

Abstract
Pancreatic Cancer (PC) is a deadly disease in need of new therapeutic options. We recently developed a novel tricarbonylmethane agent (CMC2.24) as a therapeutic agent for PC, and evaluated its efficacy in preclinical models of PC. CMC2.24 inhibited the growth of various human PC cell lines in a concentration and time-dependent manner. Normal human pancreatic epithelial cells were resistant to CMC2.24, indicating selectivity. CMC2.24 reduced the growth of subcutaneous and orthotopic PC xenografts in mice by up to 65% (p<0.02), and the growth of a human patient-derived tumor xenograft by 47.5% (p<0.03 vs vehicle control). Mechanistically, CMC2.24 inhibited the Ras-RAF-MEK-ERK pathway. Based on Ras Pull-Down Assays, CMC2.24 inhibited Ras-GTP, the active form of Ras, in MIA PaCa-2 cells and in pancreatic acinar explants isolated from Kras mutant mice, by 90.3% and 89.1%, respectively (p<0.01, for both). The inhibition of active Ras led to an inhibition of c-RAF, MEK and ERK phosphorylation by 93%, 91% and 87%, respectively (p<0.02, for all) in PC xenografts. Furthermore, c-RAF overexpression partially rescued MIA PaCa-2 cells from the cell growth inhibition by CMC2.24. In addition, downstream of ERK, CMC2.24 inhibited STAT3 phosphorylation levels at the serine 727 residue, enhanced the levels of superoxide anion in mitochondria, and induced intrinsic apoptosis as shown by the release of cytochrome c from the mitochondria to the cytosol and the further cleavage of caspase 9 in PC cells. In conclusion, CMC2.24, a potential Ras inhibitor, is an efficacious agent for PC treatment in preclinical models, deserving further evaluation.
Keywords: Kras, CMC2.24, curcumin, pancreatic cancer, Ras, ERK
INTRODUCTION
Pancreatic cancer (PC) remains a deadly disease, expected to become the second leading cause of cancer deaths by 2020 [1]. Two major contributing factors to PC’s poor prognosis are its late diagnosis and the lack of therapeutic options with durable activity [2]. For example, gemcitabine, a standard drug for PC therapy, is minimally effective, improving patient’s survival by only months [3]. Surgery, currently the only realistic option, is rarely curative, with only about 20% of PC cases being amendable to potentially curative resection [2], either due to metastasis or involvement of major blood vessels. Therefore, it is critical to develop new agents for the effective management of PC.
Mutations of the Ras family of genes occur in approximately 95% of PCs [4]. Oncogenic Kras is essential for both the initiation and maintenance of PC [5]. During all stages of pancreatic carcinogenesis, Kras upregulates inflammatory and signaling pathways that mediate paracrine interactions between epithelial and their surrounding microenvironment. These Kras-dependent pathways drive tumorigenesis [6]. Given the predominant role of Ras oncogene in pancreatic carcinogenesis, there is an intense ongoing effort to identify inhibitors of the Ras protein [5,7].
A suitable strategy in designing novel agents is the chemical modification of known drugs in order to optimize their pharmacological properties, primarily their efficacy. Curcumin (diferuloyl-methane) has been extensively studied and has shown some anticancer efficacy in various types of cancer [8], including PC [9,10]. However, due to its poor bioavailability, curcumin has only been shown to be effective in in vitro studies and is not currently utilized as an adjuvant to common chemotherapeutics for treating PC [11]. Given this problem, significant efforts have been made to modify curcumin synthetically and producing other curcumin analogs, in order to generate compounds with greater bioavailability and efficacy in vivo [12–14]. Among these efforts, our group has developed various structural analogues of curcumin (polyenolic zinc-binding agents; PEZBINs), including the current lead compound, CMC2.24 [15–20] (Fig. 1A), which has higher bioactivity, better solubility and no evidence of toxicity even at very high doses [19,20].
Figure 1: CMC2.24 inhibits pancreatic cancer growth in vitro.
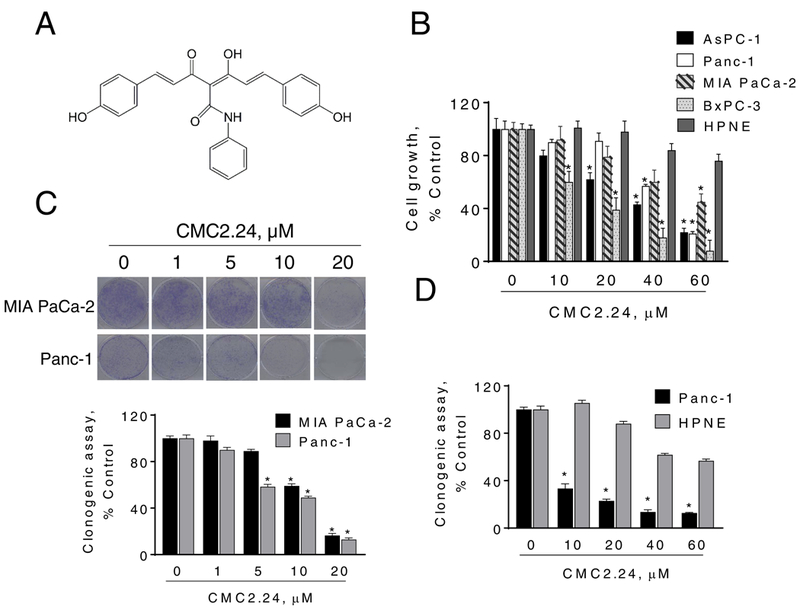
A: Chemical structure of the novel agent CMC2.24 (TRB-N0224). B: CMC2.24 inhibits PC cell growth in a concentration-dependent manner. Cell growth was determined in AsPC-1, Panc-1, MIA PaCa-2 and BxPC-3 PC cells, as well as in the human pancreatic normal epithelial cells (HPNE) after treatment with escalating concentrations of CMC2.24 for 24 h. Results are expressed as % control. (*p < 0.05, vs. control). C: CMC2.24 inhibits PC cell colony formation in a concentration-dependent manner in MIA PaCa-2 and Panc-1 cells. (*p < 0.05, vs. control). D: Colony formation assay in Panc-1 and HPNE cells. Results are expressed as the mean ± SEM (*p < 0.05, vs. control).
In the present study, we examined, for the first time, the efficacy and mechanism of action of CMC2.24 in preclinical models of PC. Our data show that CMC2.24 inhibits the growth of human PC in nude mice and in patient-derived tumor xenografts. Furthermore, CMC2.24 strongly suppresses Ras activation leading to a robust inhibition of the RAF-MEK-ERK pathway, followed by reduction in p-STAT3Ser727 levels, an increase in mitochondrial reactive oxygen species and induction of intrinsic apoptosis.
MATERIALS AND METHODS
Reagents.
Chemically-modified curcumin (CMC2.24; TRB-N0224) was a gift from Traverse Biosciences Inc. (Stony Brook, NY). Curcumin (>98% purity) was purchased from Thermo Fisher Scientific (Waltham, MA). We prepared a 100 mM stock solution of CMC2.24 and curcumin in DMSO. In all cell culture media, the final DMSO concentration was adjusted to 1% (v/v). Annexin V was purchased from Invitrogen (Carlsbad, CA). All general solvents and reagents were of HPLC grade or the highest grade commercially available.
Cell culture.
Human PC cell lines (BxPC-3, AsPC-1, Panc-1 and MIA PaCa-2) and the human pancreatic normal epithelial (HPNE) cells were from the American Type Culture Collection (Manassas, VA). FC1245 cells (KPC, murine PC cells) were a gift from Dr. David Tuveson (Cold Spring Harbor Laboratory). We have not authenticated these cell lines, however we routinely test for mycoplasma contamination in every cell line every three months. These cells were grown as monolayers in the specific medium and under conditions suggested by ATCC. All the cell lines were characterized by cell morphology and growth rate and passaged in our laboratory less than 6 months after being received.
Cell viability assay.
