

Abstract
Epithelial barrier loss is a driver of intestinal and systemic diseases. Myosin light chain kinase (MLCK) is a key effector of barrier dysfunction and a potential therapeutic target, but enzymatic inhibition has unacceptable toxicities. Here, we show that a unique domain within the MLCK splice-variant MLCK1 directs perijunctional actomyosin ring (PAMR) recruitment. Using the domain structure and multiple screens, we identified a domain-binding small molecule (Divertin) that blocks MLCK1 recruitment without inhibiting enzymatic function. Divertin blocks acute, TNF-induced MLCK1 recruitment as well as downstream MLC phosphorylation, barrier loss, and diarrhea in vitro and in vivo. Divertin corrects barrier dysfunction and prevents disease development and progression in experimental inflammatory bowel disease. Beyond applications of Divertin in gastrointestinal disease, this general approach to enzymatic inhibition by preventing access to specific subcellular sites provides a new paradigm for safely and precisely targeting individual properties of enzymes with multiple functions.
Keywords: myosin light chain kinase, tight junction, intestinal permeability, barrier function, inflammatory bowel disease, enterocolitis, occludin, actin
INTRODUCTION
Epithelial barriers are essential for survival. Barrier dysfunction, which characterizes many diseases, can occur by two distinct mechanisms. The first, cellular damage, results in catastrophic barrier loss. A second, more nuanced mechanism reflects increased permeability of the tight junctions that seal the paracellular space.1 This selectively-permeable seal is regulated by myosin light chain kinase (MLCK) and myosin II regulatory light chain (MLC) phosphorylation within the perijunctional actomyosin ring (PAMR).2,3 MLCK expressed in various epithelia and smooth muscle (visceral and vascular) are encoded by a single gene, MYLK, and share identical catalytic and calmodulin-binding regulatory domains (Fig. 1a).4,5 They differ in that ~210 kDa long MLCK expressed in epithelia includes additional 5’ exons that encode six amino-terminal immunoglobulin-cell adhesion molecule (IgCAM) domains that are absent in smooth muscle (short) MLCK (Fig. 1a).6 Intestinal epithelia express two splice variants of long MLCK, MLCK1 and MLCK2 (Fig. 1a), but do not express short MLCK.7 These variants differ by a single exon that is only present in MLCK1 and completes IgCAM3.7,8
Figure 1. Long myosin light chain kinase splice variant 1 (MLCK1) is specifically recruited to the perijunctional actomyosin ring (PAMR) in response to inflammatory stimuli.
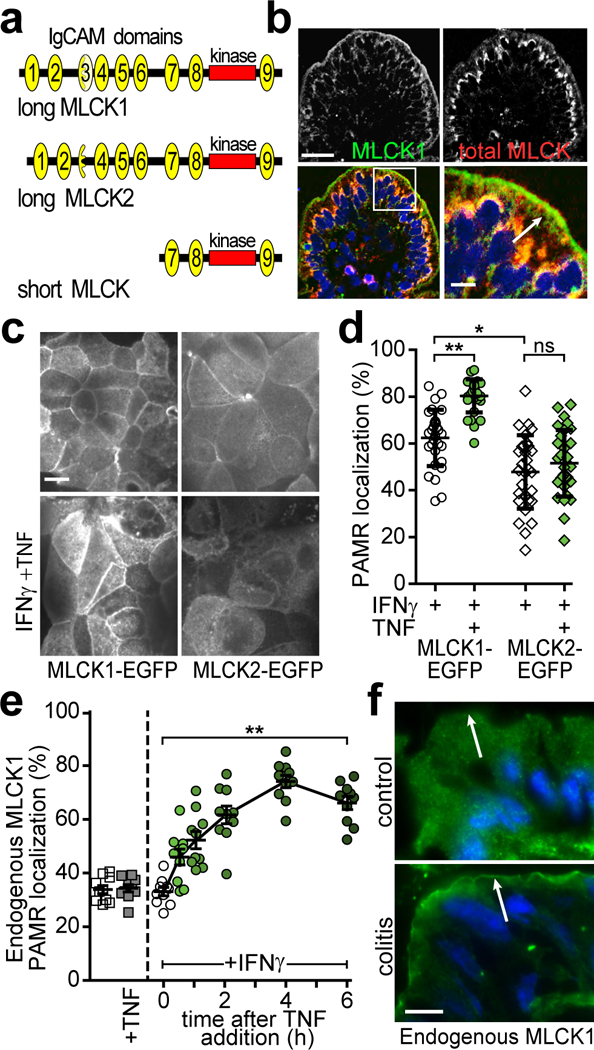
(a) Protein domain structure of MYLK gene products. IgCAM domains are numbered from the amino terminus. Long MLCK is expressed as two splice variants, MLCK1 and MLCK2, in intestinal epithelial cells. Short MLCK is expressed in smooth muscle. (b) Normal human jejunum stained for long MLCK1 (green), total MLCK (red), and nuclei (blue). Bar, 50 μm. Inset of the boxed region is shown on the bottom right and includes an arrow indicating the PAMR. Bar, 10 μm. Images are representative of more than 10 independent experiments. (c) Caco-2BBe monolayers expressing MLCK1-EGFP or MLCK2-EGFP were primed with IFNγ followed by treatment with TNF for 4 hrs. Images are representative of more than 12 independent experiments. Bar, 10 μm. (d) MLCK1-EGFP or MLCK2-EGFP colocalization with F-actin at the PAMR was determined. For this experiment, which is representative of 4 independent studies, n=8 biologically independent samples with 3 – 5 fields analyzed for each condition. *, P<0.05; **, P<0.01 by Kruskal-Wallis test with Dunn’s multiple comparison test. (e) Monolayers were treated with IFNγ and/or TNF, as indicated prior to immunostaining for endogenous MLCK1 and F-actin. PAMR localization of endogenous MLCK1 was determined. Data show the fraction of total MLCK1 localized to the PAMR and are therefore independent of absolute MLCK1 expression, which increases in response to TNF. For this experiment, which is representative of 4 independent studies, n=3 biologically independent samples with 3 – 4 fields analyzed for each condition. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. Mean ± SEM is shown. (f) Colon sections from healthy mice (control) or those with T cell transfer colitis (colitis) were stained for MLCK1 (green) and nuclei (blue). Intensity of MLCK1 staining in colonocytes from healthy control mice is enhanced to allow direct comparison with diseased colonocytes, which have increased MLCK1 expression. Arrows indicate the position of the PAMR, where distinct line of MLCK1 can be detected in colonocytes from colitic, but not control, mice. Bar, 5 μm. Images representative of more than 6 independent experiments are shown.
Long MLCK-deficient mice are viable, develop normally,9 and are protected from acute LPS- and ventilator-induced lung injury, acute tumor necrosis factor α (TNF) -induced intestinal barrier loss and diarrhea, and chronic, immune-mediated colitis.2,9,10 In contrast, short MLCK knockout mice die soon after birth.11 Thus, although long MLCK could be an attractive therapeutic target, systemic toxicities associated with enzymatic MLCK inhibition limit the utility of this approach.
Here, we report an alternative strategy for the therapeutic inhibition of long MLCK-dependent barrier loss. We show that MLCK1, but not MLCK2, is recruited to the PAMR in response to pathogenic stimuli. Using an in silico structure-based screen we have identified a small molecule that binds to IgCAM3 and prevents stimulus-induced MLCK1 recruitment to the PAMR. This molecule, termed Divertin because it diverts MLCK1 from the PAMR, does not interfere with MLCK enzymatic activity, epithelial cell wound repair, or smooth muscle contraction. It does, however, prevent MLCK-mediated intestinal barrier loss in vitro and in vivo, restore barrier function in spontaneous colitis, and attenuate experimental, immune-mediated colitis. We conclude that IgCAM3-dependent MLCK1 recruitment to the PAMR is a viable target for therapeutic preservation of epithelial barrier function in intestinal disease and may also be beneficial in pathophysiologies affecting other organs.
RESULTS
Inflammatory stimuli induce long MLCK1 trafficking to the PAMR
In human small intestine, MLCK1 expression is restricted to villous enterocytes, where it is concentrated at the PAMR and within the apical cytoplasm just beneath the brush border (Fig. 1b). MLCK2 is expressed throughout the crypt-villus axis and comprises the remainder of intestinal epithelial MLCK. The diffusely distributed cytoplasmic pool detected by antibodies against total MLCK, but not MLCK1-specific antisera, suggests that this pool represents MLCK2 (Fig. 1b).
