

Abstract
m6A is the most common form of mRNA modification. However, little is known about its role in clear cell renal cell carcinoma (ccRCC). This study aims to identify gene signatures and prognostic values of m6A regulators in ccRCC. In this study, a total of 528 ccRCC patients from TCGA database with sequencing and CNV data were included. Survival analysis was performed using log-rank tests and Cox regression model. The association between alteration of m6A regulators and clinicopathological characteristics was examined using chi-square test. The results showed that alteration of m6A regulators was associated with pathologic stage. Patients with any CNVs of the regulatory genes had worse OS and DFS than those with diploid genes. Moreover, deletion of m6A “writer” genes was an independent risk factor for OS, and copy number gain of “eraser” genes could magnify the effect in a synergistic way. Additionally, low expression of the writer gene METTL3 was related to activations of adipogenesis and mTOR pathways. Thus, we for the first time determined genetic alterations of m6A regulators in ccRCC and found a significant relationship between the alterations and worse clinical characteristics. The findings provide us clues to understand epigenetic modification of RNA in ccRCC.
Keywords: N6-Methyladenosine, m6A, renal cell carcinoma, prognosis
Introduction
Renal cell carcinoma (RCC) is the most lethal cancer in genitourinary system. The latest cancer statistic report shown that more than 65,000 new cases were diagnosed in the U.S. resulting in almost 15,000 deaths every year, which made it the sixth most common tumor site for male [1]. Clear cell renal cell carcinoma (ccRCC) is the most common type (~80%) of renal cell carcinomas. Clinically, up to 16% of patients with ccRCC are metastatic at the time of diagnosis, and the 5-year relative survival rate is only 12%. Although developments in medical oncology and surgery have revolutionized the approach to ccRCC, the prognosis has only marginally improved. As to localized ccRCC, 20% to 30% of patients experience recurrence after primary treatment and currently no approved therapies are found to reduce the risk of recurrence, progression or death [2,3]. In recent years, while target therapy has been proven to prolong survival in metastatic patients, the median survival is still less than 3 years [4]. Furthermore, drug resistance and financial burden are considerable problems in clinical practice [5]. Thus, exploring the molecular mechanism underlying the pathogenesis of ccRCC and new therapeutic targets are still challenging issues.
The genetic and epigenetic alterations of DNA and histone have been widely studied in tumor progression and led to the development of many therapeutic modalities including histone deacetylase inhibitors and drugs targeting hypoxia related pathway [6,7]. Apart from the above two molecules, the role of RNAs in diverse cellular processes attracted extensive attention and became a fast growing field in the last decade. More than one hundred of chemically modified nucleotides are found in different types of RNAs, including rRNA, tRNA, mRNA, snRNA and others [8]. The modified RNAs, especially mRNA, play critical roles in the post-transcriptional regulation of gene expression. In eukaryotes, m6A is the most common form of mRNA modification, the abundance of which has been found to be 0.1–0.4% of total adenosine residues [9–11]. m6A is proved to be widespread throughout the transcriptome, and actually it is present in the mRNAs of over 7,600 genes and in more than 300 noncoding RNAs [12]. In general, m6A is highly conserved between human and mouse, located in 3’UTRs, around stop codons and the long internal exons [13], leading to alterations of RNA stability, splicing, intracellular distribution and translation [14–17]. The cellular m6A status is mediated by a group of genes called “writers” (WTAP, METTL3 and METTL14), “erasers” (FTO and ALKBH5) and “readers” (YTHDF1, YTHDF2, YTHDF3, YTHDC1 and YTHDC2) [18]. The writers form a multisubunit methyltransferase enzyme complex and upregulate the m6A level, while erasers are m6A demethylase enzymes, which make the event reversible. Furthermore, the readers are effectors that decode the m6A methylation information and transform it into a functional signal.
m6A dysregulation is involved in diverse cellular process and causes decreased cell proliferation, impaired self-renewal capacity, developmental defects and cell death [19]. It has been reported that the alterations of m6A regulatory genes play an important role in the pathogenesis of a variety of human disease including obesity, impairment of spermatogenesis, neuronal disorders and immunological disease. More recently, the alterations of m6A regulatory genes have been shown to promote progression of both breast cancer and hematologic malignancies through cancer stem cell formation and abnormal differentiation state maintenance [20,21]. Another study also proves that METTL3, a major RNA N6-adenosine methyltransferase, promotes liver cancer progression through YTHDF2 dependent post-transcriptional silencing of SOCS2 [22]. While m6A is found to be associated with tumorigenesis in different types of cancers, little is known about the relationship between m6A-related genes and ccRCC. Hence, in this study, we analyze the clinical and sequencing data of ccRCC cohort from TCGA, and evaluate the alteration spectrum of ten m6A regulatory genes in ccRCC as well as the association between the genetic alterations and clinicopathological characteristics including survival.
