following washing, samples were mounted with ProLong Diamond Antifade Mountant (Life Technologies).
Microscopy
Slides were digitalized with Pannoramic Confocal instrument and were analysed (including morphometric measurements and counting intra and extracellular signals) with Pannoramic Viewer (v: 1.15.3) digital microscope (3DHISTECH, Budapest, Hungary). Three-dimensional (3D) reconstructions were performed with the Voloom software (v: 2.7.1; MicroDimensions, Munich, Germany) after combining 9 Z-axial confocal layers of 0.4 μm focus steps, which incorporated ~Σ = 3.6 μm section thickness. For better visibility of the fluorescent signals, contrast/brightness were edited by Microsoft PowerPoint for the entire images of Figure 3(h–j), and the Supplementary Figure 1. The HyVolution [11] and the stimulated emission depletion (STED) imaging was performed on a Leica TCS SP8 STED microscope using Leica HC PL APO ×100 (NA = 1.4) objective. For STED imaging, 660 nm depletion laser was applied. The images were restored by using Huygens Pro deconvolution, and were analysed with Leica LAS X 3.1.1 software. Quantitative image analysis was performed with the Marker Counter tool of the digital microscope. The 2,000 g EV preparations were examined by a Nikon Eclipse 80i epifluorescence microscope. We used t test to compare of examined parameters.
Figure 3.
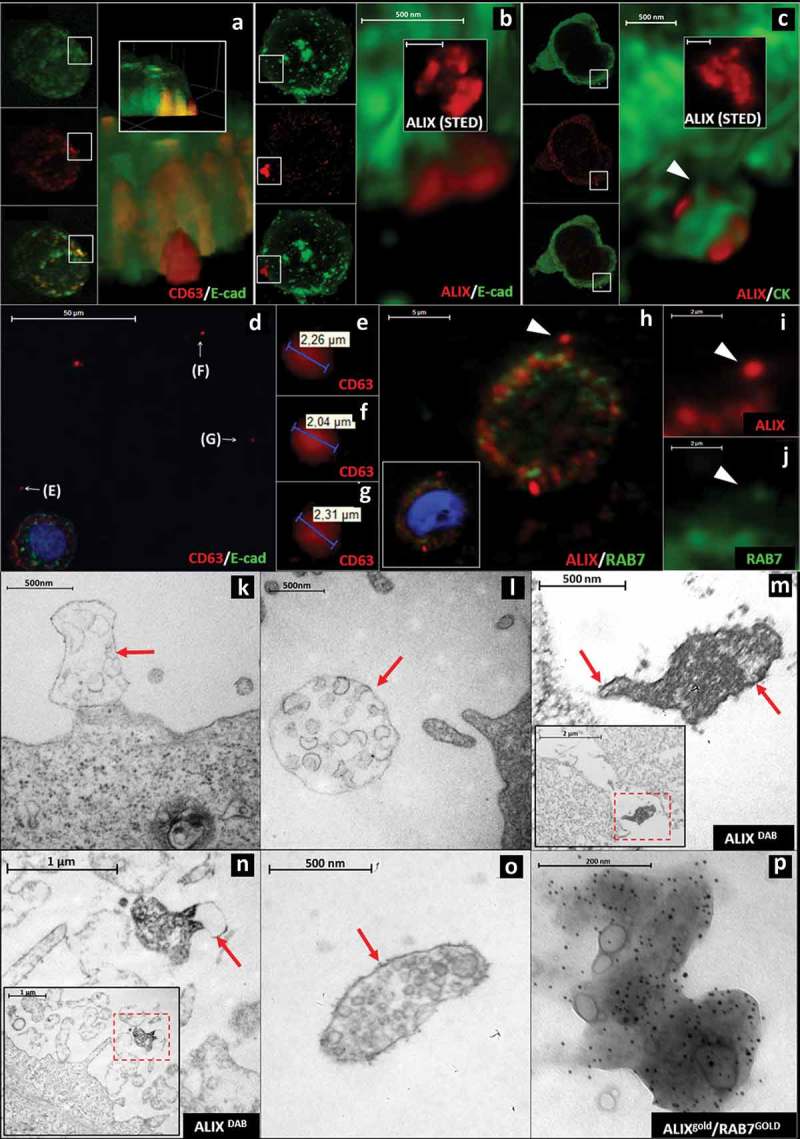
Visualization of extracellular sEVCs in HT29 colorectal carcinoma cell line (a) Confocal (CD63: red, E-cadherin: green), (b) HyVolution (ALIX: red, E-cadherin: green) and (c) HyVolution (ALIX: red, CK: green) images of HT29 cell-derived sEVCs. Arrowhead points to a cytoplasmic process in C. Scale bars: 500 nm. The inserts of (b) and (c) show STED microscopic images of the sEVCs (scale bars: 500 nm). (d–g) sEVCs in cell-free regions of HT29 culture. Scale bar: 50 µm (d). (h-j) ALIX/RAB7 co-staining of extracellular MVB-like sEVCs (arrowheads; scale bars: 5 µm (h), 2 µm (i and j)). (k, l) EM images show MVB-like sEVCs as large, membrane