

Abstract
Background
The projected rise in the incidence of type 2 diabetes mellitus (T2DM) could develop into a substantial health problem worldwide. Whether insulin secretagogues (sulphonylureas and meglitinide analogues) are able to prevent or delay T2DM and its associated complications in people at risk for the development of T2DM is unknown.
Objectives
To assess the effects of insulin secretagogues on the prevention or delay of T2DM and its associated complications in people with impaired glucose tolerance, impaired fasting blood glucose, moderately elevated glycosylated haemoglobin A1c (HbA1c) or any combination of these.
Search methods
We searched the Cochrane Central Register of Controlled Trials, MEDLINE, PubMed, Embase, ClinicalTrials.gov, the World Health Organization International Clinical Trials Registry Platform, and the reference lists of systematic reviews, articles and health technology assessment reports. We asked investigators of the included trials for information about additional trials. The date of the last search of all databases was April 2016.
Selection criteria
We included randomised controlled trials (RCTs) with a duration of 12 weeks or more comparing insulin secretagogues with any pharmacological glucose‐lowering intervention, behaviour‐changing intervention, placebo or no intervention in people with impaired fasting glucose, impaired glucose tolerance, moderately elevated HbA1c or combinations of these.
Data collection and analysis
Two review authors read all abstracts and full‐text articles/records, assessed quality and extracted outcome data independently. One review author extracted data which were checked by a second review author. We resolved discrepancies by consensus or the involvement of a third review author. For meta‐analyses we used a random‐effects model with investigation of risk ratios (RRs) for dichotomous outcomes and mean differences (MDs) for continuous outcomes, using 95% confidence intervals (CIs) for effect estimates. We carried out trial sequential analyses (TSAs) for all outcomes that could be meta‐analysed. We assessed the overall quality of the evidence by using the GRADE instrument.
Main results
We included six RCTs with 10,018 participants; 4791 participants with data on allocation to intervention groups were randomised to a second‐ or third‐generation sulphonylurea or a meglitinide analogue as monotherapy and 29 participants were randomised to a second‐generation sulphonylurea plus metformin. Three trials investigated a second‐generation sulphonylurea, two trials investigated a third‐generation sulphonylurea and one trial a meglitinide analogue. A total of 4873 participants with data on allocation to control groups were randomised to a comparator group; 4820 participants were randomised to placebo, 23 to diet and exercise, and 30 participants to metformin monotherapy. One RCT of nateglinide contributed 95% of all participants. The duration of the intervention varied from six months to five years. We judged none of the included trials as at low risk of bias for all 'Risk of bias' domains.
All‐cause and cardiovascular mortality following sulphonylurea (glimepiride) treatment were rarely observed (very low‐quality evidence). The RR for incidence of T2DM comparing glimepiride monotherapy with placebo was 0.75; 95% CI 0.54 to 1.04; P = 0.08; 2 trials; 307 participants; very low‐quality evidence. One of the trials reporting on the incidence of T2DM did not define the diagnostic criteria used. The other trial diagnosed T2DM as two consecutive fasting blood glucose values ≥ 6.1 mmol/L. TSA showed that only 4.5% of the diversity‐adjusted required information size was accrued so far. No trial reported data on serious adverse events, non‐fatal myocardial infarction (MI), non‐fatal stroke, congestive heart failure (HF), health‐related quality of life or socioeconomic effects.
One trial with a follow‐up of five years compared a meglitinide analogue (nateglinide) with placebo. A total of 310/4645 (6.7%) participants allocated to nateglinide died compared with 312/4661 (6.7%) participants allocated to placebo (hazard ratio (HR) 1.00; 95% CI 0.85 to 1.17; P = 0.98; moderate‐quality evidence). The two main criteria for diagnosing T2DM were a fasting plasma glucose level ≥ 7.0 mmol/L or a 2‐hour post challenge glucose ≥ 11.1 mmol/L. T2DM developed in 1674/4645 (36.0%) participants in the nateglinide group and in 1580/4661 (33.9%) in the placebo group (HR 1.07; 95% CI 1.00 to 1.15; P = 0.05; moderate‐quality evidence). One or more serious adverse event was reported in 2066/4602 (44.9%) participants allocated to nateglinide compared with 2089/4599 (45.6%) participants allocated to placebo. A total of 126/4645 (2.7%) participants allocated to nateglinide died because of cardiovascular disease compared with 118/4661 (2.5%) participants allocated to placebo (HR 1.07; 95% CI 0.83 to 1.38; P = 0.60; moderate‐quality evidence). Comparing participants receiving nateglinide with those receiving placebo for the outcomes MI, non‐fatal stroke and HF gave the following event rates: MI 116/4645 (2.5%) versus 122/4661 (2.6%), stroke 100/4645 (2.2%) versus 110/4661 (2.4%) and numbers hospitalised for HF 85/4645 (1.8%) versus 100/4661 (2.1%) ‐ (HR 0.85; 95% CI 0.64 to 1.14; P = 0.27). The quality of the evidence was moderate for all these outcomes. Health‐related quality of life or socioeconomic effects were not reported.
Authors' conclusions
There is insufficient evidence to demonstrate whether insulin secretagogues compared mainly with placebo reduce the risk of developing T2DM and its associated complications in people at increased risk for the development of T2DM. Most trials did not investigate patient‐important outcomes.
Plain language summary
Can the glucose‐lowering drugs insulin secretagogues prevent or delay type 2 diabetes mellitus and its associated complications in persons at increased risk of this disease?
Review question
Can the group of glucose‐lowering drugs called insulin secretagogues prevent or delay the development of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus?
Background
Insulin secretagogues are widely used to treat people with type 2 diabetes mellitus. The insulin secretagogues can be divided into two main classes of glucose‐lowering drugs, namely sulphonylureas (e.g. glibenclamide/glyburide, glipizide and gliclazide) and meglitinide analogues (nateglinide and repaglinide). Insulin secretagogues lower blood glucose by stimulating the secretion of insulin in the body, thereby increasing insulin levels in the blood. People with moderately elevated glucose levels are often said to be at an increased risk for developing type 2 diabetes (often called 'prediabetes'). Therefore, people with moderately elevated glucose levels are frequently recommended to increase exercise and lower calorie intake (behaviour changing or 'lifestyle' interventions) in order to prevent the development of type 2 diabetes. It is currently not known whether insulin secretagogues should be prescribed for people with raised blood glucose levels who do not meet the diagnostic criteria for having type 2 diabetes mellitus. We wanted to find out whether insulin secretagogues could prevent or delay the development of type 2 diabetes mellitus in people with moderately elevated glucose levels. Furthermore, we wanted to analyse the effects of insulin secretagogues on patient‐important outcomes such as complications of diabetes (for example kidney and eye disease, heart attacks, strokes), death from any cause, health‐related quality of life and side effects of the medications.
Study characteristics
We searched the medical literature and registers of ongoing trials for randomised controlled trials of at least 12 weeks' duration comparing insulin secretagogues with another glucose‐lowering drug, placebo or no intervention. Randomised controlled trials are clinical studies in which people are randomly allocated to one of two or more groups so that the effects of different interventions can be compared directly. Participants included in the studies had to have glucose levels higher than considered normal, but below the glucose levels that are used to diagnose type 2 diabetes mellitus. We combined the findings of several studies to answer our review question. We found six randomised controlled trials. A total of 10,018 participants were included. The duration of the interventions varied from six months to five years.
This evidence is up to date as of April 2016.
Key results
Few participants died following treatment with sulphonylureas. Sulphonylureas (most of the evidence was available for glimepiride) did not reduce the risk of developing type 2 diabetes mellitus compared with placebo. No study with sulphonylureas reported on serious side effects, non‐fatal heart attacks, non‐fatal stroke, heart failure, health‐related quality of life or socioeconomic effects.
Only one study reported data on a meglitinide analogue (nateglinide). This large study contributed 95% of all participants of our review. We could not establish firm evidence on the outcomes death from any cause, risk of developing type 2 diabetes mellitus or serious side effects. This study did not report on health‐related quality of life or socioeconomic effects.
Future studies should investigate patient‐important outcomes and, especially, the side effects of the medications, because we do not know for sure whether 'prediabetes' is just a condition arbitrarily defined by a laboratory measurement or is in fact a real risk factor for type 2 diabetes mellitus, which might be treatable.
Quality of the evidence
All included trials had deficiencies in the way they were conducted or how key items were reported. For the individual comparisons the number of participants was small, resulting in a high risk of random errors (play of chance).
Summary of findings
Summary of findings for the main comparison. Summary of findings (sulphonylureas).
| Insulin secretagogues for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at risk for the development of type 2 diabetes mellitus | ||||||
|
Population: people at risk for the development of type 2 diabetes mellitus Settings: outpatient Intervention: sulphonylureas (data available for glimepiride only) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Glimepiride | |||||
|
All‐cause mortality Follow‐up: mean 3.7 years |
See comment | See comment | See comment | 274 (1) | ⊕⊝⊝⊝ very lowa | 5/136 (3.7%) participants in the glimepiride group versus 2/138 (1.4%) in the placebo group |
|
Incidence of type 2 diabetes mellitus Measured as 2 consecutive fasting blood glucose values ≥ 6.1 mmol/L (NANSY 2011b) or no definition provided (Eriksson 2006) Follow‐up: 6 months and a mean of 3.7 years |
361 per 1000 | 271 per 1000 (195 to 376) | RR 0.75 (0.54 to 1.04) | 307 (2) | ⊕⊝⊝⊝ very lowc | |
| Serious adverse events | See comment | See comment | See comment | See comment | See comment | Not reported |
|
Cardiovascular mortality Follow‐up: mean 3.7 years |
See comment | See comment | See comment | 274 (1) | ⊕⊝⊝⊝ very lowa | 1/136 (0.7%) participants died due to cardiovascular disease in the sulphonylurea monotherapy group and 2/138 (1.4%) participants died in the placebo group |
| Non‐fatal myocardial infarction, non‐fatal stroke, congestive heart failure | See comment | See comment | See comment | See comment | See comment | Not reported |
| Health‐related quality of life | See comment | See comment | See comment | See comment | See comment | Not reported |
| Socioeconomic effects | See comment | See comment | See comment | See comment | See comment | Not reported |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
*Assumed risk was derived from the event rates in the comparator groups aDowngraded by three levels because of serious imprecision and possible publication bias bDiagnostic criterion for trial entry was impaired fasting glucose in the NANSY trial (baseline glycosylated haemoglobin A1c was 4.9% for both groups) and impaired glucose tolerance in Eriksson 2006. In the NANSY trial participants took glimepiride on the days when glycaemic variables were measured cDowngraded by three levels because of indirectness, serious imprecision and possible publication bias. Trial sequential analysis showed that only 4.5% of the diversity‐adjusted information size was accrued so far to detect or reject a 10% relative risk reduction
Summary of findings 2. Summary of findings (meglitinide analogues).
| Insulin secretagogues for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at risk for the development of type 2 diabetes mellitus | ||||||
|
Population: people at risk for the development of type 2 diabetes mellitus Settings: outpatients Intervention: meglitinide analogues (nateglinide) Comparison: placebo | ||||||
| Outcomes | Placebo | Nateglinide | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | Comments |
|
All‐cause mortality Follow‐up: median 6.5 years |
See comment | See comment | See comment | 9306 (1) | ⊕⊕⊕⊝ moderatea | 310/4645 (6.7%) participants died in the nateglinide group versus 312/4661 (6.7%) participants in the placebo group. Vital status was available for 95.7% of participants at the end of follow‐up. The HR was 1.00; 95% CI 0.85 to 1.17; P = 0.98 |
|
Incidence of type 2 diabetes mellitus Defined as: fasting plasma glucose ≥ 7.0 mmol/L (126 mg/dL) or a 2‐hour blood glucose after a glucose‐load test ≥ 11.1 mmol/L (200 mg/dL) or by an adjudication committeeb Follow‐up: median 5 years |
See comment | See comment | See comment | 9306 (1) | ⊕⊕⊕⊝ moderatea | Type 2 diabetes mellitus developed in 1674/4645 (36.0%) participants in the nateglinide group and in 1580/4661 (33.9%) in the placebo group. The HR was 1.07; 95% CI 1.00 to 1.15; P = 0.05 |
|
Serious adverse events Follow‐up: median 5 years |
See comment | See comment | See comment | 9306 (1) | ⊕⊕⊕⊝ moderatea | The number of participants who experienced a serious adverse events was 2066/4602 (44.9%) participants in the nateglinide group versus 2089/4599 (45.6%) participants in the placebo group |
|
Cardiovascular mortality Follow‐up: 6.5 years |
See comment | See comment | See comment | 9306 (1) | ⊕⊕⊕⊝ moderatea | The number of participants who died due to cardiovascular disease was 126/4645 (2.7%) participants in the nateglinide group versus 118/4661 (2.5%) participants in the placebo group. The HR was 1.07; 95% CI 0.83 to 1.38; P = 0.60 |
|
(a) Non‐fatal myocardial infarction (b) Non‐fatal stroke (c) Congestive heart failure Follow‐up: median 6.3 years |
See comment | See comment | See comment | 9306 (1) | (a), (b), (c): ⊕⊕⊕⊝ moderatea |
(a) The number of participants who experienced a non‐fatal myocardial infarction during the trial was 116/4645 (2.5%) participants in the nateglinide group versus 122/4661 (2.6%) participants in the placebo group (b) The number of participants who experienced a non‐fatal stroke during the trial was 100/4645 (2.2%) participants in the nateglinide group versus 110/4661 (2.4%) participants in the placebo group (c) The number of participants developing congestive heart failure was not reported. However, the number of participants hospitalised for congestive heart failure was 85/4645 (1.8%) participants in the nateglinide group versus 100/4661 (2.1%) participants in the placebo group. The HR was 0.85; 95% CI 0.64 to 1.14; P = 0.27 |
| Health‐related quality of life | See comment | See comment | See comment | See comment | See comment | Not reported |
| Socioeconomic effects | See comment | See comment | See comment | 9306 (1) | See comment | One trial specified the assessment of health economics (NAVIGATOR 2010). However, trial authors did not provide data |
| *The basis for the assumed risk (e.g. the median control group risk across trials) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDowngraded by one level because of imprecision, high risk of selective reporting and possible publication bias (see Appendix 18) bDiagnostic criterion for the NAVIGATOR trial entry was impaired glucose tolerance; baseline glycosylated haemoglobin A1c was 5.8% for both groups. Progression to diabetes was confirmed by laboratory measurements in 1587 participants in the nateglinide group (34.2%) and 1495 participants in the placebo group (32.1%). Progression to diabetes was determined by the adjudication committee in the case of 87 participants assigned to nateglinide (1.9%) and 85 assigned to placebo (1.8%)
Background
Description of the condition
'Prediabetes', 'borderline diabetes', the 'prediabetic stage', 'high risk of diabetes', 'dysglycaemia' or 'intermediate hyperglycaemia' are often characterised by various measurements of elevated blood glucose concentrations, such as isolated impaired fasting glucose (IFG), isolated impaired glucose tolerance (IGT), isolated elevated glycosylated haemoglobin A1c (HbA1c) or combinations thereof (WHO/IDF 2006). These elevated blood glucose levels, which are indicative of hyperglycaemia, are too high to be considered normal but are below the diagnostic threshold for type 2 diabetes mellitus (T2DM). Because of this continuous glycaemic spectrum from the normal to the diabetic stage, a sound evidence base is needed so that glycaemic thresholds for people at high risk of diabetes can be defined. The different terms used to describe various stages of hyperglycaemia may give rise to differing emotional reactions in affected persons. For example, a person told s/he has 'prediabetes' may take this to imply that diabetes is unavoidable, whereas someone told they are at (high) risk of diabetes may take this as meaning that they may possibly be able to avoid the disease altogether. In addition to the disputable construct of intermediate health states termed 'prediseases' (Viera 2011), many people may associate the label 'prediabetes' with dire consequences. Alternatively, any diagnosis of 'prediabetes' may be an opportunity to review, for example, eating habits and physical activity levels, thus enabling affected individuals to actively change their way of life.
The American Diabetes Association (ADA) and the World Health Organization (WHO) have established the criteria that are most commonly used today to define people with a high risk of developing T2DM. IGT was the first glycaemic measurement used by the US National Diabetes Data Group to define the prediabetic stage (NDDG 1979). It is based on the measurement of plasma glucose 2 hours after ingestion of 75 g of glucose (glucose load). The dysglycaemic range is defined as a plasma glucose level between 7.8 and 11.1 mmol/L (140 and 200 mg/dL) 2 hours after the glucose load. Studies indicate that IGT is caused by insulin resistance and defective insulin secretion (Abdul‐Ghani 2006; Jensen 2002). In 1997, the ADA, and later the WHO, introduced the IFG concept to define 'prediabetes' and intermediate hyperglycaemia (ADA 1997; WHO 1999). The initial definition of IFG was a blood glucose level of 6.1 to 6.9 mmol/L (110 to 125 mg/dL). Later, the ADA reduced the lower threshold for defining IFG to 5.6 mmol/L (100 mg/dL) (ADA 2003). However, the WHO did not endorse this lower cut‐off point for IFG for the definition of 'prediabetes' (WHO/IDF 2006). IFG seems to be associated with β‐cell dysfunction (impaired insulin secretion) and an increase in the hepatic glucose output (DeFronzo 1989). More recently, HbA1c levels have been used to identify people at high risk of developing T2DM. In 2009, the International Expert Committee (IEC) suggested that HbA1c levels ranging from 6.0% to 6.4% can be used to identify people at high risk of T2DM (IEC 2009). Shortly afterwards, the ADA redefined this HbA1c range as 5.7% to 6.4% (ADA 2010). Unlike IFG and IGT, HbA1c levels reflect longer‐term glycaemic control (i.e. a person's blood glucose levels during the preceding two to three months) (IEC 2009).
The International Diabetes Federation (IDF) estimated that, in 2010, the prevalence of IGT was 343 million people, and this is predicted to increase to 471 million people by 2035 (IDF 2013). Studies have shown poor correlations between HbA1c levels and IFG/IGT (Gosmanov 2014; Selvin 2011). Notably, the various glycaemic tests do not seem to identify the same people as there is imperfect overlap among the glycaemic modalities available to define dysglycaemia (Gosmanov 2014; Selvin 2011). A person's risk of progressing to T2DM depends on the diagnostic criteria used to identify that risk. Some people with dysglycaemia will never develop T2DM, and some people will return to normoglycaemia. IGT is often accepted as the best glycaemic variable predicting the risk of progression to T2DM (Morris 2013). However, studies indicate that less than half of the people defined as 'prediabetic' by means of IGT will develop T2DM in the following 10 years (Morris 2013). Both IFG and HbA1c levels are thought to predict a different risk spectrum for developing T2DM (Cheng 2006; Morris 2013). Most importantly, dysglycaemia is commonly an asymptomatic condition and, naturally, often remains 'undiagnosed' (CDC 2015).
It has yet to be clarified whether or not any particular intervention, especially glucose‐lowering drugs, should be recommended for people at risk for T2DM (Yudkin 2014). Trials have indicated that the progression to T2DM is reduced, or possibly just delayed, with behavioural interventions (increased physical activity, dietary changes or both) (Diabetes Prevention Program 2002; Diabetes Prevention Program FU 2009; Finnish Diabetes Prevention Study Group 2001). A recent meta‐analysis of 22 trials with interventions that changed behaviour in people at high risk of T2DM concluded that the effect of these interventions on longer‐term diabetes prevention is not clear (Dunkley 2014). Hence, more research is needed to establish optimal strategies for reducing the risk of T2DM with behavioural approaches (Dunkley 2014).
International diabetes associations and clinicians do not generally accept the prescription of pharmacological glucose‐lowering interventions for the prevention of T2DM. Several groups of pharmacological glucose‐lowering interventions have been investigated in people at risk of T2DM. Some findings indicate that the progression to T2DM is reduced or may only be delayed by such agents (Diabetes Prevention Program 2002; Diabetes Prevention Program FU 2009). However, the ADA recommends metformin in people at risk of T2DM with a body mass index (BMI) above 35 kg/m² who are aged less than 60 years, and women with prior gestational diabetes mellitus (ADA 2015).
Description of the intervention
Since the introduction of the sulphonylureas in the 1950s, this class of glucose‐lowering intervention has been a mainstay in the treatment of people with T2DM. The first of these agents to be introduced to the market were first‐generation sulphonylureas (acetohexamide, carbutamide, chlorpropamide, tolazamide and tolbutamide). Later the second‐ and third‐generations of sulphonylureas were introduced, and have now almost completely replaced the first‐generation sulphonylureas (Harrower 2000). Second‐generation sulphonylureas (e.g. glibenclamide (in the US: glybyride), glipizide and gliclazide) and third‐generation sulphonylureas (gliclazide modified release (MR), glipizide gastrointestinal therapeutic system (GITS) and glimepiride) are thought to have a better safety profile than first‐generation agents (Harrower 2000).
Another class of insulin secretagogues, meglitinide analogues, was introduced to the market in the 1990s (Black 2007). Two meglitinide analogues are currently available for clinical use in people with T2DM in Europe and the USA: repaglinide and nateglinide (ADA 2015). Another meglitinide analogue, mitiglinide, is approved for clinical use in people with T2DM in Japan (Phillippe 2013).
Sulphonylureas and meglitinide analogues can be prescribed as monotherapy in people with T2DM, usually if diet and exercise alone are not sufficient in controlling T2DM or if metformin is not tolerated or contraindicated. However, they can also be combined with other existing glucose‐lowering interventions (ADA 2015).
All sulphonylureas and meglitinide analogues are orally administered. The daily dose recommended in people with T2DM varies according to the different types of sulphonylurea or meglitinide analogue. Due to the varying half‐life of the sulphonylureas, some have to be taken once daily and others are taken twice or three times daily. The meglitinide analogues have a short half‐life and are administered in relation to meals (Blickle 2006).
For glimepiride, the recommended dose is up to 4 mg/day (Drugs.com 2016a). For gliclazide, the recommended starting dose is between 40 mg/day and 80 mg/day, but can be increased to 320 mg/day (Drugs.com 2016b).
Adverse effects of the intervention
All sulphonylureas and meglitinide analogues have the potential to cause hypoglycaemia. The risk of hypoglycaemia varies according to the type of sulphonylurea. Some sulphonylureas, such as glibenclamide, are more prone to causing prolonged hypoglycaemia than others (Harrower 2000). The risk of hypoglycaemia appears more pronounced for the first‐generation sulphonylureas compared with newer generations (Harrower 2000). Because of their short half‐life, meglitinide analogues do not cause prolonged hypoglycaemia (Scott 2012).
In 1976, the University Group Diabetes Program (UGDP) suggested that the sulphonylurea tolbutamide was associated with adverse cardiovascular effects compared with placebo and insulin in people with T2DM (UGDP 1976). More recent randomised clinical trials (RCTs) have not shown a significant increased risk of cardiovascular disease with sulphonylureas compared with other glucose‐lowering interventions in people with T2DM (ADOPT 2006; UKPDS 33 1998). Several observational studies have indicated increased risks of mortality and cardiovascular disease with sulphonylurea monotherapy compared with metformin monotherapy in people with T2DM (Roumie 2012; Schramm 2011). However, risk may vary among the different sulphonylureas (Pantalone 2012; Schramm 2011). No association between the use of meglitinide analogues and an increase in cardiovascular risk was reported in one observational study (Schramm 2011); however, some confounding factors may not have been detected in this study (Deeks 2003).
A substudy of the UK Prospective Diabetes Study (UKPDS) showed that, in participants receiving a sulphonylurea, the early addition of metformin was associated with an increased risk of mortality compared with continuation on a sulphonylurea alone (UKPDS 34 1998). The debate about the potential adverse effects of this combination therapy is ongoing.
How the intervention might work
The primary mechanism of action of the sulphonylureas and meglitinide analogues is to stimulate insulin release from the insulin‐secreting pancreatic β‐cells; hence, the term 'insulin secretagogues'. Sulphonylureas and meglitinide analogues increase pancreatic insulin release by closing the potassium‐sensitive adenosine triphosphate channels in β‐cells (Harrower 2000; Scott 2012).
The pharmacokinetic and pharmacodynamic properties of different insulin secretagogues vary, mainly as a result of differing binding affinities for sulphonylurea receptors on the β‐cell, and differing half‐lives. The meglitinide analogues exhibit a fast association/dissociation to/from the sulphonylurea receptor, and therefore mimic physiological early‐phase insulin secretion. With regard to sulphonylureas, half lives range from around 5 hours (glimepiride) to 36 hours (chlorpropamide) (McCall 2001). The half‐life of the meglitinide analogues is relatively short (1 to 1.5 hours) (Scott 2012).
It has been hypothesised that postprandial hyperglycaemia rather than fasting glucose levels is associated with cardiovascular disease (Meigs 2002). Due to the short‐acting mechanism of action of the meglitinide analogues, which primarily reduces postprandial hyperglycaemia, it has been hypothesised that meglitinide analogues could be effective in decreasing the risk of T2DM and cardiovascular disease in individuals with IGT (NAVIGATOR 2010). However, a large‐scale RCT failed to show any beneficial effect of nateglinide compared with placebo in individuals with IGT and established cardiovascular disease (or cardiovascular risk factors) after five years of intervention (NAVIGATOR 2010).
The glucagon‐like peptide‐1 (GLP‐1) and the dipeptidyl peptidase‐4 (DPP‐4) inhibitors stimulate insulin secretion by a glucose‐dependent mechanism, and inhibit glucagon secretion. These drugs increase insulin secretion indirectly by means of GLP‐1 and the glucose‐dependent insulinotropic polypeptide, two hormones that are secreted by endocrine cells located in the epithelium of the small intestine. The effects of the DPP‐4 inhibitors and the GLP‐1 receptor agonists in individuals at increased risk of developing T2DM will be evaluated in a separate Cochrane review (Hemmingsen 2016a).
Why it is important to do this review
This review is part of a series of reviews on interventions that may prevent or delay the development of T2DM and its associated complications in persons at increased risk of T2DM, which is funded by the WHO (Hemmingsen 2016a; Hemmingsen 2016b). The protocol for this review has previously been published (Hemmingsen 2016c). There has been an increased focus on the prevention or delay of T2DM with non‐pharmacological interventions and glucose‐lowering medications. Currently, several trials are ongoing to clarify whether the progression from an at‐risk status to T2DM can be stopped or postponed with glucose‐lowering compounds (ClinicalTrials.gov). However, a more important issue for people with dysglycaemia is whether or not these interventions reduce the risk of death and the complications ‐ especially cardiovascular disease ‐ related to T2DM.
Objectives
To assess the effects of insulin secretagogues on the prevention or delay of T2DM and its associated complications in people with impaired glucose tolerance, impaired fasting blood glucose, moderately elevated glycosylated haemoglobin A1c (HbA1c) or any combination of these.
Methods
Criteria for considering studies for this review
Types of studies
We included RCTs in participants at increased risk of type 2 diabetes mellitus (T2DM) comparing a second‐ or third‐generation sulphonylurea or a meglitinide analogue with another pharmacological glucose‐lowering interventions, behaviour changing intervention, placebo or no intervention, with a duration of 12 weeks or more (Hemmingsen 2016c).
Types of participants
We included individuals without a diagnosis of diabetes who were at increased risk of T2DM.
We included trials in obese people or in participants with previous gestational diabetes, provided trial investigators stated that the participants had intermediate hyperglycaemia.
Diagnostic criteria for people at risk of developing T2DM
To be consistent with changes to the classification of, and diagnostic criteria for dysglycaemia (impaired fasting glucose (IFG), impaired glucose tolerance (IGT) or elevated glycosylated haemoglobin A1c (HbA1c)) that have occurred over the years, a diagnosis should have been established using the standard criteria valid at the trial start (e.g. ADA 1997; ADA 2010; NDDG 1979; or WHO 1999). Ideally, the diagnostic criteria used in each study should have been described. We used the trial authors' definition of risk, but we contacted trial authors for additional information, if necessary. As differences in the glycaemic measurements used to define risk may introduce substantial heterogeneity, we planned to subject the diagnostic criteria used to subgroup analysis.
Types of interventions
We included trials in which a fraction of the included participants were explicitly described as having intermediate hyperglycaemia. We contacted the investigators in order to obtain separate data on the participants with intermediate hyperglycaemia.
We included a trial even if one or more of our primary or secondary outcome measures were not reported in a publication. In this case, we contacted the corresponding author for supplementary data. If no additional data were available, we present data from this trial in a supplementary table. We also list information about trials with a duration of the intervention shorter than 12 weeks in Appendix 1.
We planned to investigate the following comparisons of insulin secretagogues versus all pharmacological glucose‐lowering interventions, behaviour‐changing interventions, placebo or no intervention.
Intervention
(a) Second‐ or third‐generation sulphonylureas as monotherapy.
(b) Second‐ or third‐generation sulphonylureas as a part of combination therapy.
(c) Meglitinide analogues as monotherapy.
(d) Meglitinide analogues as a part of combination therapy.
Comparator
Any pharmacological glucose‐lowering intervention (e.g. acarbose, metformin, sodium–glucose cotransporter 2 inhibitors) compared with (a) or (c).
Any pharmacological glucose‐lowering agent (e.g. acarbose, metformin, sodium–glucose cotransporter 2 inhibitors) compared with (b) or (d) if this glucose‐lowering agent was the same in both the intervention and comparator groups (e.g. meglitinide analogue + metformin versus metformin).
Behaviour‐changing interventions (e.g. diet, exercise, diet and exercise) compared with (a) or (c).
Placebo compared with (a) or (c).
No intervention compared with (a) or (c).
Other concomitant interventions (e.g. educational programmes or additional pharmacotherapy) had to be the same in both the intervention and comparator groups in order to establish a fair comparison.
Minimum duration of intervention
We included trials that investigated the intervention for a duration of 12 weeks or more.
Specific exclusion criteria
We excluded trials in people diagnosed with the 'metabolic syndrome' as this is a special population which is not representative of people with only intermediate hyperglycaemia. Also, the composite of risk indicators, such as elevated blood lipids, insulin resistance, obesity and high blood pressure, which is termed metabolic syndrome, is of doubtful clinical usefulness and uncertain distinct disease entity. However, if we identified trials investigating participants with any definition of the metabolic syndrome, we intended to summarise some basic trial information in an additional table.
We excluded trials evaluating participants with intermediate hyperglycaemia in combination with another condition (e.g. cystic fibrosis).
We excluded trials evaluating participants with intermediate hyperglycaemia due to other medical interventions (e.g. glucocorticoids).
Types of outcome measures
Primary outcomes
All‐cause mortality
Incidence of T2DM
Serious adverse events
Secondary outcomes
Cardiovascular mortality
Non‐fatal myocardial infarction
Non‐fatal stroke
Congestive heart failure
Amputation of lower extremity
Blindness or severe vision loss
End‐stage renal disease
Non‐serious adverse events
Hypoglycaemia
Health‐related quality of life
Time to progression to T2DM
Measures of blood glucose control
Socioeconomic effects
Method and timing of outcome measurement
All‐cause mortality: defined as death from any cause. Measured at any time of the intervention and during follow‐up.
Incidence of T2DM and time to progression to T2DM: defined according to diagnostic criteria valid at the time the diagnosis was established using the standard criteria valid at the time the trial commenced (e.g. ADA 2008; WHO 1998). If necessary, we used the trial authors' definition of T2DM. Measured at the end of the intervention and the end of follow‐up.
Serious adverse events: defined according to the International Conference on Harmonization Guidelines as any event that lead to death, was life‐threatening, required inpatient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability; or any important medical event which may have jeopardised the participant or required intervention to prevent it (ICH 1997); or as reported in trials. Measured at any time of the intervention and during follow‐up.
Cardiovascular mortality, non‐fatal myocardial infarction, non‐fatal stroke, amputation of lower extremity, blindness or severe vision loss, congestive heart failure, hypoglycaemia (mild, moderate, severe/serious): defined as reported in trials. Measured at the end of the intervention and at the end of follow‐up.
End‐stage renal disease: defined as dialysis, renal transplantation or death due to renal disease. Measured at the end of the intervention and at the end of follow‐up.
Non‐serious adverse events: defined as the number of participants with any untoward medical occurrence not necessarily having a causal relationship with the intervention. Measured at any time of the intervention and during follow‐up.
Health‐related quality of life: defined as mental and physical health‐related quality of life, assessed separately or combined using a validated instrument such as Short‐Form 36. Measured at the end of the intervention and at the end of follow‐up.
Measures of blood glucose control: fasting blood glucose (FBG), blood glucose 2 hours after ingestion of 75 g glucose and HbA1c measurements. Measured at the end of the intervention and at the end of follow‐up.
Socioeconomic effects: for example costs of the intervention, absence from work, medication consumption. Measured at the end of the intervention and at the end of follow‐up.
Specification of key prognostic variables
Age
Gender
Equity issues (access to health care, social determinants)
Ethnicity
Hypertension
Cardiovascular disease
Obesity
Previous gestational diabetes
Summary of findings table
We present a 'Summary of findings' table to report the following outcomes, listed according to priority.
All‐cause mortality.
Incidence of T2DM.
Serious adverse events.
Cardiovascular mortality.
Non‐fatal myocardial infarction/stroke and congestive heart failure.
Health‐related quality of life.
Socioeconomic effects.
Search methods for identification of studies
Electronic searches
We searched the following sources from inception to the specified date, and placed no restrictions on the language of publication.
Cochrane Central Register of Controlled Trials (CENTRAL) (4 April 2016).
MEDLINE (1946 to present) (4 April 2016).
Embase (1974 to 5 April 2016) (4 April 2016).
ClinicalTrials.gov (4 April 2016).
WHO International Clinical Trials Registry Platform (ICTRP) Search Portal (http://apps.who.int/trialsearch/) (4 April 2016).