RESULTS
Mutations and CNVs of m6A regulatory genes in ccRCC patients
Among the 451 cases with sequencing data, mutations of m6A regulatory genes were found merely in 19 independent samples (Table 1); however, CNVs of the ten m6A regulatory genes were frequently observed in 528 ccRCC samples with CNV data (Figure 1A). In detail, the m6A “reader” gene YTHDC2 had the highest frequency of CNV events (55.11%, 291/528) followed by METTL3 (30.11%, 159/528), which are am6A “reader” and “writer” gene. Furthermore, we also observed frequent CNVs of VHL (89.02%) and TP53 (14.58%) in this cohort in line with published literatures [23].
Table 1. Mutations of m6A regulatory genes in 451 ccRCC patients.
| ccRCC Sample ID | ALKBH5 | FTO | METTL14 | METTL3 | WTAP | YTHDF1 | YTHDF2 | YTHDC1 | YTHDC2 | |
| TCGA-B0-5698 | S598T | |||||||||
| TCGA-CJ-6033 | D130N | |||||||||
| TCGA-A3-3346 | V519Cfs*2 | |||||||||
| TCGA-A3-3374 | D80*,S151R | |||||||||
| TCGA-A3-3382 | V871I | |||||||||
| TCGA-AS-3778 | X499_splice | |||||||||
| TCGA-A3-3362 | E775V | |||||||||
| TCGA-CJ-4636 | X506_splice, G486S,E481K |
|||||||||
| TCGA-B0-5098 | T91A | |||||||||
| TCGA-B0-5099 | Y31C | |||||||||
| TCGA-BP-4801 | M163I | |||||||||
| TCGA-BP-5199 | S256R | |||||||||
| TCGA-CJ-4905 | E483* | |||||||||
| TCGA-CZ-4853 | S351C | |||||||||
| TCGA-CZ-5459 | D499H | |||||||||
| TCGA-B0-5695 | F425S | |||||||||
| TCGA-CJ-5678 | L804* | |||||||||
| TCGA-CJ-5682 | N97H | |||||||||
| TCGA-CW-5581 | D987Y | |||||||||
Figure 1.

CNVs of m6A regulatory genes in ccRCC. (A) Percentage of ccRCC samples with CNVs of the m6A regulators based on the data from TCGA. (B) Events of copy number gain or loss of m6A regulatory genes in ccRCC samples. (C) The most common patterns of CNVs in m6A regulatory genes in ccRCC samples.
Next, we evaluated the CNV patterns in ccRCC samples and found that most of the CNV events led to loss of copy number (737/1331) (Figure 1B, Table 2), which was similar as the CNV status in AML [24]. Copy number gain of YTHDC2 was the most frequent alteration in all the CNVs of m6A regulatory genes (Figure 1C), and the simultaneous shallow deletions of METTL3 and YTHDC2 also ranked first among the concurrence of CNVs in two genes, implying the importance of m6A writer genes in the process of RNA m6A modification.
Table 2. Different CNV patterns occur in ccRCC samples (n=528).
| Diploid | Deep deletion | Shallow deletion | Copy number gain | Amplification | CNV sum | Percentage | ||
| Eraser | ALKBH5 | 457 | 0 | 45 | 26 | 0 | 71 | 13.45% |
| FTO | 408 | 0 | 22 | 98 | 0 | 120 | 22.73% | |
| Writer | METTL14 | 439 | 3 | 74 | 11 | 1 | 89 | 16.86% |
| METTL3 | 303 | 0 | 207 | 18 | 0 | 225 | 42.61% | |
| WTAP | 369 | 1 | 150 | 8 | 0 | 159 | 30.11% | |
| Reader | YTHDF1 | 408 | 0 | 0 | 119 | 1 | 120 | 22.73% |
| YTHDF2 | 424 | 0 | 95 | 9 | 0 | 104 | 19.70% | |
| YTHDC1 | 448 | 0 | 65 | 14 | 1 | 80 | 15.15% | |
| YTHDC2 | 237 | 1 | 7 | 230 | 53 | 291 | 55.11% | |
| YTHDF3 | 391 | 1 | 72 | 61 | 3 | 137 | 25.95% | |
| TP53 | 451 | 2 | 50 | 25 | 0 | 77 | 14.58% | |
| VHL | 58 | 58 | 405 | 7 | 0 | 470 | 89.02% |
Alterations of m6A regulatory genes were associated with clinicopathological and molecular characteristics
Then, we assessed the relationship between alterations (CNV and/or mutation) of m6A regulatory genes and the clinicopathological characteristics of patients. The results revealed that alterations of m6A regulatory genes were significantly associated with higher Fuhrman Nuclear Grade (Table 3). Due to the fact that VHL and TP53 play important roles in the pathogenesis of ccRCC [25], we further evaluated if the variation of m6A regulatory genes was related to the alterations of these two genes. As expected, the alterations of m6A regulatory genes were significantly correlated with VHL and TP53 alteration; in fact, only 1 sample was absent from alterations of m6A regulatory genes among the 57 patients with TP53 alteration (Table 4).