enclosed (arrows) structures close to the cell with internal vesicles in the size range of sEVs (~100–160 nm). Scale bars: 500 nm. (m, n) Membrane enclosed (arrows) MVB-like sEVCs contain ALIX positive (DAB staining) spheres close to HT29 cells (inserts). Scale bar: 500 nm and 1 µm, respectively, inserts: 2 µm and 1 µm respectively. (o) A membrane enclosed (arrow) MVB-like sEVC and (p) an ALIX (5 nm gold)/RAB7 (9 nm GOLD) positive MVB-like sEVC isolated from the conditioned media of HT29 cells. Scale bars: 500 nm and 200 nm, respectively.
Electron microscopy and immune electron microscopy
a. In the case of ultrastructural studies without immunostaining, after rinsing with PBS, the cells were post-fixed in 1% OsO4 for 15 min, rinsed with distilled water, dehydrated in graded ethanol including block-staining with 1% uranyl acetate in 50% ethanol for 15 min and embedded in Taab 812 (Aldermaston, T031).
b. Immunostaining for ALIX (HPA011905, rabbit, 1:400) was performed in situ on Lab-Tek chamber slides. The fixed HT29 cells were washed in PBS (pH 7.4), then 2.5% normal horse serum (Vector Laboratories, Burlingame, CA) for 30 min was applied to block the unspecific binding sites. Incubation in anti-ALIX was performed overnight at 4°C. As control, cells were incubated with 2.5% normal horse serum only. Universal ImmPRESS HRP Reagent kit containing anti-Rabbit/Mouse IgG (MP-7500, Vector Laboratories, Burlingame, CA) and as chromogen ImmPact DAB (SK 4105, Vector Laboratories, Burlingame, CA) were applied according to the manufacturer’s instruction. After overnight polymerization at 60°C (Taab 812), 60 nm ultrathin sections were cut by a Leica UCT ultramicrotome (Leica Microsystems, UK) in parallel with the surface of the cells. The ultrathin sections were analysed with a Hitachi7100 (Hitachi, Japan) electron microscope equipped by Veleta, a 2 k × 2 k MegaPixel side-mounted TEM CCD camera (Olympus). Contrast and brightness of electron micrographs were edited by Adobe Photoshop CS3 (Adobe Photoshop Incorporation, CA).