We continuously applied a MEDLINE (via Ovid SP) email alert service, established by the Cochrane Metabolic and Endocrine Disorders (CMED) Group, to identify newly published trials using the same search strategy as described for MEDLINE (for details on search strategies, see Appendix 2). If we identified new trials for inclusion, we intended to evaluate them, incorporate the findings into our review and resubmit another review draft (Beller 2013).
If we had detected any additional key words of relevance during any of the electronic or other searches, we intended to modify the electronic search strategies to incorporate these terms.
We obtained evaluations of all relevant non‐English articles.
Searching other resources
We attempted to identify other potentially eligible trials or ancillary publications by searching the reference lists of retrieved included trials, systematic reviews, meta‐analyses and health technology assessment reports. In addition, we contacted authors of included trials to identify any additional information about the retrieved trials and to determine whether further trials existed that we may have missed.
As none of the existing insulin secretagogues is currently approved for the treatment of persons with intermediate hyperglycaemia we did not search databases of the regulatory agencies (European Medicines Agency, US Food and Drug Administration).
Data collection and analysis
Selection of studies
Two review authors (BH and DS) independently scanned the abstract or title, or both, of every record retrieved in order to determine which trials should be assessed further. We investigated the full‐text articles of all potentially relevant articles. We resolved discrepancies through consensus or by recourse to a third review author (BR). We prepared a flow diagram of the number of trials identified and excluded at each stage, in accordance with PRISMA guidelines (Liberati 2009).
Data extraction and management
For trials that fulfilled our inclusion criteria, two review authors (BH and DS) independently extracted outcome data. Key characteristics of participants and interventions were extracted by one author (BH) and checked by another (DS). We reported data on efficacy outcomes and adverse events using standard data extraction sheets from the CMED Group. We resolved disagreements by discussion or, if required, by consultation with a third review author (BR) (for details, see Characteristics of included studies; Table 3; Appendix 1; Appendix 3; Appendix 4; Appendix 5; Appendix 6; Appendix 7; Appendix 8; Appendix 9; Appendix 10; Appendix 11; Appendix 12; Appendix 13; Appendix 14; Appendix 15).
1. Overview of trial populations.
| Intervention(s) and comparator(s) | Description of power and sample size calculation | Screened/eligible (N) | Randomised (N) | ITT (N) | Analysed (N) | Finishing trial (N) | Randomised finishing trial (%) | Follow‐up (extended follow‐up)a | |
| Eriksson 2006 | I: glipizide 2.5 mg | ‐ | b | ‐c | 17 | 16 | 16 | ‐ | 6 months (18 months) |
| C: placebo | ‐c | 17 | 17 | 16 | ‐ | ||||
| total: | 37 | 34 | 33 | 32 | ‐ | ||||
| NANSY 2011 | I: glimepiride 1.0 mg | Quote: "...assuming 3% conversion rate per year and 33% reduction of diabetes development with 5% significance and 80% statistical power" | ‐ | ‐d | 136 | 136 | ‐e | ‐ | 5 years or until diabetes developed, average follow‐up period 3.7 years |
| C: placebo | ‐d | 138 | 138 | ‐e | ‐ | ||||
| total: | 288d | 274 | 274 | 203 | 74.1 | ||||
| NAVIGATOR 2010 | I: nateglinide 60 mg, three times daily | Quote: "The sample size calculation was therefore based on a 'subadditivity / 75% additivity of effects' approach, assuming an effect size of 32% on cardiovascular outcome of the two drugs in combination. The treatment discontinuation rate was assumed to be 30% over five years, corresponding to approximately 6.9% per annum. While patients on treatment were assumed to have the full effect (i.e. 20% reduction of hazard rate if in the monotherapy group), it was assumed that patients who discontinued treatment would have only ¼ of the treatment effect remaining as carry‐over effect. Furthermore, it was expected that 75% of the patients who discontinued treatment could be followed up for events. The remaining 25% would comprise patients completely lost to follow‐up, patients who die (without reaching a primary endpoint), and those for whom events are unintentionally not reported by the investigator. Based on these assumptions, a total of 9152 patients will provide 90% testwise power to detect a treatment difference in the extended cardiovascular endpoint" | 43 502 | 4748 | 4645 | 4645 | 3726 | 78.5 | Quote: "The median follow‐up time for data on vital status was 6.5 years, and the median follow‐up times for data on the diabetes, extended cardiovascular, and core cardiovascular outcomes were 5.0, 6.3, and 6.4 years, respectively"f |
| C: placebo | 4770 | 4661 | 4661 | 3747 | 78.6 | ||||
| total: | 9518 | 9306 | 9306 | 7473 | 78.5 | ||||
| Osei 2004 | I: GITS 5 mg | ‐ | ‐ | 9 | 9 | 9 | ‐ | ‐ | 24 months (26 months) |
| C: placebo | 9 | 9 | 9 | ‐ | ‐ | ||||
| total: | 18 | 18 | 18 | ‐ | ‐ | ||||
| Page 1993 | I: gliclazide 40 mg twice daily | ‐ | ‐ | 6 | 6 | 6 | 6 | 100 | 6 months (7 months) |
| C1: placebo | 8 | 7 | 7 | 7 | 87.5 | ||||
| C2: diet + exercise | 23 | 18 | 18 | 18 | 78.2 | ||||
| total: | 37 | 31 | 31 | 31 | 83.8 | ||||
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | ‐ | ‐ | 29 | 22 | 22 | 22 | 75.9 | 2 years (2 years) |
| I2: glibenclamide 2.0 mg twice daily + placebo | 28 | 22 | 22 | 22 | 78.6 | ||||
| C1: placebo + metformin 850 mg twice daily | 30 | 23 | 23 | 23 | 76.7 | ||||
| C2: placebo | 33 | 19 | 19 | 19 | 57.6 | ||||
| total: | 120 | 86 | 86 | 86 | 71.7 | ||||
| Grand total | All interventionsh | 4820 | 3792 | ||||||
| All comparatorsh | 4873 | 3830 | |||||||
| All interventions and comparatorsi | 10,018 | 7825 j | |||||||
‐ denotes not reported
aFollow‐up under randomised conditions until end of trial or, if not available, duration of intervention; extended follow‐up refers to follow‐up of participants once the original study was terminated as specified in the power calculation bParticpants identified through screening of another trial (Botnia Study 1996). Quote: "The subjects included in the present study represented the first consecutive 37 subjects who maintained their IGT status on repeated OGTT testing during 1 year" cThe investigators described that they randomised 37 participants, and three dropped out shortly after. However, they do not describe how these three participants were allocated, but only describe that after the three participants had left 17 were allocated to each intervention group dThe investigators described that 14 randomised participants withdrew before the first occasion to establish the conversion to type 2 diabetes mellitus. All except one dropped out for administrative reasons. However, it was not specified to which intervention group these participants were allocated e71 individuals interrupted participation prematurely, however it was not described to which groups they belonged fThe trial was predefined to stop and the final analysis performed when 1374 participants have had an adjudication committee confirmed extended cardiovascular endpoint hNot all trials described the number of participants randomised to each intervention group iTwo trials did not report the number of randomised participants per intervention group. Therefore, numbers do not add up accurately jNot all trials reported the number of participants finishing the trial
C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; ITT: intention‐to‐treat; NANSY: The Nepi ANtidiabetes StudY
We planned to include information about potentially relevant ongoing trials, including the trial identifier, in a table of characteristics of ongoing studies.
For each included trial we tried to retrieve the protocol. If not available from a search of the databases, reference screening or Internet searches, we asked authors to provide a copy of the protocol. We entered predefined outcomes in a 'Matrix of trial endpoint (publications and trial documents)' (see Appendix 7).
We emailed all authors of the included trials to enquire whether they were willing to answer questions regarding their trials. We present the results of this survey in Appendix 16. We sought relevant missing information on the trials from the primary author(s) of the articles, if possible.
Dealing with duplicate and companion publications
In the event of duplicate publications, companion documents or multiple reports of a primary trial, we maximised the information by collating all available data and used the most complete data set aggregated across all known publications. We list duplicate publications, companion documents or multiple reports of a primary trial as secondary references under the primary reference of the included or excluded trial.
Assessment of risk of bias in included studies
Two review authors (BH and DS) independently assessed the risk of bias of each included trial. We resolved any disagreements by consensus, or by consultation with a third review author (BR). If adequate information was not available from the trial publication, trial protocol or both, we contacted trial authors for missing data on 'Risk of bias' items.
We used the Cochrane 'Risk of bias' assessment tool (Higgins 2011a; Higgins 2011b) and judged 'Risk of bias' criteria as being 'low', 'high', or 'unclear', evaluating individual bias items as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
Random sequence generation (selection bias due to inadequate generation of a randomised sequence) ‐ assessment at trial level
We assessed for each included trial whether the method used to generate the allocation sequence was described in sufficient detail to allow an assessment of whether it should produce comparable groups.
Low risk of bias: sequence generation was achieved using computer random number generation or a random number table. Drawing of lots, tossing a coin, shuffling cards or envelopes and throwing dice were adequate if performed by an independent person not otherwise involved in the trial. Use of the minimisation technique was considered as equivalent to being random.
Unclear risk of bias: insufficient information about the sequence generation process.
High risk of bias: the sequence generation method was non‐random (e.g. sequence generated by: odd or even date of birth, some rule based on date (or day) of admission, some rule based on hospital or clinic record number; allocation by judgement of the clinician; allocation by preference of the participant; allocation based on the results of a laboratory test or a series of tests; allocation by availability of the intervention). We excluded such trials from our review.
Allocation concealment (selection bias due to inadequate concealment of allocations prior to assignment) ‐ assessment at trial level
We described for each included trial the method used to conceal allocation to interventions prior to assignment, and assessed whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment.
Low risk of bias: central allocation (including telephone, interactive voice‐recorder, web‐based and pharmacy‐controlled randomisation); sequentially numbered drug containers of identical appearance; sequentially numbered, opaque, sealed envelopes.
Unclear risk of bias: insufficient information about the allocation concealment.
High risk of bias: use of: an open random allocation schedule (e.g. a list of random numbers); assignment envelopes without appropriate safeguards; alternation or rotation; date of birth; case record number; any other explicitly unconcealed procedure. We excluded such trials from our review.
We also evaluated trial baseline data so as to incorporate an assessment of baseline imbalance into the 'Risk of bias' judgement for selection bias (Corbett 2014; Egbewale 2014; Riley 2013). Chance imbalances might also affect judgements on the risk of attrition bias. In case of unadjusted analyses we distinguished between trials rated as at low risk of bias on the basis of both randomisation methods and baseline similarity, and trials rated as at low risk of bias on the basis of baseline similarity alone (Corbett 2014). We reclassified judgements of unclear, low or high risk of selection bias as specified in Appendix 15.
Blinding of participants and study personnel (performance bias due to knowledge of the allocated interventions by participants and personnel during the trial) ‐ assessment at outcome level
We evaluated the risk of detection bias separately for each outcome (Hróbjartsson 2013). We noted whether outcomes were self‐reported, investigator‐assessed or adjudicated outcome measures (see below).
Low risk of bias: blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken; no blinding or incomplete blinding, but the review authors judged that the outcome was not likely to be influenced by lack of blinding.
Unclear risk of bias: insufficient information about the blinding of participants and study personnel; the trial did not address this outcome.
High risk of bias: no blinding or incomplete blinding, and the outcome was likely to be influenced by lack of blinding; blinding of trial participants and key personnel attempted, but likely that the blinding could have been broken, and the outcome was likely to be influenced by lack of blinding.
Blinding of outcome assessment (detection bias due to knowledge of the allocated interventions by outcome assessment) ‐ assessment at outcome level
We evaluated the risk of detection bias separately for each outcome (Hróbjartsson 2013). We noted whether outcomes were self‐reported, investigator‐assessed or adjudicated outcome measures (see below).
Low risk of bias: blinding of outcome assessment ensured, and unlikely that the blinding could have been broken; no blinding of outcome assessment, but the review authors judged that the outcome measurement was not likely to be influenced by lack of blinding.
Unclear risk of bias: insufficient information about the blinding of outcome assessors; the trial did not address this outcome.
High risk of bias: no blinding of outcome assessment, and the outcome measurement was likely to be influenced by lack of blinding; blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement was likely to be influenced by lack of blinding.
Incomplete outcome data (attrition bias due to amount, nature or handling of incomplete outcome data) ‐ assessment at outcome level
We described for each included trial and for each outcome the completeness of data, including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported and the numbers included in the analysis at each stage (compared with the numbers of randomised participants per intervention/comparator groups), if reasons for attrition or exclusion were reported, and whether missing data were balanced across groups or were related to outcomes. We considered the implications of missing outcome data per outcome, such as high dropout rates (e.g. above 15%) or disparate attrition rates (e.g. difference of 10% or more between trial arms).
Low risk of bias: no missing outcome data; reasons for missing outcome data unlikely to be related to true outcome (for survival data, censoring unlikely to be introducing bias); missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk not sufficient to have a clinically relevant impact on the intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes not sufficient to have a clinically relevant impact on observed effect size; appropriate methods, such as multiple imputation, used to handle missing data.
Unclear risk of bias: insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias; the trial did not address this outcome.
High risk of bias: reason for missing outcome data likely to be related to true outcome, with either imbalance in numbers or reasons for missing data across intervention groups; for dichotomous outcome data, the proportion of missing outcomes compared with observed event risk sufficient to induce clinically relevant bias in intervention effect estimate; for continuous outcome data, plausible effect size (difference in means or standardised difference in means) among missing outcomes sufficient to induce clinically relevant bias in observed effect size; ‘as‐treated’ or similar analysis carried out with substantial departure of the intervention received from that assigned at randomisation; potentially inappropriate application of simple imputation.
Selective reporting (reporting bias due to selective outcome reporting) ‐ assessment at trial level
We assessed outcome reporting bias by integrating Appendix 7 (Matrix of trial endpoints (publications and trial documents) (Boutron 2014; Mathieu 2009) with Appendix 8 (High risk of outcome reporting bias according to ORBIT [Outcome Reporting Bias In Trials]) classification) (Kirkham 2010). This analysis formed the basis for the judgement of selective reporting.
Low risk of bias: the trial protocol was available and all of the trial’s prespecified (primary and secondary) outcomes that were of interest in the review have been reported in the prespecified way; the study protocol was not available but it was clear that the published reports included all expected outcomes (ORBIT classification).
Unclear risk of bias: insufficient information about selective reporting.
High risk of bias: not all of the trial’s prespecified primary outcomes were reported; one or more primary outcomes was reported using measurements, analysis methods or subsets of the data (e.g. subscales) that were not prespecified; one or more reported primary outcome was not prespecified (unless clear justification for its reporting was provided, such as an unexpected adverse effect); one or more outcomes of interest in the review was reported incompletely so that they could not be entered in a meta‐analysis; the trial report failed to include results for a key outcome that would have been expected to have been reported for such a trial (ORBIT classification).
Other bias (bias due to problems not covered elsewhere) ‐ assessment at trial level
We assessed any other risk of bias that reflected other circumstances that may have threatened the validity of the trial.
Low risk of bias: the trial appeared to be free of other sources of bias.
Unclear risk of bias: insufficient information to assess whether an important risk of bias existed; insufficient rationale or evidence that an identified problem introduced bias.
High risk of bias: used a potential source of bias related to the specific trial design; had been claimed to have been fraudulent; had some other serious problem.
We established a 'Risk of bias' graph and a 'Risk of bias' summary figure.
We distinguished between self‐reported, investigator‐assessed and adjudicated outcome measures.
We defined the following outcomes as self‐reported.
Non‐serious adverse events.
Hypoglycaemia, if reported by participants.
Health‐related quality of life.
Blood glucose control, if measured by trial participants.
We required the following outcomes to be investigator‐assessed.
All‐cause mortality.
Incidence of T2DM.
Time to progression to T2DM.
Serious adverse events.
Cardiovascular mortality.
Non‐fatal myocardial infarction.
Non‐fatal stroke.
Congestive heart failure.
Amputation of lower extremity.
Blindness or severe vision loss.
End‐stage renal disease.
Hypoglycaemia, if measured by trial personnel.
Blood glucose control, if measured by trial personnel.
Socioeconomic effects.
Summary assessment of risk of bias
Risk of bias for a trial across outcomes: some 'Risk of bias' domains such as selection bias (sequence generation and allocation sequence concealment) may affect the risk of bias across all outcome measures in a trial. Otherwise, we did not perform a summary assessment of the risk of bias across all outcomes for a trial. If we identified a high risk of selection bias, we excluded the trial.
Risk of bias for an outcome within a trial and across domains: we assessed the risk of bias for an outcome measure including all of the entries relevant to that outcome (i.e. both trial‐level entries and outcome‐specific entries). We defined 'low' risk of bias as low risk of bias for all key domains, 'unclear' risk of bias as unclear risk of bias for one or more key domains, and 'high' risk of bias as high risk of bias for one or more key domains.
Risk of bias for an outcome across trials and across domains: these were the main summary assessments that we incorporated in our judgements about the quality of evidence in the 'Summary of findings' table(s). We defined 'low' risk of bias as most information coming from trials at low risk of bias, 'unclear' risk of bias as most information coming from trials at low or unclear risk of bias and 'high' risk of bias as a sufficient proportion of information coming from trials at high risk of bias.
Measures of treatment effect
When at least two trials were available for comparison of a given outcome, we expressed dichotomous data as risk ratios (RRs) with 95% confidence intervals (CIs) and with Trial Sequential Analysis (TSA)‐adjusted 95% CIs if the diversity‐adjusted required information size was not reached. We expressed continuous data reported using the same scale as mean differences (MDs) with 95% CIs and with TSA‐adjusted CIs if the diversity‐adjusted required information size was not reached. For trials addressing the same outcome but using different outcome measure scales, we intended to use standardised mean differences (SMDs) with 95% CIs. We planned to calculate time‐to‐event data as hazard ratios (HRs) with 95% CIs using the generic inverse variance method. Our preference would have been to use unadjusted HRs, as adjustment may have differed among the included trials. For outcomes meta‐analysed as SMDs and the generic inverse variance method, we were unable to conduct TSA and adjust the 95% CIs.
Some scales measuring health‐related quality of life increase in value with improved health‐related quality of life, whereas other scales decrease in value with improved health‐related quality of life. To adjust for this, we planned to multiply by –1 the scales that report better health‐related quality of life with decreasing values.
Unit of analysis issues
We intended to take into account the level at which randomisation occurred, for example in cross‐over trials, cluster‐randomised trials and multiple observations for the same outcome. If more than one comparison from the same trial was eligible for inclusion in the same meta‐analysis, we would have either combined groups to create a single pair‐wise comparison or appropriately reduced the sample size so that the same participants did not contribute multiply (splitting the 'shared' group into two or more groups). While the latter approach offers some solution to adjusting the precision of the comparison, it does not account for correlation arising from the same set of participants being used in multiple comparisons (Higgins 2011a).
We planned to reanalyse cluster‐randomised trials that did not appropriately adjust for potential clustering of participants within clusters in their analyses. We intended to inflate the variance of the intervention effects using a design effect (DEFF). Calculation of a DEFF involves estimation of an intra‐cluster correlation (ICC). We planned to obtain estimates of ICCs through contact with authors, or by imputing them either using estimates from other included studies that report ICCs or using external estimates from empirical research (e.g. Bell 2013). We planned to examine the impact of clustering using sensitivity analyses.
Dealing with missing data
We attempted to obtain missing data from trial authors and carefully evaluated important numerical data such as numbers screened and randomised, as well as intention‐to‐treat (ITT), as‐treated and per‐protocol populations.
We investigated attrition rates (e.g. dropouts, losses to follow‐up, withdrawals) and critically appraised issues concerning missing data and imputation methods (e.g. last observation carried forward).
We converted standard errors and CIs to standard deviations (SDs) (Higgins 2011a). When no differences in means and SDs from baseline were reported, we used end of follow‐up values (Higgins 2011a). Where means and SDs for outcomes were not reported and we did not receive the information required from trial authors, we calculated the SDs from standard errors, if possible. Otherwise we planned to impute the values by assuming the SDs of the missing outcome to be the average of the SDs from the trials that reported this information.
We planned to investigate the impact of imputation on meta‐analyses by performing sensitivity analyses.
Assessment of heterogeneity
In the event of substantial clinical or methodological heterogeneity, we planned not to report trial results as the pooled effect estimate in a meta‐analysis.
We investigated heterogeneity (inconsistency) by visually inspecting the forest plots and by using a standard Chi² test with a significance level of α = 0.1. In view of the low power of this test, we also considered the I² statistic, which quantifies inconsistency across trials to assess the impact of heterogeneity on the meta‐analysis (Higgins 2002; Higgins 2003), where an I² statistic ≥ 75% indicated a considerable level of heterogeneity (Higgins 2011a).
Assessment of reporting biases
If we included 10 or more trials investigating a particular outcome, we planned to use funnel plots to assess small‐trial effects. Several explanations may account for funnel plot asymmetry, including true heterogeneity of effect with respect to trial size, poor methodological design (and hence bias of small trials) and publication bias. Therefore, we planned to interpret the results carefully (Sterne 2011).
Data synthesis
Unless good evidence showed homogeneous effects across trials, we primarily summarised data at low risk of bias using a random‐effects model (Wood 2008). We interpreted random‐effects meta‐analyses taking into consideration the whole distribution of effects, ideally by presenting a prediction interval (Higgins 2009). A prediction interval specifies a predicted range for the true treatment effect in an individual trial (Riley 2011). In addition, we performed statistical analyses according to the statistical guidelines contained in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
TSA
In a single trial sparse data and interim analyses increase the risk of type I and type II errors. To avoid type I errors, group sequential monitoring boundaries are applied to decide whether a trial could be terminated early because of a sufficiently small P value (i.e. the cumulative Z‐curve crosses the monitoring boundaries) (Lan 1983). Likewise, before reaching the planned sample size of a trial, the trial may be stopped due to futility if the cumulative Z‐score crosses the futility monitoring boundaries (Higgins 2011a). Sequential monitoring boundaries for benefit, harm or futility can be applied to meta‐analyses as well (termed trial sequential monitoring boundaries) (Higgins 2010; Wetterslev 2008). In TSA, the addition of each trial in a cumulative meta‐analysis is regarded as an interim meta‐analysis and helps to clarify whether significance or futility is reached, or whether additional trials are needed (Wetterslev 2008).
TSA combines a calculation of the diversity‐adjusted required information size (cumulated meta‐analysis sample size to detect or reject a specific relative intervention effect) for meta‐analysis with the threshold of data associated with statistics. We performed TSA on all outcomes (Brok 2009; Pogue 1997; Wetterslev 2008).
The idea in TSA is that if the cumulative Z‐curve crosses the boundary for benefit or harm before a diversity‐adjusted required information size is reached, a sufficient level of evidence for the anticipated intervention effect has been reached with the assumed type I error and no further trials may be needed. If the cumulative Z‐curve crosses the boundary for futility before a diversity‐adjusted required information size is reached, the assumed intervention effect can be rejected with the assumed type II error and no further trials may be needed. If the Z‐curve does not cross any boundary, then there is insufficient evidence to reach a conclusion. To construct the trial sequential monitoring boundaries, the required information size is needed and is calculated as the least number of participants needed in a well‐powered single trial and subsequently adjusted for diversity among the included trials in the meta‐analysis (Brok 2009; Wetterslev 2008). We applied TSA as it decreases the risk of type I and II errors due to sparse data and multiple updating in a cumulative meta‐analysis, and it provides us with important information in order to estimate the risks of imprecision when the required information size is not reached. Additionally, TSA provides important information regarding the need for additional trials and the required information size of such trials (Wetterslev 2008).
We applied trial sequential monitoring boundaries according to an estimated clinically important effect. We based the required information size on an a priori effect corresponding to a 10% relative risk reduction (RRR) for beneficial effects of the interventions and a 30% relative risk increase for harmful effects of the interventions.
TSA for continuous outcomes was performed with MDs, by using trials applying the same scale to calculate the required sample size. For continuous outcomes we tested the evidence for the achieved differences in cumulative meta‐analyses.
For adjustment of heterogeneity of the required information size we used the diversity (D²) estimated in the meta‐analyses of included trials. When diversity was zero in a meta‐analysis, we performed a sensitivity analysis using an assumed diversity of 20% when future trials are included, possibly changing future heterogeneity among trials.
Quality of evidence
We presented the overall quality of the evidence for each outcome according to the GRADE approach, which takes into account issues relating not only to internal validity (risk of bias, inconsistency, imprecision, publication bias) but also to external validity, such as directness of results. Two review authors (BH and DS) independently rated the quality of evidence for each outcome. We present a summary of the evidence in the Table 1. This provides key information about the best estimate of the magnitude of the effect, in relative terms and as absolute differences, for each relevant comparison of alternative management strategies, the numbers of participants and trials addressing each important outcome, and rates the overall confidence in effect estimates for each outcome. We created the 'Summary of findings' table on the basis of methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a) by means of the table editor in Review Manager (RevMan 2014), and include two appendices (Appendix 17; Appendix 18) providing checklists as guides to the consistency and reproducibility of GRADE assessments (Meader 2014) to help with the standardisation of the 'Summary of findings' tables. Alternatively, we would have used the GRADEproGDT software (GRADEproGDT 2015) and presented evidence profile tables as an appendix. We present results for the outcomes as described in the Types of outcome measures section. If meta‐analysis was not possible, we present the results in a narrative format in the 'Summary of findings' table. We justify all decisions to downgrade the quality using footnotes, and we make comments to aid the reader's understanding of the review where necessary.
Subgroup analysis and investigation of heterogeneity
We expected the following characteristics to introduce clinical heterogeneity, and planned to carry out subgroup analyses with investigation of interactions.
Type of sulphonylurea and type of meglitinide analogue.
Trials with long duration (two or more years) versus trials with short duration (less than two years).
Diagnostic criteria (IFG, IGT, moderately elevated HbA1c).
Age, depending on data.
Ethnicity, depending on data.
Comorbid conditions, such as hypertension, obesity, or both.
Participants with previous gestational diabetes mellitus.
Sensitivity analysis
We planned to perform sensitivity analyses to explore the influence of the following factors (when applicable) on effect sizes by restricting the analysis to the following.
Published trials.
Taking into account risk of bias, as specified in the 'Assessment of risk of bias in included studies' section.
Very long or large trials to establish the extent to which they dominate the results.
Trials using the following filters: diagnostic criteria, imputation, language of publication, source of funding (industry versus other) or country.
We also planned to test the robustness of results by repeating the analyses using different measures of effect size (RR, odds ratio (OR), etc.) and different statistical models (fixed‐effect and random‐effects models).
Results
Description of studies
For a detailed description of trials, see Table 3, Characteristics of included studies, Characteristics of excluded studies and Characteristics of ongoing studies.
Results of the search
The initial search of the databases identified 2262 records after duplicates were removed. We excluded most of the references on the basis of their titles and abstracts because they clearly did not meet the inclusion criteria (Figure 1). We evaluated 53 references further. After screening the full texts, six RCTs published in 16 records met our inclusion criteria. We excluded a total of 39 references after full‐text evaluation.
1.
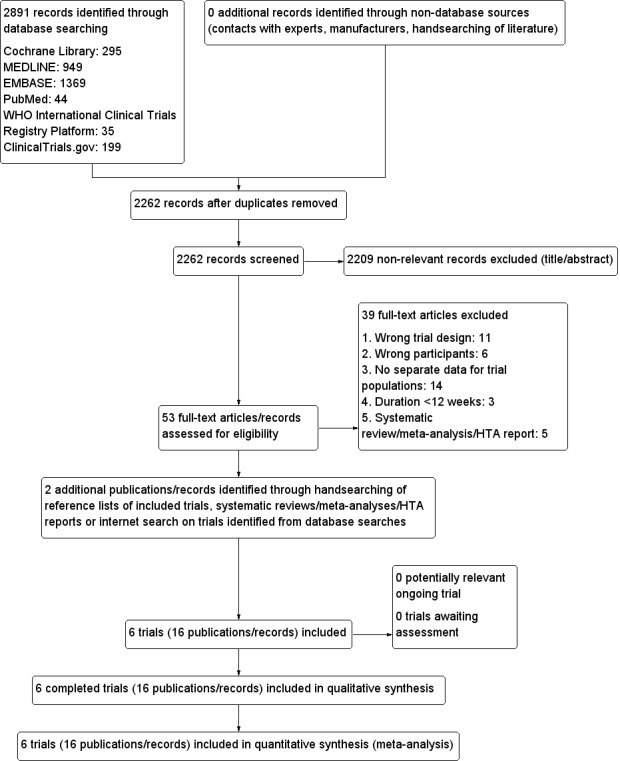
Trial flow diagram.
We identified no health technology assessment reports, systematic reviews or meta‐analyses focusing on sulphonylureas or meglitinide analogues in people at increased risk for the development of T2DM. However, four systematic reviews published in five records included a sulphonylurea or meglitinide analogue as a comparator in participants with intermediate hyperglycaemia (Anderson 2005; Bhardwaj 2010; Hopper 2011; Phung 2012; Van de Laar 2006). We evaluated all these systematic reviews but did not identify additional trials.
From the main publication of one of the included trials we identified an additional reference describing the same trial (Papoz 1978). We retrieved an additional trial protocol through an Internet search on the Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications (NAVIGATOR) trial (NAVIGATOR 2010).
We did not find any ongoing trials investigating our research question.
We sent all trial authors of the included trials a list of references and a request for information on additional trials of relevance. The trial authors did not provide additional trials or any supplementary information on our included trials.
Included studies
See Characteristics of included studies; Table 3 and Appendix 3; Appendix 4; Appendix 5; Appendix 6; Appendix 7; Appendix 8; Appendix 9; Appendix 10; Appendix 11; Appendix 12; Appendix 13; and Appendix 14.
Overview of trial populations
Only one trial reported the number of participants screened (NAVIGATOR 2010). Two trials did not report the number of participants randomised to each intervention group upon trial initiation (Eriksson 2006; NANSY 2011). A total of 4791 participants were randomised to a second‐ or third‐generation sulphonylurea or meglitinide analogue as monotherapy and 29 participants were randomised to a second‐generation sulphonylurea plus metformin (Papoz 1978). Three trials had a second‐generation sulphonylurea in the intervention arm (Eriksson 2006; Page 1993; Papoz 1978), two trials investigated a third‐generation sulphonylurea (NANSY 2011; Osei 2004) and one trial a meglitinide analogue, nateglinide (NAVIGATOR 2010). A total of 4873 participants were randomised to a comparator group; 4820 participants were randomised to placebo (Eriksson 2006; NANSY 2011; NAVIGATOR 2010; Osei 2004; Page 1993; Papoz 1978), 23 participants to diet and exercise (Page 1993) and 30 participants to metformin monotherapy (Papoz 1978).
Two publications provided information about sample size and power calculations (NANSY 2011; NAVIGATOR 2010).
Trial design
All the included trials were parallel randomised controlled clinical trials (Eriksson 2006; NANSY 2011; NAVIGATOR 2010; Osei 2004; Page 1993; Papoz 1978). All trials had performed blinding of the participants and investigators. However, in one trial only the comparison of gliclazide versus placebo was blinded, whereas the comparison of gliclazide versus diet and exercise was not (Page 1993). One RCT had a factorial design (NAVIGATOR 2010). The NAVIGATOR trial assigned participants to receive valsartan plus placebo, nateglinide plus placebo, nateglinide plus valsartan, or placebo plus placebo. Tests of interaction for the factorial allocation were provided for progression to T2DM and the two primary cardiovascular outcomes (NAVIGATOR 2010). None of the tests of interaction showed a relevant impact of the factorial design (NAVIGATOR 2010).
The duration of the intervention in the included trials varied from six months to five years. In four trials the duration of the intervention was two years or more (NANSY 2011; NAVIGATOR 2010; Osei 2004; Papoz 1978). Two trials included an extended follow‐up period after the intervention period had stopped (Eriksson 2006; Page 1993). One trial had a duration of intervention of six months, and thereafter a 12 month follow‐up period (Eriksson 2006). Another trial followed the participants one month after the end of the intervention period (i.e. for a total of seven months) (Page 1993).
The number of participants varied from 18 (Osei 2004) to 9518 (NAVIGATOR 2010). One trial contributed 97.4% of all randomised participants (NAVIGATOR 2010). Two trials were multicentre trials (NANSY 2011; NAVIGATOR 2010), three trials were single‐centre trials (Eriksson 2006; Osei 2004; Papoz 1978) and one trial did not provide any description of the number of centres involved (Page 1993).
All trials were performed in outpatient settings.
Five of the included trials stated that they had received grants from a pharmaceutical company (Eriksson 2006; NANSY 2011; NAVIGATOR 2010; Osei 2004; Page 1993), and one of these explicitly acknowledged several individuals employed by a pharmaceutical company for their contribution to the trial (NAVIGATOR 2010).
Participants
Two trials reported the ethnicity of participants; one trial included mainly white participants (NAVIGATOR 2010) and the other only black Americans (Osei 2004). Only one trial included participants from low‐income countries (NAVIGATOR 2010). In one trial all participants fulfilled the diagnostic criteria for IGT 12 months prior to randomisation and this was confirmed 12 months later after the participants were randomised (Eriksson 2006). One trial had a treatment‐free run‐in period (NAVIGATOR 2010). One trial included only males (Papoz 1978) and one trial did not report the gender of the participants (Osei 2004). For the remaining trials, authors provided gender information. The age of included participants varied from 39 to 60.4 years (Appendix 5).
All trials reported fasting glucose values at baseline, which reported plasma glucose values from 4.8 mmol/L to 6.1 mmol/L (NAVIGATOR 2010; Osei 2004). Four trials reported 2‐hour glucose values after a glucose‐load test at baseline, which varied from 7.6 mmol/L to 9.2 mmol/L (NAVIGATOR 2010; Papoz 1978). HbA1c values were reported at baseline in two trials (NANSY 2011; NAVIGATOR 2010). One trial did not report BMI at baseline (Papoz 1978). In the other trials all participants had at baseline a mean BMI over 25 kg/m2. Two trials had participants with a mean BMI over 30 kg/m2 at baseline (NAVIGATOR 2010; Osei 2004). Only one trial reported the number of participants with previous cardiovascular diseases at baseline (NAVIGATOR 2010).