Table 3. Clinical pathological parameters of ccRCC patients with or without mutation/CNV of m6A regulatory genes.
| With mutation and/or CNV* | Without mutation and CNV* | P | ||
| Age | <=60 | 32 | 218 | 0.325 |
| >60 | 25 | 225 | ||
| Gender | Female | 26 | 148 | 0.069 |
| Male | 31 | 295 | ||
| Pathological Stage | I | 36 | 206 | 0.123 |
| II | 7 | 48 | ||
| III | 8 | 113 | ||
| IV | 6 | 73 | ||
| Discrepancy | 0 | 3 | ||
| Historical Grade | G1 | 5 | 6 | <0.001 |
| G2 | 33 | 175 | ||
| G3 | 14 | 182 | ||
| G4 | 4 | 74 | ||
| x | 1 | 4 | ||
| N/A | 0 | 2 | ||
| T stage | T1 | 36 | 212 | 0.104 |
| T2 | 8 | 57 | ||
| T3 | 12 | 164 | ||
| T4 | 1 | 10 | ||
| N stage | N0 | 25 | 201 | 0.683 |
| N1 | 1 | 17 | ||
| Nx | 31 | 225 | ||
| M stage | M0 | 51 | 353 | 0.133 |
| M1 | 6 | 71 | ||
| Mx | 0 | 19 |
*With mutation or CNV: Cases have mutant or CNV or mutant+CNV, confirmed through TCGA database. Without mutant and CNV: Cases with neither mutant nor CNV, confirmed through TCGA database. Ambiguous variables (Nx, Mx, N/A, discrepancy and Gx) were excluded from chi-square test or non-parametric test
Table 4. Relationship between molecular characteristics and m6A regulatory genes alteration in ccRCC patients.
| Without mutation or CNV* | With mutation and CNV* | χ2 | P | ||
| VHL | wt | 8 | 25 | 4.362 | 0.037 |
| n=495 | alteration | 49 | 413 | ||
| TP53 | wt | 56 | 316 | 12.041 | 0.001 |
| n=456 | alteration | 1 | 83 |
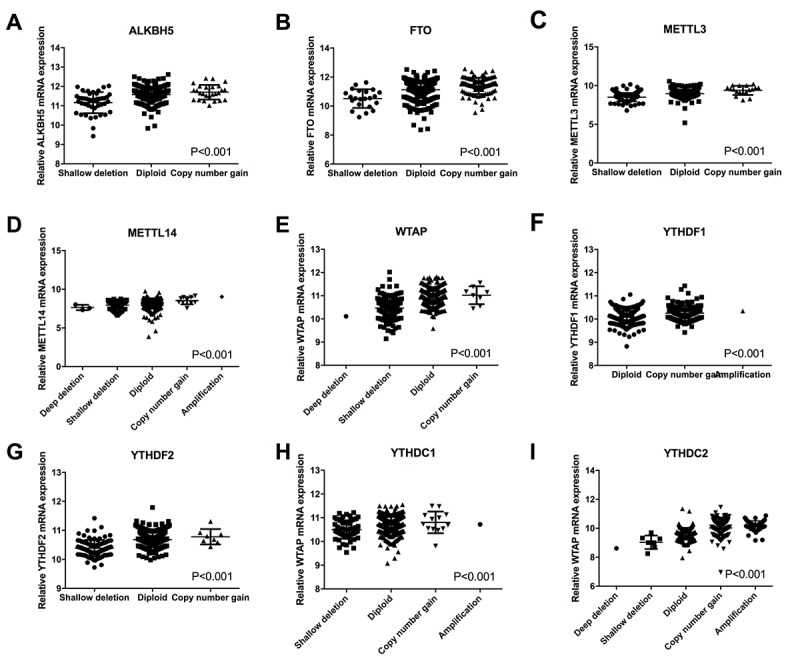
The effects of alterations in m6A regulatory genes on the mRNA expression were next evaluated. The results showed that the mRNA expression levels were significantly associated with the diverse CNV patterns in 525 ccRCC samples. For all the ten genes, copy number gains were related to higher mRNA expression; while, deep deletions or shallow deletions resulted in a decline of mRNA expression (Figure 2). Furthermore, we have done some immunohistochemistry staining for METTL3 and METTL14 protein in tissue arrays containing about 130 pairs of ccRCC tissue and normal tissue to confirm the findings. The results were in line with our analysis, which showed METTL3 and METTL14 were highly expressed in ccRCC tissues than normal (P<0.05) (Figure S1). However, due to the lack of prognosis data, we couldn’t perform the survival analysis in this study.
Figure 2. Correlation between different CNV patterns and mRNA expression levels of ten m6A regulatory genes respectively.