c. For immune EM of released EVs, intact EVs were enriched by 2,000 g centrifugation. During sample preparation, we have followed the basic protocol described previously by Visnovitz et al. [12] with minor modifications. Two to three µL of EV sample in 0.22 µm filtered PBS was applied onto the surface of a 300 mesh formvar-coated Ni grid and was incubated for 10 min at room temperature. After removal of the residual solution, EVs were fixed with 0.22 µm filtered 4% paraformaldehyde in PBS (10 min at RT). Following three washes (5 min, RT) with PBS, 5% of BSA (Sigma Aldrich) in 0.22 µm filtered PBS was used for blocking (1 h, RT). Grids were incubated overnight at 4°C with anti-RAB7 (R8779, mouse) and anti-ALIX (HPA011905, rabbit) (both in 1:50 in 0.22 µm filtered 5% BSA in PBS). This was followed by washes (three times 5 min with 0.22 µm filtered 5% BSA).) Secondary antibodies (polyclonal goat anti-rabbit IgG 10 nm gold pre-adsorbed (Abcam, Cambridge, UK) and polyclonal goat anti-mouse IgG 5 nm gold pre-adsorbed (Sigma Aldrich) were next applied (1:400) for 2 h at RT followed by another washes (three times 5 min with 0.22 µm filtered 5% BSA). Samples were then post-fixed with 2% glutaraldehyde followed by intensive washes (three times for 5 min with 0.22 µm filtered 5% BSA, three times for 5 min with 0.22 µm filtered PBS and three times for 5 min with purified water). Background contrasting was skipped. Before examination by JEOL 1011 transmission electron microscope grids, were washed three times with ultrapure water and dried.
d. Immunoelectron microscopy of HT29 cells. Fixation and embedding of HT29 cells were performed as described previously by Lőrincz et al. [13]. Cells were fixed with 2%formaldehyde, 0.05% glutaraldehyde and 0.2% picric acid in PBS (20 min, RT). Following an intensive wash with 50 mM ammonium chloride and 50 mM glycine, the cells were post-fixed with 1% uranyl acetate dissolved in 0.05M maleate buffer (3 h, RT). Cells were dehydrated and embedded in LR white resin (Sigma Aldrich) according to the manufacturer’s instructions. Ultrathin sections were treated with 1% H2O2 (1 min, RT), 0.1% NaBH4, dissolved in TBS (10 min, RT) and 50 mM glycine dissolved in TBS (30 min, RT). Sections were blocked with 10% FBS (1 h RT) and primary anti-RAB7 (R8779, mouse) and anti-E-cadherin (EP700Y, rabbit) were applied in 1:50 dilutions in 10% FSB in TBS at 4°C, overnight. Following three intensive washes (5 min, RT) with 10% FBS in TBS secondary antibodies, polyclonal goat anti-rabbit IgG 10 nm gold pre-adsorbed (Abcam, Cambridge, UK) and polyclonal goat anti-mouse IgG 5 nm gold pre-adsorbed (Sigma Aldrich) were applied in 2% FBS in TBS (2 h, RT). After washes (2% FBS in TBS three times 5 min and TBS three times 5 min), the samples were post-fixed with 2% glutaraldehyde in ultrapure water and stained with 4% uranyl acetate in 50% methanol (for 15 min) and lead citrate (for 1 min). Grids were analysed by JEOL 1011 electron microscope.
Results and discussion
In situ tissue detection of extracellular sevcs in CRC FFPE sections
Earlier we identified granular ALIX positive cytoplasmic structures in CRC histological samples as MVBs [14]. Here we primarily focused on individual, migrating CRC cells, because they were easily distinguishable by the detection of their cytokeratin (CK) intermediate filaments in the CK negative microenvironment [15]. Furthermore, these cells play a primary role in metastatic progression [16,17]. Surprisingly, CK positive, migrating tumour cells (Figure 1a and 1d) show CD63 and ALIX positive structures protruding from the cytoplasm into the extracellular space (Figure 1(a–f)). Figure 1(c) shows the 3D reconstruction of the surface of a migrating tumour cell also shown in Figure 1a and 1b. In this 3D reconstruction, the CD63 positive structures are partially covered by a thin CK positive layer (Figure 1(c)). Another 3D reconstruction (Figure 1(f)) reveals the protrusion of ALIX positive structures from the migrating tumour cell which is also shown in Figure 1d and 1e. This phenomenon was also detectable in the marginal cells of the tumour mass (Supplementary Figure 2A–D). Importantly, some of the CD63 and ALIX positive structures were also detected extracellularly, adjacent to the migrating CRC cells (Figure 1(f)). The 3D examinations confirmed that 85.32% (n = 93/109) of migrating CRC cells showed the presence of ALIX positive structures partially or completely outside (but adjacent to) of the CK positive cytoplasm. Further substantiation of the transit of these structures through the plasma membrane came from double labelling for ALIX or CD63 and epithelial (E)-cadherin. This revealed that the ALIX or CD63 positive structures were captured on both sides of the E-cadherin positive plasma membrane and among E-cadherin molecules (Supplementary Figure 3A–D). In some cases, a membrane curvature was detectable either above or below the ALIX or CD63 positive structures at the plasma membrane-stroma interface (Supplementary Figure 3E–I).