To directly compare MLCK1 and MLCK2 localization, MLCK1-EGFP and MLCK2-EGFP were expressed in Caco-2BBe human intestinal epithelial monolayers. Both splice-variants were present in the cytoplasm and in association with lateral membranes (Fig. 1c). However, MLCK1-EGFP demonstrated significantly greater localization within the apical PAMR relative to MLCK2 (Fig. 1d, P<0.05). Moreover, TNF, which activates MLCK-dependent phosphorylation of perijunctional MLC,2,12 markedly increased PAMR-associated pools of MLCK1, but not MLCK2 (Fig. 1c,d; P<0.01). Analysis of non-transfected Caco-2BBe monolayers confirmed recruitment of endogenous MLCK1 that began within 0.5 hr of TNF addition and continued to 4 hrs (Fig. 1e). Notably, MLCK1 redistribution did not occur in the absence of interferon-γ (IFNγ) pre-treatment, which is necessary to induce TNF receptor expression in these monolayers.13 Thus, endogenous MLCK1 and MLCK1-EGFP, but not MLCK2-EGFP, demonstrate TNF-induced MLCK1 recruitment to the PAMR in intestinal epithelial monolayers in vitro.
To determine if MLCK1 recruitment to the PAMR occurs in vivo, intestines of immunodeficient mice with colitis induced by adoptive transfer of CD4+CD45RBhi naïve effector T cells14 were examined. In colonocytes from these mice, MLCK1 was most concentrated within the apical cytoplasm where it formed a distinct line at the level of the PAMR (Fig. 1f, lower panel, arrow). In contrast, MLCK1 was distributed diffusely within colonocytes of healthy mice (Fig. 1f, upper panel). Thus, MLCK1 is recruited to the PAMR during chronic immune-mediated disease.
Identification and analysis of potential MLCK1 IgCAM3-targeted small molecules
The 69 amino acids encoded by exon 8, which complete IgCAM3, are the only difference between MLCK1 and MLCK2. We therefore hypothesized that a small molecule that bound to IgCAM3 might interfere with MLCK1 recruitment to the PAMR. To identify such a molecule, we solved the crystal structure of IgCAM3 to 2.5Å (Fig. 2a, Supplementary Table 1). Overall, IgCAM3 formed a Greek key β sandwich that is characteristic of the IgCAM superfamily.15 The overall structure was also similar to that reported for telokin, which forms IgCAM9 of long and short MLCK.16
Figure 2. Identification and preliminary characterization of small molecules that bind to IgCAM3.
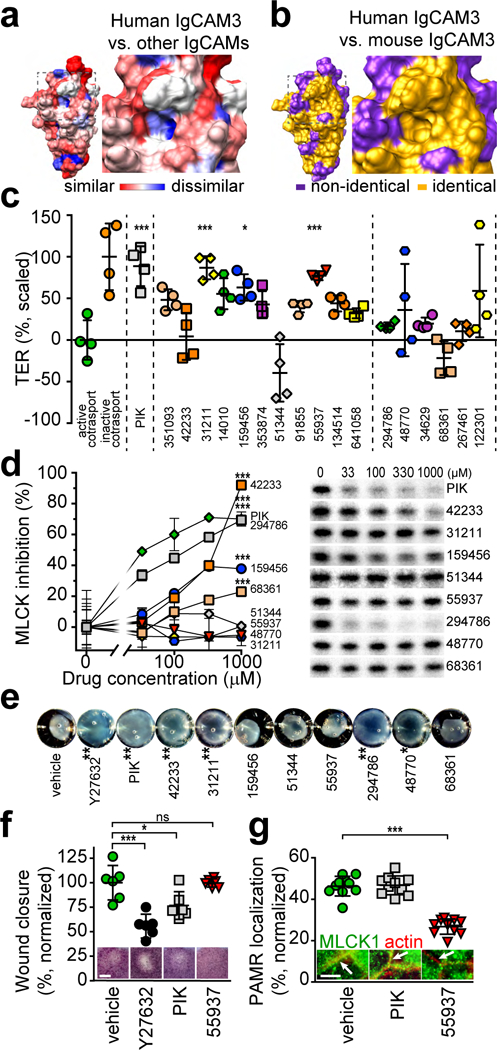
(a) Crystal structure of human IgCAM3. Colors indicate low (blue) or high (red) degree of amino acid similarity to other human long MLCK1 IgCAM domains. The box encloses the putative small molecule binding pocket, which includes a hydrophobic region that is poorly conserved between IgCAMs (Extended Data 1a). (b) Crystal structure of human IgCAM3. Colors indicate identical (gold) or non-identical (purple) residues relative to mouse MLCK1 IgCAM3. The putative binding pocket is identical in human and mouse. (c) Transepithelial resistance (TER) of Caco-2BBe monolayers with active (green circles) or inactive (orange circles) Na+-glucose cotransport. The MLCK pseudosubstrate peptide membrane Permeant Inhibitor of MLCK, PIK (150 μM, gray squares) served as a positive control for enzymatic inhibition. Small molecules are arranged from lowest to highest predicted ΔG (left to right) and separated (by dashed lines) into those with lowest (left) and intermediate (right) values. NSC numbers are indicated on the x-axis. Each small molecule was tested in 4 independent experiments, with each point representing the mean scaled TER of 3 monolayers within each experiment. Mean ± SEM. *, P<0.05; ***, P<0.001 by ANOVA with Dunn’s multiple comparison test. (d) Representative autoradiograms of in vitro MLCK activity shown as γ32P-ATP phosphorylation of recombinant MLC in the presence of PIK or small molecules. n = 3 biologically independent samples within this experiment, which is representative of 3 independent experiments. Mean ± SD. ***, P<0.001 by ANOVA with Dunn’s multiple comparison test. (e) Human aortic smooth muscle cells embedded in rat tail collagen were treated with 20 μM Y27632, 150 μM PIK, or 250 μM small molecule drugs. Gel contraction was quantified after 24 hrs. Data are representative of n = 4 biologically independent samples. Mean ± SD. *, P<0.05; **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (f) Wound recovery in confluent Caco-2BBe monolayers. Bar, 500 μm. Micrographs shown are of representative n = 5 biologically independent samples in each of 3 independent experiments. Mean ± SD. *, P<0.05; ***, P<0.001 by ANOVA with Dunn’s multiple comparison test. (g) Confluent Caco-2BBe monolayers treated with 150 μM PIK or 250 μM NSC55937 were stained for MLCK1 (green) and F-actin (red). Arrows within the images indicate the perijunctional actomyosin ring. Bar, 5 μm. The fraction of total MLCK1 localized to the PAMR is shown. For this experiment, which is representative of 4 independent studies, n=8 biologically independent samples with 3 – 5 fields analyzed for each condition. Mean ± SD. ***, P<0.001 by ANOVA with Dunn’s multiple comparison test.
Structural and sequence data from all IgCAMs of human and mouse MLCK1 were used to select a potential drug binding pocket that: i) was comprised of residues that are unique to IgCAM3 (Fig. 2a); ii) was conserved between mouse and human MLCK1 (Fig. 2b); and iii) had a suitable, surface accessible, hydrophobic core (Extended Data 1a). A library of ~140,000 molecules was docked in silico and the compounds with the lowest predicted binding energy were obtained for functional testing (Extended Data 1b).
As an initial, medium-throughput biological screen, the ability of small molecules to reverse physiological, MLCK-dependent tight junction permeability increases was assessed.17 Activation of Na+-glucose cotransport reduced transepithelial electrical resistance (TER), a sensitive measure of paracellular permeability, in Caco-2BBe monolayers (Fig. 2c). A highly-specific MLCK inhibitor, PIK,18 reversed the effects of Na+-glucose cotransport on barrier function. Small molecules with very low (left group) and intermediate (right group) predicted ΔG of binding to IgCAM3 were tested. Three molecules from the very low ΔG group, NSC31211, NSC159456, and NSC55937, but none from the intermediate ΔG group, increased TER of monolayers with active Na+-glucose cotransport to an extent similar to PIK (Fig. 2c, Supplementary Table 2). In addition to identifying three molecules that passed this screen, this result suggests that the ΔGs predicted by the in silico model are relevant in vitro.
Enzymatic MLCK inhibition could be a simple explanation for the effects of small molecules on TER. To evaluate this, effects on intestinal epithelial MLCK activity were tested in a cell-free assay. Several small molecules, including one of the three that increased TER (NSC159456) inhibited MLCK activity (Fig. 2d, Supplementary Table 2). Thus, of the small molecules that enhanced barrier function, only NSC31211 and NSC55937 passed this screen.