Most trials excluded participants with other endocrine conditions, or hepatic or kidney disease.
The diagnostic criteria used for identifying intermediate hyperglycaemia varied in the included trials: in one trial IFG was the only inclusion criterion. This trial defined IFG as two overnight consecutive FBG values ≥ 5.6 mmol/L with a mean between 5.6 and 6.0 mmol/L (NANSY 2011). Five trials included participants with IGT (Eriksson 2006; NAVIGATOR 2010; Osei 2004; Page 1993; Papoz 1978). One trial evaluated intermediate hyperglycaemia by FPG levels < 7 mmol/L and by 2‐hour plasma glucose levels after a glucose‐load test ≥ 7.8 mmol/L and < 11.1 mmol/L (Eriksson 2006). The NAVIGATOR trial required FPG levels between 5.3 and 7.0 mmol/L (NAVIGATOR 2010). One trial required FPG levels < 7.8 mmol/L (Osei 2004). Three trials required 2‐hour plasma glucose values after a glucose‐load test ≥ 7.8 mmol/L and < 11.1 mmol/L (Eriksson 2006; NAVIGATOR 2010; Osei 2004). Two trials explicitly stated that they applied the criteria for IFG as recommended by WHO at the time of screening (Eriksson 2006; Osei 2004). One trial applied FPG levels > 5.6 mmol/L and 60‐minute plasma glucose levels during a continuous infusion of glucose > 9.3 mmol/L, which was stated to be equivalent to the WHO criteria for IGT (Page 1993). One trial applied the European Diabetes Epidemiology Study Group 1970 criteria and required two separate tests. Particiants had to have 2‐hour blood glucose values after a glucose‐load test ≥ 6.6 mmol/L but < 8.3 mmol/L or FBG levels ≥ 5.5 mmol/L up to 7.2 mmol/L on the second test (Papoz 1978).
Interventions
All the participants in the included trials were treatment‐naïve with regard to pharmacological glucose‐lowering interventions. Three trials included a second‐generation sulphonylurea in the sulphonylurea‐intervention arms (Eriksson 2006; Page 1993; Papoz 1978). None of the trials investigated the same second‐generation sulphonylurea; one trial investigated glipizide 2.5 mg once daily (Eriksson 2006); one trial investigated gliclazide 40 mg twice daily (Page 1993); and one trial investigated glibenclamide 2.0 mg twice daily (Papoz 1978). One trial combined a second‐generation sulphonylurea (glibenclamide 2.0 mg twice daily) with metformin (850 mg twice daily) (Papoz 1978). Two trials investigated a third‐generation sulphonylurea (NANSY 2011; Osei 2004), one trial investigated glipizide GITS 5 mg once daily (Osei 2004) and one trial investigated glimepiride 1.0 mg once daily (NANSY 2011). One trial investigated the meglitinide analogue, nateglinide, 60 mg three times daily (NAVIGATOR 2010). All the included trials were placebo controlled. In addition, one trial included a comparator arm with an active pharmacological glucose‐lowering agent (metformin) (Papoz 1978), and one had diet and exercise as a comparator (Page 1993). We judged placebo as well as diet and exercise as adequate comparators to establish fair comparisons (Appendix 3).
In two trials participants did not take the study drug the day glycaemic tests were performed (Osei 2004; Page 1993). In one trial the participants took the study drug after the glycaemic test had been performed (NAVIGATOR 2010). In two trials the participants took the study drug on the morning before testing (NANSY 2011; Papoz 1978). In one trial participants had stopped the study drug 15 days prior to the last assessment of glycaemic variables (Papoz 1978). One trial did not specify whether the study drug was taken on the day that glycaemic variables were measured (Eriksson 2006); however, an additional glucose assessment was performed 12 months after the study drug was stopped (Eriksson 2006).
None of the included trials, except one (NAVIGATOR 2010), described the intervention strategy for the participants that progressed to type 2 diabetes mellitus (T2DM). The first phase of the NAVIGATOR trial investigated the impact of intensified lifestyle interventions with diet and exercise. If these were insufficient metformin could be added. Finally, a second non‐insulin secretagogue could be added or bedtime insulin was started (NAVIGATOR 2010).
Outcomes
Three trials explicitly specified primary outcomes but did not define secondary outcomes (NANSY 2011; NAVIGATOR 2010; Papoz 1978). The remaining trials did not specify primary or secondary outcomes (Eriksson 2006; Osei 2004; Page 1993) (see Appendix 7). Only one trial was registered on ClinicalTrials.gov (NAVIGATOR 2010), where 14 documented changes were tracked, the last change date being 28 June 2011 (NAVIGATOR 2010). Two coprimary outcomes were predefined for the NAVIGATOR trial: incident T2DM and an extended composite cardiovascular outcome (death from a cardiovascular cause, non‐fatal myocardial infarction, non‐fatal stroke, hospitalisation for heart failure, arterial revascularisation, hospitalisation for unstable angina). One of these primary outcomes (death from a cardiovascular cause, non‐fatal myocardial infarction, non‐fatal stroke or hospitalisation for heart failure) was initially designed to be assessed as a secondary outcome (NAVIGATOR 2010).
Three trials reported one or more of the primary outcomes of relevance for this review (Eriksson 2006; NANSY 2011; NAVIGATOR 2010). Three assessed the incidence of T2DM as an outcome (Eriksson 2006; NANSY 2011; NAVIGATOR 2010). One trial defined T2DM as FPG ≥ 7.0 mmol/L or a 2‐hour blood glucose after a glucose‐load test ≥ 11.1 mmol/L, confirmed by two separate measurements (NAVIGATOR 2010). One trial reported T2DM as FBG values ≥ 6.1 mmol/L on two separate measurements (NANSY 2011). One trial did not report the diagnostic criteria for T2DM, but defined IGT according to WHO 1999 criteria (Eriksson 2006). It is therefore likely, that the WHO recommendation was also applied to the diagnosis of T2DM. One trial predefined the assessment of mortality and cardiovascular complications (NAVIGATOR 2010).
The reporting of adverse events was lacking in most trials. Only one trial reported all non‐serious and serious adverse events experienced during the trial (NAVIGATOR 2010) (see Appendix 11; Appendix 12; Appendix 13; Appendix 14).
All the included trials reported on one or more of the glycaemic variables that we had predefined to assess in our review. None of the included trials reported on microvascular outcomes, health‐related quality of life or socioeconomic effects.
Source of data
We attempted to contact all authors or investigators via email; however, no additional data were provided (see Appendix 16).
Excluded studies
We excluded a total of 39 articles after full‐text evaluation (Figure 1). These references are listed in Characteristics of excluded studies and some are detailed in 'Trials with a duration less than 12 weeks' (Appendix 1).
We excluded 11 trials published in 10 references as they did not allocate participants to sulphonylureas or meglitinide analogues by randomisation. One of the trials was an RCT in which participants were randomised to diet and exercise versus control (Cederholm 1985). The participants randomised to diet and exercise were offered glipizide and were therefore not randomised to this intervention.
We excluded six trials as they did not include participants of relevance to this review. For one of the trials we were unable to judge whether participants with intermediate hyperglycaemia were included after full‐text evaluation (Gudipaty 2014). We made contact with the trial authors and they confirmed that all participants had T2DM (Gudipaty 2014). Two trials included pregnant participants with gestational diabetes (NCT00744965; NCT01563120).
We excluded four trials published in 14 records as it was not possible to obtain separate data on the participants of interest for our review, neither from the publication nor through correspondence with the investigators. We contacted two authors of one trial but did not receive a reply (Igata 2014). We also did not receive a reply from the primary investigator of the DIAbetes and diffuse coronary NArrowing study (DIANA 2012). The corresponding author of another trial responded and asked which additional data we needed (Major‐Pedersen 2008). We sent the requested information but did not receive additional data, even after sending a reminder. We were also unable to obtain additional data on the participants with intermediate hyperglycaemia in the Fasting Hyperglycaemia Study (The Fasting Hyperglycaemia Study 1997a).
We excluded three trials because of the trial duration was less than 12 weeks (Lindblad 2001; Saloranta 2002; Schmoelzer 2006) and four systematic reviews published in five references (Anderson 2005; Bhardwaj 2010; Hopper 2011; Phung 2012; Van de Laar 2006).
Risk of bias in included studies
For details on the risk of bias of the included trials, see Characteristics of included studies.
For an overview of review authors' judgements about each risk of bias item for individual trials and across all trials, see Figure 2 and Figure 3.
2.
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included trials (blank cells indicate that the particular outcome was not measured in some trials).
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included trial (blank cells indicate that the trial did not measure that particular outcome).
None of the included trials reported on microvascular outcomes, health‐related quality of life or socioeconomic effects.
Allocation
We judged only one trial to be at low risk of selection bias with regard to the methods of randomisation and allocation concealment (NAVIGATOR 2010). The remaining trials reported that participants were randomised but provided no any further description (Eriksson 2006; NANSY 2011; Osei 2004; Page 1993; Papoz 1978). We therefore judged these trials as at unclear risk of bias regarding randomisation and allocation concealment.
We evaluated trial baseline data for our predefined prognostic baseline variables. Only one trial reported all the prognostic baseline variables of interest, which were all balanced between the intervention groups (NAVIGATOR 2010). The remaining trials reported only some of our predefined key prognostic variables of interest (Eriksson 2006; NANSY 2011; Osei 2004; Page 1993; Papoz 1978). One of the trials reporting key prognostic variables showed important differences between the intervention groups for several variables (Page 1993) (see Appendix 4; Appendix 5; Appendix 6). However, the uneven distribution of these key prognostic variables was not in favour of any particular intervention group.
Blinding
All trials explicitly reported the blinding of participants and investigators. The blinding of participants was ensured by the use of placebo tablets. However, one trial had an addition to the double‐blinded placebo arm ‐ a diet and exercise arm ‐ for which no blinding of the investigators or participants was described (Page 1993). One trial mentioned that a blinded outcome committee evaluated mortality, incidence of T2DM, cardiovascular outcomes, serious adverse events and severe hypoglycaemia (NAVIGATOR 2010). None of the remaining trials reported that a blinded outcome committee was instituted to assess any of the reported outcomes.
Where measured, all primary outcomes of this review were to be investigator assessed, and we judged these to be at low risk of performance and detection bias. All the included trials reported blood glucose measurements performed by the investigators and we judged these outcomes measures to be at low risk of performance and detection bias.
Non‐serious adverse events and mild hypoglycaemia were partly or exclusively self‐reported in all trials. Only one trial reported non‐serious adverse events other than mild hypoglycaemia (NAVIGATOR 2010). One trial reported that the only adverse events observed were hypoglycaemic symptoms (Eriksson 2006). The number of participants with mild hypoglycaemia was reported in four trials (Eriksson 2006; NAVIGATOR 2010; Osei 2004; Page 1993). However, two trials reported that no participants experienced mild hypoglycaemia (Osei 2004; Page 1993). In one of these trials one of the intervention arms (diet and exercise) was not blinded (Page 1993). Overall, we considered the risk of performance bias and detection bias to be low or unclear for our secondary outcomes.
Incomplete outcome data
We considered overall risk of attrition bias to be unclear for most of our outcomes.
Only three trials reported the numbers of participants randomised and finishing the trial (NAVIGATOR 2010; Page 1993; Papoz 1978). The percentages of randomised participants completing the trials varied from 58% to 100%. Two trials did not describe how many participants were originally randomised but reported the number analysed (Eriksson 2006; NANSY 2011). In the NANSY trial, the authors stated that 71 participants interrupted participation prematurely, the reason being death for seven of these participants. Unfortunately, the trial authors did not describe numbers per allocated group. In another trial, three participants dropped out early but the trial authors did not specify to which intervention these participants belonged to (Eriksson 2006). The NAVIGATOR trial reported that 163 participants in the nateglinide group and 143 participants in the placebo group withdrew, but did not provide details (NAVIGATOR 2010). One trial gave a detailed description of the participants who did not complete the trial (Page 1993). Another trial reported on how many participants were lost to follow‐up but did not provide reasons (Papoz 1978). One trial did not mention whether any participants withdrew or were lost to follow‐up (Osei 2004).
In one trial, two participants in the glipizide group withdrew due to hypoglycaemia (Eriksson 2006). As these two dropouts could have had a substantial impact on the effect estimate for hypoglycaemia, we therefore judged the risk of attrition bias to be high for this outcome.
Selective reporting
Only one trial had a published protocol (NAVIGATOR 2010). We judged five of the included trials to be at high risk of reporting bias on one or more of the outcomes of relevance for our review (Eriksson 2006; NANSY 2011; NAVIGATOR 2010; Page 1993; Papoz 1978). One trial had an unclear risk of reporting bias (Osei 2004). For more details see Appendix 7 and Appendix 8.
Other potential sources of bias
Five of the included trials stated that they had received grants from a pharmaceutical company (Eriksson 2006; NANSY 2011; NAVIGATOR 2010; Osei 2004; Page 1993) and one explicitly acknowledged several individuals employed by a pharmaceutical company for their contribution to the trial (NAVIGATOR 2010). It is known that trials receiving funding or provision of free drug or devices from a pharmaceutical company lead to more favourable results and conclusions than trials sponsored by other sources (Lundh 2012). Therefore, we judged only one trial to be free of other sources of bias (Papoz 1978); for the rest we judge other sources of bias to be unclear.
Effects of interventions
Baseline characteristics
For details of baseline characteristics, see Appendix 4; Appendix 5; Appendix 6
Sulphonylureas as monotherapy versus any pharmacological glucose‐lowering intervention
One trial compared sulphonylurea monotherapy with another pharmacological glucose‐lowering intervention. This trial had four intervention arms: glibenclamide plus metformin, glibenclamide plus placebo, metformin plus placebo and placebo only (Papoz 1978). This trial reported FBG and blood glucose values 2 hours after an oral glucose load. The mean fasting glucose value at the end of intervention in the glibenclamide plus placebo group was 5.0 (SD 0.5) mmol/L measured in 22 participants versus 5.3 (SD 0.5) mmol/L in 23 participants in the metformin plus placebo group. The 2‐hour blood glucose values showed a mean of 6.3 (SD 1.6) mmol/L in 22 participants versus 6.4 (SD 1.3) mmol/L in 23 participants, respectively. For both glycaemic variables we converted data from mg/dL into mmol/L and calculated SDs from reported standard errors (Papoz 1978).
Sulphonylureas as monotherapy versus behaviour‐changing interventions
One trial compared sulphonylurea monotherapy with diet and exercise (Page 1993). This trial reported fasting glucose. The mean fasting glucose value at the end of intervention in the gliclazide group showed a mean of 5.1 (SD 0.8) mmol/L measured in six participants versus 5.6 (SD 0.7) mmol/L in 18 participants in the diet and exercise group. None of the participants experienced mild or severe hypoglycaemia.
Sulphonylureas as monotherapy versus placebo
Five trials compared a sulphonylurea monotherapy with placebo (Eriksson 2006; NANSY 2011; Osei 2004; Page 1993; Papoz 1978). Two trials included a third‐generation sulphonylurea (NANSY 2011; Osei 2004), the others included a second‐generation sulphonylurea (Eriksson 2006; Page 1993; Papoz 1978). Two trials had a follow‐up period without any pharmacological intervention after the intervention period was stopped (Eriksson 2006; Page 1993).
A description of the outcomes for this comparison is listed in the Table 1.
Primary outcomes
All‐cause mortality
One trial reported all‐cause mortality (NANSY 2011). The participants of this trial were allocated to glimepiride or placebo. A total of 5/136 (3.7%) participants in the glimepiride group died compared with 2/138 (1.4%) in the placebo group. In the glimepiride group, one person died from cardiovascular disease, two persons from cancer, one person from suicide and one person from drowning. In the placebo group two participants died of cardiovascular causes.
Incidence of type 2 diabetes mellitus
Two trials comparing a sulphonylurea monotherapy with placebo reported data on the incidence of T2DM (Eriksson 2006; NANSY 2011). In the NANSY trial, participants took the trial drug (glimepiride) on the days when glycaemic variables were measured. A diagnosis of T2DM was defined as two consecutive FBG values ≥ 6.1 mmol/L (NANSY 2011). The other trial reporting the incidence of T2DM did not state whether the participants took any study drug (glipizide) on the days when glycaemic measurements were performed but retested glycaemic variables 12 months after the trial drug was stopped (Eriksson 2006). The trial authors did not provide any specific glycaemic value for the diagnosis of T2DM but stated that IGT was defined according to WHO recommendations. It is therefore likely that WHO 1999 criteria were also used to define T2DM (Eriksson 2006).
One of the trials had an intervention period of six months and thereafter an extended follow‐up period of 12 additional months (Eriksson 2006). The other trial had a duration of five years or until T2DM developed (average follow‐up 3.7 years) (NANSY 2011). One trial contributed 96/97 events (NANSY 2011) and only one participant developed T2DM during the intervention period in the other trial (Eriksson 2006). The RR for the incidence of T2DM comparing glimepiride monotherapy with placebo was 0.75 (95% CI 0.54 to 1.04; P = 0.08; 2 trials; 307 participants; very low‐quality evidence; Analysis 1.1).
1.1. Analysis.

Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 1 Incidence of type 2 diabetes.
TSA showed that 4.5% of the diversity‐adjusted information size was accrued so far to detect or reject a 10% RRR. Diversity was zero, but we applied a diversity of 20% when calculating the diversity‐required information size as heterogeneity is likely to increase when future trials are included. As only a minor fraction of the diversity‐adjusted required information size to detect or reject a 10% RRR was accrued so far, the TSA‐adjusted 95% CIs could not be calculated with a diversity at 20%. However, when we set diversity to 0%, the TSA‐adjusted 95% CIs were 0.20 to 2.84.
Serious adverse events
We did not identify trials with data on serious adverse events for this comparison. However, one trial described that all reported adverse effects, with the exception of hypoglycaemia, were mild (Eriksson 2006). It is therefore likely that this trial collected data on serious adverse events but did not identify any events (Eriksson 2006).
Secondary outcomes
Cardiovascular mortality
One trial reported that in the glimepiride group 1/136 (0.7%) participant died of cardiovascular reasons compared with 2/138 (1.4%) participants in the placebo group (NANSY 2011).
Non‐fatal myocardial infarction
We did not identify trials with data on non‐fatal myocardial infarction for this comparison.
Non‐fatal stroke
We did not identify trials with data on non‐fatal stroke for this comparison.
Congestive heart failure
We did not identify trials with data on congestive heart failure for this comparison.
Amputation of lower extremity
We did not identify trials with data on amputation of lower extremity for this comparison.
Blindness or severe vision loss
We did not identify trials with data on blindness or severe vision for this comparison.
End‐stage renal disease
We did not identify trials with data on end‐stage renal disease for this comparison.
Non‐serious adverse events
We did not identify trials with data on non‐serious adverse events for this comparison. However, we considered one trial at high risk of selective outcome reporting for non‐serious adverse events (Eriksson 2006). The publication stated that side effects were mild (Eriksson 2006). However, no details were published.
Hypoglycaemia
Three trials reported data on mild hypoglycaemia (Eriksson 2006; Osei 2004; Page 1993). Two of the trials reported that no participants experienced hypoglycaemia (Osei 2004; Page 1993). Eriksson 2006 reported that 7/16 participants in the sulphonylurea group compared with 5/17 participants in the placebo group experienced mild hypoglycaemic events (Analysis 1.2; Analysis 1.3).
1.2. Analysis.
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 2 Mild hypoglycaemia.
1.3. Analysis.
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 3 Severe hypoglycaemia.
Two participants withdrew from one of the trials because of hypoglycaemia (Eriksson 2006). Whether this was due to repetitive mild hypoglycaemia or severe hypoglycaemia could not be determined from the publication (Eriksson 2006).
Health‐related quality of life
We did not identify trials with data on health‐related quality of life for this comparison.
Time to progression to T2DM
We did not identify trials with data on time to progression to T2DM for this comparison.
Measures of blood glucose control
Fasting blood glucose
Four trials comparing a sulphonylurea monotherapy with placebo reported fasting glucose values at the end of the intervention (Eriksson 2006; Osei 2004; Page 1993; Papoz 1978). In two trials the participants did not take the trial drug on the days glycaemic variables were measured (Osei 2004; Page 1993). One trial applied the trial drug on the day of glycaemic testing after 2 and 14 months; however, the last glycaemic measurements at 26 months were performed 15 days after the trial drug was stopped (Papoz 1978). One trial did not report whether the participants took any study drug on the days of glycaemic measurements, but performed a retesting of glycaemic variables 12 months after the trial drug was stopped (Eriksson 2006). Comparing sulphonylurea monotherapy with placebo showed a MD in FBG of ‐0.31 mmol/L (95% CI ‐0.59 to ‐0.02; P = 0.03; 4 trials; 105 participants; overall low or unclear risk of bias; Analysis 1.4). The termination of the trial drug 15 days before measurement at the end of the intervention in Papoz 1978 may explain why this trial had the highest fasting glucose level at the end of intervention. Unfortunately, the largest trial comparing a sulphonylurea monotherapy with placebo (274 participants) did not report fasting glucose values, even though it was obviously measured (NANSY 2011). One trial measured glucose in whole blood (Papoz 1978). In one trial it was not clearly reported how glucose was measured, but according to the diagnostic criteria for intermediate hyperglycaemia it might have been plasma glucose (Eriksson 2006). In the remaining trials it was clearly stated that glucose was measured in plasma (Osei 2004; Page 1993). Whole blood glucose values were converted to plasma glucose values (diabetes.co.uk 2016a).
1.4. Analysis.
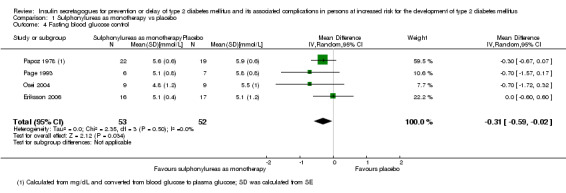
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 4 Fasting blood glucose control.
Subgroup analyses for FBG: a subgroup analysis according to the type of sulphonylurea (third‐generation versus second‐generation) showed no interaction between these subgroups (Analysis 1.5 and Appendix 19). All trials comparing a sulphonylurea monotherapy with placebo included participants with IGT. Two trials had a duration of two years or more (Osei 2004; Papoz 1978). The subgroup analysis according to the duration of the intervention showed no interaction between these subgroups (Analysis 1.6 and Appendix 19). Two trials applied diagnostic criteria for IGT as recommended by WHO (Eriksson 2006; Osei 2004). Subgroup analysis according to diagnostic criteria showed no interaction between these subgroups (Analysis 1.7 and Appendix 19). The participants included in Eriksson 2006 were about 10 to 15 years older (mean age 56.5 years) than the participants in the other trials comparing sulphonylurea monotherapy with placebo (Osei 2004; Page 1993; Papoz 1978). Subgroup analysis for age showed no marked interaction (Analysis 1.8 and Appendix 19). We could not perform subgroup analyses according to ethnicity, comorbidity or previous gestational diabetes mellitus because of lack of reporting in the included trials.
1.5. Analysis.
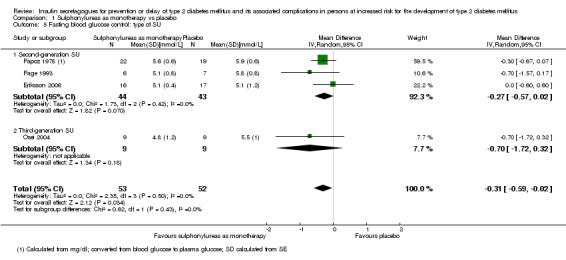
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 5 Fasting blood glucose control: type of SU.
1.6. Analysis.
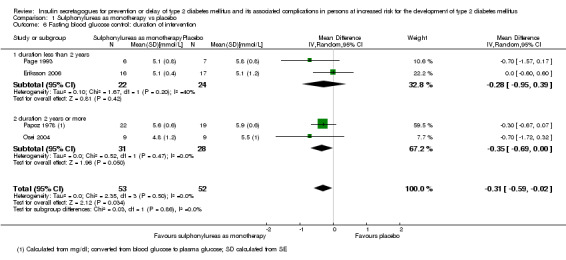
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 6 Fasting blood glucose control: duration of intervention.
1.7. Analysis.
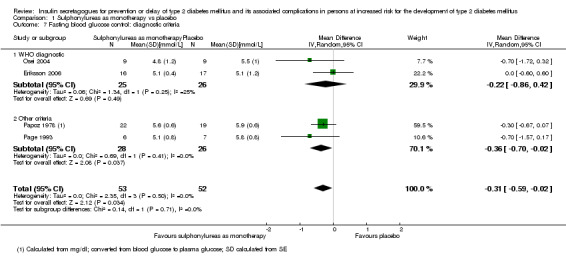
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 7 Fasting blood glucose control: diagnostic criteria.
1.8. Analysis.
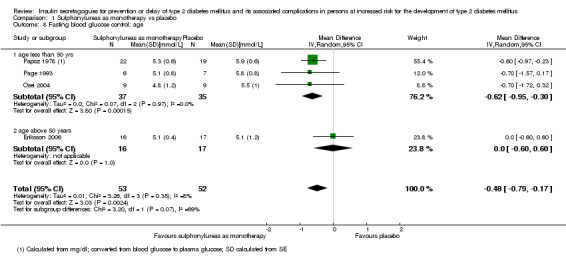
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 8 Fasting blood glucose control: age.
Sensitivity analyses for FBG: a sensitivity analysis excluding trials with a duration of less than two years (Osei 2004; Papoz 1978) showed a MD in fasting glucose of ‐0.28 mmol/L (95% CI ‐0.95 to 0.39). In comparison, trials with a duration of two years or longer showed a MD in fasting glucose of ‐0.35 mmol/L (95% CI ‐0.69 to 0.00) (Analysis 1.6). Only one trial did not receive funding from a pharmaceutical company (Papoz 1978). Excluding this trial resulted in a MD in fasting glucose of ‐0.34 mmol/L (95% CI ‐0.84 to 0.15). The predefined sensitivity analyses regarding publication status, risk of bias, language of publication, imputation or country could not be performed. TSA showed that 23.3% of the diversity‐adjusted information size has been accrued so far to detect or reject a mean difference of ‐0.31 mmol/L. Diversity was zero, but we applied a diversity of 20% as heterogeneity might increase when future trials are added. The TSA‐adjusted 95% CI was ‐0.94 to 0.33.
Two trials reported fasting glucose values after the intervention periods were stopped and observed participants without any intervention (Eriksson 2006; Page 1993). Comparing sulphonylurea monotherapy with placebo, the MD in FBG was ‐0.08 mmol/L (95% CI ‐1.04 to 0.89; Analysis 1.9). TSA showed that 0.17% of the diversity‐adjusted information size had been accrued so far to detect or reject a mean difference of ‐0.08 mmol/L. Diversity was 70.7%. As only a minor fraction of the diversity‐adjusted required information size was accrued so far, the TSA‐adjusted 95% CI could not be calculated.
1.9. Analysis.
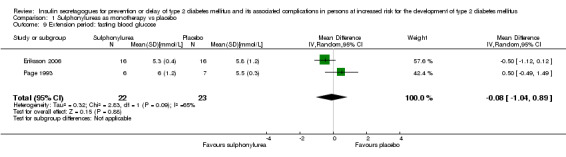
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 9 Extension period: fasting blood glucose.
Blood glucose 2 hours after an oral glucose load
Glucose values 2 hours after an oral glucose load at the end of the intervention period were reported in three trials (Eriksson 2006; Osei 2004; Papoz 1978). Sulphonylurea monotherapy compared with placebo showed a MD in 2‐hour blood glucose of ‐0.42 mmol/L (‐1.28 to 0.43; P = 0.33; 3 trials; 92 participants; overall low or unclear risk of bias; Analysis 1.10). None of the subgroup analyses showed an interaction between the subgroups (Appendix 19).
1.10. Analysis.
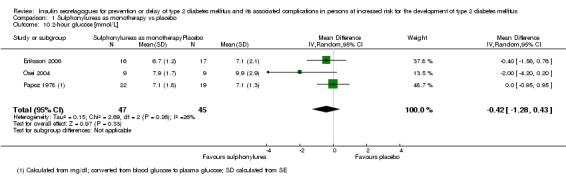
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 10 2‐hour glucose [mmol/L].
Subgroup analyses for type of sulphonylurea (Analysis 1.11), duration of intervention (Analysis 1.12) and diagnostic criteria (Analysis 1.13) showed no interaction between subgroups. We could not perform subgroup analyses according to ethnicity, comorbidity and previous gestational diabetes mellitus because of lack of reporting in the included trials.
1.11. Analysis.
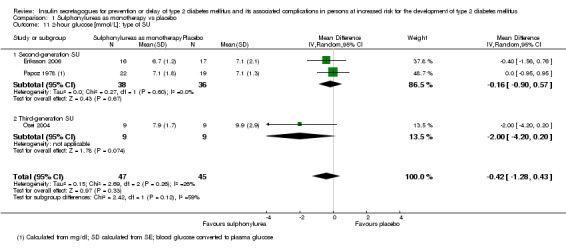
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 11 2‐hour glucose [mmol/L]: type of SU.
1.12. Analysis.
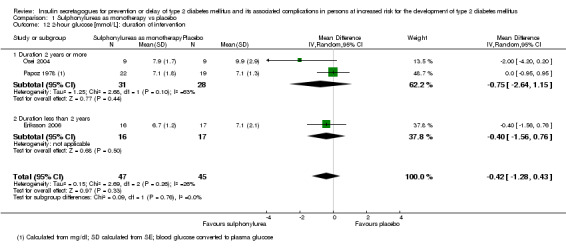
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 12 2‐hour glucose [mmol/L]: duration of intervention.
1.13. Analysis.
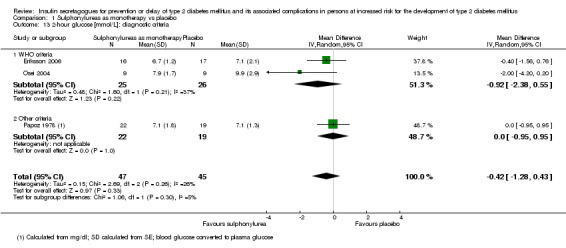
Comparison 1 Sulphonylureas as monotherapy vs placebo, Outcome 13 2‐hour glucose [mmol/L]: diagnostic criteria.
One trial reported glucose values 2 hours after an oral glucose load one month after the intervention period had ended (Eriksson 2006). The glucose values 2 hours after oral glucose load had a mean of 7.0 mmol/L (SD 1.6) in 16 participants originally allocated to sulphonylurea monotherapy and 8.6 mmol/L (SD 2.4) in 16 participants originally allocated placebo.
HbA1c
One trial clearly stated that HbA1c was measured but reported only that no statistically significant changes were found (NANSY 2011).
Socioeconomic effects
We did not identify trials with data on socioeconomic effects for this comparison.
Sulphonylureas as monotherapy versus no intervention
We did not identify trials comparing sulphonylurea monotherapy with no intervention.
Sulphonylureas as a part of combination therapy versus any pharmacological glucose‐lowering agent
One trial compared sulphonylureas as a part of combination therapy with another pharmacological glucose‐lowering agent. This trial had four intervention arms: glibenclamide plus metformin, glibenclamide plus placebo, metformin plus placebo and placebo only (Papoz 1978). This trial reported fasting glucose and glucose values 2 hours after an oral glucose load. Glucose values were reported as blood glucose values. The FBG value at the end of intervention in the glibenclamide plus metformin group was 5.2 (SD 0.8) mmol/L measured in 22 participants versus 5.3 (SD 0.5) mmol/L in 23 participants in the metformin plus placebo group. The blood glucose value 2 hours after an oral glucose load was 6.4 (SD 1.0) mmol/L in 22 participants in the glibenclamide plus metformin group versus 6.4 (SD 1.3) mmol/L in 23 participants in the metformin plus placebo group. For both glycaemic variables we converted data from mg/dL into mmol/L and calculated SDs from reported standard errors (Papoz 1978).
Meglitinide analogues as monotherapy versus any pharmacological glucose‐lowering intervention
We did not identify trials comparing meglitinide analogues as monotherapy with other pharmacological glucose‐lowering interventions.
Meglitinide analogues as monotherapy versus behaviour‐changing interventions
We did not identify trials comparing meglitinide analogues as monotherapy with behaviour changing interventions.
Meglitinide analogues as monotherapy versus placebo
One trial compared a meglitinide analogue as monotherapy (nateglinide) with placebo (NAVIGATOR 2010). The trial had a factorial design, as the participants were also randomised to valsartan or placebo (NAVIGATOR 2010). A narrative description of the outcomes is listed in the Table 2. Because we identified only one trial with meglitinide analogues we could not perform meta‐analyses or TSA.
Primary outcomes
All‐cause mortality
A total of 310 of 4645 (6.7%) participants allocated to nateglinide versus 312 of 4661 (6.7%) participants allocated to placebo died during the trial. Participants who were considered lost to follow‐up remained in the trial until trial end or until death, if known. We determined vital status at each visit and at trial end by searching public records. Vital status was available for 95.7% of the possible participants at the end of follow‐up. HR for all‐cause mortality was 1.00 (95% CI 0.85 to 1.17; P = 0.98).
Incidence of T2DM
The two main criteria for defining T2DM in NAVIGATOR trial were a FPG level ≥ 7.0 mmol/L or a 2‐hour post challenge glucose ≥ 11.1 mmol/L. A confirmatory oral glucose tolerance test had to be performed within 12 weeks after measurement of an elevated glucose level. The date of onset of T2DM was specified as the date of the first elevated glucose measurement. Furthermore, an independent committee adjudicated cases where diabetes was diagnosed by other means (e.g. cases suggestive of diabetes where the glycaemic test‐based definition was not available because of missing central laboratory measurements or repeat tests outside the 12‐week time limit). The committee also adjudicated cases where diabetes was diagnosed by a primary care physician (possibly based on local laboratory assessments), initiation of glucose‐lowering interventions or both. On testing days the participants were told to take the trial drug after the glucose tests.