Association between CNVs of m6A regulatory genes and survival of ccRCC patients
To explore the prognostic value of CNVs in the m6A regulatory genes, we analyzed the effects of CNVs on the overall survival (OS) and disease-free survival (DFS) among ccRCC patients, and found that individuals with or without m6A regulatory genes’ CNVs didn’t have any correlation with OS and DFS (Figure 3A-B). Furthermore, separate analysis of the ten genes revealed that patients affected by deletions of YTHDC1, METTL14 or METTL3 (one reader and two writer genes of m6A) had poorer OS and DFS (Figure 3C-H); while, no significant difference was observed between different subgroups based on the CNVs of the other ten m6A regulatory genes (Figure S2). Multivariate Cox regression analyses demonstrated that alteration of m6A regulator genes was an independent risk factor for overall survival (Table 5). Considering the writer genes are a group of methyltransferase enzymes and the most important part in m6A regulation process, the results implied that the down-regulation of m6A level might be associated with poor patient survival.
Figure 3.
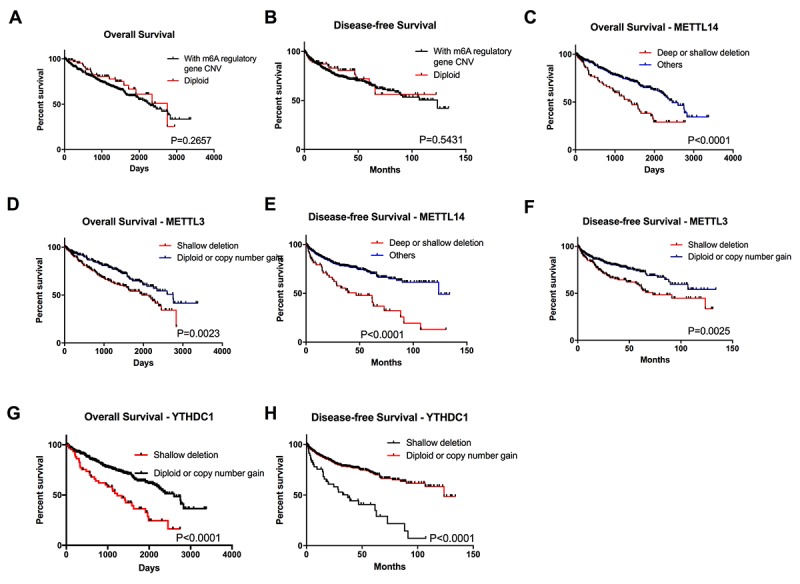
Overall survival of ccRCC patients with CNVs of m6A regulatory genes. (A-B) OS and DFS of patients with any CNVs of m6A regulatory genes or with diploid genes. (C-H) OS and DFS for patients with different CNV types of METTL3, METTL14 and YTHDC1.
Table 5. Univariate and Multivariate COX regression analysis of m6A regulatory genes for ccRCC patients' overall survival (OS) and disease-free survival (DFS)*.
| OS | DFS | ||||||||
| Variable | Univariate | Multivariate | Univariate | Multivariate | |||||
| HR (95% CI) | P | HR | P | HR (95% CI) | P | HR | P | ||
| Age (>60 vs <=60) | 1.787(1.294-2.467) | 0.000 | 1.714(1.096-2.68) | 0.018 | 1.419(0.997-2.018) | 0.052 | |||
| Gender (male vs female) | 1.078(0.782-1.485) | 0.648 | 1.398(0.945-2.069) | 0.094 | |||||
| Stage (I-II vs III-IV) | 4.370(3.116-6.128) | 0.000 | 1.313(0.54-3.192) | 0.548 | 6.426(4.327-9.545) | 0.000 | 4.499(1.92-10.541) | 0.001 | |
| M (M1 vs M1) | 4.400(3.185-6.080) | 0.000 | 2.701(1.594-4.575) | 0.000 | 7.876(5.411-11.463) | 0.000 | 3.133(1.819-5.394) | 0.000 | |
| N (N1 vs N0) | 2.880(1.524-5.442) | 0.001 | 1.468(0.735-2.933) | 0.277 | 3.851(1.888-7.855) | 0.000 | 2.078(1.01-4.276) | 0.047 | |
| T (T3-T4 vs T1-T2) | 3.645(2.643-5.028) | 0.000 | 1.608(0.719-3.596) | 0.247 | 4.413(3.063-6.359) | 0.000 | 0.734(0.354-1.526) | 0.408 | |
| Grade (3-5 vs 1-2) | 2.842(1.972-4.098) | 0.000 | 2.037(1.205-3.442) | 0.008 | 3.337(2.21-5.039) | 0.000 | 1.577(0.926-2.688) | 0.094 | |
| TP53 (altered vs diploid) | 1.300(0.853-1.980) | 0.222 | 1.193(0.732-1.946) | 0.479 | |||||
| VHL (altered vs diploid) | 0.945(0.555-1.610) | 0.835 | 1.130(0.608-2.103) | 0.699 | |||||
| m6A regulator alteration (Writer loss + Eraser gain vs others) | 1.495(1.043-2.142) | 0.028 | 1.69(1.006-2.84) | 0.047 | 1.654(1.102-2.485) | 0.015 | 1.463(0.819-2.614) | 0.199 | |
*Ambiguous variables (Nx, Mx, N/A, discrepancy and Gx) were excluded.