Following the treatment with various concentrations of CMC2.24 for 24 h, the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye (MTT), was determined according to the manufacture’s protocol (Promega, Madison, WI, USA).
Clonogenic assay.
This was performed as previously described [21]. Briefly, MIA PaCa-2, Panc-1 and HPNE cells, plated in 6-well plates (1,000 cells/well), were treated with CMC2.24 for 24 or 48 h. Following the treatment, cells were grown for 10 days, with their media replaced every 3 days. The cells were then stained with 1% crystal violet in Borate Buffer saline (0.1 M, pH 9.3) and 0.02% ethanol. Following lysis with 1% SDS the absorbance was read at 570 nm.
Cytokinetic analysis.
For apoptosis, human PC cells or HPNE cells were seeded and treated with various concentrations of CMC2.24, for 24 h, trypsinized and then stained with Annexin V-FITC (100× dilution; Invitrogen, Carlsbad, CA) and propidium iodide (0.5 μg/ml), and then analyzed by FACScaliber (BD Biosciences, San Jose, CA). Cell-cycle phase distribution was analyzed by flow cytometry. The percentage of cells in G0/G1, S and G2/M was determined from the DNA content histograms, as previously described [22].
Determination of mitochondrial ROS levels.
MIA PaCa-2 cells were treated with various concentrations of CMC2.24 for 3h, stained with 10 μM MitoSOX Red for 30 min at 37°C and fluorescence intensity was analyzed by FACScaliber.
Western blot analysis.
Total cell fractions were obtained as previously described [23]. Aliquots of total fractions containing 25–40 μg protein were separated by reducing 10–12.5% (w/v) polyacrylamide gel electrophoresis and electroblotted to nitrocellulose membranes. The membranes were probed overnight with antibodies against c-RAF (Cat number # 9422), p-c-RAF Ser259 (Cat number # 9422), p-ERK1/2 Thr202/Tyr204 (Cat number # 4376), ERK1/2 (Cat number # 4370), p-MEK1/2 Ser217/221 (Cat number # 9154), MEK1/2 (Cat number # 9122), Cytochrome C (Cat number # 4272), cleaved caspase-9 (Cat number # 7237), STAT3 (Cat number # 9139), COX IV (Cat number # 4850) (all from Cell Signaling Technologies) and p-STAT3Ser727 (Cat # sc-135649; Santa Cruz Biotechnologies) antibodies (1:1,000 dilution). β-actin (Cat number # A1978; Sigma-Aldrich) or β-tubulin (Cat number # 2146) was used as the loading control. After incubation, for 90 min at room temperature, in the presence of the secondary antibody (HRP-conjugated; 1:5,000 dilution), the conjugates were visualized by chemiluminescence.
Immunofluorescence.
Panc-1 cells, seeded overnight in chamber slide dishes, were treated with CMC2.24 1x and 1.5x-IC50 for 4 h. Following the treatment, Panc-1 cells were washed three times with PBS and fixed with paraformaldehyde (4% w/v in PBS). Immunostaining was performed using rabbit anti-p-ERK or p-STAT3 antibodies. After PBS washing, sections were incubated with a fluorophore-linked secondary antibody (Alexa Fluor 488–anti-rabbit IgG; Life Technologies). After staining, slides were mounted in VECTASHIELD with DAPI (VECTOR Laboratories, Burlingame, CA) and photographed under a Nikon ECLIPSF 90i microscope with a digital camera.
Ras pull-down assays.
MIA PaCa-2 cells or primary pancreatic acinar explants were treated with CMC2.24 for 3 h, after which total cell lysates were prepared. Total amounts of Ras were determined in samples of each lysate by SDS-PAGE followed by immunoblotting with pan-Ras Ab (Thermo Scientific, Rockford, IL). The amounts of Ras-GTP in the lysates were determined by the glutathione-S-transferase (GST)-RBD (Ras-binding domain of Raf) pull-down assay, as previously described [24]. The pulled-down Ras-GTP was subjected to SDS-PAGE followed by immunoblotting with pan-Ras Ab. Protein bands were visualized by enhanced chemiluminescence and quantified by densitometry using the Image J computer software (National Institutes of Health, Bethesda, MD).
cRAF overexpression.
MIA PaCa-2 cells were transiently transfected with nonspecific control plasmid (Control cDNA) or with 100 nM cRAF expression plasmid (cRAF cDNA; Cat # SC118323; OriGene, Rockville, MD) for 48 h using Lipofectamine 2000 following the manufacturer’s instructions. Following transfection, cells were replated and treated with CMC2.24 for up to 24 h.
Animal studies.
All animal studies were approved by the Institutional Animal Care and Use Committee at Stony Brook University.
Pancreatic Epithelial Explants Culture.
Pancreatic epithelial explants from 6–8 week-old KrasLSL-G12D/+;Ptf1aCre/+ mice were established following previously published protocols (22). Briefly, whole pancreatic tissues from mice were surgically removed and rinsed multiple times with HBSS media (additional pentamycin at 0.01ml/1ml HBSS) before being minced and digested with collagenase P (Sigma-Aldrich) for 30 min at 37 °C. Tissue debris were separated using 100μm Nylon mesh, and the acinar cells were separated and plated from the tissue using collagenase enzymatic digestion of tissue at 37 C for 30 min. After multiple rinsing in HBSS (Corning cellgo®; Mediateck, Herndon, VA), the acinar cell solution was pelleted and plated in Waymouth’s Medium (Thermo Fischer Scientific). Acinar cells were then treated with CMC2.24 55 and 80 μM for 3 h, and following treatment the Ras Pull-down assay was performed, as described above.
Toxicity of CMC2.24 in mice:
We evaluated in mice the potential acute toxicity of CMC2.24. Groups of 6 week-old female BALB/c mice (5 mice/group) were given by oral gavage, once a day, with 0, 50, 100, 250 and 350 mg/kg CMC2.24 for 3 weeks. To note, CMC2.24 350 mg/kg was the highest dose tested due to drug solubility concerns. Body weights were recorded twice weekly. On day 21, animals were euthanized, blood was drawn and serum was collected and a liver-kidney function panel was performed.
Efficacy study in nude mouse xenografts:
We performed three studies as follows:
Panc-1 subcutaneous xenografts:
Female immune deficient BALB/c nude mice at 6 weeks of age were purchased from Charles Rivers (Wilmington, MA), and were maintained in pathogen-free conditions and fed irradiated chow. Animals were bilaterally, s.c. injected with 2.5 × 106 Panc-1 cells/tumor in 0.1 ml sterile PBS. When Panc-1 cells reached palpable tumors, mice (n=7/group) were divided randomly into two groups receiving control, or CMC2.24 (50 mg/kg) in corn oil was given orally to mice five times per week during 17 days. Only corn oil was given to the untreated control mice. The dose of CMC2.24 was chosen based on our published reports [25,26]. Body weight was measured once a week whereas tumors were measured twice weekly. Tumor sizes were calculated by the formula: [length x width x (length + width/2) x 0.56] in millimeters. At the end of the experiments, animals were euthanized by CO2 asphyxiation and tumor weights were measured after their careful resection. Tumor tissue was retained for analysis.
Orthotopic xenografts model:
Male and female C57BL/6 mice (9–10 weeks old) were anaesthetized and in each a small incision was made in the left abdomen to expose the pancreas. Then, mouse KPC cells (1.0 × 105 cells suspended in 50 μL of PBS) were injected into the pancreas with a 28G insulin needle. After surgery, the mice were randomized into vehicle control (receiving PBS) and CMC2.24 (50 mg/kg/d, intraperitoneally once a day, five times per week, during 9 days) treatment groups. At the end of the treatment period, the animals were euthanized and their pancreas (cancerous and non-cancerous tissues) were excised and weighed.