Figure 1.
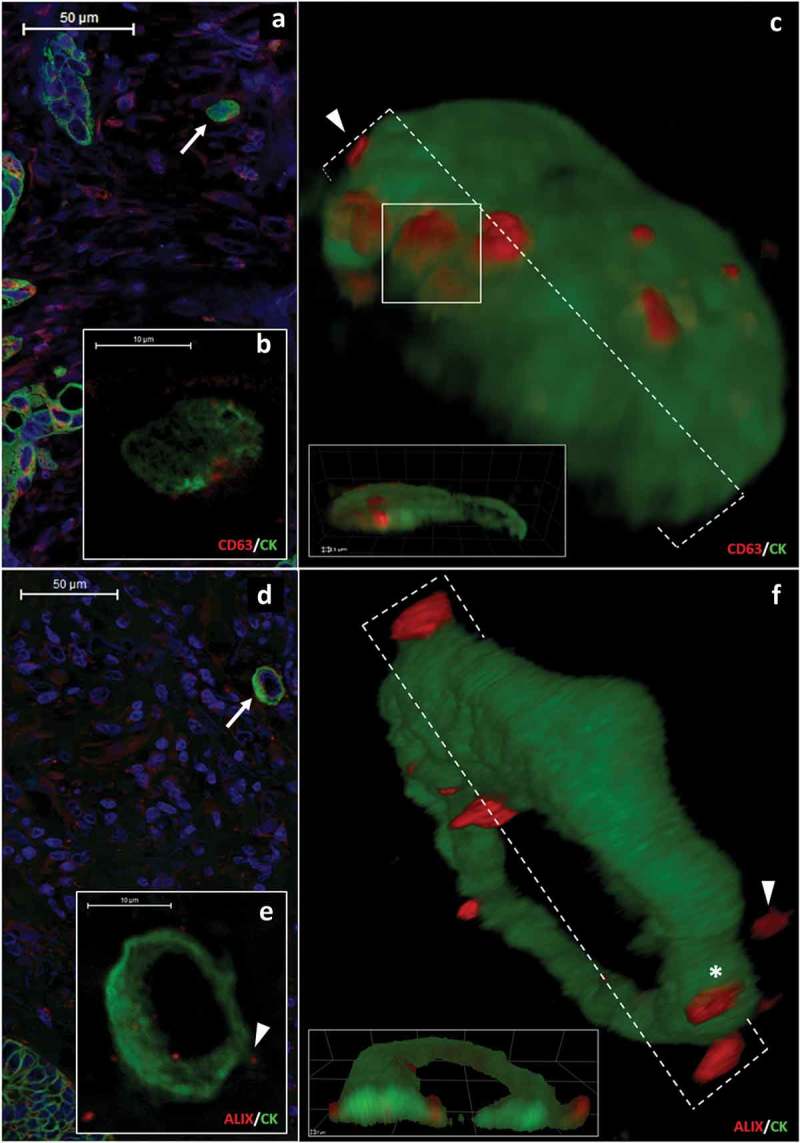
CD63 and ALIX positive protrusions in the cytoplasmic border of migrating colorectal cancer cells (a-b) show the same cytokeratin (CK, green) positive migrating CRC cell. (b) shows granular CD63 (red) protein expression in the cortical cytoplasm. Scale bars: 50 μm (a) and 10 μm (b). (c) 3D reconstruction of the same cell as in (a) and (b). CD63 positive structures are present either partially outside of the cytoplasm (arrowhead) or covered with a thin CK positive layer (white box). (d and e) ALIX (red)/CK (green) co-immunostaining in another migrating CRC cell. Scale bars: 50 μm (d) and 10 μm (e). (f) shows a 3D reconstruction of the cell same as in Figure 1(d) and 1(e) with ALIX positive structures (red) which are partially (asterisk) or completely (arrowhead) outside of CK positive cytoplasm.