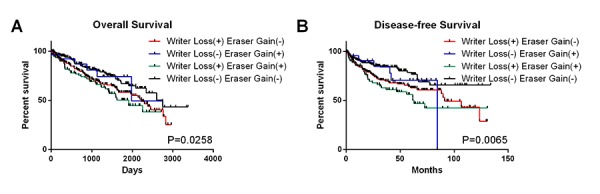
In order to confirm the above conclusion, we next tested it among patients who were affected by two kinds of CNVs (deletions of writer genes and copy number gain of eraser genes). As the result showed, patients with deletions of writer genes in combination with copy number gain of eraser genes had worse OS and DFS than those with only deletions of writer genes (Figure 4A-B). This provided more evidence for the relationship between down-regulated m6A level and poor survival.
Figure 4. OS and DFS of ccRCC patients with simultaneous alterations of writer genes and eraser genes.
Enrichment analysis of METTL3 loss of function
Given the importance of METTL3 in the methylation process and the interesting results we found, we determined to explore the role of m6A dysregulation in the pathogenesis of ccRCC. We examine the enriched gene sets in samples with different METTL3 mRNA expression levels. The GSEA analysis suggested that low METTL3 expression was associated with some critical biological processes including adipogenesis, mTOR pathway and reactive oxygen species (ROS) (Table 6 and Figure 5), giving a clue of the underlying mechanism in the pathogenesis of ccRCC. To validate our findings, we examined the expressions of genes related to the pathways above. We found that several genes of adipogenesis and mTORC1 signaling pathways were upregulated in RCC tissues (Figure S2H), which partially validate the GSEA results. Besides, several studies have found that METTL3 could participate in adipogenesis and mTORC1 signaling pathways, which are consistent with our results [26,27]. Further studies are needed to illustrate the specific effect of METTL3 and METTL14 on the regulations of the downstream genes.
Table 6. Gene sets enrichment of low METTL3 mRNA expression level in the ccRCC cohort.
| GS DETAILS | SIZE | ES | NES | NOM p-val | FDR q-val |
| HALLMARK_ADIPOGENESIS | 190 | 0.48 | 1.72 | 0.028 | 0.227 |
| HALLMARK_MTORC1_SIGNALING | 194 | 0.48 | 1.70 | 0.036 | 0.174 |
| HALLMARK_XENOBIOTIC_METABOLISM | 194 | 0.48 | 1.61 | 0.027 | 0.232 |
| HALLMARK_REACTIVE_OXIGEN_SPECIES_PATHWAY | 44 | 0.49 | 1.57 | 0.048 | 0.228 |
Figure 5.
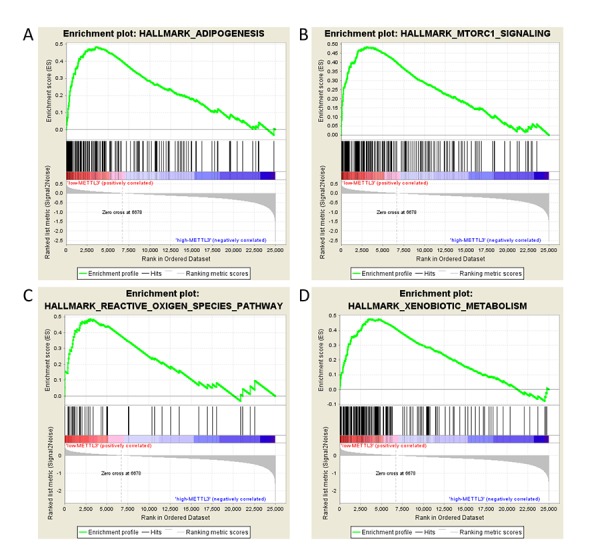
GSEA results of different expression level of METTL3. Gene set enrichment plots of (A) adipogenesis, (B) mTORC1 signaling, (C) reactive oxygen species, and (D) xenobiotic metabolism pathways related to low METTL3 mRNA level in the ccRCC samples.