Patient-derived tumor xenografts (PDTXs):
Female NOD/SCID mice (5–6 weeks old) were used for PDTX modeling. Pancreatic tumor tissue was obtained from the Cooperative Human Tissue Network (CHTN). The sample and clinical metadata were de-identified, assigned a patient code and a sample code prior to release to the researchers. Once received, the PC tissue was immediately transplanted in to the mice. When the tumor size reached about 10 mm in diameter, tumors were collected, cut into small pieces (2 × 2 × 2 mm), and single pieces were subcutaneously implanted into the right and left flanks of additional NOD/SCID mice, as described elsewhere [27]. Once the tumor reached a size of 1500 mm3, mice (n=8/group) were divided randomly into two groups; one group receiving PBS (vehicle control), and the other group receiving CMC2.24 (50 mg/kg) suspended in PBS given intraperitoneally to mice five times per week during 28 days. Body weight was measured once a week while tumors were measured twice weekly. Tumor sizes were calculated by the formula: [length x width x (length + width/2) x 0.56] in millimeters. At the end of the study, the animals were euthanized by CO2 asphyxiation and tumor weights were measured after their careful resection. Tumor tissue was collected for analysis.
Immunohistochemical analysis.
Immunohistochemical staining for Ki-67 (Cat # sc-15402, Santa Cruz Biotechnology; Santa Cruz, CA), and p-ERK1/2 (Cat number # 4376, Cell Signaling Technology; Beverly, MA) was performed on human PC xenograft tissue samples, as previously described [28]. Scoring: At least 5 fields per sample (at magnification x200) were scored. We calculated the percentage of positive cells (brown staining) by dividing the number of labeled cells by the number of cells in each field and multiplying by 100.
Statistical Analysis.
The data, obtained from at least three independent experiments, were expressed as the mean ± SEM. Statistical evaluation was performed by one-factor analysis of variance (ANOVA) followed by the Tukey test for multiple comparisons. P<0.05 was regarded as being statistically significant.
RESULTS
CMC2.24 inhibits the growth of human PC cells in culture
We initially determined the effect of CMC2.24 on the growth of human PC cells and compared it to that of the human pancreatic normal epithelial cells (HPNE). For this purpose, we treated a panel of four human PC cell lines as well as the HPNE cells with or without escalating concentrations of CMC2.24 (10–60 μM) for 24 h. As shown in Fig. 1B, CMC2.24 reduced PC cell growth in a concentration-dependent manner and more potently in all PC cells compared to HPNE cells. For instance, at 24 h, CMC2.24 60 μM reduced cell growth in AsPC-1, MIA PaCa-2 and Panc-1 cells by 78%, 55% and 79% (p<0.05), respectively. In contrast, under the same experimental conditions, CMC2.24 60 μM for 24 h had minimal effect on HPNE cells, with 76% of them being viable (Fig. 1B).
We then examined the effect of CMC2.24 on colony formation in two PC cell lines, and compared the effect to that of HPNE cells. CMC2.24 inhibited colony formation in a concentration-dependent manner (Fig. 1C). CMC2.24 at 20 μM reduced colony formation by 85% and 88% (p<0.01, for both), in MIA PaCa-2 and Panc-1 cells, respectively (Fig. 1C). In contrast, under the same experimental conditions, CMC2.24 20 μM only reduced colony formation by only 12% in HPNE cells (Fig. 1D).
Safety of CMC2.24 in mice
Prior to evaluating the efficacy of CMC2.24 in vivo, we examined the toxicity of CMC2.24 in mice. Six week-old female BALB/c mice (5 mice/group) were given by oral gavage once a day for 3 weeks vehicle control or CMC2.24 at 50, 100, 250 and 350 mg/kg. Body weights were recorded twice weekly. All CMC2.24-treated animals showed no signs of toxicity and were alive and healthy throughout the study. The body weights of vehicle- and CMC2.24-treated animals were comparable throughout the study. A liver-kidney function panel was performed in the serum from control and CMC2.24 350 mg/kg groups. All the parameters measured, including liver enzymes, were in normal range and there was no significant differences between the groups, suggesting that CMC2.24 350 mg/kg does not affect liver and kidney functions (Supplemental Figure 1). Therefore, we determined that the (sub-chronic) maximum tolerated dose of CMC2.24 (3-wk period of observation) is at least 350 mg/kg/d.
CMC2.24 inhibits the growth of pancreatic tumor xenografts
We assessed the in vivo chemotherapeutic potential of CMC2.24 using three animal models: a human PC subcutaneous xenograft model in nude mice, a syngeneic mouse KPC orthotopic model in immunocompetent mice and a PDTX model in SCID mice.
We initially evaluated the chemotherapeutic effect of CMC2.24 in pancreatic xenografts. For this purpose, Panc-1 cells were subcutaneously injected into athymic nude mice and gave rise to exponentially growing tumors. Once the tumors reached ~200 mm3, the mice were treated orally either with CMC2.24 50 mg/kg or with vehicle. On day 17 of treatment, the tumor volumes (mean±SEM) for the vehicle control and CMC2.24 50 mg/kg/d group were 452.9±69.7 and 291.8±50.3, respectively. CMC2.24 treatment reduced the rate of growth over baseline by 66.9% (p<0.05; Fig. 2A).
Figure 2: CMC2.24 inhibits the growth of pancreatic cancer xenografts.
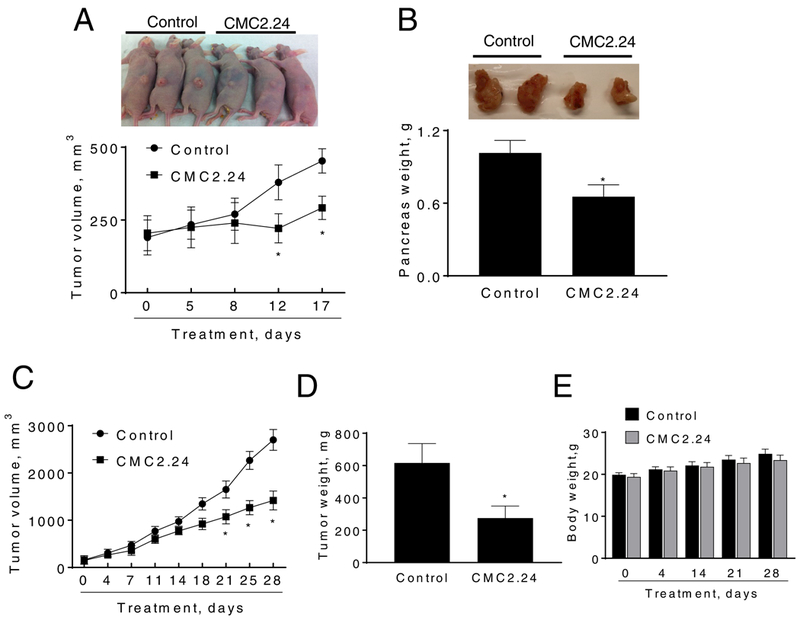
A: CMC2.24 inhibits Panc-1 tumor growth in nude mice. Panc-1 cells were injected subcutaneously in the flank areas of nude mice and when palpable tumors were observed, the mice received CMC2.24 50 mg/kg/d by oral gavage in corn oil or just corn oil (control) for 17 days. B: Mouse KPC cells were orthotopically implanted in immunocompetent mice, which were then treated without (control) or with CMC2.24 50 mg/kg for 9 days. Pancreas weight at sacrifice. Results are expressed as the mean ± SEM (*p < 0.05, vs. control). C: CMC2.24 reduces the growth of a PDTX in SCID mice. D: Tumor weight at sacrifice. Results are expressed as the mean ± SEM (*p < 0.05, vs. control). E: Mice body weight over time for control and CMC2.24 treated groups. Results are expressed as the mean ± SEM.
To rule out a cell-line specific effect, we investigated the effect of CMC2.24 in an orthotopic syngeneic model of PC. Wild-type mice bearing orthotopic FC1245 xenografts were treated with CMC2.24 (50mg/kg/d, i.p.) for 9 days. In this aggressive model, CMC2.24 significantly reduced tumor growth (35% inhibition, p<0.02; Figure 2B).