T2DM developed in 1674 of 4645 (36.0%) participants in the nateglinide group and in 1580 of 4661 (33.9%) in the placebo group. The test of interaction for the factorial allocation to valsartan and placebo did not show any important influence on the effect estimate (P = 0.5). The HR for the incidence of T2DM was 1.07 (95% CI 1.00 to 1.15; P = 0.05). The incidence of T2DM was confirmed by laboratory measurements in 1587 participants in the nateglinide group and 1495 participants in the placebo group. The incidence of T2DM was determined by the adjudication committee in 87 participants in the nateglinide group and 85 participants in the placebo group.
The trial authors also investigated the influence of several factors on the risk of developing T2DM by means of subgroup analyses, such as age, sex, ethnicity, region, fasting plasma glucose, 2‐hour postprandial glucose, BMI, waist circumference, blood pressure control, hypertension, history of cardiovascular disease and angiotensin‐converting enzyme (ACE) inhibitor treatment. All reported HRs refer to the development of T2DM.
A diagnosis on the basis of FPG resulted in a HR of 0.87 (95% CI 0.79 to 0.96; P = 0.005) in favour of nateglinide. A diagnosis on the basis of plasma glucose levels 2 hours after a glucose challenge resulted in a HR of 1.24 (95% CI 1.13 to 1.36; P < 0.001) in favour of placebo.
Participants aged < 60 years had a HR of 0.99 (95% CI 0.88 to 1.12), participants aged 60 to 67 years showed a HR of 1.19 (95% CI 1.06 to 1.35) and participants aged ≥ 67 years had a HR of 1.04 (95% CI 0.91 to 1.18) in favour of placebo.
Male participants had a HR of 0.96 (95% CI 0.87 to 1.06) and female participants a HR of 1.21 (95% CI 1.10 to 1.34) in favour of placebo.
Participants from Asian regions had a HR of 1.20 (95% CI 0.90 to 1.61), participants from European regions had a HR of 1.08 (95% CI 0.98 to 1.19), participants from Latin America had a HR of 1.02 (95% CI 0.86 to 1.21) and participants from North America had a HR of 1.05 (95% CI 0.91 to 1.22). Participants from various other regions had a HR of 1.26 (95% CI 0.84 to 1.91).
Participants with a FPG ≤ 6.1 mmol/L had a HR of 1.17 (95% CI 1.04 to 1.30) in favour of placebo. Participants with a FPG > 6.1 mmol/L had a HR of 1.01 (95% CI 0.92 to 1.11).
Participants with a 2‐hour plasma glucose value ≤ 9.0 mmol/L had a HR of 1.07 (95% CI 0.95 to 1.19). Participants with 2‐hour plasma glucose > 9 mmol/L had a HR of 1.07 (95% CI 0.97 to 1.17).
Participants with a BMI ≤ 25 kg/m2 had a HR of 1.14 (95% CI 0.92 to 1.42). Participants with a BMI 25 kg/m2 to 30 kg/m2 had a HR of 1.16 (95% CI 1.03 to 1.30) in favour of placebo. Participants with a BMI 30 kg/m2 to 35 kg/m2 had a HR of 1.00 (95% CI 0.88 to 1.13). Participants with a BMI > 35 kg/m2 had a HR of 1.01 (95% CI 0.86 to 1.18).
Participants with a waist circumference < 88 cm (women) and < 102 cm (men) had a HR of 1.17 (95% CI 1.03 to 1.33) in favour of placebo. Participants with a waist circumference ≥ 88 cm (women) and ≥ 102 cm (men) had a HR of 1.03 (95% CI 0.95 to 1.13).
Participants with systolic blood pressure < 140 mmHg or diastolic blood pressure < 90 mmHg had a HR of 1.11 (95% CI 1.00 to 1.23). Participants with a systolic blood pressure > 140 mmHg or a diastolic blood pressure > 90 mmHg or taking antihypertensive drugs had a HR of 1.05 (95% CI 0.95 to 1.15).
Participants with a history of cardiovascular disease had a HR of 1.04 (95% CI 0.91 to 1.20). Participants without a history of cardiovascular disease had a HR of 1.09 (95% CI 1.00 to 1.18).
Participants with ACE inhibitor treatment had a HR of 0.90 (95% CI 0.70 to 1.15) and participants without ACE inhibitor treatment a HR of 0.90 (95% CI 0.70 to 1.15).
Serious adverse events
Serious adverse events were reported in 2066/4602 (44.9%) participants in the nateglinide group versus 2089/4599 (45.4%) participants in the placebo group
Secondary outcomes
Cardiovascular mortality
The numbers of participants who died due to cardiovascular disease during the trial were 126/4645 (2.7%) allocated to nateglinide versus 118/4661 (2.5%) allocated to placebo. The HR for death due to cardiovascular disease was 1.07 (95% CI 0.83 to 1.38; P = 0.60).
Non‐fatal myocardial infarction
The numbers of participants who experienced a non‐fatal myocardial infarction during the trial were 116/4645 (2.5%) in the nateglinide group versus 122/4661 (2.6%) in the placebo group.
Non‐fatal stroke
The numbers of participants who experienced a non‐fatal stroke during the trial were 100/4645 (2.2%) in the nateglinide group versus 110/4661 (2.4%) in the placebo group.
Congestive heart failure
The numbers of participants developing congestive heart failure were not reported. However, the numbers of participants hospitalised for congestive heart failure were 85/4645 (1.8%) in the nateglinide group versus 100/4661 (2.1%) allocated to placebo. The HR was 0.85 (95% CI 0.64 to 1.14; P = 0.27).
Amputation of lower extremity
No data on amputation of the lower extremity were reported.
Blindness or severe vision loss
One participant developed blindness during the trial. This participant was allocated to the placebo group.
End‐stage renal disease
No data on end‐stage renal disease were reported. However, it is very likely that data on this outcome had been collected.
Non‐serious adverse events
The numbers of participants who experienced a non‐serious adverse event were 3921/4602 (85.2%) allocated to nateglinide group versus 3866/4599 (84.1%) allocated to placebo.
Hypoglycaemia
A mild hypoglycaemic event was experienced by 676/4645 (14.6%) participants allocated to nateglinide versus 411/4661 (8.8%) participants allocated to placebo. Severe hypoglycaemia was experienced by 21/4645 (0.5%) allocated to nateglinide versus 12/4661 (0.3%) allocated to placebo.
Health‐related quality of life
No data on health‐related quality of life were reported.
Time to progression to T2DM
Please see section 'Incidence of T2DM' above.
Measures of blood glucose control
Fasting glucose values were lower in participants in the nateglinide group than in those in the placebo group during the trial (MD 0.03 mmol/L; 95% CI 0.003 to 0.05; P = 0.03). However, no statistically significant difference between the intervention groups was apparent at the end of follow‐up. FBG values and SDs at the end of follow‐up were not reported, but we estimated these from the published figure. We estimated FPG measurements at the end of follow‐up of 6.2 mmol/L (SD 1.7) in the nateglinide group versus 6.3 mmol/L (SD 2.6) in the placebo group. We were not able to estimate how many participants were included in the analyses of glucose measurements at the end of follow‐up from the figure in the publication.
Two hours after an oral glucose load glucose values were higher in the nateglinide group than in the placebo group (MD 0.24 mmol/L; 95% CI 0.16 to 0.33; P < 0.001). According to the figure in the publication, glucose values 2 hours after an oral glucose load were 9.5 mmol/L (SD 3.4) in the nateglinide group and 9.2 mmol/L (SD 3.4) in the placebo group. However, we were not able to estimate how many participants were included in the analyses of glucose measurements at the end of follow‐up from the figure in the publication.
It was clearly stated in the publication that HbA1c was measured. However, trial authors reported HbA1c values only at the time the diagnosis of T2DM was established (nateglinide 6.1% (SD 0.6) versus placebo 6.3% (SD 0.6)).
Socioeconomic effects
No data on socioeconomic effects were reported. However, trial authors stated that health economics assessments would be performed.
Meglitinide analogues as monotherapy versus no intervention
We did not identify trials comparing meglitinide analogues as monotherapy with other glucose‐lowering interventions.
Meglitinide analogues as a part of combination therapy versus any pharmacological glucose‐lowering agent
We did not identify trials comparing meglitinide analogues as monotherapy with other glucose‐lowering interventions.
Subgroup analyses
We did not perform subgroups analyses for most comparisons because there were insufficient trials to estimate effects in various subgroups. However, we performed subgroup analyses for the comparison sulphonylureas as monotherapy versus placebo (Appendix 19).
Sensitivity analyses
We did not perform sensitivity analyses for most comparisons because there were insufficient trials to explore the influence of our predefined factors on effect sizes. However, we performed sensitivity analyses for the comparison sulphonylureas as monotherapy versus placebo (Appendix 19).
Assessment of reporting bias
We did not draw funnel plots due to limited number of trials.
Ongoing trials
We did not identify any ongoing RCTs.
Discussion
Summary of main results
This Cochrane review is the first systematic review investigating the effects of insulin secretagogues versus other pharmacological glucose‐lowering interventions, placebo, diet and exercise or no intervention in people at increased risk for the development of type 2 diabetes mellitus (T2DM). We included six trials with a total of 10,018 participants. We judged all trials as at unclear or high risk of bias in one or more 'Risk of bias' domains. The amount of evidence on patient‐important outcomes was limited. The single meta‐analysis comparing sulphonylurea monotherapy (glimepiride) with placebo on the incidence of T2DM after the end of the intervention established neither an advantage nor a disadvantage for glimepiride treatment on the development of T2DM. Here, as well as for all‐cause mortality and cardiovascular mortality, we judged the quality of evidence as very low. All of the included trials reported one or more glycaemic variables. However, for all variables the diversity‐adjusted required information size to confirm the findings from the meta‐analyses was not reached.
One large trial investigated a meglitinide analogue (nateglinide). There was moderate‐quality evidence for the outcomes all‐cause and cardiovascular mortality, incidence of T2DM, serious adverse events, non‐fatal myocardial infarction or stroke and congestive heart failure. Overall, we observed firm evidence neither for or against nateglinide treatment.
Overall completeness and applicability of evidence
We conducted an extensive search for trials, including publications in all languages, and tried to obtain additional data on all trials. However, no additional data were provided. We looked for additional trials and cross‐checked our data with the data from other meta‐analyses and Cochrane reviews of relevance (Anderson 2005; Bhardwaj 2010; Hopper 2011; Phung 2012; Van de Laar 2006).
The diagnosis of intermediate hyperglycaemia varied among trials and some trials used a definition which may have included participants judged to be euglycaemic or having T2DM. Detailed information about the participants was lacking in most trials, and only one trial reported the number of participants with previous cardiovascular disease at baseline (NAVIGATOR 2010). The included trials applied different types of sulphonylurea at varying doses (Eriksson 2006; NANSY 2011; Osei 2004; Page 1993; Papoz 1978). Only one trial included a meglitinide analogue (NAVIGATOR 2010).
A potential selection bias exists as more healthy and motivated people may participate in a clinical trial. However, a Cochrane systematic review observed that clinical outcomes in people participating in RCTs are comparable to similar individuals outside trials (Vist 2008).
Quality of the evidence
None of the six included trials in our review was classified as at low risk of bias on all 'Risk of bias' domains. In general, the description of randomisation and allocation in the included studies was insufficient. Most trials had insufficient reporting of one or more outcomes of relevance to our review and were classified as at high risk of bias for selective outcome reporting bias. We were able to assess one or more of our predefined outcomes in all included trials.
For the sulphonylureas (glimepiride) we judged the quality of evidence to be very low because of the risk of bias and very limited data resulting in imprecision. For the meglitinide analogues (nateglinide), we judged the quality of evidence to be moderate, mainly due to imprecision.
Certain potential limitations of this review warrant special consideration, one being that we were dealing with a heterogeneous group of trials. Our meta‐analyses are limited by an inability to use individual patient data to assess whether distinct clinical characteristics may have influenced the effect estimates of the interventions. We would have explored heterogeneity using sensitivity analyses for our patient‐important outcomes, if possible. However, only two meta‐analyses (both on glycaemic variables) provided sufficient data to perform subgroup and sensitivity analyses. Many of the included trials were not designed or powered to detect our predefined patient‐important outcomes.
Some trials required the participants to take the study drug on the days the glycaemic variables were measured, whereas others did not. This may have influenced the glucose measurements in these trials, as well as the incidence of T2DM (which is based on glycaemic measurements) making it difficult to compare incidence rates.
Some of the trials reported glucose values in whole blood whereas other reported glucose values in plasma. We converted values of whole blood glucose to plasma glucose values in order to make the data comparable. However, it is well known that such conversions might be inaccurate (WHO/IDF 2006).
Most of the included trials had a relatively small number of participants and the information sizes in the meta‐analyses were equally small. This increases the risk of unrealistic estimates of the intervention effects due to bias (systematic errors) and chance (random errors) (Wetterslev 2008; Wood 2008). We have attempted to clarify systematic errors. We contacted all trial authors for clarification if one of the bias domains was not adequately reported. However, trial authors provided no additional information. To reduce the risk of random errors, we conducted trial sequential analyses on all predefined outcomes, whenever possible.
All trials except one (Papoz 1978) received funding from the pharmaceutical industry. It is known that trials receiving funding or provision of free drug or devices from a pharmaceutical company lead to more favourable results and conclusions than trials sponsored by other sources (Lundh 2012).
Potential biases in the review process
Despite an extensive search and attempts to contact the authors of the included trials we did not retrieve any additional trials. We were unable to draw funnel plots in order to assess small study bias due to lack of data. If more data had been available and more meta‐analyses could have been performed we would have tried to investigate heterogeneity and the potential reasons for it. We excluded several trials as they did not provide separate data on participants with intermediate hyperglycaemia (DIANA 2012; Igata 2014; Major‐Pedersen 2008; The Fasting Hyperglycaemia Study 1997a). In all cases we approached the investigators in order to request separate data. None of the investigators provided additional data.
We excluded studies investigating first‐generation sulphonylureas because of the current very limited use of these drugs,
Several trials were published in more than one publication, which for some trials made it difficult to separate the primary publication from companion papers (for details see Included studies).
We made a concerted effort to obtain additional data from trial authors. As most of the included trials were relatively old, we found it difficult to identify contact information for some trials. However, if we were unable to retrieve contact information for the corresponding author, we attempted to contact one of the other coauthors. For all trials we identified contact information for one or more authors.
We excluded trials including participants with IGT due to other conditions (e.g. cystic fibrosis or glucocorticoid treatment).
We included trials with a minimum duration of 12 weeks in order to be able to detect clinically relevant differences for the predefined outcomes. We identified three trials with a duration of less than 12 weeks. Unfortunately, the reporting of long‐term data in the trials included in our review was poor.
We excluded studies assessing composite macrovascular outcomes from our review because composite outcomes are often problematic due to varying definitions in composite outcome measures. One of the trials (NAVIGATOR 2010) reported two composite cardiovascular outcomes (an extended composite cardiovascular outcome; HR 0.93; 95% CI 0.83 to 1.03; P = 0.16) and a core composite cardiovascular outcome (HR 0.94; 95% CI 0.82 to 1.09; P = 0.43).
Data extraction was carried out by two review authors. However, the review authors extracting the data were not blinded as to which trial they were extracting data from.
Agreements and disagreements with other studies or reviews
Several RCTs have assessed the effects of different pharmacological glucose‐lowering interventions for the prevention of T2DM (ACT NOW 2011; Diabetes Prevention Program FU 2009; DREAM 2008). The first RCT to investigate whether an insulin secretagogue could be effective in people at high risk of T2DM was the Bedford trial (Bedford 1982). The trial randomised participants to tolbutamide versus placebo. The trial found no statistically significant difference in the incidence of T2DM between the intervention groups after 8.5 years of follow‐up (Bedford 1982). Later, the Malmö trial randomised participants with IGT to tolbutamide versus diet, placebo, both, or no intervention and followed participants for 10 years (Sartor 1980). More than 50% of the participants in the tolbutamide group dropped out and data on T2DM prevention were inconclusive (Sartor 1980).
A pharmacological approach to the prevention of T2DM is appealing to both the clinician and the pharmaceutical industry. However, although a reduction in the incidence of T2DM is important, the major public health impact of prevention trials will be determined by whether the prevention ‐ or a delay ‐ in the development of T2DM will translate into a reduction in diabetes‐specific macro‐ and microvascular complications.
The results of the factorial NAVIGATOR RCT were largely inconclusive. Neither nateglinide (nor the combination of nateglinide and valsartan) definitely reduced the incidence of T2DM or the events of the two coprimary cardiovascular disease outcomes. The only positive result was a reduction in the incidence of T2DM with valsartan (HR 0.86; 95% CI 0.80 to 0.92; P < 0.001).
Unfortunately, our review could not clarify whether insulin secretagogues reduce the incidence of T2DM and its associated complications in individuals at high risk of T2DM. None of the trials showed statistically significant reductions in blood glucose values 2 hours after an oral glucose load at the end of the intervention compared with placebo. This was also the case for the NAVIGATOR trial, which might seem surprising, taking the mechanism of action of the meglitinide analogues into account (NAVIGATOR 2010). The authors of the NAVIGATOR trial describe this paradoxical finding as a rebound effect, since nateglinide was not administered on the mornings when the oral glucose tolerance tests were performed (NAVIGATOR 2010). Another trial did also not administer the trial drug (GITS) on the mornings when the glycaemic measurements were performed (Osei 2004). However, in this trial both fasting blood glucose (FBG) and 2‐hour glucose levels were higher in the placebo group than in the GITS group. Nevertheless, no statistically significant difference between the groups was seen. If such a rebound effect really exists, it remains to be proven.
When all the trials investigating sulphonylureas were combined FBG was reduced compared with placebo. In the NAVIGATOR trial, FBG was lower in participants allocated to nateglinide than in those receiving placebo. None of the trials reported end‐of study HbA1c levels, which can partly be explained by the age of some of the trials. However, although the NAVIGATOR trial measured HbA1c it reported this measurement only for participants who developed T2DM. For the participants who developed T2DM in the NAVIGATOR trial, HbA1c was lower in those allocated to nateglinide than in those receiving placebo. No increased risk of hypoglycaemia was reported in the trials comparing a sulphonylurea with placebo. However, the trials rarely reported hypoglycaemia as an outcome; when they did, such reporting was often insufficient (Eriksson 2006; Osei 2004; Page 1993). The only outcome which seemed to be influenced by nateglinide in the NAVIGATOR trial was hypoglycaemia; there was an increased risk with nateglinide versus placebo (NAVIGATOR 2010).
We did not identify any ongoing trials investigating the effects of an insulin secretagogue in people at increased risk of the development of T2DM. This reflects a lack of interest from the scientific community and the pharmaceutical companies as their focus now is towards newer and more expensive pharmacological glucose‐lowering interventions (Hemmingsen 2016a).
Authors' conclusions
Implications for practice.
There is insufficient evidence to demonstrate whether insulin secretagogues compared with pharmacological glucose‐lowering interventions, placebo, behaviour‐changing interventions or no intervention influence the risk of type 2 diabetes mellitus and its associated complications. The evidence on patient‐important outcomes such as mortality, macro‐ and microvascular complications is sporadic and sparsely addressed in the existing trials. We are currently not able to provide a reliable benefit:risk ratio for this type of intervention in preventing or delaying the development of type 2 diabetes mellitus.
Implications for research.
Even though it remains to be clarified whether there are any beneficial or harmful effects of insulin secretagogues in people at high risk of type 2 diabetes mellitus, no ongoing trials are investigating this issue. If new randomised controlled trials are to be performed in the future, they should focus on patient‐important outcomes. In addition, future trials should be reported according to the CONSORT (CONsolidated Standards of Reporting Trials) statement.
Notes
Portions of the background and methods sections, the appendices, additional tables and figures 1 to 3 of this review are based on a standard template established by the Cochrane Metabolic and Endocrine Disorders Group.
Acknowledgements
Thanks to Erika Ota for evaluating the Japanese references.
Appendices
Appendix 1. Trials with a trial duration of less than 12 weeks
| Intervention(s) and comparator(s) | Design | Duration of intervention | Randomised (N) | Description of participants | Outcomes reported | Outcomes reported with interest for this review | Stated purpose of study | |
| Schmoelzer 2006 | I: repaglinide 2 mg | Assuming single centre, single blinded (blinded to investigators), cross‐over trial | One administration; participants were investigated once (previous to 1 dose nateglinide); ˜ 1 week between phases | 12 | Impaired glucose tolerance (fasting blood glucose < 7.0 mmol/L
and 2‐hour blood glucose
≥ 7.8 mmol/L and < 11.1 mmol/L) Mainly males |
Flow mediated dilatation during oral glucose tolerance test, plasma insulin during oral glucose tolerance test, fasting blood glucose, 2‐hour blood glucose after oral glucose tolerance test | Fasting blood glucose, 2‐hour blood glucose after oral glucose tolerance test | Quote: "Consequently, the aim of our study was to investigate, whether reduction of post‐challenge glucose‐excursions with repaglinide, a short‐acting non‐sulfonylurea insulin secretagogue influences endothelial function during an oGTT in patients with IGT" |
| C: no intervention | ||||||||
| Saloranta 2002 | I1: nateglinide 30 mg | Multicentre (32 centres in six countries), double‐blind, parallel group | 8 weeks | 83 | Impaired glucose tolerance (fasting blood glucose < 7.0 mmol/L and 2‐hour post challenge values between 7.8 to 11.1 mmol/L) | Mild hypoglycaemia, severe hypoglycaemia (none occurred), adverse effects, serious adverse effects, deaths (none occurred), insulin response, fasting plasma glucose, fasting insulin response, HbA1c, fructosamine levels | Mild hypoglycaemia, severe hypoglycaemia, adverse effects, serious adverse effects, deaths, fasting plasma glucose, HbA1c | Quote: "The purpose of this study was to evaluate the metabolic effectiveness, safety, and tolerability of nateglinide in subjects with impaired glucose tolerance (IGT) and to identify a dose appropriate for use in a diabetes prevention study" |
| I2: nateglinide 60 mg | 76 | |||||||
| I3: nateglinide 120 mg | 86 | |||||||
| C1: placebo | 43 | |||||||
| Lindblad 2001 | I1: glimepiride 0.5 mg | Muliticentre (7 centres in Sweden), double‐blind, cross‐over pilot study to NANSY (The NEPI [The Network for Pharmacoepidemiology] Antidiabetes Study) | The participants completed four periods of one week intervention in each intervention group. One to three weeks washout period | 25 | Impaired fasting glucose (defined by a mean of three consecutive fasting blood glucose values in the range of 5.6 to 6.0 mmol/L) | Area under the curve for blood glucose, 2‐hour post challenge blood glucose, fasting blood glucose, insulin, proinsulin, mild hypoglycaemia, serious hypoglycaemia | 2‐hour post challenge blood glucose, fasting blood glucose, mild hypoglycaemia, serious hypoglycaemia | Quote: "This pilot study was conducted to find the optimum dose of glimepiride in NANSY" |
| I2: glimepiride 1.0 mg | ||||||||
| I3: glimepiride 2.0 mg | ||||||||
| C1: placebo | ||||||||
| C: comparator; HbA1c: glycosylated haemoglobin A1c; I: intervention; IGT: impaired glucose tolerance; oGTT: oral glucose tolerance test | ||||||||
Appendix 2. Search strategies
| MEDLINE (Ovid SP) |
|
Block 1: Prediabetes 1. Prediabetic state/ 2. Glucose Intolerance/ 3. (prediabet* or pre diabet*).tw. 4. intermediate hyperglyc?emi*.tw. 5. ((impaired fasting adj2 glucose) or IFG or impaired FPG).tw. 6. glucose intolerance.tw. 7. ((impaired glucose adj (tolerance or metabolism)) or IGT).tw. 8. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) adj4 (diabetes or T2D* or NIDDM or "type 2" or "type II")).tw. 9. or/1‐8 Block 2: SU or Glinides 10. exp Sulfonylurea Compounds/ 11. (sulfon?lurea* or sulphon?lurea*).tw. 12. (gl?benclamid* or glyburid* or HB 419 OR HB419 or HB 420 OR HB420).tw. 13. (gl?bornurid* or Ro 6 4563 or Ro 4563 or gluborid*).tw. 14. (glipizid* or gl?diazinamide or glypidizine or K 4024 or K4024 or melizide or napizide).tw. 15. (gliquidon* or AR DF 26 or ARDF 26 or ARDF26).tw. 16. (glisoxepid* or RP 22410 or BS 4231).tw. 17. gl?clopyramid*.tw. 18. (glimepirid* or HOE 490).tw. 19. (gl?clazid* or gl?cazid* or S 1702 or S1702 or S 852 OR S852).tw. 20. glinide.mp. 21. (nateglinid* or senaglinid* or IPCCPA or AY4166 or AY 4166 or DJN 608 or DJN608 or A 4166 or A4166 or YM 026 or YM026).mp. 22. (repaglinid* or AG EE 388 or AG EE 623 or AG EE388 or AG EE623).mp. 23. (mitiglinid* or S 21403 or S21403 or KAD 1229 or KAD1229).mp. 24. or/10‐23 Block 1 and block 2 and RCT/SR‐filter 25. 9 and 24 [26‐36: Cochrane Handbook 2008 RCT filter ‐ sensitivity max. version] 26. randomized controlled trial.pt. 27. controlled clinical trial.pt. 28. randomi?ed.ab. 29. placebo.ab. 30. drug therapy.fs. 31. randomly.ab. 32. trial.ab. 33. groups.ab. 34. or/26‐33 35. exp animals/ not humans/ 36. 34 not 35 37. 25 and 36 [38:Wong 2006a– systematic reviews filter – SensSpec version] 38. meta analysis.mp,pt. or review.pt. or search*.tw. 39. 25 and 38 40. 37 or 39 |
| Cochrane Central Register of Controlled Trials (Cochrane Register of Studies Online) |
| 1. MESH DESCRIPTOR Prediabetic state 2. MESH DESCRIPTOR Glucose Intolerance 3. (prediabet* or pre diabet*):TI,AB,KY 4. (intermediate hyperglyc?emi*):TI,AB,KY 5. ((impaired fasting ADJ2 glucose) or IFG or impaired FPG):TI,AB,KY 6. glucose intolerance:TI,AB,KY 7. ((impaired glucose ADJ (tolerance or metabolism)) or IGT):TI,AB,KY 8. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) ADJ4 (diabetes or T2D* or NIDDM or "type 2" or "type II")):TI,AB,KY 9. #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 10. MESH DESCRIPTOR Sulfonylurea Compounds EXPLODE ALL TREES 11. (sulfon?lurea* or sulphon?lurea*):TI,AB,KY 12. (gl?benclamid* or glyburid* or HB 419 OR HB419 or HB 420 OR HB420):TI,AB,KY 13. (gl?bornurid* or Ro 6 4563 or Ro 4563 or gluborid*):TI,AB,KY 14. (glipizid* or gl?diazinamide or glypidizine or K 4024 or K4024 or melizide or napizide):TI,AB,KY 15. (gliquidon* or AR DF 26 or ARDF 26 or ARDF26):TI,AB,KY 16. (glisoxepid* or RP 22410 or BS 4231):TI,AB,KY 17. gl?clopyramid*:TI,AB,KY 18. (glimepirid* or HOE 490):TI,AB,KY 19. (gl?clazid* or gl?cazid* or S 1702 or S1702 or S 852 OR S852):TI,AB,KY 20. glinide:TI,AB,KY 21. (nateglinid* or senaglinid* or IPCCPA or AY4166 or AY 4166 or DJN 608 or DJN608 or A 4166 or A4166 or YM 026 or YM026):TI,AB,KY 22. (repaglinid* or AG EE 388 or AG EE 623 or AG EE388 or AG EE623):TI,AB,KY 23. (mitiglinid* or S 21403 or S21403 or KAD 1229 or KAD1229):TI,AB,KY 24. #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 25. #9 AND #24 |
| Embase (Ovid SP) |
|
Block 1: Prediabetes 1. (prediabet* or pre diabet*).tw. 2. intermediate hyperglyc?emi*.tw. 3. ((impaired fasting adj2 glucose) or IFG or impaired FPG).tw. 4. glucose intolerance.tw. 5. ((impaired glucose adj (tolerance or metabolism)) or IGT).tw. 6. ((risk or progress* or prevent* or inciden* or conversion or develop* or delay*) adj4 (diabetes or T2D* or NIDDM or "type 2" or "type II")).tw. 7. or/1‐6 Block 2: SU or Glinides 8. gliamilide/ or glibenclamide/ or glibornuride/ or glicaramide/ or gliclazide/ or glicondamide/ or gliflumide/ or glimepiride/ or glipalamide/ or glipentide/ or glipizide/ or gliquidone/ or glisamuride/ or glisolamide/ or glisoxepide/ or glucosulfa/ or glybuthiazol/ or glybuzole/ or glycyclamide/ or glyhexamide/ or glyoctamide/ or glyparamide/ or glypinamide/ or glyprothiazol/ or glysobuzole/ 9. (sulfon?lurea* or sulphon?lurea*).tw. 10. (gl?benclamid* or glyburid* or HB 419 OR HB419 or HB 420 OR HB420).tw. 11. (gl?bornurid* or Ro 6 4563 or Ro 4563 or gluborid*).tw. 12. (glipizid* or gl?diazinamide or glypidizine or K 4024 or K4024 or melizide or napizide).tw. 13. (gliquidon* or AR DF 26 or ARDF 26 or ARDF26).tw. 14. (glisoxepid* or RP 22410 or BS 4231).tw. 15. gl?clopyramid*.tw. 16. (glimepirid* or HOE 490).tw. 17. (gl?clazid* or gl?cazid* or S 1702 or S1702 or S 852 OR S852).tw. 18. glinide.tw. 19. nateglinide/ 20. repaglinide/ 21. mitiglinide/ 22. (nateglinid* or senaglinid* or IPCCPA or AY4166 or AY 4166 or DJN 608 or DJN608 or A 4166 or A4166 or YM 026 or YM026).tw. 23. (repaglinid* or AG EE 388 or AG EE 623 or AG EE388 or AG EE623).tw. 24. (mitiglinid* or S 21403 or S21403 or KAD 1229 or KAD1229).tw. 25. or/8‐24 Block 1 and block 2 and sound treatment studies‐filter 26. 7 and 25 [27: Wong 2006b "sound treatment studies" filter ‐ SDSSGS version] 27. random*.tw. or clinical trial*.mp. or exp treatment outcome/ 28. 26 and 27 |
| ClinicalTrials.gov (Expert search) |