DISCUSSION
The transcriptome-wide mapping of m6A introduced the concept of epitranscriptome which focuses on investigating the landscapes and functions of the reversible RNA modifications in the last decade. Due to the complicated technology (m6A-Seq and m6A MeRIP) detecting the m6A level, lots of studies choose an alternative way to evaluate the genetic alterations of m6A regulatory genes and indirectly explore the association between m6A status and human diseases. Due to the tissue specificity of the “writers”, “erasers” and “readers”, the genes involved in m6A dysregulation may be different in distinct tumors. In this ccRCC cohort, the frequency of alterations of the ten m6A related genes was much higher than that reported in AML, implying that dysregulation of m6A might play a more important role in ccRCC tumorigenesis compared with AML [24]. Furthermore, there was a high frequency of concurrent alterations of two regulatory genes, indicating that m6A writer gene and reader gene may play synergistic roles in the process of RNA m6A modification. In addition, the “writers” METTL3 and METTL14 were more predisposed to mutation or CNV than the other genes in ccRCC, while alterations of the “erasers” FTO and ALKBH5 were proved to be more important in breast cancer, glioblastoma and hematological malignancies [21,28,29]. The differences of involved genes between different tumor types gave us a clue that the regulation of m6A in cellular level was complicated, and future studies focusing on the m6A “writers” are needed to further clarify the regulatory mechanism of m6A in ccRCC.
Similar as CNV status in AML, most of the observed CNVs in writer genes resulted in loss of function with down-regulation of the corresponding genes, while CNVs of the eraser genes were mainly gain of function leading to up-regulation of the corresponding genes. Considering the opposite effect on m6A status for the two gene groups, these alterations eventually decreased the m6A level in ccRCC. In accordance to our results, many studies on other solid tumors, like breast cancer and glioblastoma, also observed the down regulated m6A level [21,30]. This may be explained by the connection between m6A and differentiation pathways that control cancer stem cell fate [31]. The activation of hypoxic pathway was a hallmarker in tumorigenesis of the above three solid tumors. A study showed that hypoxia was related to increased breast cancer stem cell formation directly through upregulating ALKBH5 or indirectly through ZNF217/METTL3-METTL14-complex pathway. The consequent decreased level of NANOG mRNA m6A modification resulted in the upregulation of NANOG expression, which was a key transcription factor that is associated with pluripotency. Interestingly, overexpression of NANOG was also observed in ccRCC tissue compared with normal tissue [32]. Considering the high frequency of inactivation of von Hippel-Lindau (VHL) gene and the upregulated hypoxia induced factor-α (HIF- α) in ccRCC, we hypothesize that there may exist a m6A regulatory pathway (VHL-HIF-ZNF217-METTL3/METTL14) in ccRCC cells, leading to the formation and maintenance of ccRCC cancer stem cells. More work is needed to be done to verify the pathway.
We also evaluated the effect of m6A regulatory gene alterations on survival of ccRCC patients. In line with the characteristics of genetic alterations of m6A related genes, the writer genes METTL3 and METTL14 are the only two genes among the ten regulators that are associated with the overall and disease-free survival, which reconfirms that the writers were main regulators of m6A in ccRCC. Although the effect of METTL3 became insignificant in the multivariate Cox regression model, we should take it into consideration that METTL3 and METTL14 need to form a complex to function as RNA methyltransferases, and thus the two molecules are related to each other, possibly resulting in the statistically insignificance. A worst overall survival in patients with writer gene loss of function in combination with eraser gene gain of function was observed, making it clear that decreased level of m6A plays a significant role in ccRCC progression. However, we failed to reach any significant results referred to interactions between the combined genetic alteration and DFS, possibly because of the limited number of patients with writer gene loss of function in combination with eraser gene gain of function. Direct detecting the m6A level and evaluating its effect on ccRCC survival in a new cohort is needed to illustrate this contradictory phenomenon.
Series of cancer related pathways are dysregulated in ccRCC development. We found in this ccRCC cohort that low METTL3 mRNA expression level was associated with activated adipogenesis and mTOR pathway, which are two very important cellular processes in ccRCC development. Similar as our results, a recent study showed knockdown of METTL3 in ccRCC cell lines led to obvious upregulation of PI3k, AKT and mTOR expression and patients with positive METTL3 expression had obviously longer survival time than those with negative METTL3 expression [26], implying that the mRNAs of molecules in mTOR pathway may be the target mRNA of m6A modification. Moreover, Kobayashi et al. found that WTAP-METTL3-METTL14 complex played a role in the mechanism for adipogenesis, which is consistent with our GSEA results [27]. In addition, we observed the alteration of m6A regulatory genes was significantly associated with VHL mutation and TP53 alteration. These two genes are the most important tumor suppressor genes in ccRCC. It has been reported in a human liver cancer cell line that loss of METTL3 leads to alternative splicing and gene expression changes of more than 20 genes involved in the p53 signaling pathway including MDM2, MDM4, and P21 [13]. Also, Liu et al. found that reduced METTL3 expression could lead to reductions in m6A methylation and have an effect on AKT signaling in human endometrial cancer [33]. Thus, it is likely that genetic alterations of m6A regulators, VHL-mediated hypoxia pathway and p53-mediated cell processes act in a synergistic way to promote the pathogenesis and progress of ccRCC.