To evaluate the efficacy of CMC2.24 further, we used a PDTX model. A patient pancreatic tumor was implanted in a SCID mouse and then expanded to multiple mice. Once the expanded tumors reached a size of 150 mm3, mice were divided randomly into two groups receiving PBS (vehicle control), or CMC2.24 (50 mg/kg) suspended in PBS given i.p to mice five times per week during 28 days. At sacrifice, CMC2.24 treatment reduced the rate of growth over baseline by 50.1% (p<0.01; Fig. 2C), and tumor weight by 55% (p<0.01; Fig. 2D). Regarding its safety, CMC2.24 was well tolerated, with the mice showing no weight loss or other signs of toxicity during treatment (Fig. 2E). For instance, on the day of sacrifice, the mean body weight in both groups of mice was as follows: Control group = 24.9±1.5 g; and CMC2.24 group = 23.4±1.2 g.
CMC2.24 inhibits the growth of human PC through a strong cytokinetic effect
CMC2.24 inhibited tumor growth through a potent cytokinetic effect. In vitro treatment of AsPC-1 cells with CMC2.24 for 24h led to a concentration-dependent induction of apoptosis (Fig. 3A). This was also observed in MIA PaCa-2, Panc-1 and BxPC-3 cells, where CMC2.24 1.5xIC50 induced apoptosis by 9.8, 12.5 and 5.5-fold, respectively (Fig. 3B). Of significance, CMC2.24 55μM induced apoptosis in Panc-1 by 10.6-fold, whereas only by 3.2-fold in HPNE cells (Fig. 3C). These results further indicate that CMC2.24 decreases cell growth preferentially in PC cells compared to those of HPNE.
Figure 3: The cell kinetic effect of CMC2.24 in pancreatic cancer cells.
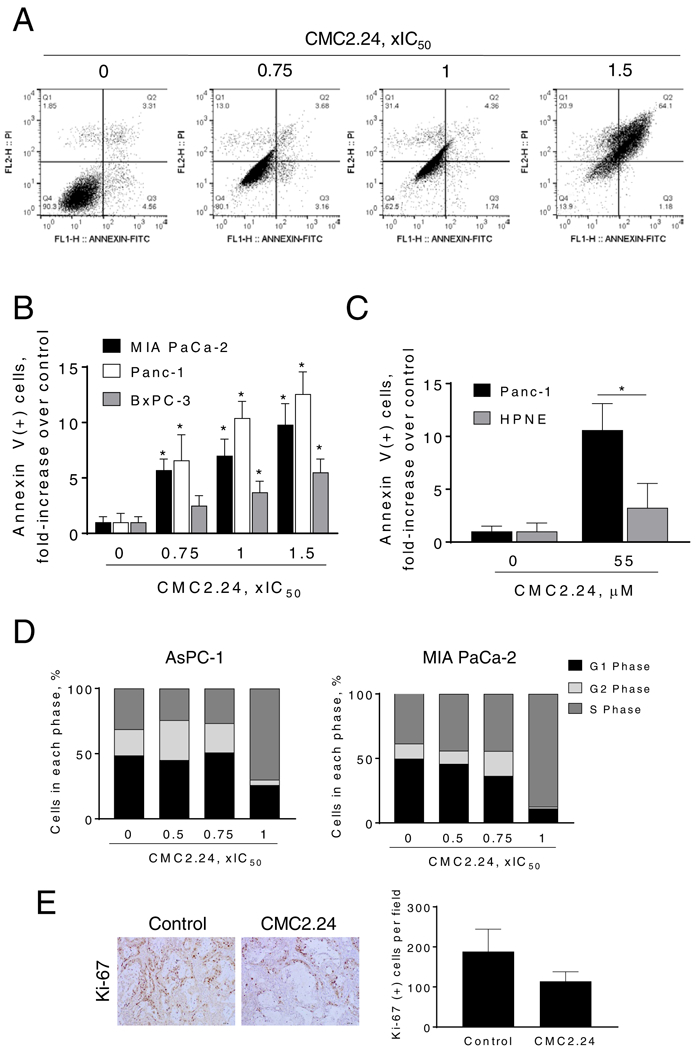
A: AsPC-1 cells treated with CMC2.24 for 24 h were stained with Annexin V/propidium iodide, and the percentage of apoptotic cells was determined by flow cytometry. B: MIA Paca-2, Panc-1 and BxPC-3 cells treated with various concentrations of CMC2.24 for 24 h were stained with Annexin V/propidium iodide, and the percentage of apoptotic cells was determined by flow cytometry. Results are expressed as fold-increase compared with the percentages of apoptotic cells in the control cells. *p<0.05, vs. control. C: Differential cytotoxic effect of CMC2.24 in Panc-1 cells compared with that in HPNE. Apoptosis was determined by flow cytometry in HPNE and Panc-1 cells incubated with or without 55 μM of CMC2.24 for 24h. Results are expressed as fold-increase compared with the percentages of apoptotic cells in the control cells. D: CMC blocks the S/G2 cell cycle phase transition after 24 h treatment in human AsPC-1 and MIA PaCa-2 cells, determined by flow cytometry following propidium iodide staining. E: The percentage of proliferating cells in vehicle or CMC2.24-treated PDTX xenografts were determined by Ki-67 staining. Representative images (x20) of tissue sections from PDTX tumors treated with either vehicle (control) or CMC2.24 and stained for Ki-67 expression (proliferation marker). The proliferation indices of xenograft tumors were determined and expressed as the mean ± SEM.
In addition, CMC2.24 inhibited the S to G2 cell cycle phase transition in MIA PaCa-2 and AsPC-1 cells (Fig. 3D), and inhibited cell proliferation as observed in vivo. In the PDTX model, CMC2.24 inhibited cell proliferation by 40%, as shown by Ki-67 immunohistochemical staining (Fig. 3E).
CMC2.24 inhibits ERK signaling pathway in PC cells and xenografts
To elucidate the mechanism of action of CMC2.24, we initially evaluated various key pathways, commonly active in PC, related to cell growth and cell-cycle control, and compared its effect to that of curcumin at same potency (1xIC50). In contrast to curcumin, CMC2.24 had only minor effects on the NF-κB pathway in MIA PaCa-2 cells (Supplemental Figure 2). However, CMC2.24 strongly affected the ERK pathway, a critical pathway in PC [29], and this effect by CMC2.24 tended to be stronger than the one exerted by curcumin, even though it did not reach significance between the groups (p = 0.08; Fig. 4A). CMC2.24 treatment led to a significant time-dependent inhibition of ERK1/2 phosphorylation in MIA PaCa-2 and Panc-1 cells, with nearly 90% reduction in MIA PaCa-2 cells, and 70% reduction in Panc-1 cells after 4 h treatments (Fig. 4B). CMC2.24 also reduced ERK1/2 phosphorylation levels in a concentration-dependent manner in mouse KPC cells, as determined by immunoblotting (Fig. 4C). These findings were confirmed by immunofluorescence. CMC significantly reduced p-ERK1/2 levels in Panc-1 cells by 48% (p<0.05; Fig. 4D).
Figure 4: CMC2.24 inhibits ERK1/2 activation in pancreatic cancer cells and xenografts.