In selected cases, the 3D reconstruction-based image analysis defined the exact localization of ALIX and CD63 positive structures in relation to the cytoplasm (Supplementary Video 1).
In FFPE samples we did not find significant difference between the diameters of the ALIX- and CD63 positive structures at least partial extracellular localization. However, one has to be aware of the limitations and inaccuracy of fluorescent measurements [18]. The diameters ranged for ALIX from 0.62 to 1.94 μm, mean±S.D.: 1.17 ± 0.34 μm and for CD63 from 0.55 to 2.24 μm, mean±S.D.: 1.20 ± 0.50 μm, respectively (p = 0.27) (Figure 2a and 2b). Similarly, we could not find a difference between the number of extracellular (ALIX mean±S.D.: 2.57 ± 1.62 and CD63 mean±S.D.: 2.18 ± 1.79; p = 0.08; Figure 2d) and intracellular (ALIX mean±S.D.: 9.50 ± 5.55 and CD63 mean±S.D.: 9.20 ± 7.65, p = 0.74; Figure 2e) structures, respectively. Further analysis using HyVolution microscopy revealed that the ALIX positive structures were uneven, showing small lumps emerging from their surface (Figure 2f and Supplementary Video 2). STED-microscopy also confirmed that these structures were composed of smaller, distinguishable ALIX-positive spheroids of 98–150 nm diameter (mean±S.D.: 128.96 ± 16.73 nm; Figure 2a and 2g–j) falling into the size range of sEVs [1]. Based on these properties, we identified these structures as sEVCs.
Figure 2.
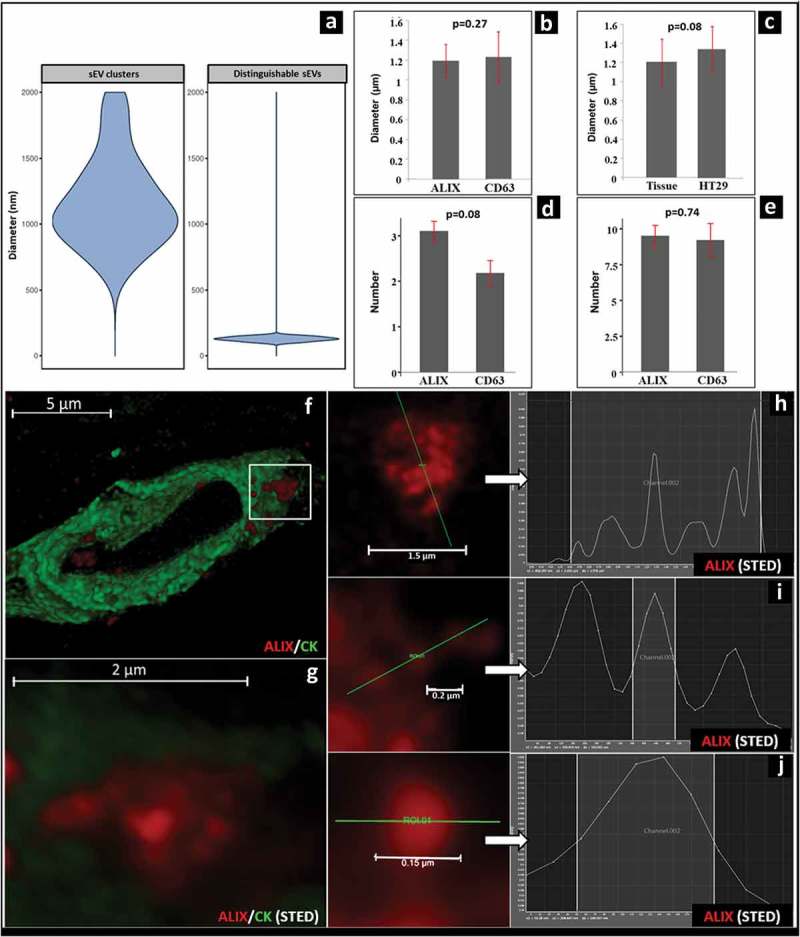
Morphology and size distribution of ALIX/CD63 positive sEVs and sEVCs in CRC tissue. (a) Violin plot shows the diameter distribution of extracellular sEVCs (left) and distinguishable ALIX-positive spheroids inside and close to the sEVCs (right) (measured with Pannoramic Viewer 1.15.3 and Leica LAS X 3.1.1 software). (b) Comparison of the diameters of the at least partially extracellularly localized ALIX/CD63 positive structures in migrating CRC cells. (c) Comparison of the diameters of similarly localized CD63-positive structures in CRC tissues and in HT29 culture. (d) Average number of the at least partially extracellular ALIX-positive or CD63-positive signals in migrating CRC cells (tissue). (e) Average number of intracellularly localized ALIX-positive or CD63-positive signals in migrating CRC cells (tissue). B-E shows mean±S.D. of n = 40 cells. HyVolution (f) and STED (g) microscopic images of an ALIX positive (red) sEVC (white box) which is partially embedded within the CK positive cytoplasm (green) of the migrating CRC cell. Scale bars: 5 μm and 2 μm respectively. (h) Fluorescence intensity profile of ALIX positive sEVC (red) crossed by a selected line (green). Scale bar: 1.5 μm. (i) Fluorescence intensity profile and measured Full-Width at Half-Maximum (FWHM) values of a small group ALIX positive sEVs and (j) a single ALIX positive sEV. Scale bars: 0.2 µm and 0.15 μm (200 nm and 150 nm) respectively. FWHM values: 141 and 149 nm, respectively.