In conclusion, we for the first time determine the genetic alterations of m6A regulatory genes in ccRCC and find an obvious relationship between the alterations resulting in decreased m6A level and worse clinical characteristics including survival. It is plausible that VHL-HIF-METTL3/14 pathways are involved in the m6A regulation in ccRCC cancer cells, and PI3K-mTOR as well as p53 signaling pathways are possible downstream targets of m6A in ccRCC. To further clarify the definite target mRNA of the m6A modification during ccRCC initiation and progression, future studies in another ccRCC cohort with m6A-Seq and m6A MeRIP will be helpful to confirm our findings.
MATERIALS AND METHODS
Ethics statement
All of these clinical data, CNV, mutation, mRNA expression data were retrieved from TCGA program by cBioportal platform [34] and TCGA-assembler [35] which are open to the public under some guidelines. So it is confirmed that all written informed consent was achieved.
Data processing
Within the TCGA database, we identified 528 ccRCC patients with CNV data and pathology reports [36]. For CNV, the loss and gain levels of copy-number changes have been identified using segmentation analysis and GISTIC algorithm. To investigate the clinicopathological significance of the status of CNV and/or mutation, this ccRCC cohort was divided into two subgroups; “with mutation and/or CNV of these ten m6A regulatory genes” and “without CNV and mutation”. The mRNA expression data were calculated from RNA-Seq V2 RSEM release, and being applied log scale before analyzing the relationship between mRNA expression and CNV.
Gene set enrichment analysis (GSEA)
GSEA was provided by the JAVA program with MSigDB v6.1 and downloaded from the website of Broad Institute [37]. In this study, cases were divided into two groups according to the first and fourth quartile of METTL3 expression level. Finally, 18419 genes were enrolled into the GSEA process. Hallmark gene set “h.all.v6.0.symbols.gmt” was used in this study [38]. Gene sets with normalized p-value <0.05, and the false discovery rate (FDR) <0.25 were considered to be significantly enriched.
Statistical analysis
All statistical data and figures were analyzed by using SPSS 20.0 (IBM, Chicago, USA) and GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA). The association between m6A regulatory genes’ CNV and clinicopathological characteristics were analyzed with chi-square test or Mann-Whitney U test. Kaplan-Meier curve and log-rank test were used to evaluate the prognosis value of m6A regulatory gene’s alteration. Cox proportional hazard regression model was performed using SPSS. All statistical results with a p-value <0.05 were considered to be significant.
SUPPLEMENTARY MATERIAL
ACKNOWLEDGMENTS
We would like to express our sincere gratitude to TCGA program, which provides the high quality genomic data. Without the help of cBioPortal platform, we couldn’t complete this study. We thank Dr. Dingfang Bu, from Peking University First Hospital Medical Experiment Center, for his suggestions to this manuscript.
Abbreviations
- m6A
methylation of N6 adenosine
- ccRCC
renal cell carcinoma
- TCGA
the Cancer Genome Atlas
- CNV
copy number variation
- OS
overall survival
- DFS
disease-free survival
- rRNA
ribosomal RNA: tRNA: transfer RNA: mRNA: messenger RNA
- snRNA
small nuclear RNA
- GIST
Genomic Identification of Significant Targets in Cancer
- AML
acute myelocytic leukemia
- ROS
reactive oxygen species
- HIF-α
hypoxia induced factor-α
- VHL
von Hippel-Lindau
- GSEA
gene set enrichment analysis
Footnotes
CONFLICTS OF INTEREST: The authors declare that they have no competing interests.
FUNDING: This study was funded by the National Natural Science Foundation of China (No. 81572506 and 81872081)), and the Fundamental Research Funds for the Central Universities (No. BMU2018JI002).