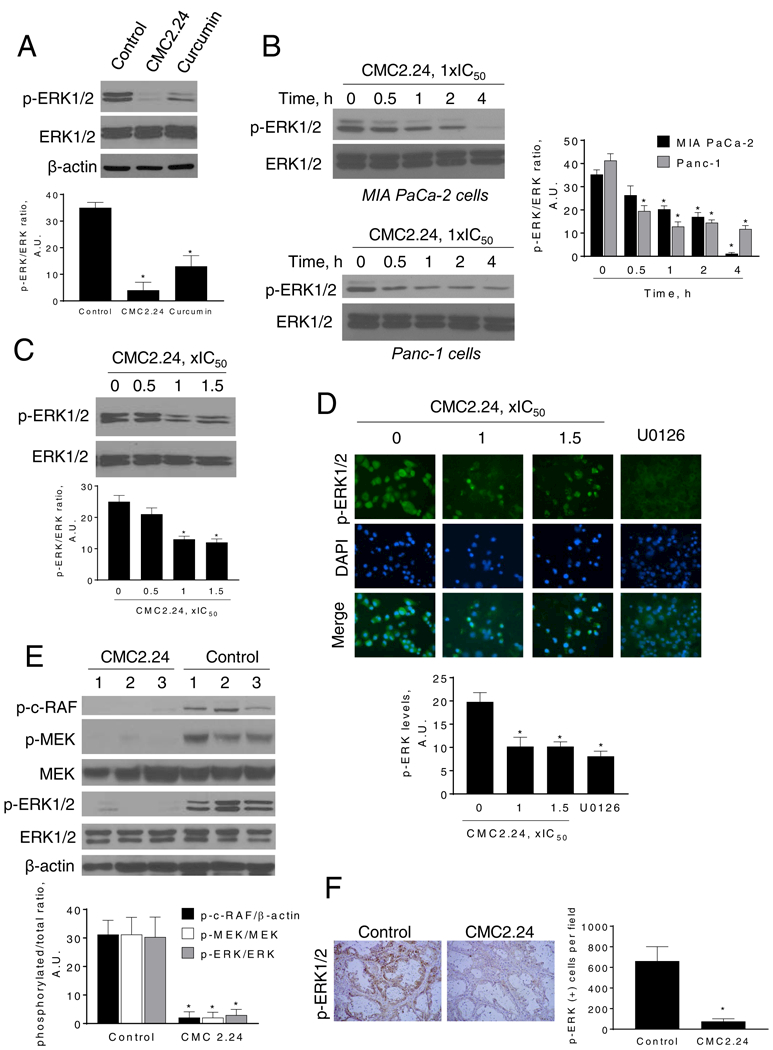
A: Immunoblots of p-ERK and ERK in MIA PaCa-2 cells treated with CMC2.24 or curcumin at 1xIC50 for 4 h. Results we quantified as the ration between p-ERK/ERK; *p<0.05 versus control. B: Immunoblots of p-ERK and ERK in MIA PaCa-2 and Panc-1 total cell extracts treated with CMC2.24 1xIC50 for up to 4 h. Bands were quantified and results expressed as the p-ERK/ERK ratio. *p<0.05 versus time zero. C: Immunoblots of p-ERK and ERK in mouse KPC cells treated with CMC2.24 at 0.5x, 1x or 1.5x IC50 and quantified p-ERK values. *p<0.05 versus control. D: Immunofluorescence staining for p-ERK1/2 in Panc-1 cells treated with CMC2.24 (1x and 1.5xIC50) or U0126 (10 μM) for 3 h. Representative images in each group from three independent experiments are shown (original magnification, x20). The immunofluorescence intensity in each group was quantified. *p<0.05 versus control. E: Panc-1 xenograft tumor lysates were analyzed for p-c-RAF, p-MEK, MEK, pERK1/2 and ERK1/2 by immunoblotting. Loading control: β-actin. Each lane represents a different tumor sample. Bands were quantified and results are expressed as the ratio of phospho/total expression levels for each protein; *p<0.05 vs. control. F: Representative images (x20) of tissue sections from PDTX tumors treated with either vehicle (control) or CMC2.24 and stained for p-ERK1/2. The percentage of p-ERK1/2-positive cells per field was determined and expressed as mean ± SEM. *p <0.01 vs. control.
Moreover, CMC2.24 reduced p-ERK1/2 levels in vivo. In total tissue homogenates, isolated form Panc-1 xenografts, CMC2.24 reduced ERK1/2 phosphorylation by 87% (p<0.02), compared to controls (Fig. 4E). This effect was also observed in the PDTX model. Immunostaining of PDTX tissue sections revealed that CMC2.24 reduced the expression of p-ERK1/2 by 89%, compared to control (p<0.01; Fig. 4F).
We next evaluated whether CMC2.24 could affect the activation of proteins upstream of ERK1/2. For this purpose, we explored the effect of CMC2.24 on the phosphorylation status of MEK, and c-RAF in Panc-1 tumor xenografts. CMC2.24 treatment reduced c-RAF and MEK phosphorylation levels by 93% and 91%, respectively (p<0.05) compared to controls (Fig. 4E). The strong effect of CMC2.24 on ERK1/2, MEK and c-RAF phosphorylation led us to explore the direct effect of CMC2.24 on Ras.
CMC2.24 inhibits Ras activation
We examined whether or not the PC growth inhibitory effect of CMC2.24 was associated with down regulation of Ras signaling. For this purpose, we used the Ras-GTP pull-down assay. In MIA PaCa-2 cells, CMC2.24 significantly (p<0.01) inhibited active Ras (Ras-GTP) by 90% (Fig. 5A).
Figure 5: CMC inhibits Ras activation in pancreatic cancer.
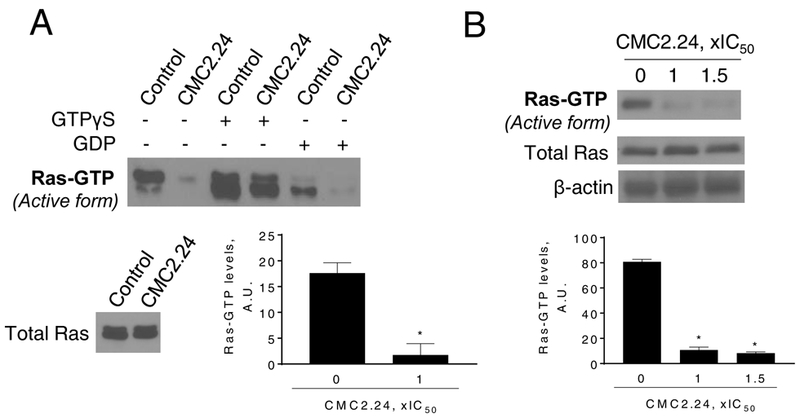
A: Immunoblots of Ras-GTP (active-Ras) and total K-Ras in MIA PaCa-2 cells treated without or with CMC2.24, as indicated, for 3 h. B: Immunoblots of Ras-GTP (active Ras) in cell protein extracts from primary acinar explants isolated from Kras-active mice treated with CMC2.24 1x and 1.5xIC50, for 3 h. *p <0.05 vs. control.
The inhibition of Ras by CMC2.24 was confirmed in primary acinar explants. We used the Ras-GTP pull-down assay to test the capacity of CMC2.24 to inhibit active Ras in fresh protein lysates from PC acinar cells isolated from active Kras mice. Compared to controls, CMC2.24 at 55 μM or 80 μM reduced Ras activation in the primary acinar explants by 87 and 90 %, respectively (p<0.01, for both; Fig. 5B).
Taken together, these in vivo results, in agreement with our in vitro results, suggest that Ras is a critical molecular target of CMC2.24, likely accounting for its PC growth inhibitory effect.
Overexpression of cRAF reduces, in part, the anticancer effect of CMC2.24
To confirm that the Ras → c-RAF→ ERK signaling is a key pathway of CMC2.24, we generated MIA PaCa-2 cells overexpressing c-RAF (c-RAF cDNA), using as controls MIA PaCa-2 cells transfected with scramble cDNA (control cDNA). The overexpression of c-RAF in MIA PaCa-2 cells was confirmed by immunoblotting (Fig. 6A). While CMC2.24 reduced p-c-RAF levels in MIA PaCa-2 cells, this effect was prevented by c-RAF overexpression (Fig. 6A). Moreover, c-RAF overexpression abrogated, in part, the growth inhibitory effect of CMC2.24. For instance, treatment of MIA PaCa-2 cells with CMC2.24 60 μM for 24h reduced cell growth by 57%. In contrast, overexpression of c-RAF partially prevented the reduction in cell growth induced by CMC2.24 60 μM (70% of viable cells in c-RAF overexpressing cells; Fig. 6B).
Figure 6: c-RAF overexpression ameliorates, in part, the cell growth inhibition by CMC2.24.