Detection of the release of sEVCs in vitro
Figure 3a and 3b show HT29 cell-derived ALIX and CD63 positive sEVCs localized partly in the cytoplasm, and partly in the extracellular space. Figure 3c shows a rare example where a sEVCs hovers on a cytoplasmic process. Diameters of the at least partially extracellularly localized, CD63 positive HT29-derived sEVCs ranged from 0.73 to 2.44 μm (mean±S.D.: 1.34 ± 0.46 μm) and showed an agreement with our in situ FFPE measurements (p = 0.08) (Figure 2c). Similarly, the STED microscopic 3D reconstruction proved that the HT29-derived ALIX positive extracellular structures were sEVCs by demonstration that they were composed of spheres with ~100-~150 nm in diameter (Figure 3b and 3c inserts). Importantly, individual sEVCs were also observed in cell-free zones at a distance from cultured HT29 cells (Figure 3(d–g)). This suggested that these structures were released by cells into the extracellular space. Furthermore, some of these sEVCs also show positivity for RAB7 (Figure 3(h–j)), a small GTP-ase which is required for the maturation of late endosomes/MVBs [19,20] and is also involved in MVB-derived sEV release [21,22].
EM images also showed that large (from ~500 to ~1500 nm), limiting membrane enclosed sEVCs (sEV diameter: ~100–170 nm) were associated with the plasma membrane (Figure 3k) or they were completely separated from it and were found at different distances from the cells (Figure 3l). Immune EM of cultures of HT29 cells showed that sEVCs were covered by a limiting membrane and showed ALIX positivity (Figure 3m and 3n). In 2,000 g pellets of the conditioned medium of HT29 cells, limiting membrane-enclosed sEVCs were detectable (Figure 3(o)), which also were detected as ALIX/RAB7 double positive sac-like structures (Figure 3(p)). Based on the presence of a limiting membrane and RAB7 positivity, we identified these sEVCs as MVB-like sEVCs.
In our experimental settings, the directionality of MVB-like sEVC transit through the plasma membrane of CRC cells often could not be determined. Thus, theoretically the MVB-like sEVCs crossing the plasma membrane could be released either by the tumour or by stromal cells. Importantly, our in vitro studies with HT29 CRC cell line (in the absence of stromal cells) showed MVB-like sEVCs in the conditioned medium providing evidence for the release of these structures by CRC cells.