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018; 68:7–30. 10.3322/caac.21442 [DOI] [PubMed] [Google Scholar]
- 2.Hakimi AA, Chen L, Kim PH, Sjoberg D, Glickman L, Walker MR, Russo P. The impact of metformin use on recurrence and cancer-specific survival in clinically localized high-risk renal cell carcinoma. Can Urol Assoc J. 2013; 7:E687–91. 10.5489/cuaj.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De P, Otterstatter MC, Semenciw R, Ellison LF, Marrett LD, Dryer D. Trends in incidence, mortality, and survival for kidney cancer in Canada, 1986-2007. Cancer Causes Control. 2014; 25:1271–81. 10.1007/s10552-014-0427-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greef B, Eisen T. Medical treatment of renal cancer: new horizons. Br J Cancer. 2016; 115:505–16. 10.1038/bjc.2016.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shih YC, Chien CR, Xu Y, Pan IW, Smith GL, Buchholz TA. Economic burden of renal cell carcinoma in the US: part II--an updated analysis. Pharmacoeconomics. 2011; 29:331–41. [DOI] [PubMed] [Google Scholar]
- 6.Morris MR, Latif F. The epigenetic landscape of renal cancer. Nat Rev Nephrol. 2017; 13:47–60. 10.1038/nrneph.2016.168 [DOI] [PubMed] [Google Scholar]
- 7.la Rosa AH, Acker M, Swain S, Manoharan M. The role of epigenetics in kidney malignancies. Cent European J Urol. 2015; 68:157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 2011; 39:D195–201. 10.1093/nar/gkq1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perry RP, Kelley DE, Friderici K, Rottman F. The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′ terminus. Cell. 1975; 4:387–94. 10.1016/0092-8674(75)90159-2 [DOI] [PubMed] [Google Scholar]
- 10.Dubin DT, Taylor RH. The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res. 1975; 2:1653–68. 10.1093/nar/2.10.1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975; 4:379–86. 10.1016/0092-8674(75)90158-0 [DOI] [PubMed] [Google Scholar]
- 12.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012; 149:1635–46. 10.1016/j.cell.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012; 485:201–06. 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- 14.Maity A, Das B. N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. 2016; 283:1607–30. 10.1111/febs.13614 [DOI] [PubMed] [Google Scholar]
- 15.Pan T. N6-methyl-adenosine modification in messenger and long non-coding RNA. Trends Biochem Sci. 2013; 38:204–09. 10.1016/j.tibs.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niu Y, Zhao X, Wu YS, Li MM, Wang XJ, Yang YG. N6-methyl-adenosine (m6A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013; 11:8–17. 10.1016/j.gpb.2012.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE Jr, Darnell RB. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017; 31:990–1006. 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X, Bhattarai DP, Zhao YL, Sun BF, Yang YG. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017; 27:444–47. 10.1038/cr.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N, Pan T. N6-methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol. 2016; 23:98–102. 10.1038/nsmb.3162 [DOI] [PubMed] [Google Scholar]
- 20.Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, Weitman S, Tomlinson GE, Rao MK, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014; 28:1171–74. 10.1038/leu.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, Semenza GL. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016; 7:64527–42. 10.18632/oncotarget.11743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, Wong CC, Ng IO, Wong CM. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018; 67:2254–70. 10.1002/hep.29683 [DOI] [PubMed] [Google Scholar]
- 23.Turajlic S, Larkin J, Swanton C. SnapShot: renal cell carcinoma. Cell. 2015; 163:1556–1556.e1. 10.1016/j.cell.2015.11.026 [DOI] [PubMed] [Google Scholar]
- 24.Kwok CT, Marshall AD, Rasko JE, Wong JJ. Genetic alterations of m6A regulators predict poorer survival in acute myeloid leukemia. J Hematol Oncol. 2017; 10:39. 10.1186/s13045-017-0410-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harlander S, Schönenberger D, Toussaint NC, Prummer M, Catalano A, Brandt L, Moch H, Wild PJ, Frew IJ. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat Med. 2017; 23:869–77. 10.1038/nm.4343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Tang J, Huang W, Wang F, Li P, Qin C, Qin Z, Zou Q, Wei J, Hua L, Yang H, Wang Z. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017; 8:96103–16. 10.18632/oncotarget.21726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi M, Ohsugi M, Sasako T, Awazawa M, Umehara T, Iwane A, Kobayashi N, Okazaki Y, Kubota N, Suzuki R, Waki H, Horiuchi K, Hamakubo T, et al. The RNA Methyltransferase Complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol Cell Biol. 2018; 38:e00116-18. 10.1128/MCB.00116-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, Majumder S, He C, Huang S. m 6 A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017; 31:591–606.e6. 10.1016/j.ccell.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, Qin X, Tang L, Wang Y, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N 6-methyladenosine RNA demethylase. Cancer Cell. 2017; 31:127–41. 10.1016/j.ccell.2016.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG, Riggs AD, He C, Shi Y. m 6 A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Reports. 2017; 18:2622–34. 10.1016/j.celrep.2017.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, Ben-Haim MS, Eyal E, Yunger S, et al. Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015; 347:1002–06. 10.1126/science.1261417 [DOI] [PubMed] [Google Scholar]
- 32.Yu B, Cai H, Xu Z, Xu T, Zou Q, Gu M. Expressions of stem cell transcription factors Nanog and Oct4 in renal cell carcinoma tissues and clinical significance. Artif Cells Nanomed Biotechnol. 2016; 44:1818–23. 10.3109/21691401.2015.1105238 [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, Huang JT, Chen SM, Xu ZG, et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018; 20:1074–83. 10.1038/s41556-018-0174-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y, Qiu P, Ji Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat Methods. 2014; 11:599–600. 10.1038/nmeth.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49. 10.1038/nature12222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005; 102:15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011; 27:1739–40. 10.1093/bioinformatics/btr260 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.