As inhibition of MLCK-dependent smooth muscle contraction, which is essential for normal gastrointestinal motility and regulation of vascular tone,19 would represent an unacceptable toxicity, the effect of small molecules on contraction of human smooth muscle cells was assessed. Consistent with the observed MLCK enzymatic inhibition, NSC42233 and NSC294786 limited contraction (Fig. 2e and Supplementary Table 2). NSC31211 and NSC48770, which did not inhibit MLCK enzymatic activity, also reduced smooth muscle-dependent gel contraction. NSC55937 was therefore the only small molecule that increased TER without inhibiting MLCK enzymatic activity or smooth muscle contraction (Supplementary Table 2). Further, NSC55937 did not interfere with epithelial wound closure (Fig. 2f), which requires intact MLCK function.
Consistent with our initial hypothesis that a small molecule bound to IgCAM3 might prevent MLCK1 recruitment, NSC55937 markedly reduced the PAMR-associated MLCK1 fraction (Fig. 2g). In contrast, MLCK enzymatic inhibition did not affect MLCK1 localization. Because NSC55937 diverted MLCK1 from the PAMR, it was designated Divertin.
To directly measure Divertin binding, we took advantage of the single tryptophan (W447) buried within IgCAM3 (Extended Data 2a). Divertin caused a dose-dependent red shift in peak tryptophan fluorescence (Extended Data 2b) and quenched overall fluorescence (Extended Data 2c), consistent with movement of W447 to a more polar environment. Mutation of Leu449, Gln457, and Asp481, which line the putative drug-binding pocket, eliminated Divertin-induced changes in tryptophan fluorescence (Extended Data 2d). These data indicate that Divertin binds specifically to MLCK1 IgCAM3 via an interaction at or near the putative drug-binding pocket.
Divertin reverses TNF-induced MLC phosphorylation and barrier loss in vitro
The data above suggest that Divertin-mediated inhibition of MLCK1 recruitment to the PAMR may be able to reduce MLCK-dependent MLC phosphorylation and prevent or reverse TNF-induced tight junction barrier defects. Consistent with this hypothesis, Divertin blocked TNF-induced MLCK1 recruitment to the PAMR (Fig. 3a,b). Moreover, Divertin restored barrier function in TNF-treated Caco-2BBe monolayers in a manner that was similar to enzymatic MLCK inhibition (Fig. 3c). The rapidity of this TER restoration strongly suggests that, consistent with previous work, the effect of Divertin reflects regulation of paracellular, rather than transcellular, transport. Finally, Divertin reversed TNF-induced MLC phosphorylation (Fig. 3d). Thus, although cell free assays showed that Divertin is not a direct inhibitor of MLCK enzymatic activity, it is as effective as an enzymatic inhibitor in correcting TNF-induced MLC phosphorylation and restoring barrier function. Together, these data suggest that Divertin acts by preventing MLCK1 from gaining the physical proximity required for phosphorylation of perijunctional MLC (Fig. 3e).
Figure 3. Divertin reverses acute TNF-induced MLC phosphorylation and barrier dysfunction in vitro.
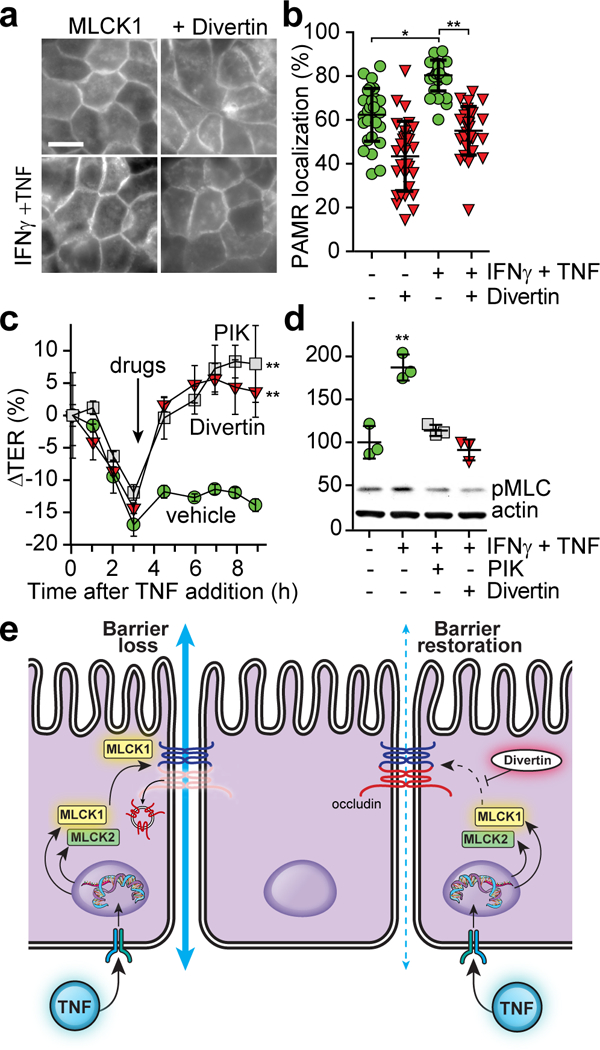
(a) Monolayers expressing MLCK1-EGFP were primed with IFNγ followed by TNF for 4 hrs (bottom row). 250 μM Divertin (NSC55937) was added for 1 hr prior to fixation (right column). Bar, 10 μm. (b) Colocalization of MLCK1-EGFP with F-actin for the monolayers shown in panel a. For this experiment, which is representative of 5 independent studies, n=8 biologically independent samples with 3 – 5 fields analyzed for each condition. Mean ± SD. *, P<0.05; **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (c) TER of IFNγ-primed monolayers treated with TNF for indicated times. After 3 hrs (arrow), vehicle, 150 μM PIK, or 150 μM Divertin was added to the apical media. For this experiment, which is representative of 3 independent studies, n=3 biologically independent samples for each condition. Mean ± SD. **, P<0.01 by repeated measures ANOVA with Bonferroni correction. (d) Immunoblots of phosphorylated MLC (pMLC) and actin from monolayers treated with IFNγ for 16 hrs and TNF for 4 hrs. 150 μM PIK or 150 μM Divertin were added at 3 hrs. For this experiment, which is representative of 3 independent studies, n=3 biologically independent samples for each condition. Mean ± SD. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (e) Proposed mechanism of Divertin action.
Divertin prevents acute TNF-induced MLC phosphorylation, barrier loss, and diarrhea in vivo
The in silico screen was designed to identify small molecules capable of interacting with both human and mouse MLCK1 to allow analysis of Divertin efficacy in vivo. Mouse jejunal segments were perfused in situ as described previously (Fig. 4a).2,20 Intraperitoneal (i.p.) TNF administration reversed the direction of fluid flow to net secretion (Fig. 4b), induced barrier loss (Fig. 4c), and enhanced intestinal epithelial MLC phosphorylation (Fig. 4d). Addition of Divertin to the perfusate in a manner topologically analogous to oral administration restored fluid absorption, prevented barrier loss, and blocked TNF-induced increases in MLC phosphorylation. Divertin did not interfere with basal fluid transport or barrier function in the absence of TNF, suggesting that it is unlikely to negatively impact epithelial homeostasis or intestinal transport under basal conditions.
Figure 4. Divertin prevents acute TNF-induced barrier dysfunction in mouse jejunum in vivo and human jejunal mucosae ex vivo.