Potential mechanisms of MVB-like sEVCs release
Our data suggest different possible mechanisms by which MVB-like sEVCs can be released by CRC cells. A potential mechanism for MVB-like sEVC release is captured in Supplementary Figure 4A and 4B. Here we found evidence for the release of blister-like large, membrane enclosed structures which also contained an MVB (Supplementary Figure 4B). Supplementary Figure 4D and E suggest another mechanism for MVB-like sEVC secretion. A cellular process contains at least two MVBs in Supplementary Figure 4E. This cell process has a relatively narrow stalk which may break off to release MVB-like EVCs along with a portion the cytoplasm surrounded by plasma membrane. Supplementary Figure 4G–J show plasma membrane protrusions containing sEVs. Furthermore, Supplementary Figure 4K shows a RAB7/E-cadherin double positive cellular protrusion. Most of the surface of this structure is covered by a membrane, which gives a smooth contour to the small EV cluster. Interestingly, on one side, membrane enclosed small EVs are clearly visible suggesting the discontinuity/rupture of the limiting membrane. Schematic representations of the possible release mechanisms are shown in Supplementary Figures 4C, 4F and 4L.
Plasma membrane rupture as a proposed MVB secretion mechanism [23] was not detected in our EM samples. From among the recently described novel types of EVs [6,7,9], the MVB-like sEVCs described here show strongest resemblance to the multivesicular spheres [8]. Of note, overlap of the RAB7 with LC3 protein staining in sEV preparations isolated from conditioned media (Supplementary Figure 5) raises the possibility that sEVCs may be released from HT29 cells by secretory autophagy.
It is obvious that the examined sEVCs are heterogeneous in terms of their origin, size and type of their secretion, thus, further experiments are necessary in order to classify their subgroups.
In conclusion, the present study demonstrates in situ (in tissue sections of patients with CRC) for the first time that migrating carcinoma cells release large, MVB-like sEVCs. In CRC, MVB-like sEVCs are likely to contribute to the paracrine regulation of cancer, which effect may differ from that mediated by conventionally secreted individual sEVs. Uptake of MVB-like sEVCs may have a substantially stronger effect on recipient cells than individual sEVs. In fact, it has been shown recently that in a somewhat similar scenario, vesicle-cloaked viruses had a disproportionately larger contribution to infectivity than free individual virus particles [24]. In addition, due to the possible disruption of the limiting membrane of the released MVB-like sEVC, the release of sEVs might lead to a delayed effect to the microenvironment.
Key aspect of is study is that it may open new perspectives for fluorescent microscopic analysis of EV-based communication in archived, formalin-fixed and paraffin-embedded human tissue samples. The fixed-embedded human tissue samples provide solid evidence that the MVB-like sEVC release described in this article is a non-negligible process that takes place in human tumour tissues in vivo.
Funding Statement
This work was supported by National Research, Development and Innovation Office NKFIH, Hungary KMR-12-1-2012-0216, NVKP_16-1-2016-0004, NVKP_16-1-2016-0017, OTKA11958, OTKA120237, K115398, European Regional Development Fund and the Hungarian Government TRAIN-EV H2020-MSCA-ITN-722148 by the European Commission, VEKOP-2.3.2-16-2016-00002 and VEKOP-2.3.3-15-2016-00016, as well as STIA_17. The study that served as a basis of this publication was also supported by the Institutional Higher Education Excellence Program of the Ministry of Human Capacities within the framework of the “Molecular biology” and the “Therapeutic development” thematic programs. The study also supported by Bolyai Fellowship (590/2015), MTA Premium postdoctoral research grant PPD-222/2018, Momentum Program of the Hungarian Academy of Sciences (LP2012-025), Breast Cancer Research Foundation (BCRF-17-156) and Research and Technology Innovation Fund (KTIA_NAP_13-2014-0021).
Acknowledgements
We are grateful to Gabriella Kónyáné Farkas, Zita Bratu, Anna Tamási, Éva Balogh Mátrainé, Tibor Horváth, Mónika Paulusz, Titanilla Dankó, Ádám Oszvald and Györgyné Vidra for their support. The authors thank Gábor Juhász (Eötvös Loránd University, Budapest and Biological Research Centre, Szeged) for the access to transmission electron microscope at Eötvös Loránd University and his financial support for the operation of the TEM core facility.