MIA PaCa-2 cells were transfected with a control (cDNA) or c-RAF-expressing plasmid for 48 h and then treated with CMC2.24 for 24 h. A: Immunoblots of p-c-RAF and c-RAF in MIA PaCa-2 total cell extracts treated with CMC2.24 1xIC50 for 4 h. Bands were quantified and results expressed as the p-c-RAF/c-RAF ratio. *p<0.05 versus control. B: Cell growth was evaluated by the MTT assay. c-RAF overexpression rescues cells from the cell growth inhibition induced by CMC2.24. Top: c-RAF expression status in whole cell protein lysates following transfection.
CMC2.24 inhibits STAT3Ser727 phosphorylation in PC cells
Because ERK1/2 can phosphorylate STAT3 at the Ser727 residue [30,31], we explored whether CMC2.24 affected STAT3 activation. In Panc-1 cells, CMC2.24 treatment reduced p-STAT3Ser727 levels in a time-dependent manner, reducing p-STAT3 levels by 60% at 4 h (Fig. 7A). This effect was confirmed by immunofluorescence. CMC2.24 1xIC50 and 1.5xIC50 significantly reduced p-STAT3Ser727 levels in Panc-1 cells by 73% and 71%, respectively (p<0.05; Fig. 7B).
Figure 7: CMC2.24 reduces STAT3Ser727 phosphorylation levels, induces mitochondrial reactive oxygen species and mitochondrial cell death in pancreatic cancer cells.
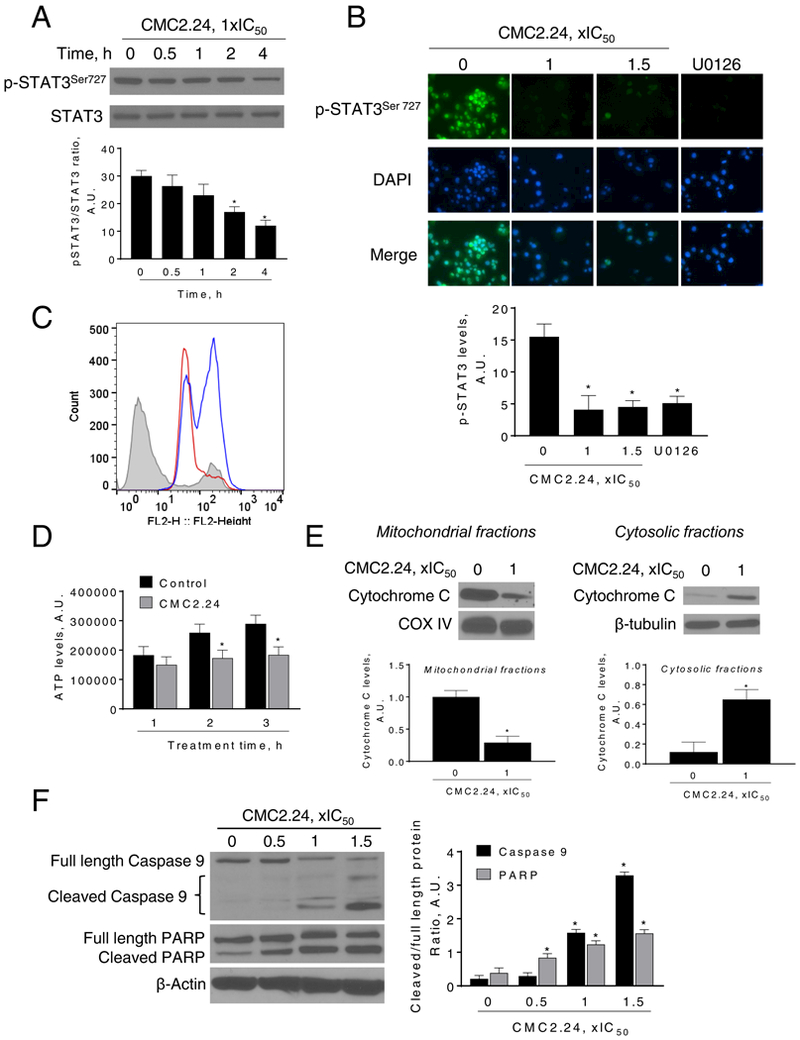
A: Immunoblots of STAT3 and phosphorylated STAT3 (STAT3Ser727) from Panc-1 cells treated with CMC2.24 for up to 4 h. Bands were quantified and results expressed as the p-STAT3Ser727/STAT3 ratio. *p<0.05 versus time zero. B: Immunofluorescence staining for p-STAT3Ser727 in Panc-1 cells treated with CMC2.24 (1x and 1.5xIC50) or U0126 (10 μM) for 3 h. Representative images in each group from three independent experiments are shown (original magnification, x20). The immunofluorescence intensity in each group was quantified. *p<0.05 versus control. C: MitoSOX Red fluorescence was measured by flow cytometry in MIA PaCa-2 cells treated with CMC2.24 for 3 h. MitoSOX Red fluorescent histograms for control (grey), CMC2.24 1xIC50 (red line) and CMC2.24 1.5xIC50 (blue line) are shown. D: ATP levels were determined in MIA PaCa-2 cells treated without (control) or with (CMC2.24 1xIC50) for up to 3 h. Results were quantified and expressed as mean ± SEM; *p<0.05 vs. control. E: CMC induces the release to the cytosol of cytochrome C. Mitochondrial fractions loading control: COX IV. Cytosolic fractions loading control: β-tubulin. Bands were quantified and results are shown as mean ± SEM; *p<0.05 vs. control. F: Immunoblots for full length and cleaved caspase 9 as well as full length and cleaved PARP in total cell protein extracts from MIA PaCa-2 cells treated with CMC2.24, as indicated, for 24 h. Loading control: β-actin. Bands were quantified and results are shown as the ratio between the cleaved/full length protein; *p<0.05 vs. control.
CMC2.24 induces mitochondrial reactive oxygen species and intrinsic apoptosis
STAT3 can play a role in the mitochondria, as a regulator of the electron transport chain (ETC) and thereby modulating mitochondrial production of ATP and reactive oxygen species [30,32]. Since phosphorylation at the serine 727 residue is critical for the translocation of STAT3 into the mitochondria [31], and CMC2.24 reduced STAT3 phosphorylation at this residue, we next evaluated the effect of CMC2.24 on mitochondrial function, by determining its effect on the induction of mitochondrial reactive oxygen species, ATP production and intrinsic apoptosis.
Treatment with increasing concentrations of CMC2.24 for 4 h led to increased levels of mitochondrial reactive oxygen species in MIA PaCa-2 cells, as determined using the MitoSOX probe. For example, CMC2.24 1xIC50 and 1.5xIC50 showed a 2.8-fold and 3.8-fold increase, respectively, in the MitoSOX Average Geo Mean when compared to the control (Fig. 7C).
CMC2.24 also reduced ATP levels in PC cells. In MIA PaCa-2 cells, CMC2.24 80 μM for 3 h reduced ATP levels by 36% when compared to controls (p<0.05; Fig. 7D). We next explored other parameters of intrinsic apoptosis by evaluating the effect of CMC2.24 on cytochrome C release from the mitochondria, followed by the activation of caspase 9 proteins. In MIA PaCa-2 cells, CMC2.24 treatment induced the release of cytochrome C to the cytosol (Fig. 7E). This led to an activation of caspases 9 and 3, as determined by PARP cleavage. For example, CMC2.24 1x and 1.5x IC50 reduced full-Caspase 9 levels by 42% and 51%, respectively, compared to the control (p<0.05). As a consequence of caspase 9 activation, levels of cleaved PARP increased in all CMC2.24 treatments (Fig. 7F).
DISCUSSION
PC remains a devastating malignancy due to the lack of efficacious treatments. Because clinical progress has been, to date, limited in effectively treating this disease, the search for new agents that can combat this disease represents a critical need. In this study, we show that the novel agent CMC2.24 is effective against PC in mice by inhibiting Ras activation and its downstream effector ERK1/2 pathway. The anti-cancer efficacy of CMC2.24, accompanied by its apparent lack of toxicity, makes it a promising drug candidate for the treatment of PC.