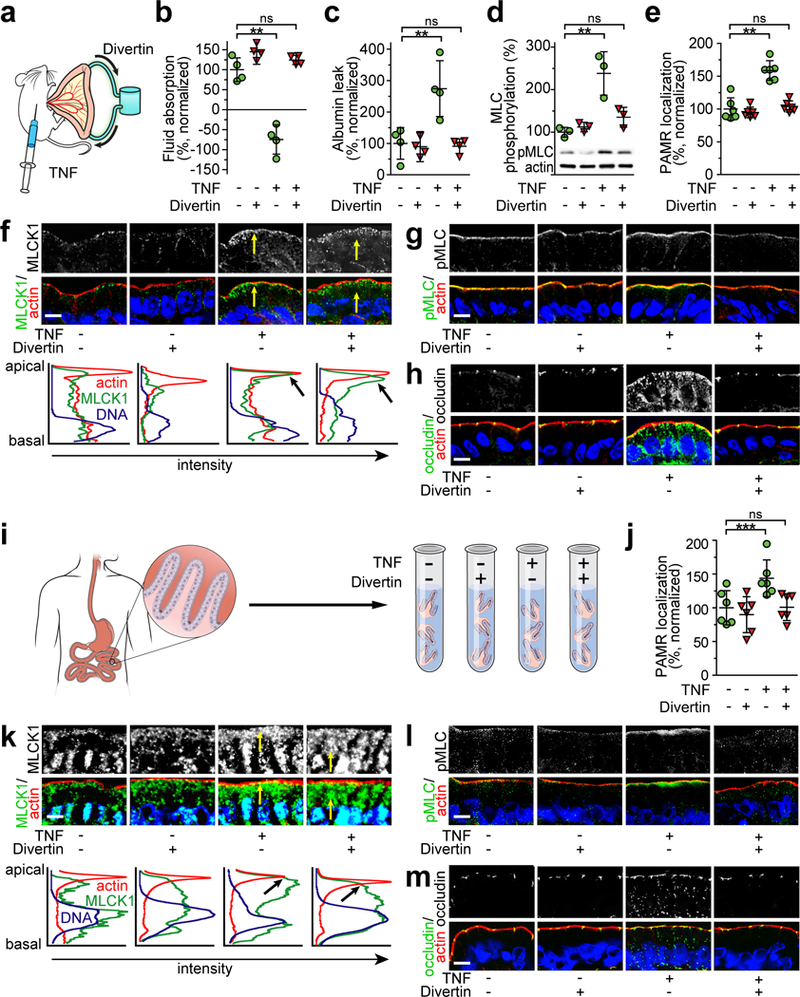
(a) Mice were injected with vehicle or 5 μg TNF and jejunal loops were perfused with either saline containing vehicle or Divertin (150 μM). (b) TNF induces net fluid secretion, but Divertin restores absorption. n=4 biologically independent samples for each condition. Mean ± SD. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (c) TNF-induced increases in albumin leak from bloodstream to gut lumen are blocked by Divertin. n=4 biologically independent samples for each condition. Mean ± SD. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (d) Divertin blocks TNF-induced MLC phosphorylation. n = 3 biologically independent samples for each condition in this experiment, which is representative of 3 independent experiments. Mean ± SD. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (e) Divertin blocks acute, TNF-induced MLCK1 recruitment to the PAMR. n = 2 fields in each of 3 independent mice for each condition in this experiment, which is representative of 3 independent experiments. Mean ± SD. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (f) Micrographs showing that Divertin blocks TNF-induced MLCK1 (green) recruitment to the PAMR. F-actin (red), nuclei (blue). Images are representative of 9 independent mice (3 in each of 3 experiments) per condition. Bar, 10 μm. Line scans show the intensity of the staining from the apical to the basal part of the cell. Arrows in the line scan graphs indicate colocalization of MLCK1 with the PAMR in TNF-treated mice. n = 2 fields in each of 3 independent mice for each condition in this experiment, which is representative of 3 independent experiments. Mean ± SD. (g) Divertin blocks TNF-induced increases in MLC phosphorylation (green) within the PAMR. F-actin (red), nuclei (blue). Bar, 10 μm. Images are representative of 9 independent mice (3 in each of 3 experiments) per condition. (h) Divertin blocks TNF-induced internalization of occludin (green). F-actin (red), nuclei (blue). Bar, 10 μm. Images are representative of 9 independent mice (3 in each of 3 experiments) per condition. (i) Human jejunal mucosal biopsies were incubated ex vivo with TNF and/or Divertin. (j) Divertin blocks TNF-induced MLCK1 recruitment to the PAMR but does not block TNF-induced increases in MLCK1 expression. n = 2 fields in each of 3 biologically independent specimens for each condition in this experiment, which is representative of 3 independent experiments. Mean ± SD. **, P<0.01 by ANOVA with Dunn’s multiple comparison test. (k) Divertin blocks TNF-induced MLCK1 (green) recruitment to the PAMR. F-actin (red), nuclei (blue). Bar, 10 μm. Images are representative of 9 biologically independent specimens (3 in each of 3 experiments) per condition. Line scans show the intensity of the staining from the apical to the basal part of the cell. n = 2 fields in each of 3 biologically independent specimens for each condition in this experiment, which is representative of 3 independent experiments. **, P<0.01. (l) Divertin prevents TNF-induced phosphorylation of PAMR-associated MLC (green). F-actin (red), and nuclei (blue). Bar, 10 μm. Images are representative of 9 biologically independent specimens (3 in each of 3 experiments) per condition. (m) Divertin blocks TNF-induced occludin internalization (green). F-actin (red), nuclei (blue). Bar, 10 μm. Images are representative of 9 biologically independent specimens (3 in each of 3 experiments) per condition.
Divertin prevented TNF-induced recruitment of MLCK1 to the PAMR (Fig. 4e,f), although it did not interfere with TNF-induced upregulation of MLCK1 expression.21 Divertin also prevented TNF-induced phosphorylation of perijunctional MLC (Fig. 4g). Previous work has shown that caveolar endocytosis of the tight junction protein occludin is both required for and is a robust marker of TNF-induced MLC phosphorylation-mediated barrier regulation.20 Consistent with this, Divertin prevented TNF-induced occludin endocytosis (Fig. 4h).20 These data indicate that Divertin-mediated disruption of MLCK1 recruitment can prevent TNF-induced tight junction barrier loss and diarrhea in vivo.
Divertin prevents TNF-induced MLCK1 trafficking, MLC phosphorylation, and tight junction reorganization in human jejunal mucosae ex vivo
To determine if Divertin is also effective in human jejunum, mucosal biopsies from normal human subjects were treated with TNF and Divertin (Fig. 4i). As in intact mouse jejunum, Divertin prevented TNF-induced MLCK1 recruitment to the PAMR in human jejunal enterocytes (Figs. 4j,k). Divertin did not, however, interfere with TNF-induced upregulation of MLCK1 expression (Fig. 4k), indicating that Divertin did not simply block TNF signaling within intestinal epithelia. Divertin also limited TNF-induced perijunctional MLC phosphorylation (Fig. 4l). Although barrier function was not measured directly, the ability of Divertin to inhibit endocytosis of tight junction-associated occludin (Fig. 4m) is strong evidence that TNF-induced barrier loss was prevented or reversed.20 Thus, similar to intact mouse jejunum, Divertin prevents TNF-induced MLCK1 trafficking, MLC phosphorylation, and occludin endocytosis in human jejunal epithelia.
Divertin does not have significant toxicity
The analyses above, in which intestinal epithelial cells were directly exposed to Divertin, show that this small molecule protects human and mouse cells and tissues from acute TNF-induced MLCK1 trafficking, MLC phosphorylation, occludin endocytosis, and barrier dysfunction in vitro, in vivo, and ex vivo. In order to determine whether Divertin could be therapeutically effective in chronic disease, we first assessed the potential toxicity of systemic administration. A previous analysis in leukemia showed that daily i.p. injections of 50 mg/kg or 100 mg/kg, for nine or three days, respectively, had no effect on survival after 30 days.22 Consistent with this, we found that daily i.p injections of 12.5 mg/kg or 25 mg/kg were without toxicity, in terms of weight loss or behavior, over 31 days (Fig. 5a). Daily 50 mg/kg doses did not affect survival or behavior but did induce weight loss (data not shown).
Figure 5. Divertin corrects increased intestinal permeability in IL-10 knockout mice without significant systemic, mucosal, or epithelial toxicities.