Competing financial interest
The authors declare no competing financial interests.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental materials data can be accessed here.
References
- [1].Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tkach M, Théry C.. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–9. [DOI] [PubMed] [Google Scholar]
- [3].Huang B, Babcock H, Zhuang X. Breaking the diffraction barrier: super-resolution imaging of cells. Cell. 2010;143:1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].György B, Szabó TG, Pásztói M, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68:2667–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Buzas EI, György B, Nagy G, et al. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. 2014;10:356–364. [DOI] [PubMed] [Google Scholar]
- [6].Ma L, Li Y, Peng J, et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015;25:24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Muratori C, Cavallin LE, Krätzel K, et al. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe. 2009;6:218–230. [DOI] [PubMed] [Google Scholar]
- [8].Junquera C, Castiella T, Muñoz G, et al. Biogenesis of a new type of extracellular vesicles in gastrointestinal stromal tumors: ultrastructural profiles of spheresomes. Histochem Cell Biol. 2016;146:557–567. [DOI] [PubMed] [Google Scholar]
- [9].Fertig ET, Gherghiceanu M, Popescu LM. Extracellular vesicles release by cardiac telocytes: electron microscopy and electron tomography. J Cell Mol Med. 2014;18:1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bosman FT, Carneiro F, Hruban RH, et al. WHO classification of tumours of the digestive system. International Agency for Research on Cancer. Lyon: IARC Press; 2010. [Google Scholar]
- [11].Borlinghaus RT, Kappel C. HyVolution-the smart path to confocal super- resolution. Nat Methods. 2016;13 DOI: 10.1038/nmeth.f.392. [DOI] [Google Scholar]
- [12].Visnovitz T, Osteikoetxea X, Sódar BW, et al. An improved 96 well plate format lipid quantification assay for standardisation of experiments with extracellular vesicles. J Extracell Vesicles. 2019;8:1565263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lorincz P, Lakatos Z, Maruzs T, et al. Atg6/UVRAG/Vps34-containing lipid kinase complex is required for receptor downregulation through endolysosomal degradation and epithelial polarity during Drosophila wing development. Biomed Res Int. 2014;2014:851349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Valcz G, Galamb O, Krenács T, et al. Exosomes in colorectal carcinoma formation: ALIX under the magnifying glass. Mod Pathol. 2016;29:928–938. [DOI] [PubMed] [Google Scholar]
- [15].Mitrovic B, Schaeffer DF, Riddell RH, et al. Tumor budding in colorectal carcinoma: time to take notice. Mod Pathol. 2012;25:1315–1325. [DOI] [PubMed] [Google Scholar]
- [16].Brabletz T, Jung A, Spaderna S, et al. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. [DOI] [PubMed] [Google Scholar]
- [17].Valcz G, Buzás EI, Szállási Z, et al. Perspective: bidirectional exosomal transport between cancer stem cells and their fibroblast-rich microenvironment during metastasis formation. NPJ Breast Cancer. 2018;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shaw PJ, Rawlins DJ. The point-spread function of a confocal microscope: its measurement and use in deconvolution of 3-D data. J Microsc. 1991;163:151–165. [Google Scholar]
- [19].Poteryaev D, Datta S, Ackema K, et al. Identification of the switch in early-to-late endosome transition. Cell. 2010;141:497–508. [DOI] [PubMed] [Google Scholar]
- [20].Hyttinen JM, Niittykoski M, Salminen A, et al. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta. 2013;1833:503–510. [DOI] [PubMed] [Google Scholar]
- [21].Baietti MF, Zhang Z, Mortier E, et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat Cell Biol. 2012;14:677–685. [DOI] [PubMed] [Google Scholar]
- [22].van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–228. [DOI] [PubMed] [Google Scholar]
- [23].Nonaka T, Wong DTW. Saliva-Exosomics in Cancer: molecular Characterization of Cancer-Derived Exosomes in Saliva. Enzymes. 2017;42:125–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Santiana M, Ghosh S, Ho BA, et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe. 2018;24:208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.