| ( prediabetes OR prediabetic OR "pre diabetes" OR "pre diabetic" OR hyperglycemia OR hyperglycaemia OR hyperglycemic OR hyperglycaemic OR "impaired glucose tolerance" OR "impaired fasting glucose" OR "glucose intolerance" OR IGT OR IFG OR ((diabetes OR "type 2" OR "type II" OR T2D OR T2DM) AND (risk OR progress OR progression OR progressed OR incident OR incidence OR conversion OR developed OR development OR develop OR delay OR delayed OR prevention OR prevent OR prevented)) ) AND ( gliamilide OR glibenclamide OR glybenclamide OR glibornuride OR glybornuride OR glicaramide OR gliclazide OR glyclazide OR glicondamide OR gliflumide OR glimepiride OR glipalamide OR glipentide OR glipizide OR glydiazinamide OR glidiazinamide OR glypidizine OR melizide OR napidizide OR gliquidone OR glisamuride OR glisolamide OR glisoxepide OR glucosulfa OR glyburide OR glybuthiazol OR gluboride OR glybuzole OR glycyclamide OR glyhexamide OR glyoctamide OR glyparamide OR glypinamide OR glyprothiazol OR glysobuzole OR gliclopyramide OR glyclopyramide OR "HB 419" OR HB419 OR "HB 420" OR HB420 OR "Ro 6 4563" OR "Ro 4563" OR "K 4024" OR K4024 OR "AR DF 26" OR "ARDF 26" OR ARDF26 OR "RP 22410" OR "BS 4231" OR "HOE 490" OR "S 1702" OR S1702 OR "S 852" OR S852 OR sulfonylurea OR sulfonilurea OR sulfonylureas OR sulfonilureas OR sulphonylurea OR sulphonilurea OR sulphonylureas OR sulphonilureas OR glinide OR nateglinide OR repaglinide OR mitiglinide OR senaglinide OR IPCCPA OR AY4166 OR "AY 4166" OR "DJN 608" OR DJN608 OR "A 4166" OR A4166 OR "YM 026" OR YM026 OR "AG EE 388" OR "AG EE 623" OR "AG EE388" OR "AG EE623" OR "S 21403" OR S21403 OR "KAD 1229" OR KAD1229 ) [TREATMENT] |
| WHO ICTRP Search Portal (Standard search) |
| 1) prediabetes AND sulfon* OR pre diabetes AND sulfon* OR impaired glucose tolerance AND sulfon* OR impaired fasting glucose AND sulfon* OR glucose intolerance AND sulfon* OR diabetes AND risk AND sulfon* OR diabetes AND prevent* AND sulfon* OR prediabetes AND sulphon* OR pre diabetes AND sulphon* OR impaired glucose tolerance AND sulphon* OR impaired fasting glucose AND sulphon* OR glucose intolerance AND sulphon* OR diabetes AND risk AND sulphon* OR diabetes AND prevent* AND sulphon* OR prediabetes AND glibenclamid* OR pre diabetes AND glibenclamid* OR impaired glucose tolerance AND glibenclamid* OR impaired fasting glucose AND glibenclamid* OR glucose intolerance AND glibenclamid* OR diabetes AND risk AND glibenclamid* OR diabetes AND prevent* AND glibenclamid* OR prediabetes AND glybenclamid* OR pre diabetes AND glybenclamid* OR impaired glucose tolerance AND glybenclamid* OR impaired fasting glucose AND glybenclamid* OR glucose intolerance AND glybenclamid* OR diabetes AND risk AND glybenclamid* OR diabetes AND prevent* AND glybenclamid* 2) prediabetes AND glyburid* OR pre diabetes AND glyburid* OR impaired glucose tolerance AND glyburid* OR impaired fasting glucose AND glyburid* OR glucose intolerance AND glyburid* OR diabetes AND risk AND glyburid* OR diabetes AND prevent* AND glyburid* OR prediabetes AND glipizid* OR pre diabetes AND glipizid* OR impaired glucose tolerance AND glipizid* OR impaired fasting glucose AND glipizid* OR glucose intolerance AND glipizid* OR diabetes AND risk AND glipizid* OR diabetes AND prevent* AND glipizid* OR prediabetes AND glimepirid* OR pre diabetes AND glimepirid* OR impaired glucose tolerance AND glimepirid* OR impaired fasting glucose AND glimepirid* OR glucose intolerance AND glimepirid* OR diabetes AND risk AND glimepirid* OR diabetes AND prevent* AND glimepirid* OR prediabetes AND gliclazid* OR pre diabetes AND gliclazid* OR impaired glucose tolerance AND gliclazid* OR impaired fasting glucose AND gliclazid* OR glucose intolerance AND gliclazid* OR diabetes AND risk AND gliclazid* OR diabetes AND prevent* AND gliclazid* 3) prediabetes AND nateglinide OR pre diabetes AND nateglinide OR impaired glucose tolerance AND nateglinide OR impaired fasting glucose AND nateglinide OR glucose intolerance AND nateglinide OR diabetes AND risk AND nateglinide OR diabetes AND prevent* AND nateglinide OR prediabetes AND repaglinide OR pre diabetes AND repaglinide OR impaired glucose tolerance AND repaglinide OR impaired fasting glucose AND repaglinide OR glucose intolerance AND repaglinide OR diabetes AND risk AND repaglinide OR diabetes AND prevent* AND repaglinide OR prediabetes AND mitiglinide OR pre diabetes AND mitiglinide OR impaired glucose tolerance AND mitiglinide OR impaired fasting glucose AND mitiglinide OR glucose intolerance AND mitiglinide OR diabetes AND risk AND mitiglinide OR diabetes AND prevent* AND mitiglinide |
| PubMed (subsets not available on Ovid) |
| 1. (prediabet*[tiab] OR pre diabet*[tiab] OR hyperglyc*[tiab] OR ("impaired fasting"[tiab] AND glucose[tiab]) OR IFG[tiab] OR "impaired FPG"[tiab] OR "glucose intolerance"[tiab] OR ("impaired glucose"[tiab] AND (tolerance[tiab] OR metabolism[tiab])) OR IGT[tiab] OR ((risk[tiab] OR progress*[tiab] OR prevent*[tiab] OR inciden*[tiab] OR conversion[tiab] OR develop*[tiab] OR delay*[tiab]) AND (diabetes[tiab] OR T2D*[tiab] OR NIDDM[tiab] OR "type 2"[tiab] OR "type II"[tiab]))) 2. (sulfonylurea*[tiab] OR sulfonilurea*[tiab] OR sulphonylurea*[tiab] OR sulphonilurea*[tiab] OR glybenclamid*[tiab] OR glibenclamid*[tiab] OR glyburid*[tiab] OR "HB 419"[tiab] OR HB419[tiab] OR "HB 420"[tiab] OR HB420[tiab] OR glybornurid*[tiab] OR glibornurid*[tiab] OR "Ro 6 4563"[tiab] OR "Ro 4563"[tiab] OR gluborid*[tiab] OR glipizid*[tiab] OR glydiazinamid*[tiab] OR glidiazinamid*[tiab] OR glypidizin*[tiab] OR "K 4024"[tiab] OR K4024[tiab] OR melizide[tiab] OR napizide[tiab] OR gliquidon*[tiab] OR "AR DF 26"[tiab] OR "ARDF 26"[tiab] OR ARDF26[tiab] OR glisoxepid*[tiab] OR "RP 22410"[tiab] OR "BS 4231"[tiab] OR glyclopyramid*[tiab] OR gliclopyramid*[tiab] OR glimepirid*[tiab] OR "HOE 490"[tiab] OR glyclazid*[tiab] OR gliclazid*[tiab] OR glycazid*[tiab] OR glicazid*[tiab] OR "S 1702"[tiab] OR S1702[tiab] OR "S 852"[tiab] OR S852[tiab] OR glinide[tiab] OR nateglinid*[tiab] OR senaglinid*[tiab] OR IPCCPA[tiab] OR AY4166[tiab] OR "AY 4166"[tiab] OR "DJN 608"[tiab] OR DJN608[tiab] OR "A 4166"[tiab] OR A4166[tiab] OR "YM 026"[tiab] OR YM026[tiab] OR repaglinid*[tiab] OR "AG EE 388"[tiab] OR "AG EE 623"[tiab] OR "AG EE388"[tiab] OR "AG EE623"[tiab] OR mitiglinid*[tiab] OR "S 21403"[tiab] OR S21403[tiab] OR "KAD 1229"[tiab] OR KAD1229[tiab]) 3. #1 AND #2 4. publisher[sb] 5. #3 AND #4 6. (random*[tiab] OR placebo[tiab] OR trial[tiab] OR groups[tiab]) OR (meta analysis[tiab] OR review[tiab] OR search*[tiab]) 7. #5 AND #6 |
Appendix 3. Description of interventions
| Intervention(s) (route, frequency, total dose/day) | Intervention(s) appropriate as applied in a clinical practice settinga (description) | Comparator(s) (route, frequency, total dose/day) | Comparator(s) appropriate as applied in a clinical practice settinga (description) | |
| Eriksson 2006 | Glipizide 2.5 mg, orally, once daily | N/CPS Low dose applied in trial, compared to maximum dose applicable in people with T2DM in clinical practice |
Placebo, orally, once daily | Placebo is an appropriate comparator |
| NANSY 2011 | Glimepiride 1.0 mg, orally, once daily. Advice about diet and exercise | N/CPS Low dose applied in trial, compared to maximum dose applicable in people with T2DM in clinical practice |
Placebo, orally, once daily; advice about diet and exercise | Placebo is an appropriate comparator |
| NAVIGATOR 2010b | Nateglinide 60 mg, orally, three times daily. Nateglinide was initiated with 30 mg daily and titrated to full dose after 2 weeks. The tablet should be taken 1‐30 minutes before each main meal of the day. All participants were required to participate in a study‐specific lifestyle modification programme |
N/CPS | Placebo, orally, thrice daily (the tablet should be taken 1‐30 minutes before each main meal of the day); all participants were required to participate in a study‐specific lifestyle modification programme | Placebo is an appropriate comparator |
| Osei 2004 | GITS 5 mg, orally, once daily | N/CPS | Placebo, orally once daily | Placebo is an appropriate comparator |
| Page 1993 | I1: gliclazide 40 mg, orally, twice daily | N/CPS | C1: placebo, orally, twice daily | Placebo is an appropriate comparator |
| C2: diet intervention aimed at increasing fibre intake, increasing carbohydrate intake to 50%‐55% of the total energy intake, decreasing fat to 30% of total energy intake; aimed a ratio of polyunsaturated fat to saturated fatty acid ratio of 1. If body mass index was > 25 mg/kg2 then reduction in energy intake was stressed, home visit of a dietician was offered. Minimum exercise three times daily provided free of charge at different sport centres |
Diet and exercise is an appropriate comparator | |||
| Papoz 1978 | I1: glibenclamide 2.0 mg, orally, twice daily and metformin 850 mg, orally, twice daily. Overweight participants were recommended calorie restriction |
N/CPS | C1: placebo, orally, twice daily plus metformin 850 mg, orally, twice daily; overweight participants were recommended calorie restriction | Metformin is not general accepted in clinical settings with intermediate hyperglycaemia |
| I2: glibenclamide 2.0 mg, orally, twice daily and placebo, orally, twice daily. Overweight participants were recommended calorie restriction | C2: placebo, orally, twice daily; overweight participants were recommended calorie restriction | Placebo is an appropriate comparator | ||
|
aThe term 'clinical practice setting' refers to the specification of the intervention/comparator as used in the course of a standard medical treatment (such as dose, dose escalation, dosing scheme, provision for contraindications and other important features)
bFor the participants who progressed to type 2 diabetes mellitus: first step was intensified lifestyle interventions with diet and exercise. If this was insufficient metformin would be added. Finally a second non‐insulin secretagogue would be added or bedtime insulin C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; N/CPS: no specification of clinical practice setting possible; T2DM: type 2 diabetes mellitus | ||||
Appendix 4. Baseline characteristics (I)
| Intervention(s) and comparator(s) | Duration of intervention (duration of follow‐up) | Description of participants | Trial period (year to year) | Country | Setting | Ethnic groups (%) | Duration of being at risk for T2DM | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 6 months (18 months) | First‐degree relatives of people with T2DM with IGT and being overweight | ‐ | Finland | Outpatients | ‐ | 1 year |
| C1: placebo | ||||||||
| NANSY 2011 | I1: glimepiride 1.0 mg | 5 years, average follow up time 3.71 years | People with IGT. Screening focused on people with at least one known component of the metabolic syndrome, and on first‐degree relatives of people with known T2DM. However, opportunistic screening was also performed | February 2000 to March 2003 | Sweden | Outpatients | ‐ | ‐ |
| C1: placebo | ||||||||
| NAVIGATOR 2010 | I1: nateglinide 60 mg three times daily | 5 years (5 years) | IGT and cardiovascular disease or cardiovascular risk factors | 2001 to 2009 | Argentina, Australia, Austria, Belgium, Brazil, Canada, Chile, China, Colombia, Czech Republic, Denmark, Ecuador Peru, Finalnd, France, Germany, Greece, Guatemala, Hong Kong, Hungary, Ireland, Italy, Malaysia, Mexico, New Zealand, Norway, Peru, Poland, Russia, Singapore, Slovakia, South Africa, Spain, Sweden, Switzerland, Taiwan, The Netherlands, Turkey, UK, Uruguay, USA (including Puerto Rico) | Outpatients | White: 83
Black: 3 Asian: 7 Other: 8 |
‐ |
| C1: placebo | White: 83
Black: 3 Asian: 7 Other: 8 |
|||||||
| Osei 2004 | I1: GITS 5 mg | 24 months (24 months) | First‐degree relatives of African American people with T2DM and IGT | NR, but initial screening was started 1996 | USA | Outpatients | Assuming included are Black Americans, as participants are first‐degree relatives to African American | ‐ |
| C1: placebo | ||||||||
| Page 1993 | I1: gliclazide 40 mg twice daily | 6 months (7 months) | IGT | ‐ | ‐ | Outpatients | ‐ | ‐ |
| C1: placebo | ||||||||
| C2: diet + exercise | ||||||||
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | 24 months (26 months) | Impaired fasting glucose, IGT, or both | Participants entered the trial from 1969 to 1971 | France | Outpatients | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | ||||||||
| C1: placebo + metformin 850 mg twice daily | ||||||||
| C2: placebo | ||||||||
| ‐ denotes not reported C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; IGT: impaired glucose tolerance; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; NR: not reported; SD: standard deviation; T2DM: type 2 diabetes mellitus | ||||||||
Appendix 5. Baseline characteristics (II)
| Intervention(s) and comparator(s) | Sex (female %) | Age (mean years (SD)) | Systolic/diastolic blood pressure (mean mmHg (SD)) | Access to health care, social determinants | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 88 | 59 (2) | 143 (21)a / 88 (8)a | Assuming equal and free (Finland) |
| C1: placebo | 59 | 54 (3) | 134 (21)a / 83 (8)a | ||
| NANSY 2011 | I1: glimepiride 1.0 mg | 35 | 60.4 (6.8) | 144 (18) / 82 (9) | Assuming equal and free (Sweden) |
| C1: placebo | 46 | 59.6 (6.7) | 141 (18) / 82 (9) | ||
| NAVIGATOR 2010 | I1: nateglinide 60 mg three times daily | 51 | 63.7 (6.8) | 140 (18) / 83 (10) | ‐ |
| C1: placebo | 50 | 63.8 (6.9) | 140 (17) / 83 (10) | ||
| Osei 2004 | I1: GITS 5 mg | ‐ | 43.3 (8.7) | ‐ | ‐ |
| C1: placebo | 41 (4.7)b | ||||
| Page 1993 | I1: gliclazide 40 mg twice daily | 33 | 44 (6) | 130 (15) / 86 (12) | ‐ |
| C1: placebo | 0 | 40 (10) | 138 (19) / 87 (9) | ||
| C2: diet + exercise | 44 | 39 (11) | 124 (15) / 77 (11) | ||
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | 0 | 44 (5.4)a | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | 0 | 43 (10.6)a | |||
| C1: placebo + metformin 850 mg twice daily | 0 | 44 (5.5)a | |||
| C2: placebo | 0 | 45 (5.7)a | |||
| ‐ denotes not reported aSD calculated from standard error bAge reported in abstract: 41.5 (5.7) years C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; SD: standard deviation | |||||
Appendix 6. Baseline characteristics (III)
| Intervention(s) and comparator(s) | Fasting plasma glucose (mean mmol/L (SD)) | 2‐hour plasma glucose (mean mmol/L (SD)) | HbA1c (mean % (SD)) | BMI (mean kg/m² (SD)) | Comedications/Cointerventions (%) | Comorbidities (%) | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 5.3 (0.4)a | 7.9 (0.8)a | ‐ | 27.9 (6.2)a | 19% (only antihypertensives reported) | ‐ |
| C1: placebo | 5.3 (0.4)a | 8.2 (1.2)a | 28.8 (4.9)a | 12% (only antihypertensives reported) | |||
| NANSY 2011 | I1: glimepiride 1.0 mg | 6.3 (0.3)c | ‐ | 4.9 (0.5) | 29.9 (4.6) | ‐ | ‐ |
| C1: placebo | 6.3 (0.4)c | ‐ | 4.9 (0.5) | 29.6 (4.2) | |||
| NAVIGATOR 2010 | I1: nateglinide 60 mg three times daily | 6.1 (0.45) | 9.2 (0.93) | 5.8 (0.5) | 30.5 (5.4) | Angiotensin‐converting enzyme inhibitor: 7.1 Angiotensin receptor blocker: 0.4 Alpha blocker: 6.2 Beta blocker: 40.3 Calcium channel blocker: 32.7 Diuretics: 31.5 Lipid modulating drug: 38.7 Antidiabetic drug: <0.1 Aspirin or other antiplatelet drugs: 36.9 |
History of cardiovascular disease: 24.5 Hypertension: 77.7 |
| C1: placebo | 6.1 (0.46) | 9.2 (0.94) | 5.8 (0.5) | 30.5 (5.4) | Angiotensin‐converting enzyme inhibitor: 7.4 Angiotensin receptor blocker: 0.6 Alpha blocker: 6.2 Beta blocker: 38.5 Calcium channel blocker: 32.0 Diuretics: 32.2 Lipid modulating drug: 38.2 Antidiabetic drug: 0.1 Aspirin or other antiplatelet drugs: 36.8 |
History of cardiovascular disease: 24.2 Hypertension: 77.4 |
|
| Osei 2004 | I1: GITS 5 mg | 4.9 (0.7)b | 8.5 (0.8)b | ‐ | 32.9 (6.3) | ‐ | ‐ |
| C1: placebo | 4.6 (0.4)b | 8.9 (1.1)b | 39.0 (4.2) | ||||
| Page 1993 | I1: gliclazide 40 mg twice daily | 5.8 (0.5) | ‐ | ‐ | 23.5 (3) | ‐ | ‐ |
| C1: placebo | 5.8 (0.8) | ‐ | 27.5 (4) | ||||
| C2: diet + exercise | 5.6 (0.6) | ‐ | 25.5 (4) | ||||
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | 6.4 (0.7)a, b, c | 8.2 (2.0)a, b, c | ‐ | ‐ | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | 6.7 (0.7)a, b, c | 8.8 (2.0)a, b, c | |||||
| C1: placebo + metformin 850 mg twice daily | 6.7 (0.7)a, b, c | 8,2 (1.7)a, b, c | |||||
| C2: placebo | 6.3 (0.7)a, b, c | 8.3 (2.1)a, b, c | |||||
| ‐ denotes not reported aSD calculated from standard error bGlucose concentrations were converted from mg/dL to mmol/L (diabetes.co.uk 2016b) cBlood glucose concentrations were converted to plasma glucose values (diabetes.co.uk 2016a) BMI: body mass index; C: comparator; HbA1c: glycosylated haemoglobin A1c; I: intervention; GITS: glipizide gastrointestinal therapeutic system; N/A: not applicable; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; SD: standard deviation; WHO: World Health Organization | |||||||
Appendix 7. Matrix of trial endpoints (publications and trial documents)
| Endpoints quoted in trial document(s) (ClinicalTrials.gov, FDA/EMA document, manufacturer's website, published design paper)a | Trial results available in trial register | Endpoints quoted in publication(s)b,c | Endpoints quoted in abstract of publication(s)b,c | |
| Eriksson 2006 | Source: N/T | N/A | Primary outcome measure(s): ‐ | Primary outcome measure(s): ‐ |
| Secondary outcome measure(s): ‐ | Secondary outcome measure(s): ‐ | |||
| Other outcome measure(s): BMI, fasting blood glucose, 2‐hour glucose, insulin, systolic blood pressure, diastolic blood pressure, cholesterol, HDL cholesterol, triglycerides, HOMA‐IR index, acute first phase insulin release (AIR), insulin sensitivity index (ISI), disposition index (DI), development of T2DM, regression to normal glucose tolerance (after 6 months and 18 months), discontinuation due to hypoglycaemic symptoms, mild side effects | Other outcome measure(s): fasting insulin, HOMA‐IR index, HDL cholesterol, fasting glucose, 2‐hour glucose, prevalence of T2DM | |||
| NANSY 2011 | Source: N/T | N/A | Primary outcome measure(s): 5‐year risk conversion to T2DM | Primary outcome measure(s): 5‐year risk conversion to T2DM |
| Secondary outcome measure(s): ‐ | Secondary outcome measure(s): ‐ | |||
| Other outcome measure(s): all‐cause mortality, cardiovascular mortality. | Other outcome measure(s): ‐ | |||
| NAVIGATOR 2010 |
Source:NCT00097786 and protocol published as supplementary material to main publication of this trial Primary outcome measure(s): 1) incident diabetes mellitus 2) extended cardiovascular endpoint (the time to first occurrence of a cardiovascular morbidity/mortality event; including cardiovascular death, non‐fatal myocardial infarction, non‐fatal stroke, revascularisation procedure, hospitalisation for congestive heart failure, hospitalisation for unstable angina) 3) core cardiovascular endpoint (the time to first occurrence of a cardiovascular morbidity/mortality event (cardiovascular death, myocardial infarction, stroke or hospitalisation for congestive heart failure) |
Yes |
Primary outcome measure(s): incidence of diabetes, core composite cardiovascular outcome, extended composite cardiovascular outcome |
Primary outcome measure(s): incidence of diabetes, core composite cardiovascular outcome, extended composite cardiovascular outcome |
|
Secondary outcome measure(s): All‐cause death (see protocol, page 23 ‐ page 78 of Appendix_nejm_navigator_1463sa2.pdf) |
Secondary outcome measure(s): ‐ | Secondary outcome measure(s): ‐ | ||
|
Other outcome measure(s): serious adverse events, adverse events, mild hypoglycaemia, hypoglycaemia, HbA1c, fasting blood glucose, 2‐hour blood glucose after OGTT, haematology, blood chemistry, biomarkers, urine creatinine and albumin, blood pressure, pulse rate, weight, waist circumference, electrocardiogram, health economics assessment; time to first occurrence of each of the individual components of the extended cardiovascular endpoint; time to all‐cause death; time to first cardiovascular‐related hospitalisation, time to first cardiovascular endpoint including suspected events which were not committee‐confirmed, time to development of microalbuminuria ; time to progression from microalbuminuria at baseline to macroalbuminuria; time to two‐fold increase from baseline in serum creatinine, time to progression to diabetes excluding adjudication committee‐confirmed cases not meeting laboratory test‐based definition Substudy 1: polymorphisms in genes |
Other outcome measure(s): fasting plasma glucose, 2 hour OGTT, weight, blood pressure, waist circumference, adverse events, hypoglycaemia, death from any cause, fatal and non‐fatal stroke, fatal and non‐fatal myocardial infarction, hospitalisation for unstable angina, hospitalisation for heart failure, arterial revascularisation, hospitalisation for cardiovascular cause | Other outcome measure(s): hypoglycaemia | ||
| History of changes: 14 documented changes; last change 28/06/2011 | ||||
| Osei 2004 | Source: N/T | N/A | Primary outcome measure(s): ‐ | Primary outcome measure(s): ‐ |
| Secondary outcome measure(s): ‐ | Secondary outcome measure(s): ‐ | |||
| Other outcome measure(s): fasting glucose levels, 2‐hour glucose levels, serum insulin, C‐peptide, acute first and second insulin release, insulin sensitivity, disposition index for insulin, hypoglycaemia, weight | Other outcome measure(s): fasting glucose levels, 2‐hour glucose levels, serum insulin, C‐peptide, acute first and second insulin release, insulin sensitivity, disposition index for insulin, hypoglycaemia, weight | |||
| Page 1993 | Source: N/T | N/A | Primary outcome measure(s): ‐ | Primary outcome measure(s): ‐ |
| Secondary outcome measure(s): ‐ | Secondary outcome measure(s): ‐ | |||
| Other outcome measure(s): body weight, blood pressure, physical fitness, glucose tolerance, hypoglycaemia, insulin and C‐peptide levels, insulin sensitivity cholesterol and lipids levels, energy intake (and how the calories were shared on different categories), HbA1c, fructosamine | Other outcome measure(s): glucose levels, plasma cholesterol, blood pressure, HDL:LDL ratio | |||
| Papoz 1978 | Source: N/T | N/A | Primary outcome measure(s): blood glucose, insulin levels | Primary outcome measure(s): blood glucose, insulin levels |
| Secondary outcome measure(s): ‐ | Secondary outcome measure(s): ‐ | |||
| Other outcome measure(s): weight | Other outcome measure(s): weight | |||
| ‐ denotes not reported aTrial document(s) refers to all available information from published design papers and sources other than regular publications (e.g. FDA/EMA documents, manufacturer's websites, trial registers) bPublication(s) refers to trial information published in scientific journals (primary reference, duplicate publications, companion documents or multiple reports of a primary trial) cOther outcome measures refer to all outcomes not specified as primary or secondary outcome measures BMI: body mass index; EMA: European Medicines Agency; FDA: US Food and Drug Administration; HbA1c: glycosylated haemoglobin A1c; HDL: high‐density lipoprotein; HOMA‐IR: homeostatic model assessment insulin resistance; LDL: low‐density lipoprotein; N/A: not applicable; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; N/T: no trial document available; OGTT: oral glucose tolerance test; T2DM: type 2 diabetes mellitus | ||||
Appendix 8. High risk of outcome reporting bias according to ORBIT classification
| Outcome | High risk of bias (category A)a | High risk of bias (category D)b | High risk of bias (category E)c | High risk of bias (category G)d | |
| Eriksson 2006 | N/D | ||||
| NANSY 2011 | HbA1c | Yes | ‐ | ‐ | ‐ |
| NAVIGATOR 2010 | End‐stage renal disease | ‐ | Yes | ‐ | ‐ |
| HbA1c | ‐ | Yes | ‐ | ‐ | |
| Socioeconomic effects | ‐ | Yes | ‐ | ‐ | |
| Osei 2004 | N/D | ||||
| Page 1993 | HbA1ce | Yes | ‐ | ‐ | ‐ |
| Papoz 1978 | Adverse events | ‐ | ‐ | ‐ | Yes |
| "‐" denotes no risk of bias detected aClear that outcome was measured and analysed; trial report states that outcome was analysed but reports only that result was not significant (Classification 'A', table 2, Kirkham 2010) bClear that outcome was measured and analysed; trial report states that outcome was analysed but reports no results (Classification 'D', table 2, Kirkham 2010) cClear that outcome was measured but was not necessarily analysed; judgement says likely to have been analysed but not reported because of non‐significant results (Classification 'E', table 2, Kirkham 2010) dUnclear whether outcome was measured; not mentioned, but clinical judgement says likely to have been measured and analysed but not reported on the basis of non‐significant results (Classification 'G', table 2, Kirkham 2010) eHbA1 was measured which would be assumed to be the approximate value of HbA1c HbA1c: glycosylated haemoglobin A1c; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; N/D: none detected; ORBIT: Outcome Reporting Bias In Trials | |||||
Appendix 9. Definition of endpoint measurementa (I)
| All‐cause mortality (AO, IO) | Incidence of T2DM (AO, IO, SO) | Severe/serious adverse events (AO, IO) | Cardiovascular mortality (AO, IO) | Non‐fatal myocardial infarction (AO, IO, SO) | Non‐fatal stroke (AO, IO, SO) | Congestive heart failure (AO, IO, SO) | Amputation of lower extremity (AO, IO, SO) | |
| Eriksson 2006 | N/I | Development of T2DM (IO) |
N/D Only reported one dropout due to adverse events: "One subject in the glipizide treatment group discontinued the study early due to hypoglycaemic symptoms" (IO) |
N/I | N/I | N/I | N/I | N/I |
| NANSY 2011 | IO | Two consecutive fasting blood glucose values ≥ 6.1 mmol/L (IO) The possible conversion to manifest diabetes was explored at the annual check‐ups, or when implied by symptoms of hyperglycaemia |
N/I | Death due to cardiovascular disease (IO) |
N/I | N/I | N/I | N/I |
| NAVIGATOR 2010 | Death from any cause AO |
"1. Endpoint definition based on laboratory tests: FPG ≥ 126 mg/dL (7.0 mmol/L) or a
2hr post‐challenge glucose (OGTT) ≥ 200 mg/dL (11.1 mmol/L) during two consecutive valid measurements that are within 12 weeks (≤ 84 days) but at least one day apart. The time to progression to diabetes is defined as the time of the original measurement (subsequently confirmed) of an FPG ≥ 126 mg/dL (7.0 mmol/L) or a 2hr post‐challenge glucose ≥ 200 mg/dL (11.1 mmol/L).
2. Adjudication by the Diabetes Endpoint Adjudication Committee: progression to
diabetes may be confirmed by the committee in cases suggestive of diabetes but where above laboratory test‐based definition does not hold (e.g. due to missing central laboratory measurements or repeat tests outside the 12‐week time limit). E.g., the Committee will adjudicate cases where diabetes has been diagnosed by a primary care physician (possibly based on local laboratory assessments) and/or where anti‐diabetic medication has been initiated; adjudication also includes deciding on the time to progression to diabetes. Details of adjudication are provided in a Diabetes Endpoint Adjudication Charter to be followed by the Committee." (IO, AO) |
Quote: "A serious adverse event is an
undesirable sign, symptom or medical condition which:
1. is fatal or life‐threatening
2. requires or prolongs hospitalization
3. results in persistent or significant disability/incapacity.
4. constitutes a congenital anomaly or a birth defect
5. are medically significant, may jeopardize the subject and may require medical or surgical
intervention to prevent one of the outcomes listed above." IO |
Quote: "This category will include the following: 1. Sudden Cardiac Death ‐ Death that occurs instantaneously or within 60 minutes of onset of symptoms and the cause of the death is unknown. Unobserved death within 60 minutes of last contact will be classified as sudden death. Sudden death may occur in the hospital. This category will also include postresuscitation death, defined as follows: Patients in whom a cardiac and/or respiratory arrest occurs within 60 minutes of the onset of cardiac or suspected cardiac symptoms but a) are resuscitated and b) do not regain normal vital functions and c) die more than 60 minutes from the onset of symptoms leading to the arrest. 2. Myocardial Infarction Death ‐ Death which occurs during the hospitalization for the MI and is related to a cardiac complication (e.g. CHF, arrhythmia, shock) of the acute event. MI is documented by clinical, electrocardiographic and enzyme criteria or angiographic or pathological findings. If patient has a documented MI then dies "suddenly" while making an otherwise normal recovery the death will be classified in this category. Probable ‐ As above but MI is documented by two of three criteria (ECG, Enzyme, Clinical Setting); or patient presentation in typical clinical setting with chest pain or other findings suggestive of Acute MI in the absence of diagnostic enzyme or ECG changes; or the attending physician states that the patient died from MI but does not provide documentation. 3. Congestive Heart Failure ‐ Death from intractable congestive heart failure (Class III or IV) not associated with an acute event. 4 Stroke‐ Death in which the primary cause is stroke. 5. Other Cardiovascular Cause ‐ Death in which there is evidence of a primary cardiovascular etiology, and does not clearly meet the criteria for the categories outlined above. This category also includes: arrhythmogenic death, cardiac rupture, and vascular death (arterial embolism, pulmonary embolism, spontaneous aortic dissection/rupture, and bleeding). 6. Presumed CV death: death occurring when the patient was last seen > 60 minutes before death and presumed to be cardiovascular." | Quote: "At least one of the following biochemical indicators for detecting
myocardial necrosis must be present:
1. CK‐MB: (preferred)
a. Maximal value of CK‐MB > 2x the
upper limit of normal on one occasion during the first 24 hours after the index
clinical event. OR
b. Maximal value of CK‐MB, preferable CK‐MB mass, > upper limit of normal on two successive samples.
2. Troponin T or I:
a. Maximal concentration of Troponin T or
I > the MI decision limit (Upper Limit of
Normal) on at least one occasion
during the first 24 hours after the index clinical event.
3. Total CK:
a. In the absence of availability of a
Troponin or CK‐MB assay, total CK > 2x the upper limit of normal, or the B
fraction of CK may be employed, but
these last two biomarkers are
considerably less satisfactory than
CKMB.
AND ONE OF THE FOLLOWING:
1. Ischemic symptoms. Ischemic symptoms
may include but are not limited to:
a. chest discomfort; or
b. unexplained nausea and vomiting; or
c. persistent shortness of breath
secondary to left ventricular failure
2. Either ST segment depression or T wave
abnormalities
3. ST‐segment elevation: New or presumed new ST segment elevation at the J point in two or more contiguous leads with the cutoff points = 0.2 mV in leads V1, V2, or V3, or = 0.1 mV in other leads, or 4. New, or presumably new, tall R wave with R/S of 1 in V1 and R/S 1.5 in V2 (true Posterior MI), or 5. The development of new Q‐waves > 40 ms (0.04s). Q wave changes must be present in any two contiguous leads, or 6. New, or presumably new, LBBB" "Special Circumstances • For patients within 24 hours post PCI, the CK‐MB (or CK if MB not available) must be > 3x upper limit of normal. • For patients within 24 hours post CABG, the CK‐MB (or CK if MB not available) must be > 5x upper limit of normal and new Q waves must be present as defined above. 6.1.3 Silent Myocardial Infarction An asymptomatic or non‐recognized myocardial infarction discovered by the development of new pathological Q waves (as defined above) in two or more contiguous leads. In case of doubt about the date of the silent MI, the date of the first qualifying ECG performed, showing the silent MI, should be taken as the date of onset for the endpoint." "Fatal vs. Non‐fatal MI A MI is considered fatal if a MI‐caused death occurs during the same calendar day. 7. Cardiovascular procedure related death: death during or within 24 hours following a surgical or percutaneous cardiovascular procedure (e.g. PCI, CABG, etc.) and considered related to the procedure." (AO) |
Quote: "An acute neurological dysfunction of vascular origin (verified or presumed) with clinical signs and/or symptoms that persist for 24 hours or more." | Quote: "CHF requiring hospitalization.
Development of the signs and symptoms of CHF not present at screening and
requiring hospital management or
previously documented CHF that worsens,
requiring hospital management.