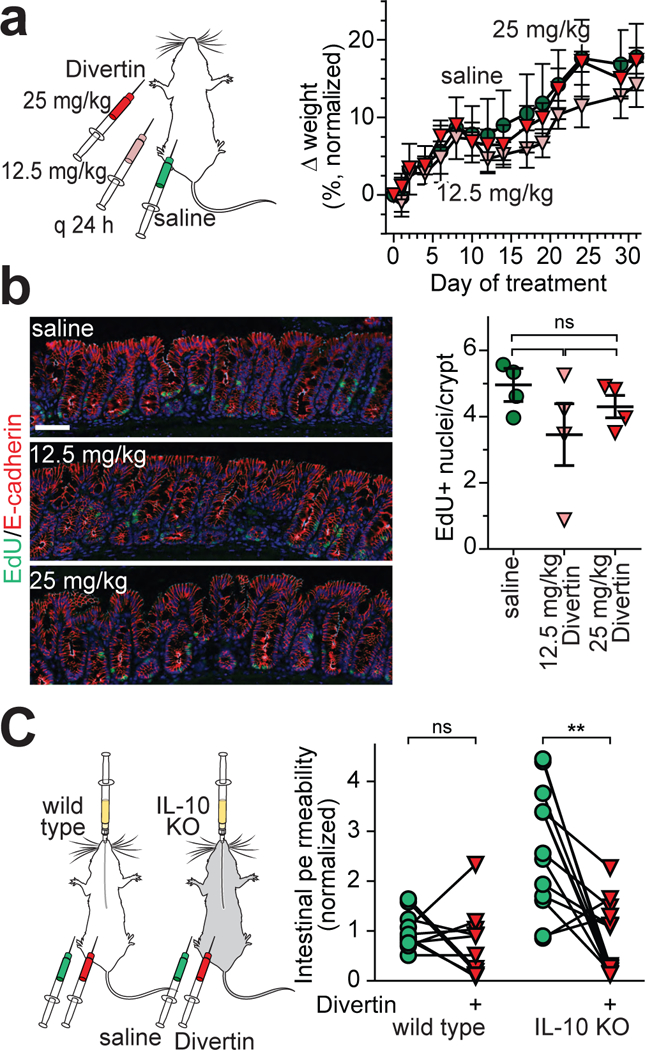
(a) Wild type mice were injected i.p. daily with saline (vehicle) or Divertin at 12.5 mg/kg or 25 mg/kg. Body weight changes, normalized to initial body weight, are shown. For this experiment, which is representative of 3 independent studies, n=5 biologically independent samples for each condition. Mean ± SD. (b) Mice were injected with EdU (green) 16 hours before sacrifice. E-cadherin (red) is shown for orientation. Each point represents the mean of 5 microscopic fields from a single mouse. Mean ± SD. n = 4 mice per condition in this representative experiment. No groups were significantly different from any other group (ANOVA with Dunn’s multiple comparison test). (c) Wild type or IL-10 knockout mice were gavaged with fluorescein and injected i.p. with vehicle. Serum fluorescein recovery at 3 hrs was used as a measure of intestinal permeability. The same mice were injected i.p. with a single 25 mg/kg dose of Divertin and gavaged with fluorescein one week later. Lines connect data from individual mice without (green) or with (red) Divertin treatment. Data are normalized to the mean of healthy wild type mice. n = 8 (wild type), 11 (IL-10 knockout). **, P<0.01 by two-sided paired t-test.
To better characterize potential toxicities, histopathology and epithelial turnover were assessed in small intestine and colon after Divertin treatment. No Divertin-induced histopathology was evident on H&E-stained sections. Moreover, Divertin had no effect on epithelial turnover, as numbers of EdU-labelled cells were similar in small intestine and colon (Fig. 5b) of mice treated with 12.5 mg/kg/day or 25 mg/kg/day of Divertin for 31 days. Thus, Divertin does not appear to have systemic, mucosal, or epithelial toxicities over this relatively short period of exposure.
Divertin limits development of experimental inflammatory bowel disease
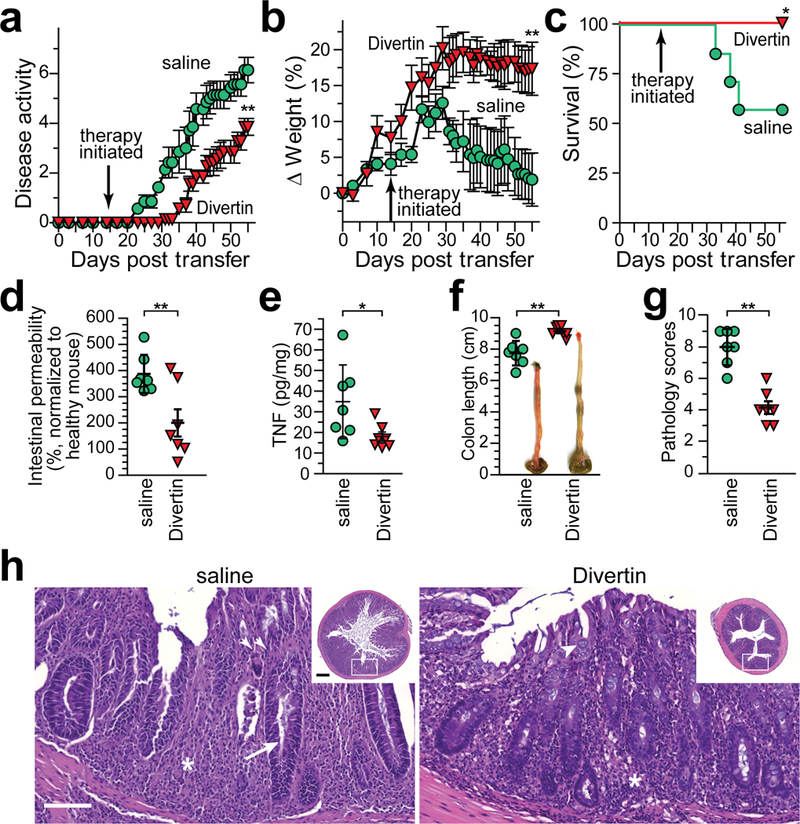
The data above indicate that Divertin can prevent acute inflammatory diarrhea when given prior to disease onset and is not toxic when given daily at a dose of 25 mg/kg. To determine if Divertin is also able to prevent initiation and subsequent progression of chronic disease, we used a T cell transfer model. We have previously reported that intestinal epithelial MLCK activation accelerates disease progression and, conversely, intestinal epithelial MLCK knockout delays onset and lessens disease severity in this IBD model.10,23 Mice were treated with daily i.p. injections of Divertin or saline (vehicle) beginning 14 days after adoptive transfer, prior to onset of symptoms (Extended Data 3a). Mice treated with saline developed disease activity and began to lose weight one week later (day 21), but this was delayed by nearly two weeks, to day 33, in mice that received Divertin (Extended Data 3a, b). Divertin also prevented mortality in this colitis model (Extended Data 3c).
As expected, disease progression in saline-treated mice was associated with increased intestinal permeability to 4 kDa dextran (28 Å diameter), but barrier function was largely preserved in Divertin-treated mice (Extended Data 3d). Consistent with reduced disease, mucosal TNF production was also lower in Divertin-treated mice (Extended Data 3e). Finally, the efficacy of Divertin in preventing disease development was evident on gross, i.e. colonic shortening and thickening (Extended Data 3f), and microscopic (Extended Data 3g, h) examination. Divertin is therefore able to limit initial development of experimental IBD.
Divertin acutely restores barrier function in spontaneous immune-mediated colitis
We used the IL-10 knockout mouse model of spontaneous colitis to determine whether Divertin could restore intestinal barrier function in chronic disease.24,25 Intestinal permeability was assessed in IL-10 knockout mice with mild disease and re-assessed one week later in the same mice following a single 25 mg/kg dose of Divertin. Intestinal permeability to fluorescein, a sensitive marker of tight junction barrier loss due to its small size (hydrodynamic diameter = 8 Å), in IL-10 knockout mice was 2.9±0.3-fold that of wild type controls, consistent with the presence of disease (Fig. 5c). The recognized variability of disease severity in IL-10 knockout mice is reflected in the significantly greater range of intestinal permeability in IL-10 knockout mice relative to wild type controls (Fig. 5c). Divertin collapsed the range of permeabilities and reduced absolute fluorescein permeabilities in IL-10 knockout mice such that they were similar to wild type mice (Fig. 5c). Overall, Divertin reduced intestinal permeability in 9 of 11 IL-10 knockout mice but only 4 of 8 wild type mice (Fig. 5c). These data indicate that Divertin can restore intestinal barrier function in a mechanistically-relevant inflammatory bowel disease model.
Divertin limits severity of experimental, chronic inflammatory bowel disease
The disease variability inherent in IL-10 knockout mice makes it difficult to use this model to test the ability of Divertin as therapy for established disease. We therefore returned to the T cell transfer model (Fig. 6a), in which disease is relatively uniform in each mouse. Moreover, like human disease, this model is responsive to anti-TNF therapy.14 By day 18 after transfer, mice that received T cells all displayed softened stool, weight loss, and modest increases in clinical scores. These mice were randomized into four treatment groups. Separate groups of mice received saline or Divertin beginning on day 19 after T cell transfer. These were compared to mice treated with anti-TNF antibodies, which are effective in T cell transfer colitis.14 We also asked if combination therapy with Divertin and anti-TNF provided any benefit relative to each agent alone.
Figure 6. Divertin corrects intestinal permeability defects and limits progression of experimental inflammatory bowel disease.