We used three complementary preclinical models to evaluate the anticancer effect of CMC2.24 in PC. These included: 1) a Panc-1 Kras mutant subcutaneous xenograft model in nude mice, 2) a mouse KPC orthotopic syngeneic model in immunocompetent mice, and 3) a patient-derived tumor xenograft in SCID mice. Based on our findings from human PC xenografts, which allowed us to evaluate the therapeutic potential of CMC2.24, the inhibition of PC growth by CMC2.24 was strong, significantly reducing the rate of growth of Panc-1 tumors by up to 66.9% in nude mice. In a more aggressive model, as is the orthotopic implantation KPC cells, CMC2.24 reduced tumor growth by 35% and in a clinically relevant model, such as the patient-derived pancreatic tumor xenograft model, the chemotherapeutic effect of CMC2.24 was also strong, reducing the rate of growth by 50% without causing any toxic side effects. In all these model, Kras mutations are important contributors to the altered tumor growth. Overall, these three preclinical models indicate that CMC2.24 may be efficacious in PC.
CMC2.24 was generated by chemically modifying curcumin in order to enhance its bioavailability and biological activity [18,19]. Previous studies by our group have shown that CMC2.24 is safe in mice and rats, and CMC2.24 has a better pharmacokinetic profile than curcumin. The innovation behind the development of CMC2.24 relies on the improvement in the acidic character of the enolic system, by introducing an electron-withdrawing group at the 4-Carbon of the curcumin molecule, resulting in a tri-ketonic zinc-binding domain. This modification significantly improves its solubility and increases its biologic activity [19,26,33]. For example, we have shown that the tri-ketonic nature of CMC2.24 provides an advantage over curcumin (diketonic) in their capacity to inhibit matrix metalloproteinase molecules [19,20], by potentially binding to the zinc atom in the catalytic domain of the matrix metalloproteinase molecules. Furthermore, the tri-ketonic motif also allows these molecules to bind more strongly to serum albumin than curcumin, for improved transport. In addition, the removal of the methoxy groups present on curcumin helps in making these molecules less toxic.
A critical characteristic of the novel agent CMC2.24 is its selectivity toward cancer cells. Ideally, anticancer drugs should target specifically the tumor and not the normal surrounding tissue. In other words, they should specifically kill the cancer cells while sparing the adjacent normal cells, to minimize toxicity. CMC2.24 displays such selectivity. Compared with several PC cell lines, HPNE cells are more resistant to CMC2.24-induced cell death and to the suppression of their growth. Such selectivity, together with lack of apparent toxicity, if broadly confirmed, will be a significant advantage of CMC2.24.
Mutations in the Kras oncogene constitute one of the key genetic alterations found in PC. Kras activity is not only essential for pancreatic tumor formation and development but, at least in mice, the requirement for Kras is continuous [5,6]. In addition, oncogenic Kras promotes metabolic reprogramming in pancreatic tumors [34]. Moreover, inactivation of oncogenic Ras in mice results in tumor shrinkage [6]. These findings strongly indicate that Ras is widely considered a therapeutic target of exceptional importance. Even though early efforts to target Ras with farnesyltransferase inhibitors failed to extend patients’ lives [35], there are currently numerous extensive efforts, including the Ras initiative from the U.S. National Cancer Institute, to develop drugs that can target Ras [24,35–40]. As our results indicate, the novel CMC2.24 has proved to be a strong inhibitor of Ras activation.
A main effector pathway, downstream of Ras, is the RAF-MEK-ERK pathway [41,42]. In response to growth factor stimulation or oncogene activation, the RAF-MEK-ERK pathway can modulate a wide variety of cellular processes such as proliferation, survival, differentiation and apoptosis [29]. CMC2.24 inhibited prominently the RAF-MEK-ERK downstream pathway. In particular, CMC2.24 strongly inhibited ERK phosphorylation both in vitro and in vivo. The centrality of the RAF-MEK-ERK pathway in the reduction of cell growth by CMC2.24 was documented by the finding that c-RAF overexpression ameliorated the anti-cancer effect of CMC2.24. Because of the requirement of RAF-MEK-ERK signaling in the aberrant behavior of the PC cell, its downregulation could explain, in part, the reduction in cell growth observed in CMC2.24-treated tumors.
The deregulation of the ERK pathway is a signature of many epithelial cancers, including PC. Thus, it is an attractive target for the development of anticancer drugs [43]. Recently, many MEK inhibitors targeting the RAF-MEK-ERK pathway have been included in clinical trials for cancer therapy [44], clearly stressing the importance of this pathway as a target for therapeutics. Therefore, our findings provide clinical relevance to the robust inhibition of ERK1/2 pathway by CMC2.24, and suggest that Ras inhibition could be beneficial in inhibiting downstream effectors such as ERK1/2.
STAT3 is overexpressed in PC and has been described as a key driver of the progression into advanced PC [45–47]. Besides its established canonical pathway in which STAT3 functions as a transcription factor, a non-canonical activation pathway of STAT3 has been discovered in which STAT3 holds key functions in the mitochondria [48,49]. Phosphorylation at the serine 727 residue is critical for the translocation of STAT3 to the mitochondria, and interestingly, ERK may phosphorylate STAT3 at this residue [31]. Given that STAT3 phosphorylation at the serine 727 residue is downstream of ERK, we postulated that CMC2.24 might affect this critical protein in PC. CMC2.24 reduced STAT3Ser727 phosphorylation levels, enhanced mitochondrial reactive oxygen species and induced intrinsic apoptosis. Because of the potential effect of STAT3 in Ras-dependent transformation [48], and the importance of ERK in its regulation, its down-regulation could explain, in part, the increased in apoptosis observed in CMC2.24-treated PC cells. At this time, the exact mechanisms of the effects of CMC2.24 on mitochondrial STAT3 still remains elusive.
In conclusion, the novel compound CMC2.24 is an effective anticancer agent in preclinical models of PC, acting primarily through the inhibition of Ras activation and its downstream effector ERK1/2. Taken together, our results point to the potential of CMC2.24, a novel Ras inhibitor, as a new agent against PC and deserves further evaluation.
Supplementary Material
Acknowledgements:
Grant Support: This work was supported in part by the Stony Brook Cancer Center, the University of California, Davis and the NIH grants CA185209, CA175699 and CA181727 to G.G.M, and CA192994 to J.L.W. The study sponsors had no role in the study design, in the collection, analysis and interpretation of data; in the writing of the manuscript; nor in the decision to submit the manuscript for publication.
Abbreviations:
- CMC2.24
Chemically-modified curcumin
- ERK
extracellular signal–regulated kinase
- PI
propidium iodide
Footnotes
Conflict of interest disclosure: LMG and FJ are listed as inventors on several issued patents on chemically-modified curcumins & these have been assigned to Stony Brook University (LMG) and to Chem-Master, Int (FJ). Moreover, both have an equity position in Traverse Biosciences, Inc, the company that provided the test compound. All other authors declare no conflict of interest.