CHF is clinically manifested by one or more of the following features:
a. Dyspnea on exertion in the
absence of new pulmonary disease
b. Paroxysmal nocturnal dyspnoea
(shortness of breath that awakens
the patient from sleep)
c. Orthopnea (sleeping on two or more pillows to facilitate breathing)
AND one or more of the following criteria:
a. Pulmonary rales >1/3 of the way up the lung fields present after coughing in the absence of chronic lung disease or respiratory
infection.
b. Pulmonary edema on chest x‐ray in absence of high suspicion for noncardiac
origin
c. New use of oral/intravenous
diuretics, intravenous inotropes,
intravenous vasodilators, or
adjustment of previous diuretic
dose
d. Oxygen desaturation (<90%) with
no evidence of acute or chronic
lung disease
e. Jugular venous distention (JVD)
f. Bilateral pedal edema
g. Cardiomegaly (cardiothoracic ratio ≥ 0.55)
h. Left ventricular ejection fraction ≤
0.40 (new or presumably new)
i. Left ventricular fractional shortening < 0.25
j. S3 gallop on auscultation k. Elevated BNP level" |
N/I |
| Osei 2004 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
| Page 1993 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
| Papoz 1978 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
|
aIn addition to definition of endpoint measurement, description who measured the outcome (AO: adjudicated outcome measurement; IO: investigator‐assessed outcome measurement; SO: self‐reported outcome measurement) BNP: brain natriuretic peptide; CABG: coronary artery bypass graft; CHF: congestive heart failure; CK: creatine kinase; CK‐MB: creatine kinase‐MB; CV: cardiovascular; ECG: electrocardiogram; FPG: fasting plasma glucose; MI: myocardial infarction; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; N/D: not defined; N/I: not investigated; OGTT: oral glucose tolerance test;PCI: percutaneous coronary intervention; T2DM: type 2 diabetes mellitus | ||||||||
Appendix 10. Definition of endpoint measurementa (II)
| Blindness or severe vision loss (AO, IO, SO) | End‐stage renal disease (AO, IO, SO) | Non‐serious adverse events (AO, IO, SO) | Hypoglycaemic events (AO, IO, SO) | Health‐related quality of life (AO, IO, SO) | Time to progression to T2DM (AO, IO, SO) | Measures of blood glucose control (AO, IO, SO) | Socioeconomic effects (AO, IO, SO) | |
| Eriksson 2006 | N/I | N/I | Quote: "All other side effects were mild" (assuming SO) |
Quote: "...hypoglycaemic symptoms (e.g. hunger, fatigue, palpitations, tremor)" (SO) |
N/I | N/I | Fasting blood glucose, 2‐hour glucose (IO) | N/I |
| NANSY 2011 | N/I | N/I | N/I | N/I | N/I | N/I | N/I | N/I |
| NAVIGATOR 2010 | Blindness SO, IO |
‐ | Quote: "An adverse event is any undesirable sign, symptom or medical condition occurring after starting study drug(s) (or therapy) even if the event is not considered to be related to study
drug (or therapy). Study drug (or therapy) includes the drug (or therapy) under evaluation, and any reference or placebo drug (or therapy) given during any phase of the trial." SO, IO |
Quote: "Confirmed hypoglycemia is defined as plasma glucose < 3.3 mmol/L (60 mg/dL) and symptoms suggestive of hypoglycemia." Mild: "THE SUBJECT DOES NOT REQUIRE THE ASSISTANCE OF ANOTHER PERSON. Adrenergic (e.g., tachycardia, palpitations, shakiness) or cholinergic (e.g., sweating) defense symptoms or the neurologic symptoms (e.g., inability to concentrate, dizziness, hunger, blurred vision, obvious impairment of motor function, confusion or inappropriate behavior but still alert enough to seek self‐treatment)" Severe: "THE SUBJECT REQUIRES THE ASSISTANCE OF ANOTHER PERSON. Episode resulting in coma, seizure, or significant neurologic impairment so that the subject is unable to initiate self‐treatment or requires the assistance of another person." SO, IO |
N/I | Quote: "Progression to diabetes was defined as either
‐ a FPG ≥ 126 mg/dL (7.0 mmol/L) or a 2hr post‐challenge glucose (OGTT) ≥ 200 mg/dL (11.1 mmol/L) confirmed by repeat testing at a different day within 12 weeks (≤ 84 days) of the initial test result, or
‐ a suspected case confirmed by the Diabetes Endpoint Adjudication Committee (DEAC)." IO, AO |
Fasting blood glucose, 2‐hour glucose, HbA1c IO | Quote: "Health economics assessment. Health economics assessments will be performed at Visit 2, every 6 months thereafter and at
the final visit. The assessment will include information on hospitalization (diagnosis, admission, discharge date), medical care in addition to this protocol, and the patient’s health state using a visual analogue scale (VAS)." SO/IO |
| Osei 2004 | N/I | N/I | N/I | Quote: "Thus, symptoms suggestive of hypoglycaemia (e.g., nervousness, excessive hunger, tremors, confusion, etc) were recorded in a logbook, ..." (SO) |
N/I | N/I | Fasting blood glucose, 2‐hour glucose (IO) | N/I |
| Page 1993 | N/I | N/I | N/I | Hypoglycaemic episodes (SO) |
N/I | N/I | Fasting blood glucose, HbA1c (IO) |
N/I |
| Papoz 1978 | N/I | N/I | N/I | N/I | N/I | N/I | Fasting blood glucose, 2‐hour glucose levels (IO) |
N/I |
|
aIn addition to definition of endpoint measurement, description who measured the outcome (AO: adjudicated outcome measurement; IO: investigator‐assessed outcome measurement; SO: self‐reported outcome measurement) FPG: fasting plasma glucose; HbA1c: glycosylated haemoglobin A1c; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; N/I: not investigated; OGTT: oral glucose tolerance test; T2DM: type 2 diabetes mellitus | ||||||||
Appendix 11. Adverse events (I)
| Intervention(s) and comparator(s) | Participants included in analysis (N) | Deaths (N) | Deaths (%) | Participants with at least one adverse event (N) | Participants with at least one adverse event (%) | Participants with at least one severe/serious adverse event (N) | Participants with at least one severe/serious adverse event (%) | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 16 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 17 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| NANSY 2011 | I1: glimepiride 1.0 mg | 136 | 5 | 3.7 | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 138 | 2 | 1.4 | ‐ | ‐ | ‐ | ‐ | |
| NAVIGATOR 2010 | I1: nateglinide 60 mg three times daily | 4645/4602* | 319 | 6.9 | 3921 | 85.2 | 2066 | 44.9 |
| C1: placebo | 4661/4599 | 312 | 6.7 | 3866 | 84.1 | 2089 | 45.4 | |
| Osei 2004 | I1: GITS 5 mg | 9 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 9 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Page 1993 | I1: gliclazide 40 mg twice daily | 6 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 8 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C2: diet + exercise | 23 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C1: placebo + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C2: placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ denotes not reported C: comparator; I: intervention: GITS: glipizide gastrointestinal therapeutic system; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications | ||||||||
Appendix 12. Adverse events (II)
| Intervention(s) and comparator(s) | Participants included in analysis (N) | Participants discontinuing trial due to an adverse event (N) | Participants discontinuing trial due to an adverse event (%) | Participants with at least one hospitalisation (N) | Participants with at least one hospitalisation (%) | Participants with at least one outpatient treatment (N) | Participants with at least one outpatient treatment (%) | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 17 | 1 | 5.8 | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 17 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| NANSY 2011 | I1: glimepiride 1.0 mg | 136 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 138 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| NAVIGATOR 2010 | I1: nateglinide 60 mg three times daily | 4645 | 520 | 11.2 | ‐a | ‐ | ‐ | ‐ |
| C1: placebo | 4661 | 485 | 10.4 | |||||
| Osei 2004 | I1: GITS 5 mg | 9 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 9 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| Page 1993 | I1: gliclazide 40 mg twice daily | 6 | 0 | 0 | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 8 | 0 | 0 | ‐ | ‐ | ‐ | ‐ | |
| C2: diet + exercise | 23 | 0 | 0 | ‐ | ‐ | ‐ | ‐ | |
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C1: placebo + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C2: placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ denotes not reported aReports hospitalisation for unstable angina, heart failure and cardiovascular cause C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications | ||||||||
Appendix 13. Adverse events (III)
| Intervention(s) and comparator(s) | Participants included in analysis (N) | Participants with a specific adverse event (description) | Participants with at least one specific adverse events (N) | Participants with at least one specific adverse event (%) | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 16 | (1) Only adverse event reported (hypoglycaemic symptoms) | (1) 7 | (1) 41 |
| C1: placebo | 17 | (1) Only adverse event reported (hypoglycaemic symptoms) | (1) 5 | (1) 32 | |
| NANSY 2011 | I1: glimepiride 1.0 mg | 136 | ‐ | ‐ | ‐ |
| C1: placebo | 138 | ‐ | ‐ | ‐ | |
| NAVIGATOR 2010a | I1: nateglinide 60 mg three times daily | 4645 | (1) Hypotension related (2) Back pain (3) Nasopharyingitis (4) Arthralgia (5) Hypertension (6) Diarrhoea (7) Influenza (8) Pain in extremity (9) Osteoarthritis (10) Upper respiratory tract infection (11) Headache (12) Cough (13) Fatigue (14) Peripheral edema (15) Bronchitis |
(1) 1855 (2) 752 (3) 807 (4) 759 (5) 797 (6) 593 (7) 602 (8) 568 (9) 576 (10) 525 (11) 559 (12) 478 (13) 462 (14) 500 (15) 477 |
(1) 40 (2) 16 (3) 17 (4) 16 (5) 17 (6) 13 (7) 13 (8) 12 (9) 12 (10) 11 (11) 12 (12) 10 (13) 10 (14) 11 (15) 10 |
| C1: placebo | 4661 | (1) Hypotension related (2) Back pain (3) Nasopharyingitis (4) Arthralgia 759 (5) Hypertension (6) Diarrhoea (7) Influenza (8) Pain in extremity (9) Osteoarthritis (10) Upper respiratory tract infection (11) Headache (12) Cough (13) Fatigue (14) Peripheral edema (15) Bronchitis |
(1) 1789 (2) 705 (3) 798 (4) 762 (5) 846 (6) 586 (7) 630 (8) 530 (9) 578 (10) 556 (11) 604 (12) 450 (13) 432 (14) 486 (15) 477 |
(1) 38 (2) 15 (3) 17 (4) 16 (5) 18 (6) 13 (7) 14 (8) 11 (9) 12 (10) 12 (11) 13 (12) 10 (13) 9 (14) 10 (15) 10 |
|
| Osei 2004 | I1: GITS 5 mg | 9 | ‐ | ‐ | ‐ |
| C1: placebo | 9 | ‐ | ‐ | ‐ | |
| Page 1993 | I1: gliclazide 40 mg twice daily | 6 | ‐ | ‐ | ‐ |
| C1: placebo | 8 | ‐ | ‐ | ‐ | |
| C2: diet + exercise | 23 | ‐ | ‐ | ‐ | |
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | ‐ | ‐ | ‐ | ‐ | |
| C1: placebo + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | |
| C2: placebo | ‐ | ‐ | ‐ | ‐ | |
| ‐ denotes not reported aEvents reported if observed in 10% or more of either treatment group C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications | |||||
Appendix 14. Adverse events (IV)
| Intervention(s) and comparator(s) | Participants included in analysis (N) | Participants with at least one hypoglycaemic episode (N) | Participants with at least one hypoglycaemic episode (%) | Participants with at least one nocturnal hypoglycaemic episode (N) | Participants with at least one nocturnal hypoglycaemic episode (% participants) | Participants with at least one severe/serious hypoglycaemic episode (N) | Participants with at least one severe/serious hypoglycaemic episode (%) | |
| Eriksson 2006 | I1: glipizide 2.5 mg | 16 | 7 | 41 | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 17 | 5 | 32 | ‐ | ‐ | ‐ | ‐ | |
| NANSY 2011 | I1: glimepiride 1.0 mg | 136 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| C1: placebo | 138 | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| NAVIGATOR 2010 | I1: nateglinide 60 mg three times daily | 4645 | 911 | 19.6 | ‐ | ‐ | 21 | 0.5 |
| C1: placebo | 4661 | 527 | 11.3 | ‐ | ‐ | 12 | 0.3 | |
| Osei 2004 | I1: GITS 5 mg | 9 | 0 | 0 | 0 | 0 | 0 | 0 |
| C1: placebo | 9 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Page 1993 | I1: gliclazide 40 mg twice daily | 6 | 0 | 0 | 0 | 0 | 0 | 0 |
| C1: placebo | 7 | 0 | 0 | 0 | 0 | 0 | 0 | |
| C2: diet + exercise | 18 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Papoz 1978 | I1: glibenclamide 2.0 mg twice daily + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| I2: glibenclamide 2.0 mg twice daily + placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C1: placebo + metformin 850 mg twice daily | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| C2: placebo | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ | |
| ‐ denotes not reported C: comparator; GITS: glipizide gastrointestinal therapeutic system; I: intervention; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications | ||||||||
Appendix 15. Selection bias decisions
| Selection bias decisions for trials reporting unadjusted analyses ‐ comparison of results obtained using method details alone with results using method details and trial baseline informationa | |||
| Reported randomisation and allocation concealment methods | ' Risk of bias' judgement using methods reporting | Information gained from study characteristics data | Risk of bias using baseline information and methods reporting |
| Unclear methods | Unclear risk | Baseline imbalances present for important prognostic variable(s) | High risk |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited or no baseline details | Unclear risk | ||
| Would generate a truly random sample, with robust allocation concealment | Low risk | Baseline imbalances present for important prognostic variable(s) | Unclear riskc |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited baseline details, showing balance in some important prognostic variablesb | Low risk | ||
| No baseline details | Unclear risk | ||
| Sequence is not truly random, or allocation concealment is inadequate | High risk | Baseline imbalances present for important prognostic variable(s) | High risk |
| Groups appear similar at baseline for all important prognostic variables | Low risk | ||
| Limited baseline details, showing balance in some important prognostic variablesb | Unclear risk | ||
| No baseline details | High risk | ||
| aTaken from Corbett 2014; judgements highlighted in bold indicate situations in which the addition of baseline assessments would change the judgement about risk of selection bias, compared with using methods reporting alone bDetails for the remaining important prognostic variables not reported cImbalance identified which appears likely to be due to chance | |||
Appendix 16. Survey of trial investigators providing information on included trials
| Date trial author contacted | Date trial author replied | Date trial author was asked for additional information (short summary) | Date trial author provided data (short summary) | |
| Eriksson 2006 | 25/4/16 | 25/04/16 | Author could not provide any additional information | N/A |
| NANSY 2011 | 04/05/16 | 04/05/16 | Author replied that he would try to answer our request as good as possible. As we did not receive an answer with regard to our request we sent a friendly reminder (13/05/16) but again did not receive a reply | N/A |
| NAVIGATOR 2010 | 28/04/16 | 29/04/16 and 05/05/16 | Two individuals employed at Novartis were contacted. The answer was that we should apply for any additional information through www.clinicalstudydatarequest.com. The request form was submitted on 05/05/16 without a reply. We sent a message through www.clinicalstudydatarequest.com asking if the request was received but again did not receive a reply | N/A |
| Osei 2004 | 27/04/16 | No reply | N/A | N/A |
| Page 1993 | 28/04/16 | No reply | No contact information could be identified for the first author. Contact information on one of the other authors was identified through an Internet search (Dr Levy). However, no reply was given | N/A |
| Papoz 1978 | 04/05/16 | No reply | No contact information could be identified for the first author. Contact information on one of the other authors was identified through an Internet search (Dr Eschwege). However, no reply was given | N/A |
| N/A: not applicable; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications | ||||
Appendix 17. Checklist to aid consistency and reproducibility of GRADE assessments ‐ sulphonylureas (glimepiride)
| (1) All‐cause mortality | (2) Incidence of type 2 diabetes mellitus | (3) Serious adverse events | (4) Cardiovascular mortality | (5) Non‐fatal myocardial infarction/stroke, congestive heart failure | (6) Health‐related quality of life | (7) Socioeconomic effects | ||
| Trial limitations (risk of bias)a | Was random sequence generation used (i.e. no potential for selection bias)? | Unclear | Unclear | N/A | Unclear | N/A | N/A | N/A |
| Was allocation concealment used (i.e. no potential for selection bias)? | Unclear | Unclear | Unclear | |||||
| Was there blinding of participants and personnel (i.e. no potential for performance bias) or outcome not likely to be influenced by lack of blinding? | Yes | Yes | Yes | |||||
| Was there blinding of outcome assessment (i.e. no potential for detection bias) or was outcome measurement not likely to be influenced by lack of blinding? | Yes | Yes | Yes | |||||
| Was an objective outcome used? | Yes | Yes | Yes | |||||
| Were more than 80% of participants enrolled in trials included in the analysis (i.e. no potential reporting bias)?e | Yes | Yes | Yes | |||||
| Were data reported consistently for the outcome of interest (i.e. no potential selective reporting)? | Yes | Yes | Yes | |||||
| No other biases reported (i.e. no potential of other bias)? | Unclear | Unclear | Unclear | |||||
| Did the trials end up as scheduled (i.e. not stopped early)? | Yes | Yes | Yes | |||||
| Inconsistencyb | Point estimates did not vary widely? | N/A | Yes | N/A | ||||
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | Substantial | N/A | N/A | |||||
| Was the direction of effect consistent? | N/A | Yes | N/A | |||||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40% to 60%), high I² > 60%)? | N/A | Low | N/A | |||||
| Was the test for heterogeneity statistically significant (P < 0.1)? | N/A | Not statistically significant | N/A | |||||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | ||||
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | |||||
| Was the included outcome not a surrogate outcome? | Yes | No (↓) | Yes | |||||
| Was the outcome timeframe sufficient? | Sufficient | Sufficient | Sufficient | |||||
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | |||||
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | N/A | No (↓) | N/A | ||||
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100 to 300 participants, low: < 100 participants)?e | High | High | High | |||||
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5 to 10 studies, small: < 5 studies)?e | Small (↓) | Small (↓) | Small (↓) | |||||
| Was the outcome a common event (e.g. occurs more than 1/100)? | Yes | Yes | No (↓) | |||||
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | ||||
| Was grey literature searched? | Yes | Yes | Yes | |||||
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | |||||
| There was no industry influence on studies included in the review? | No (↓) | No (↓) | No (↓) | |||||
| There was no evidence of funnel plot asymmetry? | N/A | N/A | N/A | |||||
| There was no discrepancy in findings between published and unpublished trials? | N/A | N/A | N/A | |||||
|
aQuestions on risk of bias are answered in relation to the majority of the aggregated evidence in the meta‐analysis rather than to individual trials
bQuestions on inconsistency are primarily based on visual assessment of forest plots and the statistical quantification of heterogeneity based on I² cWhen judging the width of the confidence interval it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished trials eDepends on the context of the systematic review area (↓): key item for potential downgrading the quality of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s); N/A: not applicable | ||||||||
Appendix 18. Checklist to aid consistency and reproducibility of GRADE assessments ‐ meglitinide analogues (nateglinide)
| (1) All‐cause mortality | (2) Incidence of type 2 diabetes mellitus | (3) Serious adverse events | (4) Cardiovascular mortality | (5) Non‐fatal myocardial infarction/stroke, congestive heart failure | (6) Health‐related quality of life | (7) Socioeconomic effects | ||
| Trial limitations (risk of bias)a | Was random sequence generation used (i.e. no potential for selection bias)? | Yes | Yes | Yes | Yes | Yes | N/A | N/A |
| Was allocation concealment used (i.e. no potential for selection bias)? | Yes | Yes | Yes | Yes | Yes | |||
| Was there blinding of participants and personnel (i.e. no potential for performance bias) or outcome not likely to be influenced by lack of blinding? | Yes | Yes | Yes | Yes | Yes | |||
| Was there blinding of outcome assessment (i.e. no potential for detection bias) or was outcome measurement not likely to be influenced by lack of blinding? | Yes | Yes | Yes | Yes | Yes | |||
| Was an objective outcome used? | Yes | Yes | Yes | Yes | Yes | |||
| Were more than 80% of participants enrolled in trials included in the analysis (i.e. no potential reporting bias)?e | Yes | Yes | Yes | Yes | Yes | |||
| Were data reported consistently for the outcome of interest (i.e. no potential selective reporting)? | No (↓) | No (↓) | No (↓) | No (↓) | No (↓) | |||
| No other biases reported (i.e. no potential of other bias)? | Yes | Yes | Yes | Yes | Yes | |||
| Did the trials end up as scheduled (i.e. not stopped early)? | Yes | Yes | Yes | Yes | Yes | |||
| Inconsistencyb | Point estimates did not vary widely? | N/A | N/A | N/A | N/A | N/A | ||
| To what extent did confidence intervals overlap (substantial: all confidence intervals overlap at least one of the included studies point estimate; some: confidence intervals overlap but not all overlap at least one point estimate; no: at least one outlier: where the confidence interval of some of the studies do not overlap with those of most included studies)? | N/A | N/A | N/A | N/A | N/A | |||
| Was the direction of effect consistent? | N/A | N/A | N/A | N/A | N/A | |||
| What was the magnitude of statistical heterogeneity (as measured by I²) ‐ low (I² < 40%), moderate (I² 40% to 60%), high I² > 60%)? | N/A | N/A | N/A | N/A | N/A | |||
| Was the test for heterogeneity statistically significant (P < 0.1)? | N/A | N/A | N/A | N/A | N/A | |||
| Indirectness | Were the populations in included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | ||
| Were the interventions in the included studies applicable to the decision context? | Highly applicable | Highly applicable | Highly applicable | Highly applicable | Highly applicable | |||
| Was the included outcome not a surrogate outcome? | Yes | No (↓) | Yes | Yes | Yes | |||
| Was the outcome timeframe sufficient? | Sufficient | Sufficient | Sufficient | Sufficient | Sufficient | |||
| Were the conclusions based on direct comparisons? | Yes | Yes | Yes | Yes | Yes | |||
| Imprecisionc | Was the confidence interval for the pooled estimate not consistent with benefit and harm? | N/A | N/A | N/A | N/A | N/A | ||
| What is the magnitude of the median sample size (high: 300 participants, intermediate: 100 to 300 participants, low: < 100 participants)?e | High | High | High | High | High | |||
| What was the magnitude of the number of included studies (large: > 10 studies, moderate: 5 to 10 studies, small: < 5 studies)?e | Small (↓) | Small (↓) | Small (↓) | Small (↓) | Small (↓) | |||
| Was the outcome a common event (e.g. occurs more than 1/100)? | Yes | Yes | Yes | Yes | Yes | |||
| Publication biasd | Was a comprehensive search conducted? | Yes | Yes | Yes | Yes | Yes | ||
| Was grey literature searched? | Yes | Yes | Yes | Yes | Yes | |||
| Were no restrictions applied to study selection on the basis of language? | Yes | Yes | Yes | Yes | Yes | |||
| There was no industry influence on studies included in the review? | No (↓) | No (↓) | No (↓) | No (↓) | No (↓) | |||
| There was no evidence of funnel plot asymmetry? | N/A | N/A | N/A | N/A | N/A | |||
| There was no discrepancy in findings between published and unpublished trials? | N/A | N/A | N/A | N/A | N/A | |||
|
aQuestions on risk of bias are answered in relation to the majority of the aggregated evidence in the meta‐analysis rather than to individual trials
bQuestions on inconsistency are primarily based on visual assessment of forest plots and the statistical quantification of heterogeneity based on I² cWhen judging the width of the confidence interval it is recommended to use a clinical decision threshold to assess whether the imprecision is clinically meaningful dQuestions address comprehensiveness of the search strategy, industry influence, funnel plot asymmetry and discrepancies between published and unpublished trials eDepends on the context of the systematic review area (↓): key item for potential downgrading the quality of the evidence (GRADE) as shown in the footnotes of the 'Summary of finding' table(s); N/A: not applicable | ||||||||
Appendix 19. Subgroup analyses; sulphonylurea monotherapy versus placebo
| Outcome | Type of sulphonylurea second‐generation versus third‐generation (P value for test of interaction) | Trials with long duration (≥ 2 years) versus trials with short duration (< 2 years) (P value for test of interaction) | WHO criteria for IGT versus other diagnostic criteria (P value for test of interaction) | Younger versus older participants (see text) (P value for test of interaction) | Ethnicity (P value for test of interaction) | Comorbidity (P value for test of interaction) | Participants with previous gestational diabetes mellitus (P value for test of interaction) |
| Fasting blood glucose | 0.43 | 0.86 | 0.71 | 0.07 | Not possible due to lack of reporting in trials | Not possible due to lack of reporting in trials | Not possible due to lack of reporting in trials |
| 2‐hour blood glucose | 0.22 | 0.76 | 0.30 | 0.76 | Not possible due to lack of reporting in trials | Not possible due to lack of reporting in trials | Not possible due to lack of reporting in trials |
| IGT: impaired glucose tolerance; WHO: World Health Organization | |||||||
Data and analyses
Comparison 1. Sulphonylureas as monotherapy vs placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Incidence of type 2 diabetes | 2 | 307 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.54, 1.04] |
| 2 Mild hypoglycaemia | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Severe hypoglycaemia | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Fasting blood glucose control | 4 | 105 | Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.59, ‐0.02] |
| 5 Fasting blood glucose control: type of SU | 4 | 105 | Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.59, ‐0.02] |
| 5.1 Second‐generation SU | 3 | 87 | Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.57, 0.02] |
| 5.2 Third‐generation SU | 1 | 18 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐1.72, 0.32] |
| 6 Fasting blood glucose control: duration of intervention | 4 | 105 | Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.59, ‐0.02] |
| 6.1 duration less than 2 years | 2 | 46 | Mean Difference (IV, Random, 95% CI) | ‐0.28 [‐0.95, 0.39] |
| 6.2 duration 2 years or more | 2 | 59 | Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.69, 0.00] |
| 7 Fasting blood glucose control: diagnostic criteria | 4 | 105 | Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.59, ‐0.02] |
| 7.1 WHO diagnostic | 2 | 51 | Mean Difference (IV, Random, 95% CI) | ‐0.22 [‐0.86, 0.42] |
| 7.2 Other criteria | 2 | 54 | Mean Difference (IV, Random, 95% CI) | ‐0.36 [‐0.70, ‐0.02] |
| 8 Fasting blood glucose control: age | 4 | 105 | Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐0.79, ‐0.17] |
| 8.1 age less than 50 yrs | 3 | 72 | Mean Difference (IV, Random, 95% CI) | ‐0.62 [‐0.95, ‐0.30] |
| 8.2 age above 50 years | 1 | 33 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.60, 0.60] |
| 9 Extension period: fasting blood glucose | 2 | 45 | Mean Difference (IV, Random, 95% CI) | ‐0.08 [‐1.04, 0.89] |
| 10 2‐hour glucose [mmol/L] | 3 | 92 | Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐1.28, 0.43] |
| 11 2‐hour glucose [mmol/L]: type of SU | 3 | 92 | Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐1.28, 0.43] |
| 11.1 Second‐generation SU | 2 | 74 | Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.90, 0.57] |
| 11.2 Third‐generation SU | 1 | 18 | Mean Difference (IV, Random, 95% CI) | ‐2.0 [‐4.20, 0.20] |
| 12 2‐hour glucose [mmol/L]: duration of intervention | 3 | 92 | Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐1.28, 0.43] |
| 12.1 Duration 2 years or more | 2 | 59 | Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐2.64, 1.15] |
| 12.2 Duration less than 2 years | 1 | 33 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐1.56, 0.76] |
| 13 2‐hour glucose [mmol/L]: diagnostic criteria | 3 | 92 | Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐1.28, 0.43] |
| 13.1 WHO criteria | 2 | 51 | Mean Difference (IV, Random, 95% CI) | ‐0.92 [‐2.38, 0.55] |
| 13.2 Other criteria | 1 | 41 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.95, 0.95] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Eriksson 2006.
| Methods | Parallel randomised controlled clinical trial, randomisation ratio 1:1, superiority design | |
| Participants |
Inclusion criteria: first‐degree relatives of people with T2DM; age 35–70 years; BMI 25–35 kg/m2; fulfilling WHO criteria for IGT on two consecutive OGTTs Exclusion criteria: other endocrine, renal, hepatic or inter‐current diseases Diagnostic criteria: WHO criteria for IGT on two consecutive OGTTs (fasting plasma glucose < 7.0 mmol/L and 2‐hour plasma glucose ≥ 7.8 mmol/L and < 11.1 mmol/L) |
|
| Interventions |
Number of study centres: 1 Run‐in period: none, but all participants had to fulfill the diagnostic criteria for IGT prior to randomisation. OGTT was performed 12 months later (e.g. study baseline and confirmed the IGT diagnosis), after which the participants were randomised Administration‐free period before testing during trial: not specified if any study drug was taken on the testing day at the end of the intervention. However, a 12 retesting was done 12 months after the end of the intervention Extension period: participants were observed 1 year after the intervention was stopped |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details |
Trial terminated early: no Trial ID: NR |
|
| Publication details |
Language of publication: English Funding: commercial funding: grants from Orion Pharmaceuticals / non‐commercial funding: grant from the Sigrid Juselius Foundation, the Finnish Diabetes Research Society Publication status: peer‐reviewed journal/full article |
|
| Stated aim for study | Quote from publication: "Given these considerations, we designed a placebo‐controlled study with the aim of assessing the effect of an insulin secretagogue, i.e. glipizide, on glucose and insulin metabolism in glucose‐intolerant first‐degree relatives of patients with type 2 diabetes" | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote from publication: "... the subject was randomized to receive either glipizide 2.5 mg or placebo once daily" Comment: method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk |
Quote from publication: "... the subject was randomized to receive either glipizide 2.5 mg or placebo once daily" Comment: method of allocation not described |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Low risk |
Quote from publication: "A similar number of subjects in both groups reported hypoglycaemic symptoms (e.g. hunger, fatigue, palpitations, tremor) during the study" Comment: self‐reported outcome measurement. Blinding of participants was ensured by placebo tablets |
| Blinding of participants and personnel (performance bias) incidence of T2DM | Low risk |
Quote from publication: "After 6 months, i.e. at termination of drug treatment, both the OGTT and the IVGTT were repeated on different occasions 1 week apart. The participants continued without study drugs and an OGTT was performed at 18 months from baseline, i.e. 12 months after discontinuation of study medication" Comment: investigator‐assessed. Blinding of investigators was ensured by placebo tablets |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk | Quote from publication: "...placebo‐controlled study with the aim of assessing..." Comment: investigator‐assessed. Blinding of participants and investigators was ensured by placebo tablets |
| Blinding of participants and personnel (performance bias) non‐serious adverse events | Low risk |
Quotes from publication: "All other side effects were mild" and "...placebo‐controlled study with the aim of assessing..." Comment: self‐reported outcome measurement. Blinding of participants and investigators was ensured by placebo tablet |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Low risk |
Quote from publication: "A similar number of subjects in both groups reported hypoglycaemic symptoms (e.g. hunger, fatigue, palpitations, tremor) during the study" Comment: self‐reported outcome measurement. Blinding of participants was ensured by use of placebo tablets |
| Blinding of outcome assessment (detection bias) incidence of T2DM | Low risk |
Quote from publication: "After 6 months, i.e. at termination of drug treatment, both the OGTT and the IVGTT were repeated on different occasions 1 week apart. The participants continued without study drugs and an OGTT was performed at 18 months from baseline, i.e. 12 months after discontinuation of study medication" Comment: investigator‐assessed. Blinding of investigators was ensured by placebo tablets |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk | Comment: investigator‐assessed at baseline and after 6 and 18 months.Blinding of investigators was ensured by placebo tablets |
| Blinding of outcome assessment (detection bias) non‐serious adverse events | Unclear risk |
Quote from publication: "All other side effects were mild" Comment: self‐reported outcome measurement. Blinding of participants was ensured by placebo tablets |
| Incomplete outcome data (attrition bias) hypoglycaemia | High risk |
Quotes from publication: "Three subjects dropped out early in the intervention – one because of hypoglycaemia, one because of an aortic aneurysm and one because of systemic lupus erythematosus and concomitant peroral corticosteroid treatment" and "One subject in the glipizide treatment group discontinued the study early due to hypoglycaemic symptoms" Comment: two persons withdrew due to hypoglycaemia in the glipizide group. The two dropouts could have a substantial impact on the effect estimate for hypoglycaemia |
| Incomplete outcome data (attrition bias) incidence of T2DM | Unclear risk |
Quotes from publication: "Three subjects dropped out early in the intervention – one because of hypoglycaemia, one because of an aortic aneurysm and one because of systemic lupus erythematosus and concomitant peroral corticosteroid treatment" and "One subject in the glipizide treatment group discontinued the study early due to hypoglycaemic symptoms" Comment: reported and reasons explained. Number of dropouts could influence this outcome |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Low risk |
Quotes from publication: "Three subjects dropped out early in the intervention – one because of hypoglycaemia, one because of an aortic aneurysm and one because of systemic lupus erythematosus and concomitant peroral corticosteroid treatment" and "One subject in the glipizide treatment group discontinued the study early due to hypoglycaemic symptoms" Comment: reported and reasons explained. Number too low to have impact on clinical relevance |
| Incomplete outcome data (attrition bias) non‐serious adverse events | Unclear risk |
Quotes from publication: "Three subjects dropped out early in the intervention – one because of hypoglycaemia, one because of an aortic aneurysm and one because of systemic lupus erythematosus and concomitant peroral corticosteroid treatment" and "One subject in the glipizide treatment group discontinued the study early due to hypoglycaemic symptoms" Comment: reported and reasons explained. However, no data for non‐serious adverse events are available, so the impact of the missing data can not be evaluated |
| Selective reporting (reporting bias) | High risk | No trial protocol available. Non‐serious adverse events are reported incompletely, so these data can not be included in this review. Two participants dropped out due to hypoglycaemia, one of these allocated to the glipizide intervention group. Not described whether it was due to repetitive mild hypoglycaemia or severe hypoglycaemia |
| Other bias | Unclear risk | Comment: commercial funding |
NANSY 2011.