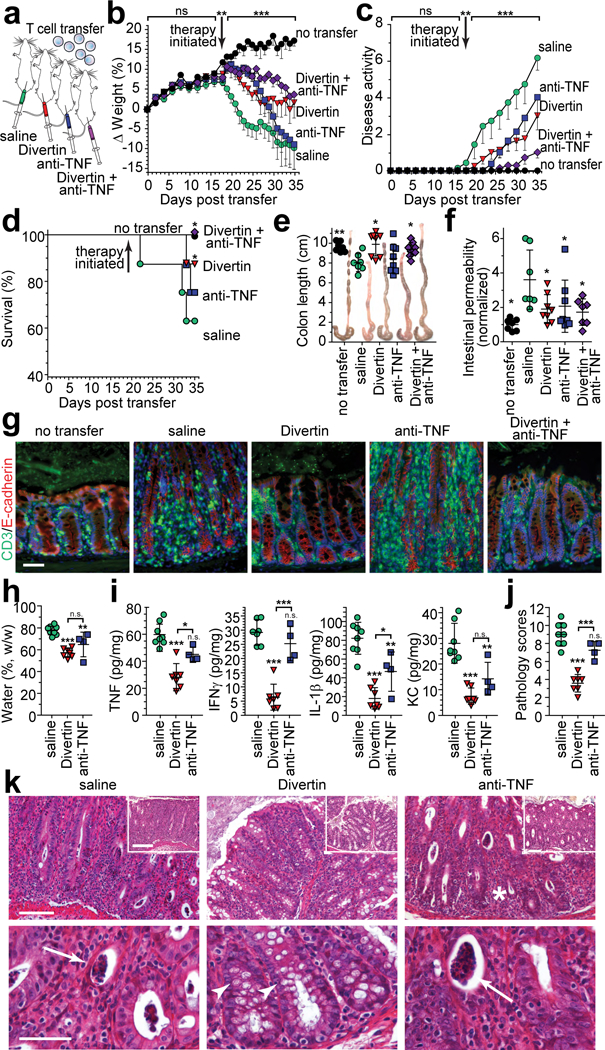
(a) Immunodeficient mice received naïve CD4+ effector T cells. Therapy by daily i.p. injection with saline (vehicle), Divertin, anti-TNF, or combined Divertin and anti-TNF was initiated after definitive features of disease were present (day 19). (b – g) Female mice were used in this experiment that included n = 7 mice without T cell transfer and n = 8 mice in each of the other 4 groups. (b, c) Normalized weight data and disease activity scores. Mean, SD. **, P<0.01 by two-tailed t test for no transfer vs. all other mice at day 18. ***, P<0.001 by ANOVA with Tukey’s multiple comparison test over the interval from 19 −35 days. (d) Survival at day 35. *, P<0.05, vs. saline-treated mice, by Gehan-Breslow-Wilcoxon test. (e, f) Colon length. Mean ± SD. *, P<0.05; **, P<0.01, vs. saline-treated mice, by ANOVA with Newman-Keuls multiple comparison test. (f) Intestinal permeability to FITC-4 kDa dextran. Data are normalized to mice that did not receive T cell transfer. Mean ± SD. *, P<0.05, vs. saline-treated mice, by ANOVA with Newman-Keuls multiple comparison test. (g) Colonic mucosa immunostained for CD3 (green) and E-cadherin (red). Bar, 50 μm. Images are representative of two independent experiments. (h - k) Male mice were used in this experiment that included n = 10 saline-treated mice and n = 7 in each of the other groups. Weight loss, disease activity, and survival were similar to the experiment using female mice (above, b – g). (h) Fecal water content of male mice after T cell transfer (from male donors; fecal water was 53% ± 4% for mice that did not receive T cell transfer and 66% ± 8% at day 18 for mice that received T cells). Mean ± SD. **, P<0.01; ***, P<0.001 by ANOVA with Bonferroni correction. (i) Indicated cytokines were determined by ELISA in distal colonic homogenates. Mean ± SD. n = 8 for saline-treated mice (n = 7 for IFNγ and IL-1β); n = 7 for Divertin-treated mice; n = 4 for anti-TNF-treated mice. *, P<0.05; **, P<0.01; ***, P<0.001 by ANOVA with Bonferroni correction. Mean cytokine levels of mice that did not receive T cell transfer or were sacrificed at 18 days after transfer were, respectively: TNF 11 pg/mg and 37 pg/mg; IFNγ undetectable and 17 pg/mg; IL-1β undetectable and 58 pg/mg; KC 2.5 pg/mg and 16 pg/mg. (j) Colonic histopathology on day 35. Mean ± SD. n = 8 for saline-treated mice; n = 7 for Divertin-treated mice; n = 4 for anti-TNF-treated mice. ***, P<0.001 by ANOVA with Bonferroni correction. Pathology scores of mice that did not receive T cell transfer or were sacrificed at 18 days after transfer were 0 and 5.5, respectively. (k) Representative histopathology of mice treated with saline (vehicle), Divertin, or anti-TNF (arrows indicate crypt abscesses). Note the goblet cell preservation in Divertin-treated mice (arrowheads). Bars, 100 μm (upper row); 200 μm (insets); 50 μm (lower row). Images are representative of 2 independent experiments with similar results.
Divertin, anti-TNF, and the combination of Divertin and anti-TNF each limited weight loss over the first week of treatment (Fig. 6b). Thereafter, weight loss accelerated in anti-TNF-treated mice and by day 35 weights of mice receiving anti-TNF or vehicle (saline) were comparable (Fig. 6b). While all three drug treatments also significantly limited disease activity, combination treatment was better than either drug alone (Fig. 6c). This trend was mirrored by survival, as combination treatment was the only intervention that resulted in 100% survival at day 35. Divertin, but not anti-TNF, treatment also significantly improved survival relative to mice that received only saline (Fig. 6d).
Similar to survival, Divertin or combination treatment, but not anti-TNF alone, significantly prevented colonic shortening and thickening (Fig. 6e). Nevertheless, all three treatments resulted in significant preservation of intestinal barrier function as assessed by 4 kDa dextran (28 Å diameter) permeability (Fig. 6f). Thus, most analyses showed Divertin to be modestly more effective than anti-TNF. Combination treatment was superior to either alone in preventing progression of clinically-evident disease activity, but, by all other measures, addition of anti-TNF provided no benefit relative to Divertin alone. In a manner that correlated directly with survival, colonic mucosal T cell infiltration in mice treated with saline or anti-TNF was markedly greater than that in mice that received Divertin alone or combination treatment (Fig. 6g).
Both Divertin and anti-TNF treatments significantly reduced fecal water content, i.e. diarrhea (Fig. 6h), but the effect of Divertin was not significantly different than that of anti-TNF. Consistent with this, colonic TNF, IFNγ, IL-1β, and KC (IL-8) production were significantly reduced in Divertin-treated, relative to saline-treated, mice. Anti-TNF treatment, in contrast, reduced only IL-1β and KC (Fig. 6i). Divertin was also more effective than anti-TNF in limiting histopathologic features of disease (Fig. 6j). Crypt loss (asterisks), crypt abscesses (arrows), goblet cell (mucin) depletion, dense lamina propria lymphoplasmacytic infiltrates, and mucosal hyperplasia were not substantially affected by anti-TNF treatment (Fig. 6k). Crypt abscesses were uncommon in Divertin-treated mice and mucin-laden goblet cells (arrowheads) were preserved. Moreover, although some crypt elongation was present in Divertin-treated mice, overall mucosal hyperplasia was substantially reduced relative to saline and anti-TNF-treated mice (Fig. 6j, k). Thus, consistent with attenuation of weight loss, colonic shortening, and TNF and IFNγ production, Divertin, but not anti-TNF, significantly reduced histopathologic features of colitis. Overall, these data indicate that Divertin can both reduce severity and limit progression of experimental IBD.
DISCUSSION
Epithelial barrier loss is a critical component of acute and chronic gastrointestinal diseases, including infectious enterocolitis, food allergy, celiac disease, and IBD.26 This relationship is further emphasized by the observations that intestinal epithelial barrier loss is linked to IBD risk alleles27–29 and that barrier loss during remission is a prognostic marker of disease reactivation in Crohn’s disease.30 Experimental models further show that intestinal epithelial barrier dysfunction precedes the onset of enterocolitis,31 that increased intestinal permeability enhances IBD progression,23,32 and that barrier restoration limits IBD pathogenesis.23,31
Although the molecular mechanisms that trigger intestinal permeability defects are incompletely defined, non-muscle MLCK activation is a convergence point for many pathophysiologic stimuli. MLCK is also implicated in barrier dysfunction in other organ systems and in vascular endothelia, and, therefore, represents a potential therapeutic target. However, the catalytic domains of non-muscle and smooth muscle MLCK are identical; thus enzymatic inhibitors that target one will target both. This would be a source of substantial toxicity, as smooth muscle MLCK inhibition results in hypotension, intestinal obstruction, and death.19 Further, the most widely-used non-muscle and smooth muscle MLCK inhibitors, ML-7 and ML-9, inhibit many other kinases, including cardiac and skeletal muscle MLCK, ROCK-II, AMPK, and DYRK1A, at concentrations that fail to inhibit smooth and non-muscle MLCK completely.33 Together with recognition that MLCK serves critical roles in epithelial migration and wound repair, among other functions, enzymatic MLCK inhibition is unlikely to be achievable without unacceptable toxicities.