REFERENCES
- 1.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74(11):2913–2921. [DOI] [PubMed] [Google Scholar]
- 2.Kleeff J, Korc M, Apte M et al. Pancreatic cancer. Nature reviews Disease primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 3.Ying JE, Zhu LM, Liu BX. Developments in metastatic pancreatic cancer: is gemcitabine still the standard? World journal of gastroenterology : WJG 2012;18(8):736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hruban RH, van Mansfeld AD, Offerhaus GJ et al. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. The American journal of pathology 1993;143(2):545–554. [PMC free article] [PubMed] [Google Scholar]
- 5.di Magliano MP, Logsdon CD. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology 2013;144(6):1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins MA, Bednar F, Zhang Y et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122(2):639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nature reviews Drug discovery 2014;13(11):828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mimeault M, Batra SK. Potential applications of curcumin and its novel synthetic analogs and nanotechnology-based formulations in cancer prevention and therapy. Chinese medicine 2011;6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunnumakkara AB, Guha S, Krishnan S, Diagaradjane P, Gelovani J, Aggarwal BB. Curcumin potentiates antitumor activity of gemcitabine in an orthotopic model of pancreatic cancer through suppression of proliferation, angiogenesis, and inhibition of nuclear factor-kappaB-regulated gene products. Cancer Res 2007;67(8):3853–3861. [DOI] [PubMed] [Google Scholar]
- 10.Diaz Osterman CJ, Gonda A, Stiff T et al. Curcumin Induces Pancreatic Adenocarcinoma Cell Death Via Reduction of the Inhibitors of Apoptosis. Pancreas 2016;45(1):101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The Essential Medicinal Chemistry of Curcumin. Journal of medicinal chemistry 2017;60(5):1620–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solano LN, Nelson GL, Ronayne CT et al. Synthesis, in vitro, and in vivo evaluation of novel functionalized quaternary ammonium curcuminoids as potential anti-cancer agents. Bioorganic & medicinal chemistry letters 2015;25(24):5777–5780. [DOI] [PubMed] [Google Scholar]
- 13.Kesharwani P, Xie L, Banerjee S et al. Hyaluronic acid-conjugated polyamidoamine dendrimers for targeted delivery of 3,4-difluorobenzylidene curcumin to CD44 overexpressing pancreatic cancer cells. Colloids and surfaces B, Biointerfaces 2015;136:413–423. [DOI] [PubMed] [Google Scholar]
- 14.Gundewar C, Ansari D, Tang L et al. Antiproliferative effects of curcumin analog L49H37 in pancreatic stellate cells: a comparative study. Annals of gastroenterology : quarterly publication of the Hellenic Society of Gastroenterology 2015;28(3):391–398. [PMC free article] [PubMed] [Google Scholar]
- 15.Botchkina GI, Zuniga ES, Rowehl RH et al. Prostate cancer stem cell-targeted efficacy of a new-generation taxoid, SBT-1214 and novel polyenolic zinc-binding curcuminoid, CMC2.24. PLoS One 2013;8(9):e69884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elburki MS, Moore DD, Terezakis NG et al. A novel chemically modified curcumin reduces inflammation-mediated connective tissue breakdown in a rat model of diabetes: periodontal and systemic effects. Journal of periodontal research 2016. [DOI] [PubMed] [Google Scholar]
- 17.Elburki MS, Rossa C, Guimaraes MR et al. A novel chemically modified curcumin reduces severity of experimental periodontal disease in rats: initial observations. Mediators of inflammation 2014;2014:959471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Golub LM, Johnson F, Wishnia A. pKa, zinc- and serum albumin-binding of curcumin and two novel biologically-active chemically-modified curcumins. Current medicinal chemistry 2012;19(25):4367–4375. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Gu Y, Lee HM et al. Design, synthesis and biological activity of new polyenolic inhibitors of matrix metalloproteinases: a focus on chemically-modified curcumins. Current medicinal chemistry 2012;19(25):4348–4358. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, McClain SA, Lee HM et al. A Novel Chemically Modified Curcumin “Normalizes” Wound-Healing in Rats with Experimentally Induced Type I Diabetes: Initial Studies. Journal of diabetes research 2016;2016:5782904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nature protocols 2006;1(5):2315–2319. [DOI] [PubMed] [Google Scholar]
- 22.Zhao W, Mackenzie GG, Murray OT, Zhang Z, Rigas B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: a redox-dependent effect. Carcinogenesis 2009;30(3):512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackenzie GG, Queisser N, Wolfson ML, Fraga CG, Adamo AM, Oteiza PI. Curcumin induces cell-arrest and apoptosis in association with the inhibition of constitutively active NF-kappaB and STAT3 pathways in Hodgkin’s lymphoma cells. Int J Cancer 2008;123(1):56–65. [DOI] [PubMed] [Google Scholar]
- 24.Mackenzie GG, Bartels LE, Xie G et al. A novel Ras inhibitor (MDC-1016) reduces human pancreatic tumor growth in mice. Neoplasia 2013;15(10):1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Ge L, Abdel-Razek O et al. Differential Susceptibility of Human Sp-B Genetic Variants on Lung Injury Caused by Bacterial Pneumonia and the Effect of a Chemically Modified Curcumin. Shock 2016;45(4):375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, McClain SA, Lee HM et al. A Novel Chemically Modified Curcumin “Normalizes” Wound-Healing in Rats with Experimentally Induced Type I Diabetes: Initial Studies. Journal of diabetes research 2016;2016:5782904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubio-Viqueira B, Jimeno A, Cusatis G et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 2006;12(15):4652–4661. [DOI] [PubMed] [Google Scholar]
- 28.Mackenzie GG, Ouyang N, Xie G et al. Phospho-sulindac (OXT-328) combined with difluoromethylornithine prevents colon cancer in mice. Cancer Prev Res (Phila) 2011;4(7):1052–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gysin S, Lee SH, Dean NM, McMahon M. Pharmacologic inhibition of RAF-->MEK-->ERK signaling elicits pancreatic cancer cell cycle arrest through induced expression of p27Kip1. Cancer Res 2005;65(11):4870–4880. [DOI] [PubMed] [Google Scholar]
- 30.Yang R, Rincon M. Mitochondrial Stat3, the Need for Design Thinking. Int J Biol Sci 2016;12(5):532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gough DJ, Koetz L, Levy DE. The MEK-ERK pathway is necessary for serine phosphorylation of mitochondrial STAT3 and Ras-mediated transformation. PLoS One 2013;8(11):e83395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meier JA, Hyun M, Cantwell M et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci Signal 2017;10(472). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elburki MS, Moore DD, Terezakis NG et al. A novel chemically modified curcumin reduces inflammation-mediated connective tissue breakdown in a rat model of diabetes: periodontal and systemic effects. Journal of periodontal research 2017;52(2):186–200. [DOI] [PubMed] [Google Scholar]
- 34.Ying H, Kimmelman AC, Lyssiotis CA et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149(3):656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future medicinal chemistry 2011;3(14):1787–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldberg L, Haklai R, Bauer V, Heiss A, Kloog Y. New derivatives of farnesylthiosalicylic acid (salirasib) for cancer treatment: farnesylthiosalicylamide inhibits tumor growth in nude mice models. Journal of medicinal chemistry 2009;52(1):197–205. [DOI] [PubMed] [Google Scholar]
- 37.Haklai R, Elad-Sfadia G, Egozi Y, Kloog Y. Orally administered FTS (salirasib) inhibits human pancreatic tumor growth in nude mice. Cancer Chemother Pharmacol 2008;61(1):89–96. [DOI] [PubMed] [Google Scholar]
- 38.Rotblat B, Ehrlich M, Haklai R, Kloog Y. The Ras inhibitor farnesylthiosalicylic acid (Salirasib) disrupts the spatiotemporal localization of active Ras: a potential treatment for cancer. Methods in enzymology 2008;439:467–489. [DOI] [PubMed] [Google Scholar]
- 39.Sun Q, Burke JP, Phan J et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl 2012;51(25):6140–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmermann G, Papke B, Ismail S et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013;497(7451):638–642. [DOI] [PubMed] [Google Scholar]
- 41.Botta GP, Reginato MJ, Reichert M, Rustgi AK, Lelkes PI. Constitutive K-RasG12D activation of ERK2 specifically regulates 3D invasion of human pancreatic cancer cells via MMP-1. Molecular cancer research : MCR 2012;10(2):183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collisson EA, Trejo CL, Silva JM et al. A Central Role for RAF->MEK->ERK Signaling in the Genesis of Pancreatic Ductal Adenocarcinoma. Cancer discovery 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milella M, Kornblau SM, Estrov Z et al. Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest 2001;108(6):851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson DB, Flaherty KT, Weber JS et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32(33):3697–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corcoran RB, Contino G, Deshpande V et al. STAT3 Plays a Critical Role in KRAS-Induced Pancreatic Tumorigenesis. Cancer Res 2011;71(14):5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fukuda A, Wang SC, Morris JPt et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011;19(4):441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lesina M, Kurkowski MU, Ludes K et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011;19(4):456–469. [DOI] [PubMed] [Google Scholar]
- 48.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009;324(5935):1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wegrzyn J, Potla R, Chwae YJ et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009;323(5915):793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.