| Methods | Parallel randomised controlled clinical trial, randomisation ratio 1:1, superiority design | |
| Participants |
Inclusion criteria: 40–70 years, IFG defined by two (10‐h overnight) consecutive fasting blood glucose values ≥ 5.6 mmol/L Exclusion criteria: myocardial infarction or stroke during the last 12 months, heart failure NYHA III‐IV, endocrine disease, impaired renal function (serum creatinine ≥ 130 mmol/L), impaired hepatic function, mental condition interfering with ability to participate, treatment with oral glucocorticoids, current or planned pregnancy or breastfeeding, progressive serious disease Diagnostic criteria: IFG defined by two (10‐h overnight) consecutive fasting blood glucose values ≥ 5.6 mmol/L with an arithmetic mean within 5.6 to 6.0 mmol/L |
|
| Interventions |
Number of study centres: 23 Run‐in period: none Administration‐free period before testing during trial: received study‐drug on the day of testing blood glucose for conversion to T2DM Extension period: no |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (unrestricted starting grant from Hoechst‐Marion‐Roussel (now Sanofi‐Aventis)) and non‐commercial funding Publication status: peer‐reviewed journal/full article (research letter) and abstract |
|
| Stated aim for study | Quote from publication: "The Nepi ANtidiabetes StudY (NANSY) is a randomized, double‐blind and placebo‐controlled prospective trial assessing whether the addition of a once‐daily low dose of SU to lifestyle changes in subjects with IFG will help to delay the conversion to T2D" | |
| Notes | Randomisation of participants was slower than expected. A NANSY‐eye subprotocol was published; however, final data are not available. Blood glucose values in this trial were reported as whole blood glucose. In the tables and result section all values are converted to plasma glucose values (diabetes.co.uk 2016a) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote from publication: "Patients were randomly assigned to glimepiride (Amaryl® from Hoechst‐Marion Roussel, now Sanofi‐Aventis) 1 mg or placebo once a day for 5 years" Comment: method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk |
Quote from publication: "Patients were randomly assigned to glimepiride (Amaryl® from Hoechst‐Marion Roussel, now Sanofi‐Aventis) 1 mg or placebo once a day for 5 years" Comment: method of allocation not described |
| Blinding of participants and personnel (performance bias) all‐cause/cardiovascular mortality | Low risk |
Quote from publication: "...a randomized, double‐blind and placebo‐controlled prospective trial assessing..." Comment: investigator‐assessed outcome measurement |
| Blinding of participants and personnel (performance bias) incidence of T2DM | Low risk |
Quote from publication: "...a randomized, double‐blind and placebo‐controlled prospective trial assessing..." Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) all‐cause/cardiovascular mortality | Low risk |
Quote from publication: "...a randomized, double‐blind and placebo‐controlled prospective trial assessing..." Comment: investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) incidence of T2DM | Low risk |
Quote from publication: "...a randomized, double‐blind and placebo‐controlled prospective trial assessing..." Comment: investigator‐assessed |
| Incomplete outcome data (attrition bias) all‐cause/cardiovascular mortality | Low risk |
Quote from publication: "Seventy‐one subjects interrupted participation prematurely; 7 interruptions were on account of death, 5 among subjects allocated to glimepiride and 2 among those allocated to placebo" Comment: only reported, but it is assumed that mortality status was known on the participants who left the trial |
| Incomplete outcome data (attrition bias) incidence of T2DM | Unclear risk |
Quote from publication: "Seventy‐one subjects interrupted participation prematurely..." Comment: only reported. Not known to which intervention group the participants who stopped prematurely were randomised or the reasons. Assuming the diabetes status was unknown for the participants who dropped out |
| Selective reporting (reporting bias) | High risk | Comment: clear that fasting blood glucose was measured, however, no data were reported. HbA1c reported as no significant difference between the intervention groups (see Appendix 8) |
| Other bias | Unclear risk | Comment: commercial funding |
NAVIGATOR 2010.
| Methods | Factorial randomised controlled clinical trial, randomisation ratio 1:1, superiority design | |
| Participants |
Inclusion criteria: written informed consent to participate; females must be surgically sterile or postmenopausal; 50 years or older; presence of cardiovascular risk factors (see notes) or cardiovascular disease(s) (see notes) at visit 1 (age 50 to 54 years ‐ 1 or more cardiovascular disease(s); age 55 years and older ‐ 1 or more cardiovascular risk factors or 1 or more cardiovascular disease(s)); fasting plasma glucose > 5.3 mmol/L and < 7.0 mmol/L at visit 1; 2 hour post‐challenge glucose (after a 75‐g OGTT) ≥ 7.8 mmol/L but < 11.1 mmol/L at visit 1; ability and willingness to comply with all study requirements Exclusion criteria: failure to provide written informed consent; evidence of hepatic disease defined as serum glutamic oxaloacetic transaminase or serum glutamic‐pyruvic transaminase > 2 times the upper limit of normal at visit 1; renal failure with a serum creatinine > 221 μmol/L at visit 1; clinically significant laboratory abnormalities that may interfere with the assessment of safety, efficacy, or both of the study drug, other than hyperglycaemia, hyperinsulinaemia and glycosuria; individuals requiring thyroid hormone replacement who have been on their current medication dosage for < 3 months prior to month –1; history of malignancy including leukaemia or lymphoma (but not basal cell skin cancer) within the past 5 years; individuals on an angiotensin‐converting enzyme inhibitor for hypertension who are unable or unwilling to discontinue the medication under supervision of their physician at least 4 weeks prior to screening and during the full course of double‐blind treatment. A subject may be washed out from an angiotensin‐converting enzyme inhibitor only, if in the investigator’s opinion, it is in the best interest of the subject and/or the subject has been intolerant to the angiotensin‐converting enzyme inhibitor. For those individuals taken off an angiotensin‐converting enzyme inhibitor with the intent to enter the individual in the study, written informed consent will be obtained at the time of discontinuation of medication (at least 4 weeks prior to the screening visit); individuals on an angiotensin receptor blocker who are unable or unwilling to discontinue the medication under supervision of their physician at least 4 weeks prior to screening and during the full course of double‐blind treatment. A subject may be washed out from an angiotensin receptor blocker only, if in the investigator’s opinion, it is in the best interest of the subject. For those individuals taken off an angiotensin receptor blocker with the intent to enter the individual in the study, written informed consent will be obtained at the time of discontinuation of medication (at least 4 weeks prior to the screening visit). It is the physician’s responsibility to appropriately monitor the individual during this period; use of oral antidiabetics or insulin within the past 5 years; history of clinically significant autoimmune disorders such as systemic lupus erythematosus; history of active substance or alcohol abuse within the past year; known hypersensitivity or contraindication to nateglinide or valsartan; history of non‐compliance to medical regimens and/or individuals who are considered potentially unreliable; previous investigational drug treatment within the past month, unless local health authorities mandate a longer period; congestive heart failure NYHA class 3 or 4; presence of any concomitant condition which, in the opinion of the investigator or the sponsor, could interfere with the interpretation of efficacy and safety data gathered in this trial; chronic (> 7 days) concomitant use of oral corticosteroids within 1 month prior to screening; myocardial infarction, diagnosis of stable or unstable angina, multivessel percutaneous transluminal coronary angioplasty or coronary artery bypass grafting, limb bypass surgery or percutaneous angioplasty, non traumatic limb or foot amputation, or stroke of atherosclerotic origin within 4 weeks prior to visit 1 and the time period between visit 1 and visit 2 Diagnostic criteria: fasting plasma glucose ≥ 5.3 mmol/L and < 7.0 mmol/L at visit 1 and 2‐hour post‐challenge glucose (after a 75‐g OGTT) ≥ 7.8 mmol/L but < 11.1 mmol/L at visit 1 |
|
| Interventions |
Number of study centres: 806 Run‐in period: 4 weeks (at visit 1, eligible people entered a treatment‐free run‐in period for up to 4 weeks) Administration‐free period before testing during trial: participants were asked not to take study drug on the testing days until after the glucose tests had been performed Extension period: no |
|
| Outcomes |
Composite outcome measures reported: yes Two composite primary cardiovascular outcomes are reported: ‐ extended cardiovascular endpoint (the time to first occurrence of a cardiovascular morbidity/mortality event (including cardiovascular death, non‐fatal myocardial infarction, non‐fatal stroke, revascularisation procedure, hospitalisation for congestive heart failure, hospitalisation for unstable angina) ‐ core cardiovascular endpoint (the time to first occurrence of a cardiovascular morbidity/mortality event (cardiovascular death, myocardial infarction, stroke or hospitalisation for congestive heart failure) The 'core' cardiovascular endpoint was initially a secondary outcome, but was added during the trial |
|
| Study details |
Trial terminated early: no Trial ID: NCT00097786 |
|
| Publication details |
Language of publication: English Funding: commercial funding (Novartis Pharma) Publication status: peer‐reviewed journal |
|
| Stated aim for study | Quote from publication: "The aim of the Nateglinide and Valsartan in Impaired Glucose Tolerance Outcomes Research (NAVIGATOR) trial11 was to determine whether the risk of diabetes and cardiovascular events could be reduced in this population" | |
| Notes | Cardiovascular risk factors: family history of premature coronary heart disease (definite myocardial infarction or sudden death before 55 years of age in father or other male first‐degree relative, or before 65 years of age in mother or other female first‐degree relative); current cigarette smoking (defined as smoking at least 10 cigarettes/day on a regular basis for at least 5 years prior to inclusion in the study; if the individual has quit smoking, s/he will be considered a smoker if s/he stopped less than 12 months before inclusion); hypertension (> 140 mmHg systolic or 90 mmHg diastolic or on antihypertensive medication); low HDL cholesterol (< 1.0 mmol/L); high LDL (≥ 4.1 mmol/L) or high non‐HDL (> 4.9 mmol/L) if triglycerides are > 2.3 mmol/L or on lipid‐lowering therapy; left ventricular hypertrophy with strain pattern defined as per electrocardiogram (Sokolow and Lyon criteria or Cornell criteria); known microalbuminuria (> 30 mg/g creatinine) Cardiovascular disease(s): previous myocardial infarction (> 1 month ago); stable angina or unstable angina (> 1 month ago) each with documented multivessel coronary disease; > 50% stenosis in at least two major coronary arteries, or positive stress test; multivessel percutaneous transluminal coronary angioplasty > 1 month ago; multivessel coronary artery bypass grafting > 4 years ago or with angina; previous limb bypass surgery or percutaneous angioplasty; previous non‐traumatic limb or foot amputation; history of intermittent claudication with ankle:arm blood pressure ratio of < 0.80 in at least one side; significant peripheral stenosis (> 50%) documented by angiography; stroke of atherosclerotic origin > 1 month ago The trial had a factorial design randomising participants to 1) nateglinide + valsartan; 2) nateglinide + valsartan‐placebo; 3) nateglinide‐placebo + valsartan; 4) nateglinide‐placebo + valsartan‐placebo |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from publication: "We used a computerized, interactive voice‐response telephone randomization system involving concealed study‐group assignments to randomly assign patients to valsartan or matching placebo (and nateglinide or matching placebo) in a 2‐by‐2 factorial design. Randomization was stratified according to center, with a block size of eight within each center" |
| Allocation concealment (selection bias) | Low risk | Quote from publication: "We used a computerized, interactive voice‐response telephone randomization system involving concealed study‐group assignments to randomly assign patients to valsartan or matching placebo (and nateglinide or matching placebo) in a 2‐by‐2 factorial design. Randomization was stratified according to center, with a block size of eight within each center" |
| Blinding of participants and personnel (performance bias) all‐cause/cardiovascular mortality | Low risk |
Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: adjudicated outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) amputation, blindness/severe vision loss, end‐stage renal disease | Low risk |
Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: adjudicated/investigator‐assessed. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Low risk |
Quote from publication: "A qualified person must review diary entries with the patient at each visit to determine if the signs and symptoms are consistent with hypoglycemia" Comment: investigator‐assessed/self‐reported outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) incidence of T2DM | Low risk |
Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: adjudicated/investigator‐assessed outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk | Quotes from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" and "The fasting plasma glucose level or the plasma glucose level 2 hours after a glucose challenge was measured at the closeout visit or during the final 6 months of..." Comment: adjudicated/investigator‐assessed outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Low risk | Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: adjudicated outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) non‐serious adverse events | Low risk | Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: self‐reported/investigator‐assessed outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) serious adverse events | Low risk | Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: adjudicated/investigator‐assessed outcome measurement. Blinding of participants and investigators ensured |
| Blinding of participants and personnel (performance bias) time to progression to T2DM | Low risk | Quote from publication: "The double‐blinding of the randomized study medication will be maintained by the use of identical placebo and active tablets and capsules for nateglinide and valsartan, respectively" Comment: adjudicated/investigator‐assessed outcome measurement. Blinding of participants and investigators ensured |
| Blinding of outcome assessment (detection bias) all‐cause/cardiovascular mortality | Low risk |
Quote from publication: "Occurrence of a suspected morbidity/mortality endpoint will be reported by the investigator and adjudicated by the Cardiovascular Endpoint Committee. Only Adjudication Committee‐confirmed events can be considered for this primary endpoint" Comment: adjudicated/investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) amputation, blindness/severe vision loss, end‐stage renal disease | Low risk | Comment: adjudicated/investigator‐assessed. Only blindness reported |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Low risk |
Quote from publication: "A qualified person must review diary entries with the patient at each visit to determine if the signs and symptoms are consistent with hypoglycemia" Comment: adjudicated/investigator‐assessed/self‐reported outcome measurement. Blinding ensured for participant, investigators and outcome assessors |
| Blinding of outcome assessment (detection bias) incidence of T2DM | Low risk |
Quotes from publication: "Adjudication by the Diabetes Endpoint Adjudication Committee: progression to diabetes may be confirmed by the Committee in cases suggestive of diabetes but where above laboratory test‐based definition does not hold (e.g. due to missing central laboratory measurements or repeat tests outside the 12‐week time limit). E.g., the Committee will adjudicate cases where diabetes has been diagnosed by a primary care physician (possibly based on local laboratory assessments) and/or where anti‐diabetic medication has been initiated; adjudication also includes deciding on the time to progression to diabetes" and "The Diabetes Endpoint Adjudication Committee will be responsible for an independent and blinded assessment of all suspected cases of diabetes" Comment: adjudicated, investigator‐assessed, or both |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk |
Quotes from publication: "The progression to diabetes endpoint is defined through (a) an algorithm based on central laboratory measurements of FPG and/or 2hr OGTT, or (b) adjudication by the Diabetes Endpoint Adjudication Committee as follows..." and "The fasting plasma glucose level or the plasma glucose level 2 hours after a glucose challenge was measured at the closeout visit or during the final 6 months of..." Comment: adjudicated/investigator‐assessed |
| Blinding of outcome assessment (detection bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Low risk |
Quotes from publication: "Occurrence of a suspected morbidity/mortality endpoint will be reported by the investigator and adjudicated by the Cardiovascular Endpoint Committee. Only Adjudication Committee‐confirmed events can be considered for this primary endpoint" and "...to provide an independent and blinded assessment of the cardiovascular efficacy endpoints as defined..." Comment: adjudicated outcome measurement |
| Blinding of outcome assessment (detection bias) non‐serious adverse events | Low risk | Quote from publication: "Information about all adverse events, whether volunteered by the subject, discovered by investigator questioning, or detected through physical examination, laboratory test or other means, will be collected and recorded in the patient’s source documents and CRF and followed as appropriate" Comment: self‐reported outcome measurement/investigator‐assessed |
| Blinding of outcome assessment (detection bias) serious adverse events | Low risk | Comment: adjudicated/investigator‐assessed outcome measurement |
| Blinding of outcome assessment (detection bias) time to progression of T2DM | Low risk |
Quote from publication: "The progression to diabetes endpoint is defined through (a) an algorithm based on central laboratory measurements of FPG and/or 2hr OGTT, or (b) adjudication by the Diabetes Endpoint Adjudication Committee" Comment: adjudicated/investigator‐assessed outcome measurement |
| Incomplete outcome data (attrition bias) all‐cause/cardiovascular mortality | Unclear risk |
Quotes from publication: "Patients who are considered lost to follow‐up at one point in time will remain in the study until trial end or patient’s death, if known. At every scheduled visit until trial end or patient’s death, if known, every possible effort should be made to contact the patient (or patient’s relatives) to obtain visit information. At least vital status information, as a matter of public record in most countries, must be followed at every scheduled visit to the end of the study" and "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups. However, reasons for missing data were not reported for the 163 persons in the nateglinide group and 143 persons in the placebo group |
| Incomplete outcome data (attrition bias) amputation, blindness/severe vision loss, end‐stage renal disease | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) hypoglycaemia | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) incidence of T2DM | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) non‐fatal myocardial infarction/stroke, congestive heart failure | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) non‐serious adverse events | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) serious adverse events | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Incomplete outcome data (attrition bias) time to progression to T2DM | Unclear risk |
Quotes from publication: "After randomization, 212 participants at 10 sites were excluded when the sites were closed owing to deficiencies in the adherence to Good Clinical Practice guidelines, leaving 9306 participants whose data were included in the final analyses (Fig. 1)" and "A total of 609 participants in the nateglinide group (13.1%) and 602 in the placebo group (12.9%) were lost to follow‐up or withdrew from the study; however, because many of these participants were lost to follow‐up or withdrew consent late in the study, information on vital status was available for 95.7% of the possible follow‐up time in both groups" Comment: missing outcomes balanced between intervention groups with similar reasons for missingness. However, unclear why 163 in the nateglinide group and 143 in the placebo group withdrew participation. Also, the overall attrition rate was high |
| Selective reporting (reporting bias) | High risk | Comment: no reporting of HbA1c, end‐stage renal disease and health economics, even though it was stated these outcomes would be measured (see Appendix 8) |
| Other bias | Unclear risk | Comment: commercial funding |
Osei 2004.
| Methods | Parallel randomised controlled clinical trial, randomisation ratio 1:1, superiority design | |
| Participants |
Inclusion criteria: first‐degree relatives (offspring and siblings) of African American individuals with T2DM; IGT during OGTT Exclusion criteria: diabetes, liver, heart, lung and kidney disease, participants taking part in endurance exercise or competitive sports Diagnostic criteria: IGT was defined as fasting serum glucose < 7.8 mmol/L and 2‐hour serum glucose after a 75‐g oral glucose challenge > 7.8 but < 11.1 mmol/L (WHO 1985) |
|
| Interventions |
Number of study centres: 1 Run‐in period: none, but the participants were instructed to follow specific meals at least 3 days before the test Administration free period before testing during trial: participants were asked not to take the study drug on the mornings on which glycaemic measurements were performed. Extension period: no |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (Pfizer Pharmaceutical Inc. supplied glipizide GITS) and non‐commercial funding Publication status: peer‐reviewed journal |
|
| Stated aim for study | Quote from publication: "Therefore, we tested the hypothesis that SU could reverse the early beta‐cell dysfunction and ultimately improve glucose homeostasis in high‐risk African‐American patients with IGT" | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote from publication: "After satisfying study entry requirements and baseline studies, the subjects were randomized in a double blind, placebo‐controlled manner to receive either GITS (5 mg/d or identical placebo for 24 months" Comment: method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk |
Quote from publication: "After satisfying study entry requirements and baseline studies, the subjects were randomized in a double blind, placebo‐controlled manner to receive either GITS (5 mg/d or identical placebo for 24 months" Comment: method of allocation not described |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Low risk |
Quote from publication: "Thus, symptoms suggestive of hypoglycaemia (e.g., nervousness, excessive hunger, tremors, confusion, etc) were recorded in a logbook..." Comment: self‐reported outcome measurement. Blinding of participants was ensured by placebo tablets |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk | Comment: investigator‐assessed. Blinding of participants was ensured by placebo tablets |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Low risk |
Quote from publication: "Thus, symptoms suggestive of hypoglycaemia (e.g., nervousness, excessive hunger, tremors, confusion, etc) were recorded in a logbook..." Comment: self‐reported outcome measurement. Blinding of participants was ensured by use of placebo tablets |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk | Comment: investigator‐assessed.Blinding of participants was ensured by placebo tablets |
| Incomplete outcome data (attrition bias) hypoglycaemia | Unclear risk | Comment: described that no participants had symptoms suggestive of hypoglycaemia. Not reported if any data were missing |
| Selective reporting (reporting bias) | Unclear risk | Comment: no trial protocol available |
| Other bias | Unclear risk | Comment: commercial funding |
Page 1993.
| Methods | Parallel randomised controlled clinical trial, randomisation ratio 1:1:3 (see notes), superiority design | |
| Participants |
Inclusion criteria: family history of T2DM, previously gestational diabetes or a previously raised plasma glucose (5.6 to 6.6 mmol/L) with hyperglycaemia on two separate glucose tolerance tests Exclusion criteria: fasting plasma glucose > 7 mmol/L, diastolic blood pressure > 105 mmHg, treatment with thiazide diuretics, beta‐blockers or calcium antagonists, angina or previously myocardial infarction, lung disease and inability to participate in VO2 measurements, pregnancy, vigorous exercise three or more times per week for a period of 30 minutes or more or participating in regular manual work, BMI < 20 kg/m2, coexisting chronic disease requiring therapy which would prevent participation Diagnostic criteria: fasting plasma glucose > 5.6 mmol/L and (or age‐corrected) achieved plasma glucose during continuous infusion of glucose ( 60 minutes) with model assessment above 9.3 mmol/L. According to the authors this value has been show to be equivalent to the WHO's criteria for impaired glucose tolerance |
|
| Interventions |
Number of study centres: NR Run‐in period: none Administration‐free period before testing during trial: participants were asked not to take the study drug on the morning of the glycaemic measurement at 6 weeks and at 6 months. Participants were retested 4 weeks after the end of the intervention period Extension period: yes |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: commercial funding (Servier) and non‐commercial funding Publication status: peer‐reviewed journal |
|
| Stated aim for study | Quote from publication: "The objectives of this study were: to determine whether when given access to exercise classes, dietary advice and verbal support it is possible to alter the lifestyle of subjects with IGT; to determine whether regular exercise and aiming for an ideal body weight by a high fibre, low fat, high carbohydrate diet will improve plasma glucose levels and cardiovascular risk factors in subjects with impaired glucose tolerance; and to determine whether sulphonylurea therapy with gliclazide 40 mg twice daily is more or less effective than healthy living in improving glucose tolerance and cardiovascular risk factors" | |
| Notes | For the initial 6 weeks, the healthy living group was divided into three subgroups; diet only; exercise only; and diet and exercise. After 6 weeks all three healthy living intervention groups participated in both diet and exercise for the remaining 6 months of the trial | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quotes from publication: "Subjects were randomized into three main groups..." and "Despite randomization, the groups showed disparities in baseline characteristics, and this may lead to error in the interpretation of the results" Comment: method of randomisation not described. However, important differences exist in some baseline characteristics, but the uneven distribution does not appear to be in favour of any of the intervention groups |
| Allocation concealment (selection bias) | Unclear risk |
Quotes from publication: "Subjects were randomized into three main groups..." and "Despite randomization, the groups showed disparities in baseline characteristics, and this may lead to error in the interpretation of the results" Comment: method of allocation not described. However, important differences exist in some baseline characteristics, but the uneven distribution does not appear to be in favour of any of the intervention groups |
| Blinding of participants and personnel (performance bias) hypoglycaemia | Unclear risk | Quote from publication: "No hypoglycaemic episodes were reported in any group" Comment: self‐reported outcome measurement. For the comparison of gliclazide and placebo the participants in both groups and investigators were blinded. However, the participants and investigators were aware of the allocation to the diet and exercise intervention |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk | Comment: investigator‐assessed outcome measurement. For the comparison of gliclazide and placebo the participants in both groups and investigators were blinded. However, the participants and investigators were aware of the allocation to the diet and exercise intervention. The outcome is unlikely to be influenced by the lack of blinding |
| Blinding of outcome assessment (detection bias) hypoglycaemia | Unclear risk | Comment: self‐reported outcome measurement. For the comparison of gliclazide and placebo the participants in both groups and investigators were blinded. However, the participants and investigators were aware of the allocation to the diet and exercise intervention |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk | Comment: investigator‐assessed outcome measurement. For the comparison of gliclazide and placebo the participants in both groups and investigators were blinded. However, the participants and investigators were aware of the allocation to the diet and exercise intervention. The outcome is unlikely to be influenced by the lack of blinding |
| Incomplete outcome data (attrition bias) hypoglycaemia | Unclear risk |
Quote from publication: "One subject in the placebo withdrew before 6‐week assessment due to prolonged holiday and non‐compliance, and three subjects withdrew from the healthy living groups before this time: one became pregnant, one found the time commitment too great and one was apprehensive about blood test. After 6 weeks no subject withdrew from the 'healthy living' group, due to the commitment required to participate in the study" Comment: reasons reported and explained. It is not possible to assess whether the missing data are likely to introduce bias |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Unclear risk |
Quote from publication: "One subject in the placebo withdrew before 6‐week assessment due to prolonged holiday and non‐compliance, and three subjects withdrew from the healthy living groups before this time: one became pregnant, one found the time commitment too great and one was apprehensive about blood test. After 6 weeks no subject withdrew from the 'healthy living' group, due to the commitment required to participate in the study" Comment: reasons reported and explained. It is not possible to assess whether the missing data are likely to introduce bias |
| Selective reporting (reporting bias) | High risk | Comment: according to the publication HbA1c was measured and analysed, but the result is reported only as a non‐significant change (see Appendix 6) |
| Other bias | Unclear risk | Comment: commercial funding |
Papoz 1978.
| Methods | Parallel randomised controlled clinical trial, randomisation ratio 1:1, superiority design | |
| Participants |
Inclusion criteria: male, 25 to 55 years, 'borderline' diabetes (see criteria in the section 'diagnostic criteria') Exclusion criteria: NR Diagnostic criteria: fasting blood glucose ≥ 5.6 mmol/L and < 7.2 mmol/L or 2‐hour blood glucose after a 75‐g oral glucose challenge ≥ 6.7 mmol/L and < 8.3 mmol/L; when these criteria for intermediate hyperglycaemia were fulfilled, a second test was performed: blood glucose concentrations were determined fasting at 15, 30, 60, 120, 80, 240 and 300 minutes after an oral glucose load. Eligible individuals had 2‐hour blood glucose concentrations ≥ 6.7 mmol/L but < 8.3 mmol/L or fasting blood glucose concentrations ≥ 5.6 mmol/L and < 7.2 mmol/L; blood glucose after 30 minutes ≥ 8.9 mmol/L and < 12.2 mmol/L; blood glucose after 60 minutes ≥ 8.9 mmol/L and < 12.2 mmol/L (the European Diabetes Epidemiology Study Group 1970 criteria) |
|
| Interventions |
Number of study centres: 1 Run‐in period: none Administration‐free period before testing during trial: participants received study‐drug on the day of testing blood glucose at 2 months and 14 months. However, the last glycaemic measurements were performed 15 days after the study drug was stopped Extension period: none |
|
| Outcomes | Composite outcome measures reported: no | |
| Study details | Trial terminated early: no | |
| Publication details |
Language of publication: English Funding: non‐commercial funding Publication status: peer‐reviewed journal |
|
| Stated aim for study | Quote from publication: "A double blind controlled clinical trial was undertaken to test the effectiveness of oral hypoglycaemic drugs in improving blood glucose and plasma insulin levels of borderline diabetic patients" | |
| Notes | Blood glucose values in this trial were reported as whole blood glucose. In the tables and result section all values are converted to plasma glucose values (diabetes.co.uk 2016a) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote from publication: "They were randomized into 4 groups according..." Comment: method of randomisation not described |
| Allocation concealment (selection bias) | Unclear risk |
Quote from publication: "They were randomized into 4 groups according..." Comment: method of allocation not described |
| Blinding of participants and personnel (performance bias) measures of blood glucose control | Low risk |
Quote from publication: "A double blind controlled clinical trial was undertaken..." Comment: investigator‐assessed, double‐blinding |
| Blinding of outcome assessment (detection bias) measures of blood glucose control | Low risk |
Quote from publication: "A double blind controlled clinical trial was undertaken..." Comment: investigator‐assessed, double‐blinding |
| Incomplete outcome data (attrition bias) measures of blood glucose control | Unclear risk |
Quote from publication: "Thirty four patients (24 during the first year, 10 during the second year of the study) were lost to follow‐up; they came equally from the four different treatment groups and exhibited similar baseline characteristics to the follow‐up patients. Their removal from the trial did not introduce any bias into the study" Comment: the number of participants lost to follow‐up are reported, but no reasons explained |
| Selective reporting (reporting bias) | High risk | Comment: likely that adverse events have been assessed, as the investigators describe no pathological symptoms occurred during the trial (see Appendix 6) |
| Other bias | Low risk | Comment: the trial appeared to be free of other sources of bias |
Note: where the judgement is 'unclear' and the description is blank, the trial did not report that particular outcome.
BMI: body mass index; FPG: fasting plasma glucose; GITS: glipizide gastrointestinal therapeutic system; HbA1c: glycosylated haemoglobin A1c; HDL: high density lipoprotein; IFG: impaired fasting glucose; IGT: impaired glucose tolerance; IVGTT: intravenous glucose tolerance test; LDL: low density lipoprotein; NANSY: The Nepi ANtidiabetes StudY; NAVIGATOR: Nateglinide+Valsartan to Prevent or Delay Type 2 Diabetes Mellitus and Cardiovascular Complications; NR: not reported; NYHA: New York Heart Association; OGTT: oral glucose tolerance test; SU: sulphonylurea; T2D(M): type 2 diabetes mellitus; VO2: maximum rate of oxygen consumption; WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bavirti 2003 | Not a randomised clinical trial |
| Bitzen 1988 | Not a randomised clinical trial |
| Cederholm 1985 | Participants not randomised to treatment with glipizide |
| Cederholm 1986 | Not a randomised clinical trial |
| DIANA 2012 | Not possible to get separate data on the participants with impaired glucose tolerance (investigator did not reply) |
| Gudipaty 2014 | All participants had a history of type 2 diabetes mellitus |
| Hirose 2002 | Not a randomised clinical trial |
| Igata 2014 | Not possible to get separate data on the participants with impaired glucose tolerance (investigators did not reply) |
| Inoue 1997 | Not a randomised clinical trial |
| Johanson 2005 | Did not have impaired fasting glucose, impaired glucose tolerance or moderately elevated HbA1c as an inclusion criterion |
| Katahira 2005 | Not a randomised clinical trial |
| Lindblad 2001 | Duration of the intervention less than 12 weeks |
| Major‐Pedersen 2008 | Not possible to get separate data on the participants with impaired glucose tolerance (investigators contacted, and replied, but no data were provided) |
| NCT00744965 | Includes pregnant women with gestational diabetes |
| NCT01563120 | Includes pregnant women with gestational diabetes |
| Pontiroli 1991 | Not a randomised clinical trial |
| Ratzmann 1981 | Not a randomised clinical trial |
| Ratzmann 1983 | Not a randomised clinical trial |
| Saloranta 2002 | Duration of the intervention less than 12 weeks |
| Schmoelzer 2006 | Duration of the intervention less than 12 weeks |
| The Fasting Hyperglycaemia Study 1997a | Not possible to get separate data on the participants with impaired glucose tolerance (Rury Holman was asked and replied) |
Differences between protocol and review
No major differences.
Contributions of authors
All review authors read and approved the final review draft.
Bianca Hemmingsen (BH): protocol draft, search strategy development, acquisition of trial reports, trial selection, data extraction, data analysis, data interpretation, review of drafts and future review updates.
David Peick Sonne (DS): protocol draft, acquisition of trial reports, trial selection, data extraction, data analysis, data interpretation, and future review updates.
Maria‐Inti Metzendorf (MIM): protocol draft, search strategy development, review of drafts, and future review updates.
Bernd Richter (BR): protocol draft, search strategy development, data interpretation, review of drafts, and future review updates.
Sources of support
Internal sources
No sources of support supplied
External sources
-
World Health Organization, Other.
This review is part of a series of reviews on interventions for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus which is funded by the WHO (Hemmingsen 2016b; Hemmingsen 2016a).
Declarations of interest
BH: this review is part of a series of reviews on interventions for the prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus, which is funded by the WHO (Hemmingsen 2016a; Hemmingsen 2016b; Hemmingsen 2016c).
DS: none known.
MIM: none known.
BR: none known.
New
References
References to studies included in this review
Eriksson 2006 {published data only}
- Eriksson JG, Lehtovirta M, Ehrnstrom B, Salmela S, Groop L. Long‐term beneficial effects of glipizide treatment on glucose tolerance in subjects with impaired glucose tolerance. Journal of Internal Medicine 2006;259(6):553‐60. [PUBMED: 16704555] [DOI] [PubMed] [Google Scholar]
NANSY 2011 {published data only}
- Lindblad U, Lindberg G, Mansson NO, Ranstam J, Tyrberg M, Jansson S, et al. Can sulphonylurea addition to lifestyle changes help to delay diabetes development in subjects with impaired fasting glucose? The Nepi ANtidiabetes StudY (NANSY). Diabetes, Obesity & Metabolism 2011; Vol. 13, issue 2:185‐8. [PUBMED: 21199271] [DOI] [PubMed]
- Melander A, Lindblad U, Ranstam J. The NEPI Antidiabetes Study (NANSY), a Scandinavian Complement to the DPP Study. Diabetes 1999; Vol. SA351.
- Tyrberg M, Melander A, Lovestam‐Adrian M, Lindblad U. Retinopathy in subjects with impaired fasting glucose: the NANSY‐Eye baseline report. Diabetes, Obesity & Metabolism 2008;10(8):646‐51. [PUBMED: 17645554] [DOI] [PubMed] [Google Scholar]
NAVIGATOR 2010 {published data only}
- Novartis clinical result database ‐ NAVIGATOR. www.novctrd.com/CtrdWeb/displaypdf.nov?trialresultid=3223 (accessed 28 April 2016).
- Bethel MA, Chacra AR, Deedwania P, Fulcher GR, Holman RR, Jenssen T, et al. Estimating diabetes risk in patients with impaired glucose tolerance and cardiovascular disease (CVD) or risk factors for CVD. Diabetes 2011;60(0):A357‐8. [Google Scholar]
- Califf RM, Boolell M, Haffner SM, Bethel M, McMurray J, Duggal A, et al. Prevention of diabetes and cardiovascular disease in patients with impaired glucose tolerance: rationale and design of the Nateglinide And Valsartan in Impaired Glucose Tolerance Outcomes Research (NAVIGATOR) Trial. American Heart Journal 2008;156(4):623‐32. [PUBMED: 18946890] [DOI] [PubMed] [Google Scholar]
- Califf RM, Chiang FT, Gaciong Z, Giles TD, Laakso M, Leiter LA, et al. Stroke in people with impaired glucose tolerance. Circulation 2011;124(21). [Google Scholar]
- NAVIGATOR Study Group. Effect of nateglinide on the incidence of diabetes and cardiovascular events. New England Journal of Medicine 2010;362(16):1463‐76. [DOI: 10.1056/NEJMoa1001122.] [DOI] [PubMed] [Google Scholar]
- NAVIGATOR Study Group. Effect of valsartan on the incidence of diabetes and cardiovascular events. New England Journal of Medicine 2010;362(16):1477‐90. [DOI: 10.1056/NEJMoa1001121] [DOI] [PubMed] [Google Scholar]
- NCT00097786. Long‐term study of nateglinide+valsartan to prevent or delay type II diabetes mellitus and cardiovascular complications (Navigator). clinicaltrials.gov/ct2/show/NCT00097786 (accessed 22 April 2016).