Here, we demonstrate an alternative therapeutic strategy for preventing MLCK-dependent barrier dysregulation in acute and chronic intestinal disease. We show that a specific long MLCK splice variant, MLCK1, is recruited to the PAMR where it mediates TNF-induced intestinal epithelial tight junction regulation. Taking advantage of the unique IgCAM3 within MLCK1, we screened for compounds that bind to this domain. Secondary screens identified molecules that could enhance barrier function and eliminated those that inhibited MLCK enzymatic activity or smooth muscle contraction. One small molecule, termed Divertin, binds to this unique IgCAM domain and prevents MLCK1 recruitment to the PAMR without interfering with epithelial migration and wound repair.
To assess the effects of Divertin in intact tissue, we used an established in vivo mouse model of TNF-induced, MLCK-dependent diarrhea2,20,34 and acute TNF-induced MLCK activation leading to tight junction reorganization in human jejunal mucosae ex vivo. In both tissues, Divertin prevented MLCK1 recruitment to the PAMR and subsequent phosphorylation of perijunctional MLC. Further, Divertin prevented TNF-induced diarrhea and restored net fluid absorption in mouse jejunum in vivo. This functional preservation correlated directly with maintenance of occludin at the tight junction pools, i.e. inhibition of endocytosis. These data confirm the necessity of depleting occludin from the tight junction to drive TNF-induced barrier loss and diarrhea; previous studies have shown that occludin overexpression, which augments tight junction occludin pools, or direct, genetic or pharmacologic, inhibition of occludin endocytosis limits TNF-induced barrier loss and diarrhea in mice.20 Thus, while we were unable to measure transport, the efficacy of Divertin in blocking occludin endocytosis in human intestinal biopsies can be taken as de facto evidence that paracellular barrier function and transepithelial fluid transport were preserved. Thus, Divertin is equally effective in an in vivo mouse model and an ex vivo human model of acute, TNF-induced tight junction barrier loss. Moreover, unlike the in vitro studies of cultured monolayers, these data indicate that Divertin has no effect on epithelial morphology, tight junction structure, barrier function, or fluid transport in the absence of stimuli but can prevent development of TNF-induced barrier loss. Finally, when given prior to clinical presentation, Divertin limited experimental IBD development. These data indicate that Divertin might be safely used as a prophylactic therapy, for example as a maintenance therapy in IBD.
In vitro, Divertin reversed both morphological and functional sequelae of acute TNF-induced MLCK activation, including perijunctional MLC phosphorylation, tight junction reorganization, and barrier loss. Divertin had a similar effect in vivo, where a single dose was able to restore intestinal barrier function in colitic IL-10 knockout mice. These data suggest that continuous Divertin treatment might be effective in established disease. To assess this, mice received T cell transfer, but therapy was not initiated until day 19, after weight loss, disease activity, cytokine production, and histopathologic features of experimental IBD were established. In this model, Divertin was compared directly to treatment with anti-TNF antibodies, which have revolutionized therapy in human IBD. Consistent with previous work,14 anti-TNF provided only partial protection from T cell transfer colitis-associated weight loss and mortality. In contrast, however, Divertin limited weight loss and mortality. Thus, by these measures, Divertin was superior to anti-TNF. Because they have different mechanisms of action, we hypothesized that Divertin and anti-TNF might have additive effects. However, combination therapy was not significantly superior to Divertin alone by any measure.
The efficacy with which Divertin prevents barrier loss and restores function suggests that it may also be useful in other diseases with epithelial and vascular endothelial tight junctions dysfunction, including celiac disease,29,35 atopic dermatitis,36 pulmonary infection and acute respiratory distress syndrome,37,38 graft versus host disease,39 and multiple sclerosis.40,41 Moreover, the molecularly-targeted approach defined here may have much broader relevance, as recruitment to specific subcellular locations is essential for function of proteins involved in diverse cellular activities including mitosis, membrane traffic, cytoskeletal regulation, and ion transport.42–44 Disruption of site-specific recruitment may allow certain processes to be blocked without affecting other essential functions. Our characterization of Divertin therefore demonstrates a novel means of enhancing therapeutic specificity without many of the toxicities that often plague less precisely-targeted inhibitors.
In summary, we have shown that the long MLCK splice variant MLCK1 is recruited to the intestinal epithelial tight junction in response to inflammatory stimuli; used a structure-based drug discovery approach to prevent MLCK1 recruitment; and identified a small molecule that blocks recruitment, corrects acute inflammatory barrier loss and diarrhea, and prevents chronic immune-mediated colitis. In addition to the potential of this specific agent, this approach to kinase inhibition by limiting substrate accessibility, rather than enzymatic activity, represents a new paradigm for enhancing specificity of small molecule therapies.
METHODS
Cloning and vector construction.
The human MLCK1 IgCAM3 domain sequence was selected based on the previously solved structure of IgCAM9, also known as telokin (PDB code 1tlk).16 Oligonucleotides were designed based on the human MLCK1 cDNA sequence (GenBank accession No. AY424270): forward, 5’-ATGGAGGGCCAGAGGGATTCA-3’, and reverse, 5’-CTATTCCACTTGGAGGGTCCAGCTACAG-3’. These primers correspond to nucleotides 1215–1517 of the cDNA sequence. In addition to the native MLCK1 cDNA sequence, the reverse primer included an artificial stop codon (in antisense) at its 5’ end. The final peptide encoded by the PCR amplicon represents the entire IgCAM3 domain of MLCK1, i.e. amino acids 405–506 of the complete protein (GenBank No. AAR29062). The PCR products were ligated into pETBlue-1 (Novagen) and sequenced for confirmation. Amino acid substitution to create mutant IgCAM3 (IgCAM3 Leu449Arg, Gln457Lys, Asp481Val) was done by mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene). Oligonucleotides were Leu449Arg sense 5′- TGAAGTGGCCTGGTTCAGAGAAGGCACCCCCGTGA −3′, Leu449Arg antisense 5′- TCACGGGGGTGCCTTCTCTGAACCAGGCCACTTCA −3′, Gln457Lys sense 5′- CCCCCGTGAGGAGAAAGGAAGGCAGCATTG −3′, Gln457Lys antisense 5′- CAATGCTGCCTTCCTTTCTCCTCACGGGGG −3′, Asp481Val sense 5′- GAAAGCCCGGACCAGGGTAAGTGGGACATACAGCTG −3′, Asp481Val antisense 5′- CAGCTGTATGTCCCACTTACCCTGGTCCGGGCTTTC −3′. MLCK1-EGFP and MLCK2-EGFP fusion protein constructs were generated using MLCK1 (AY424270.1) and MLCK2 (AY424269.1) cDNA cloned from Caco-2BBe and fused at the C-terminus to monomeric EGFP (after removing the stop codon). These were cloned into piggyBAC-TREtight plasmids and stably-expressed in Caco-2BBe cells, as described.45 All constructs were verified by direct sequencing.
Recombinant protein expression and purification.
Constructs were transformed into BL21-CodonPlus (DE3)-RIPL Competent E. coli (Stratagene) for IPTG-induced expression. Cultures were grown to an OD600 of 0.5–0.9 at 37°C and IPTG was added to a final concentration of 1 mM. Cultures were grown for an additional 3 hrs at 37°C. After induction, bacteria were collected by centrifugation (6000 x g), lysed (50 mM Tris pH 7.5, 150 mM NaCl, 0.05% NP-40) and sonicated. SDS-PAGE confirmed expression. Solubilized protein was initially passed through a series of centrifugal filtration devices (Amicon) in order to concentrate proteins in the 3–30 kDa size range. These preparations were further purified through Bio-Gel P-30 size-exclusion column (1.5 × 50 cm) chromatography using a BioLogic Lp chromatography system (Bio-Rad). The Bio-Gel column was equilibrated with 10 mM Tris pH 8.0, 50 mM NaCl and the protein sample was fractionated at a rate of 1 ml min-1. SDS-PAGE confirmed the positive fractions and these fractions were pooled and concentrated to 2 mg/ml with a purity of >95% using centrifugal filtration.