Osei 2004 {published data only}
- Osei K, Rhinesmith S, Gaillard T, Schuster D. Beneficial metabolic effects of chronic glipizide in obese African Americans with impaired glucose tolerance: implications for primary prevention of type 2 diabetes. Metabolism: Clinical and Experimental 2004;53(4):414‐22. [PUBMED: 15045685] [DOI] [PubMed] [Google Scholar]
Page 1993 {published data only}
- Page RC, Harnden KE, Cook JT, Turner RC. Can life‐styles of subjects with impaired glucose tolerance be changed? A feasibility study. Diabetic Medicine 1992;9(6):562‐6. [PUBMED: 1643806] [DOI] [PubMed] [Google Scholar]
- Page RC, Harnden KE, Walravens NK, Onslow C, Sutton P, Levy JC, et al. 'Healthy living' and sulphonylurea therapy have different effects on glucose tolerance and risk factors for vascular disease in subjects with impaired glucose tolerance. The Quarterly Journal of Medicine 1993;86(3):145‐54. [PUBMED: 8483989] [PubMed] [Google Scholar]
Papoz 1978 {published data only}
- Eschwege E. French Study Group for Diabetes Epidemiology (A.F.E.D.I.A.). Short term effects (2 years) of oral treatment for diabetes in the borderline impairment of oral glucose tolerance test. Diabetologia 1974;10(363):Abstract 31. [Google Scholar]
- Papoz L, Job D, Eschwege E, Aboulker JP, Cubeau J, Pequignot G, et al. Effect of oral hypoglycaemic drugs on glucose tolerance and insulin secretion in borderline diabetic patients. Diabetologia 1978;15(5):373‐80. [PUBMED: 104897] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bavirti 2003 {published data only}
- Bavirti S, Tayek JA. Low‐dose oral glyburide reduces glucose production rates in patients with impaired fasting glucose. Metabolism: Clinical and Experimental 2003;52(4):407‐12. [PUBMED: 12701050] [DOI] [PubMed] [Google Scholar]
Bitzen 1988 {published data only}
- Bitzen PO, Melander A, Schersten B, Wahlin‐Boll E. The influence of glipizide on early insulin release and glucose disposal before and after dietary regulation in diabetic patients with different degrees of hyperglycaemia. European Journal of Clinical Pharmacology 1988;35(1):31‐7. [PUBMED: 3065086] [DOI] [PubMed] [Google Scholar]
Cederholm 1985 {published data only}
- Cederholm J. Short‐term treatment of glucose intolerance in middle‐aged subjects by diet, exercise and sulfonylurea. Upsala Journal of Medical Sciences 1985;90(3):229‐42. [PUBMED: 4095819] [DOI] [PubMed] [Google Scholar]
Cederholm 1986 {published data only}
- Cederholm J, Wibell L. The impact of treatment on insulin release and relative peripheral resistance during the oral glucose tolerance test. A study of noninsulin‐dependent diabetes mellitus and glucose intolerance. Diabete & Metabolisme 1986;12(1):10‐5. [PUBMED: 3516748] [PubMed] [Google Scholar]
DIANA 2012 {published data only}
- Kataoka Y, Yasuda S, Miyamoto Y, Sase K, Kosuge M, Kimura K, et al. Effects of voglibose and nateglinide on glycemic status and coronary atherosclerosis in early‐stage diabetic patients. Circulation Journal 2012;76(3):712‐20. [PUBMED: 22240597] [DOI] [PubMed] [Google Scholar]
Gudipaty 2014 {published data only}
- Gudipaty L, Fuller C, Rosenfeld N, Rickels MR. No effect of exenatide or sitagliptin therapy on beta‐cell secretory capacity in early type 2 diabetes. Diabetes 2013;62:A288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipaty L, Rosenfeld NK, Fuller CS, Gallop R, Schutta MH, Rickels MR. Effect of exenatide, sitagliptin, or glimepiride on beta‐cell secretory capacity in early type 2 diabetes. Diabetes Care 2014;37(9):2451‐8. [PUBMED: 24969577] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00775684. Effect of exenatide, sitagliptin or glimepiride on functional ß‐cell mass. clinicaltrials.gov/ct2/show/NCT00775684 (accessed 3 March 2016).
Hirose 2002 {published data only}
- Hirose T, Mizuno R, Yoshimoto T. The effects of nateglinide following oral glucose load in impaired glucose tolerance subjects: rapid insulin stimulation by nateglinide in IGT subjects. Endocrine Journal 2002;49(6):649‐52. [PUBMED: 12625415] [DOI] [PubMed] [Google Scholar]
Igata 2014 {published data only}
- Igata S, Tahara N, Tahara A, Nitta Y, Honda A, Kodama N, et al. Visceral fat metabolic activity is independently associated with coronary artery inflammation in patients with impaired glucose intolerance or type 2 diabetes. Circulation 2014;130. [Google Scholar]
- Kodama N. Effects of pioglitazone versus glimepiride on abdominal fat glucose metabolism in patients with impaired glucose tolerance and/or type 2 diabetes mellitus. Circulation 2012;126(21). [Google Scholar]
- Kodama N, Tahara N, Tahara A, Honda A, Nitta Y, Mizoguchi M, et al. Effects of pioglitazone on visceral fat metabolic activity in impaired glucose tolerance or type 2 diabetes mellitus. Journal of Clinical Endocrinology and Metabolism 2013;98(11):4438‐45. [PUBMED: 24030946] [DOI] [PubMed] [Google Scholar]
- Mizoguchi M, Tahara N, Tahara A, Nitta Y, Kodama N, Oba T, et al. Pioglitazone attenuates atherosclerotic plaque inflammation in patients with impaired glucose tolerance or diabetes a prospective, randomized, comparator‐controlled study using serial FDG PET/CT imaging study of carotid artery and ascending aorta. JACC. Cardiovascular Imaging 2011;4(10):1110‐8. [PUBMED: 21999871] [DOI] [PubMed] [Google Scholar]
- NCT00722631. Anti‐inflammatory effects of pioglitazone. clinicaltrials.gov/ct2/show/NCT00722631 (accessed 22 April 2016).
- Nitta Y, Tahara N, Honda A, Tahara A, Kodama N, Mizoguchi M, et al. Effects of pioglitazone on coronary arterial inflammation evaluated by serial FDG‐PET/CT in patients with impaired glucose tolerant or type 2 diabetes. Circulation. Conference: American Heart Association. 2012; Vol. 126, issue 21.
- Nitta Y, Tahara N, Tahara A, Honda A, Kodama N, Mizoguchi M, et al. Pioglitazone decreases coronary artery inflammation in impaired glucose tolerance and diabetes mellitus: evaluation by FDG‐PET/CT imaging. JACC. Cardiovascular Imaging 2013;6(11):1172‐82. [PUBMED: 24229770] [DOI] [PubMed] [Google Scholar]
- Tahara N, Yamagishi S, Mizoguchi M, Tahara A, Imaizumi T. Pioglitazone decreases asymmetric dimethylarginine levels in patients with impaired glucose tolerance or type 2 diabetes. Rejuvenation Research 2013;16(5):344‐51. [PUBMED: 23777507] [DOI] [PubMed] [Google Scholar]
Inoue 1997 {published data only}
- Inoue I, Takahashi K, Noji S, Awata T, Negishi K, Katayama S. Acarbose controls postprandial hyperproinsulinemia in non‐insulin dependent diabetes mellitus. Diabetes Research and Clinical Practice 1997;36(3):143‐51. [PUBMED: 9237780] [DOI] [PubMed] [Google Scholar]
Johanson 2005 {published data only}
- Johanson EH, Jansson PA, Gustafson B, Sandqvist M, Taskinen MR, Smith U, et al. No acute effect of nateglinide on postprandial lipid and lipoprotein responses in subjects at risk for type 2 diabetes. Diabetes/Metabolism Research and Reviews 2005;21(4):376‐81. [PUBMED: 15724236] [DOI] [PubMed] [Google Scholar]
Katahira 2005 {published data only}
- Katahira H, Ishida H. [Effects of mitiglinide in treatment of impaired glucose tolerance]. Nihon Rinsho. Japanese Journal of Clinical Medicine 2005;63 Suppl 2:444‐50. [PUBMED: 15779420] [PubMed] [Google Scholar]
Lindblad 2001 {published data only}
- Lindblad U, Lindwall K, Sjostrand A, Ranstam J, Melander A. The NEPI antidiabetes study (NANSY). 1: short‐term dose‐effect relations of glimepiride in subjects with impaired fasting glucose. Diabetes, Obesity & Metabolism 2001;3(6):443‐51. [PUBMED: 11903417] [DOI] [PubMed] [Google Scholar]
Major‐Pedersen 2008 {published data only}
- Major‐Pedersen A, Ihlemann N, Hermann TS, Christiansen B, Kveiborg B, Dominguez H, et al. Effects of acute and chronic attenuation of postprandial hyperglycemia on postglucose‐load endothelial function in insulin resistant individuals: is stimulation of first phase insulin secretion beneficial for the endothelial function?. Hormone and Metabolic Research 2008;40(9):607‐13. [PUBMED: 18792871] [DOI] [PubMed] [Google Scholar]
- NCT00259168. Insulin resistance and vessel function after meals: does early intervention make a difference?. clinicaltrials.gov/ct2/show/NCT00259168?term=NCT00259168&rank=1 (accessed 22 April 2016).
NCT00744965 {published data only}
- NCT00744965. Treatment of mild gestational diabetes with glyburide versus placebo. clinicaltrials.gov/ct2/show/NCT00744965 (accessed 17 May 2016).
NCT01563120 {published data only}
- NCT01563120. A comparison between two oral hypoglycemics ‐ metformin and glybenclamide for the treatment of gestational diabetes mellitus. clinicaltrials.gov/show/NCT01563120 (accessed 17 March 2016).
Pontiroli 1991 {published data only}
- Pontiroli AE, Perfetti MG, Pozza G. Acute effect of glipizide on glucose tolerance in obesity and diabetes mellitus (NIDDM). European Journal of Clinical Pharmacology 1991;40(1):23‐6. [PUBMED: 2060541] [DOI] [PubMed] [Google Scholar]
- Pontiroli AE, Perfetti MG, Pozza G. Improvement of glucose tolerance by minimal doses of glipizide in obese subjects with different degrees of glucose intolerance. Hormone and Metabolic Research. Supplement Series 1992;26:32‐4. [PUBMED: 1490690] [PubMed] [Google Scholar]
Ratzmann 1981 {published data only}
- Ratzmann KP, Witt S, Schulz B, Jahr D, Heinke P. [Effect of glibenclamide therapy on carbohydrate and lipid metabolism and insulin secretion in patients with glucose tolerance disorders : a 5‐year study] [Einfluss einer Glibenclamid‐Therapie auf Kohlenhydrat‐ und Fettstoffwechsel sowie Insulinsekretion bei Personen mit gestörter Glukosetorleranz: Verlaufsbeobachtungen bis zu 5 Jahren]. Zeitschrift fur die Gesamte Innere Medizin und ihre Grenzgebiete 1981;36(23):913‐7. [PUBMED: 6805141] [PubMed] [Google Scholar]
Ratzmann 1983 {published data only}
- Ratzmann KP, Witt S, Schulz B. The effect of long‐term glibenclamide treatment on glucose tolerance, insulin secretion and serum lipids in subjects with impaired glucose tolerance. Diabète & Métabolisme 1983;9(2):87‐93. [PUBMED: 6413265] [PubMed] [Google Scholar]
Saloranta 2002 {published data only}
- Saloranta C, Guitard C, Pecher E, Pablos‐Velasco P, Lahti K, Brunel P, et al. Nateglinide improves early insulin secretion and controls postprandial glucose excursions in a prediabetic population. Diabetes Care 2002;25(12):2141‐6. [PUBMED: 12453951] [DOI] [PubMed] [Google Scholar]
Schmoelzer 2006 {published data only}
- Schmoelzer I, Wascher TC. Effect of repaglinide on endothelial dysfunction during a glucose tolerance test in subjects with impaired glucose tolerance. Cardiovascular Diabetology 2006;5:9. [PUBMED: 16606452] [DOI] [PMC free article] [PubMed] [Google Scholar]
The Fasting Hyperglycaemia Study 1997a {published data only}
- Dyson PA, Hammersley MS, Morris RJ, Holman RR, Turner RC. The Fasting Hyperglycaemia Study: II. Randomized controlled trial of reinforced healthy‐living advice in subjects with increased but not diabetic fasting plasma glucose. Metabolism: Clinical and Experimental 1997;46(12 Suppl 1):50‐5. [PUBMED: 9439560] [DOI] [PubMed] [Google Scholar]
- Hammersley MS, Meyer LC, Morris RJ, Manley SE, Turner RC, Holman RR. The Fasting Hyperglycaemia Study: I. Subject identification and recruitment for a non‐insulin‐dependent diabetes prevention trial. Metabolism: Clinical and Experimental 1997;46(12 Suppl 1):44‐9. [PUBMED: 9439559] [DOI] [PubMed] [Google Scholar]
- Karunakaran S, Hammersley MS, Morris RJ, Turner RC, Holman RR. The Fasting Hyperglycaemia Study: III. Randomized controlled trial of sulfonylurea therapy in subjects with increased but not diabetic fasting plasma glucose. Metabolism: Clinical and Experimental 1997;46(12 Suppl 1):56‐60. [PUBMED: 9439561] [DOI] [PubMed] [Google Scholar]
Additional references
Abdul‐Ghani 2006
- Abdul‐Ghani MA, Jenkinson CP, Richardson DK, Tripathy D, DeFronzo RA. Insulin secretion and action in subjects with impaired fasting glucose and impaired glucose tolerance: results from the Veterans Administration Genetic Epidemiology Study. Diabetes 2006;55(5):1430‐5. [PUBMED: 16644701] [DOI] [PubMed] [Google Scholar]
ACT NOW 2011
- DeFronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. New England Journal of Medicine 2011;364(12):1104‐15. [PUBMED: 21428766] [DOI] [PubMed] [Google Scholar]
ADA 1997
- The Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 1997;20(7):1183‐97. [DOI] [PubMed] [Google Scholar]
ADA 2003
- Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Report of the expert committee on the diagnosis and classification of diabetes mellitus. Diabetes Care 2003;26(Suppl 1):S5‐20. [DOI] [PubMed] [Google Scholar]
ADA 2008
- American Diabetes Association. Standards of medical care in diabetes ‐ 2008. Diabetes Care 2008;31(Suppl 1):S12‐54. [PUBMED: 18165335] [DOI] [PubMed] [Google Scholar]
ADA 2010
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010;33(Suppl 1):S62‐9. [PUBMED: 20042775] [DOI] [PMC free article] [PubMed] [Google Scholar]
ADA 2015
- American Diabetes Association. Standards of medical care in diabetes‐2015. Diabetes Care 2015;38(Suppl 1):S1‐93. [PUBMED: 24357209] [PubMed] [Google Scholar]
ADOPT 2006
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. New England Journal of Medicine 2006;355(23):2427‐43. [DOI] [PubMed] [Google Scholar]
Anderson 2005
- Anderson DC Jr. Pharmacologic prevention or delay of type 2 diabetes mellitus. Annals of Pharmacotherapy 2005;39(1):102‐9. [PUBMED: 15562143] [DOI] [PubMed] [Google Scholar]
Bedford 1982
- Keen H, Jarrett RJ, McCartney P. The ten‐year follow‐up of the Bedford survey (1962‐1972): glucose tolerance and diabetes. Diabetologia 1982;22(2):73‐8. [PUBMED: 7060852] [DOI] [PubMed] [Google Scholar]
Bell 2013
- Bell ML, McKenzie JE. Designing psycho‐oncology randomised trials and cluster randomised trials: variance components and intra‐cluster correlation of commonly used psychosocial measures. Psycho‐oncology 2013;22:1738‐47. [DOI] [PubMed] [Google Scholar]
Beller 2013
- Beller EM, Chen JK, Wang UL, Glasziou PP. Are systematic reviews up‐to‐date at the time of publication?. Systematic Reviews 2013;2:36. [2046‐4053: (Electronic)] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhardwaj 2010
- Bhardwaj A, Agarwal V, Phung OJ, Baker WL, Tongbram V, Coleman CI. Can oral hypoglycemic agents restore normoglycemia in individuals with insulin resistance?. Diabetes. Conference: 70th Scientific Sessions of the American Diabetes Association Orlando, FL United States. 2010; Vol. 0, issue 0.
Black 2007
- Black C, Donnelly P, McIntyre L, Royle P, Shepherd JJ, Thomas S. Meglitinide analogues for type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD004654.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Blickle 2006
- Blickle JF. Meglitinide analogues: a review of clinical data focused on recent trials. Diabetes & Metabolism 2006;32(2):113‐20. [PUBMED: 16735959] [DOI] [PubMed] [Google Scholar]
Botnia Study 1996
- Groop L, Forsblom C, Lehtovirta M, Tuomi T, Karanko S, Nissen M, et al. Metabolic consequences of a family history of NIDDM (the Botnia study): evidence for sex‐specific parental effects. Diabetes 1996;45(11):1585‐93. [PUBMED: 8866565] [DOI] [PubMed] [Google Scholar]
Boutron 2014
- Boutron I, Altman DG, Hopewell S, Vera‐Badillo F, Tannock I, Ravaud P. Impact of spin in the abstracts of articles reporting results of randomized controlled trials in the field of cancer: the SPIIN randomized controlled trial. Journal of Clinical Oncology 2014;32:4120‐6. [DOI] [PubMed] [Google Scholar]
Brok 2009
- Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta‐analyses may be inconclusive ‐ Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta‐analyses. International Journal of Epidemiology 2009;38(1):287‐98. [PUBMED: 18824466] [DOI] [PubMed] [Google Scholar]
CDC 2015
- Centers for Disease Control and Prevention. 2014 National Diabetes Statistics Report. www.cdc.gov/diabetes/data/statistics/2014statisticsreport.html (accessed 15 October 2015).
Cheng 2006
- Cheng C, Kushner H, Falkner BE. The utility of fasting glucose for detection of prediabetes. Metabolism: Clinical and Experimental 2006;55(4):434‐8. [PUBMED: 16546472] [DOI] [PubMed] [Google Scholar]
ClinicalTrials.gov
- clinicaltrials.gov/ct2/results?term=prediabetes+drugs&Search=Search (accessed 12 February 2016).
Corbett 2014
- Corbett MS, Higgins JP, Woolacott NF. Assessing baseline imbalance in randomised trials: implications for the Cochrane risk of bias tool. Research Synthesis Methods 2014;5:79‐85. [DOI] [PubMed] [Google Scholar]
Deeks 2003
- Deeks JJ, Dinnes J, D'Amico R, Sowden AJ, Sakarovitch C, Song F, et al. Evaluating non‐randomised intervention studies. Health Technology Assessment (Winchester, England) 2003;7(27):iii‐x, 1‐173. [PUBMED: 14499048] [DOI] [PubMed] [Google Scholar]
DeFronzo 1989
- DeFronzo RA, Ferrannini E, Simonson DC. Fasting hyperglycemia in non‐insulin‐dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism: Clinical and Experimental 1989;38(4):387‐95. [PUBMED: 2657323] [DOI] [PubMed] [Google Scholar]
Diabetes Prevention Program 2002
- Knowler WC, Barrett‐Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. New England Journal of Medicine 2002;346(6):393‐403. [PUBMED: 11832527] [DOI] [PMC free article] [PubMed] [Google Scholar]
Diabetes Prevention Program FU 2009
- Diabetes Prevention Program Research Group, Knowler WC, Fowler SE, Hamman RF, Christophi CA, Hoffman HJ, Brenneman AT, et al. 10‐year follow‐up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009;374(9702):1677‐86. [DOI: 10.1016/S0140-6736(09)61457-4; PUBMED: 19878986] [DOI] [PMC free article] [PubMed] [Google Scholar]
diabetes.co.uk 2016a
- Diabetes.co.uk. Convert whole blood results to plasma readings. www.diabetes.co.uk/whole‐blood‐readings‐to‐plasma‐converter.html (assessed 22 August 2016).
diabetes.co.uk 2016b
- Diabetes.co.uk. Blood sugar converter. www.diabetes.co.uk/blood‐sugar‐converter.html (assessed 22 August 2016).
DREAM 2008
- DREAM Trial Investigators, Dagenais GR, Gerstein HC, Holman R, Budaj A, Escalante A, et al. Effects of ramipril and rosiglitazone on cardiovascular and renal outcomes in people with impaired glucose tolerance or impaired fasting glucose: results of the Diabetes REduction Assessment with ramipril and rosiglitazone Medication (DREAM) trial. Diabetes Care 2008;31(5):1007‐14. [DOI: 10.2337/dc07-1868%3C/body%3E%3C/html%3E] [DOI] [PubMed] [Google Scholar]
Drugs.com 2016a
- Drugs.com. Glimepiride. www.drugs.com/pro/glimepiride.html (accessed 5 October 2016).
Drugs.com 2016b
- Drugs.com. Gliclazide 80 mg tablets. www.drugs.com/uk/gliclazide‐80‐mg‐tablets‐leaflet.html (accessed 5 October 2016).
Dunkley 2014
- Dunkley AJ, Bodicoat DH, Greaves CJ, Russell C, Yates T, Davies MJ, et al. Diabetes prevention in the real world: effectiveness of pragmatic lifestyle interventions for the prevention of type 2 diabetes and of the impact of adherence to guideline recommendations: a systematic review and meta‐analysis. Diabetes Care 2014;37(4):922‐33. [PUBMED: 24652723] [DOI] [PubMed] [Google Scholar]
Egbewale 2014
- Egbewale BE, Lewis M, Sim J. Bias, precision and statistical power of analysis of covariance in the analysis of randomized trials with baseline imbalance: a simulation study. BMC Medical Research Methodology 2014;14:49. [DOI: 10.1186/1471-2288-14-49] [DOI] [PMC free article] [PubMed] [Google Scholar]
Finnish Diabetes Prevention Study Group 2001
- Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne‐Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. New England Journal of Medicine 2001;344(18):1343‐50. [PUBMED: 11333990] [DOI] [PubMed] [Google Scholar]
Gosmanov 2014
- Gosmanov AR, Wan J. Low positive predictive value of hemoglobin A1c for diagnosis of prediabetes in clinical practice. American Journal of the Medical Sciences 2014;348(3):191‐4. [PUBMED: 24556928] [DOI] [PubMed] [Google Scholar]
GRADEproGDT 2015 [Computer program]
- GRADE Working Group, McMaster University. GRADEproGDT. Hamilton (ON): GRADE Working Group, McMaster University, 2015.
Harrower 2000
- Harrower AD. Comparative tolerability of sulphonylureas in diabetes mellitus. Drug Safety 2000;22(4):313‐20. [PUBMED: 10789825] [DOI] [PubMed] [Google Scholar]
Hemmingsen 2016a
- Hemmingsen B, Krogh J, Metzendorf MI, Richter B. Dipeptidyl‐peptidase (DPP)‐4 inhibitors or glucagon‐like peptide (GLP)‐1 analogues for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2016, Issue 5. [DOI: 10.1002/14651858.CD012204] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hemmingsen 2016b
- Hemmingsen B, Krogh J, Metzendorf MI, Richter B. Sodium‐glucose cotransporter (SGLT) 2 inhibitors for prevention or delay of type 2 diabetes mellitus and its associated complications in people at risk for the development of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD012106.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta‐analysis. Statistics in Medicine 2002;21:1539‐58. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analysis. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2009
- Higgins JPT, Thompson SG, Spiegelhalter DJ. A re‐evaluation of random‐effects meta‐analysis. Journal of the Royal Statistical Society: Series A (Statistics in Society) 2009;172(1):137‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2010
- Higgins JP, Whitehead A, Simmonds M. Sequential methods for random‐effects meta‐analysis. Statistics in Medicine 2011;30(9):903‐21. [PUBMED: 21190240] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:d5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hopper 2011
- Hopper I, Billah B, Skiba M, Krum H. Prevention of diabetes and reduction in major cardiovascular events in studies of subjects with prediabetes: meta‐analysis of randomised controlled clinical trials. European Journal of Cardiovascular Prevention and Rehabilitation 2011;18(6):813‐23. [PUBMED: 21878448] [DOI] [PubMed] [Google Scholar]
Hróbjartsson 2013
- Hróbjartsson A, Thomsen AS, Emanuelsson F, Tendal B, Hilden J, Boutron I, et al. Observer bias in randomized clinical trials with measurement scale outcomes: a systematic review of trials with both blinded and nonblinded assessors. Canadian Medical Association Journal 2013;185(4):E201‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice //1997 CFR & ICH Guidelines. PA 19063‐2043. USA: Barnett International/PAREXEL, 1997.
IDF 2013
- International Diabetes Federation. IDF Diabetes Atlas. 6th Edition. Brussels: International Diabetes Federation, 2013. [Google Scholar]
IEC 2009
- International Expert Committee. International Expert Committee report on the role of the A1C assay in the diagnosis of diabetes. Diabetes Care 2009;32(7):1327‐34. [PUBMED: 19502545] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jensen 2002
- Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE. Beta‐cell function is a major contributor to oral glucose tolerance in high‐risk relatives of four ethnic groups in the U.S. Diabetes 2002;51(7):2170‐8. [PUBMED: 12086947] [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:c365. [DOI: 10.1136/bmj.c365] [DOI] [PubMed] [Google Scholar]
Lan 1983
- Lan KKG, DeMets DL. Discrete sequential boundaries for clinical trials. Biometrika 1983;70:659‐63. [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic and meta‐analyses of studies that evaluate interventions: explanation and elaboration. PLoS Medicine 2009;6(7):1‐28. [DOI: 10.1371/journal.pmed.1000100] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
Mathieu 2009
- Mathieu S, Boutron I, Moher D, Altman DG, Ravaud P. Comparison of registered and published primary outcomes in randomized controlled trials. JAMA 2009;302:977‐84. [DOI] [PubMed] [Google Scholar]
McCall 2001
- McCall AL. Clinical review of glimepiride. Expert Opinion on Pharmacotherapy 2001;2(4):699‐713. [PUBMED: 11336617] [DOI] [PubMed] [Google Scholar]
Meader 2014
- Meader N, King K, Llewellyn A, Norman G, Brown J, Rodgers M, et al. A checklist designed to aid consistency and reproducibility of GRADE assessments: development and pilot validation. Systematic Reviews 2014;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meigs 2002
- Meigs JB, Nathan DM, D'Agostino RB Sr, Wilson PW. Fasting and postchallenge glycemia and cardiovascular disease risk: the Framingham Offspring Study. Diabetes Care 2002;25(10):1845‐50. [PUBMED: 12351489] [DOI] [PubMed] [Google Scholar]
Morris 2013
- Morris DH, Khunti K, Achana F, Srinivasan B, Gray LJ, Davies MJ, et al. Progression rates from HbA1c 6.0‐6.4% and other prediabetes definitions to type 2 diabetes: a meta‐analysis. Diabetologia 2013;56(7):1489‐93. [PUBMED: 23584433] [DOI] [PubMed] [Google Scholar]
NDDG 1979
- National Diabetes Data Group. Classification and diagnosis of diabetes mellitus and other categories of glucose intolerance. National Diabetes Data Group. Diabetes 1979;28(12):1039‐57. [PUBMED: 510803] [DOI] [PubMed] [Google Scholar]
Pantalone 2012
- Pantalone KM, Kattan MW, Yu C, Wells BJ, Arrigain S, Jain A, et al. Increase in overall mortality risk in patients with type 2 diabetes receiving glipizide, glyburide or glimepiride monotherapy versus metformin: a retrospective analysis. Diabetes, Obesity & Metabolism 2012;14(9):803‐9. [PUBMED: 22486923] [DOI] [PubMed] [Google Scholar]
Phillippe 2013
- Phillippe HM, Wargo KA. Mitiglinide for type 2 diabetes treatment. Expert Opinion on Pharmacotherapy 2013;14(15):2133‐44. [PUBMED: 23992284] [DOI] [PubMed] [Google Scholar]
Phung 2012
- Phung OJ, Baker WL, Tongbram V, Bhardwaj A, Coleman CI. Oral antidiabetic drugs and regression from prediabetes to normoglycemia: a meta‐analysis. Annals of Pharmacotherapy 2012;46(4):469‐76. [PUBMED: 22474136] [DOI] [PubMed] [Google Scholar]
Pogue 1997
- Pogue JM, Yusuf S. Cumulating evidence from randomized trials: utilizing sequential monitoring boundaries for cumulative meta‐analysis. Controlled Clinical Trials 1997;18(6):580‐93; discussion 661‐6. [PUBMED: 9408720] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Riley 2011
- Riley RD, Higgins JP, Deeks JJ. Interpretation of random effects meta‐analyses. BMJ 2011;342:d549. [DOI] [PubMed] [Google Scholar]
Riley 2013
- Riley RD, Kauser I, Bland M, Thijs L, Staessen JA, Wang J, et al. Meta‐analysis of randomised trials with a continuous outcome according to baseline imbalance and availability of individual participant data. Statistics in Medicine 2013;32(16):2747‐66. [DOI] [PubMed] [Google Scholar]
Roumie 2012
- Roumie CL, Hung AM, Greevy RA, Grijalva CG, Liu X, Murff HJ, et al. Comparative effectiveness of sulfonylurea and metformin monotherapy on cardiovascular events in type 2 diabetes mellitus: a cohort study. Annals of Internal Medicine 2012;157(9):601‐10. [PUBMED: 23128859] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sartor 1980
- Sartor G, Schersten B, Carlstrom S, Melander A, Norden A, Persson G. Ten‐year follow‐up of subjects with impaired glucose tolerance: prevention of diabetes by tolbutamide and diet regulation. Diabetes 1980;29(1):41‐9. [PUBMED: 7380107] [DOI] [PubMed] [Google Scholar]
Schramm 2011
- Schramm TK, Gislason GH, Vaag A, Rasmussen JN, Folke F, Hansen ML, et al. Mortality and cardiovascular risk associated with different insulin secretagogues compared with metformin in type 2 diabetes, with or without a previous myocardial infarction: a nationwide study. European Heart Journal 2011;32(15):1900‐8. [PUBMED: 21471135] [DOI] [PubMed] [Google Scholar]
Scott 2012
- Scott LJ. Repaglinide: a review of its use in type 2 diabetes mellitus. Drugs 2012;72(2):249‐72. [PUBMED: 22268393] [DOI] [PubMed] [Google Scholar]
Selvin 2011
- Selvin E, Steffes MW, Ballantyne CM, Hoogeveen RC, Coresh J, Brancati FL. Racial differences in glycemic markers: a cross‐sectional analysis of community‐based data. Annals of Internal Medicine 2011;154(5):303‐9. [PUBMED: 21357907] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JA, Sutton AJ, Ioannidis JP, Terrin N, Jones DR, Lau J, et al. Recommendations for examining and interpreting funnel plot asymmetry in meta‐analyses of randomised controlled trials. BMJ 2011;343:d4002. [DOI] [PubMed] [Google Scholar]
UGDP 1976
- The University Group Diabetes Program. A study of the effects of hypoglycemic agents on vascular complications in patients with adult‐onset diabetes. VI. Supplementary report on nonfatal events in patients treated with tolbutamide. Diabetes 1976;25:1129‐53. [DOI] [PubMed] [Google Scholar]
UKPDS 33 1998
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood‐glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352(9131):837‐53. [PUBMED: 9742976] [PubMed] [Google Scholar]
UKPDS 34 1998
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998;352(9131):854‐65. [PUBMED: 9742977] [PubMed] [Google Scholar]
Van de Laar 2006
- Laar FA, Lucassen PL, Akkermans RP, Lisdonk EH, Grauw WJ. Alpha‐glucosidase inhibitors for people with impaired glucose tolerance or impaired fasting blood glucose. Cochrane Database of Systematic Reviews 2006;4:CD005061. [PUBMED: 17054235] [DOI] [PubMed] [Google Scholar]
Viera 2011
- Viera AJ. Predisease: when does it make sense?. Epidemiologic Reviews 2011;33:122‐34. [DOI] [PubMed] [Google Scholar]
Vist 2008
- Vist GE, Bryant D, Somerville L, Birminghem T, Oxman AD. Outcomes of patients who participate in randomized controlled trials compared to similar patients receiving similar interventions who do not participate. Cochrane Database of Systematic Reviews 2008, Issue 3. [DOI: 10.1002/14651858.MR000009.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [PUBMED: 18083463] [DOI] [PubMed] [Google Scholar]
WHO 1998
- Alberti KM, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part I: diagnosis and classification of diabetes mellitus. Provisional report of a WHO consultation. Diabetic Medicine 1998;15(7):539‐53. [DOI] [PubMed] [Google Scholar]
WHO 1999
- World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1. Diagnosis and classification of diabetes mellitus. WHO, 1999. [Google Scholar]
WHO/IDF 2006
- World Health Organization/ International Diabetes Federation. Definition and diagnosis of diabetes mellitus and intermediate hyperglycaemia: report of a WHO/IDF consultation. www.idf.org/webdata/docs/WHO_IDF_definition_diagnosis_of_diabetes.pdf (accessed 10 October 2015).
Wong 2006a
- Wong SS, Wilczynski NL, Haynes RB. Comparison of top‐performing search strategies for detecting clinically sound treatment studies and systematic reviews in MEDLINE and EMBASE. Journal of the Medical Library Association 2006;94(4):451‐5. [PMC free article] [PubMed] [Google Scholar]
Wong 2006b
- Wong SSL, Wilczynski NL, Haynes RB. Developing optimal search strategies for detecting clinically sound treatment studies in EMBASE. Journal of the Medical Library Association 2006;94(1):41‐7. [PMC free article] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yudkin 2014
- Yudkin JS, Montori VM. The epidemic of pre‐diabetes: the medicine and the politics. BMJ 2014;349:g4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Hemmingsen 2016c
- Hemmingsen B, Sonne DP, Metzendorf MI, Richter B. Insulin secretagogues for prevention or delay of type 2 diabetes mellitus and its associated complications in persons at increased risk for the development of type 2 diabetes mellitus. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD012151] [DOI] [PMC free article] [PubMed] [Google Scholar]