Crystallization and data collection
The purified MLCK1 IgCAM3 domain was concentrated to 6 mg/mL. Crystals of MLCK1 IgCAM3 domain were obtained from the Nextal PEGS screen condition H7 (200 mM ammonium dihydrogen phosphate, 20% PEG3350) at 4°C. Crystals were mounted in a nylon loop and frozen in liquid nitrogen. Data were collected at The Advanced Light Source, beamline 5.02, wavelength 1.0Å, at 100°K. The X-ray structure was determined by molecular replacement using MrBump46 as implemented in the CCP4 7.0.048 program suite.47 Refinement and model building were performed with BUSTER 2.11.748 and COOT 0.8.8,49 respectively, using data to 2.5Å, with 98.2% in preferred and allowed regions of the Ramachandran plot. Data collection and refinement statistics appear in Supplementary Table 1.
Molecular docking and acquisition of potential IgCAM3 interacting molecules.
The NCI Developmental Therapeutics Program chemical library of was filtered for compounds that met the Lipinski rules. The three-dimensional coordinates for the NCI/DTP set of 139,735 plated compounds was obtained from ZINC.50 All docking calculations were performed with DOCK6 (University of California, San Francisco) and run by parallel processing on 16 CPU of a Linux cluster at the University of Florida High Performance Computing Center. The general features of DOCK include rigid orienting of ligands to receptor spheres, AMBER energy scoring, GB/SA solvation scoring, contact scoring, internal nonbonded energy scoring, ligand flexibility, and both rigid and torsional simplex minimization. To prepare the site for docking, all water molecules were removed. Protonation of receptor residues was performed with DockPrep in Chimera (University of California, San Francisco). The structure was explored using sets of spheres to describe potential binding pockets using SPHGEN_CPP. The number of orientations per molecule was 100. Intermolecular AMBER energy scoring (vdw + columbic), contact scoring, and bump filtering were implemented in DOCK6. Chimera was used to generate molecular graphic images. Top scoring nonproprietary compounds, designated compounds were obtained from the Developmental Therapeutics Program, National Cancer Institute, National Institutes of Health. Each compound was solubilized in DMSO at 10 mM and stored at −20 °C.
Synthesis of Divertin.
An aqueous formalin solution (0.2 mol, 37% w/v) was added dropwise to hydrazine (0.75 equiv, 0.15 mol) at 0o C. Stirring was continued for 20 min at room temperature, and then the reaction mixture was left to stand at room temperature for 3 days. The crude product was filtered and washed with hot propan-2-ol to yield the product, octahydro-[1,2,4,5]tetrazino[1,2-a][1,2,4,5]tetrazine, as a white powder. Yield, 18%. CHN Analysis: C 32.70 %, H 8.28 %, N 56.60 %. 1H NMR (500 MHz, D2O): 3.61 (d, J = 11.5 Hz, 2H), 3.32 (d, J = 11.5 Hz, 2H).
Epithelial cell culture and TER measurement.
Caco-2BBe cells were grown as monolayers on collagen-coated polycarbonate membrane Transwell supports (Corning-Costar, Acton, MA) and used 17–20 days after confluence, as described previously.17. Electrophysiological measurements were made using agar bridges and Ag-AgCl calomel electrodes. A 50-μA current was passed across the monolayers using a model 558 voltage clamp (Univ. of Iowa Bioengineering, Iowa City, IA) to measure potential differences before and during application of a 50-μA current. Transepithelial short-circuit current (Isc) and transepithelial resistance (TER) were calculated with Ohm’s law, as described.17
For drug screens (Fig. 2c), it was not possible to test all drugs in a single experiment. All trials included monolayers with active Na+-glucose cotransport (in which MLCK is active)17,51, inhibited Na+-glucose cotransport (which blocks MLCK activation and typically produces an ~20% increase in TER)17,52. After measuring TER, small molecules (250 μM) were added to the apical surface (in the presence of active Na+-glucose cotransport) and TER measured after 60 minutes. TER values for each monolayer after drug treatment were normalized to those before drug addition. To control for differences in monolayer responsivity between experiments, a scale was developed using monolayers with active Na+-glucose cotransport (set to 0%) and inhibited Na+-glucose cotransport (set to 100%) for two-point calibration. The enzymatic MLCK inhibitor PIK (which increases TER similarly to inhibition of Na+-glucose cotransport at the 150 μM dose used) was included in each experiment. Scaled values shown for active and inhibited Na+-glucose cotransport, PIK treatment, and drug treatments are taken from multiple experiments.
For experiments examining cytokine-treated monolayers (Fig. 3c), IFN-γ (10 ng/ml; Peprotech, Rocky Hill, NJ) was added to the basal chamber 16 hours before TNF addition in order to induce TNF receptor expression.13 Preliminary experiments showed that this did not affect TER. TER was then measured and TNF (1 or 2.5 ng/ml; Peprotech) was added to the basal chamber (at t=0) without manipulating the apical media, as described.13,53 Drugs were added to the apical chamber only. TER of each monolayer was normalized, at each time point, to the starting TER (before TNF addition) of that monolayer.
Tryptophan fluorescence.
N-acetyl-tryptophanamide (NATA), recombinant IgCAM3, or mutant IgCAM3 was added to phosphate buffer (10 mM potassium phosphate, pH 6.7, 130 mM NaCl) in a quartz cuvette. Samples were then analyzed on a FluoroMax-3 scanning fluorimeter at 25 °C using an excitation wavelength of 296 nm (1-nm bandwidth) and recording the emission spectra from 315 to 400 nm.
Animals.
All experiments were performed in an Association of Assessment and Accreditation of Laboratory Animal Care–accredited facility under protocols approved by The University of Chicago, Brigham and Women’s Hospital, Boston Children’s Hospital, and Soochow University Animal Care and Use Committees. All studies are in compliance with all relevant ethical regulations. For acute studies, 7–10-wk-old C57BL/6 mice (Jackson Laboratory) received i.p. injections of vehicle or 5 μg recombinant mouse TNF (Peprotech) as described previously.20 In vivo perfusion analyses were performed as described previously.2,20 Sections of jejunum were harvested and snap-frozen in optimal cutting temperature (OCT) media.
129S1/SvImJ and 129(B6)-Il10tm1Cgn/J mice (Jackson Laboratory) were used as controls and IL-10 knockouts, respectively, at 7–10-weeks of age. Mice received i.p. injections of vehicle or Divertin (25 mg/kg) followed by gavage with 250 μl of 10 mg/ml fluorescein or 80 mg/ml FITC-4 kDa dextran solution. Mice were bled from the tail vein 3 hours later and recovery was assessed using a fluorescent platereader. For analysis of epithelial turnover, mice received 0.1 mg 5-ethynyl-2’-deoxyuridine (EdU)54 16 hrs before sacrifice.
Adoptive transfer colitis was established in 6-wk-old C57BL/6J Rag1−/− or C57BL/6J Rag1−/− xIl10rb−/− mice (Jackson Laboratory) by i.v. injection of 500,000 CD4+C25- or CD4+CD45RBhi T cells. Disease activity was scored from 0–2 each for motor activity, fur texture, posture, and diarrhea (0–8). For preventative experiments, Divertin was given 14 days after T cell transfer of FACS-sorted CD4+CD45RBhi T cells. For experiments assessing effects on established disease, mice received column-purified naïve CD4+ T cells (CD4+C25-) splenocytes (Miltenyi). By 18 days after adoptive transfer mice had reproducible disease characterized by reduced weight (relative to mice that did not receive T cells), increased stool water, and other clinically-evident features of disease. The latter were generally too mild to trigger clinical score increases. Mice were then randomized into groups to be sacrificed at that time (to confirm disease) or to receive daily, 200 μl i.p. injections of saline or drugs beginning on day 19. Drug schedules were 25 mg/kg Divertin each day or 150 μg rat anti-mouse TNF clone XT3.11 (BioXCell) every three days. Mice receiving only anti-TNF received saline on other days. For mice receiving combination treatment, Divertin and anti-TNF were combined in a single i.p injection on days when both were given. Cytokines were measured in homogenates of distal colon by multiplex ELISA (Quansys). Histopathology was scored on well-oriented colonic cross-sections stained by H&E and scored semiquantitatively from 0 to 3 each for lymphoid infiltrates, polymorphonuclear infiltrates, mucosal hyperplasia, and ulceration (total 0–12), as described.10
Human tissue.
Mucosa was removed from normal small intestinal tissue excised during bariatric surgery and incubated in HBSS with 300 ng/ml recombinant human TNF (Peprotech), Divertin (250 μM), or both, as indicated, for 60 min. Tissue was then snap-frozen in OCT media. De-identified, discarded human tissues were used under an exempted protocol that did not require informed consent. All procedures involving human tissues were approved by The University of Chicago Institutional Review Board and all relevant ethical regulations were followed.
Immunoblotting.
Lysates of Caco-2BBe monolayers or jejunal epithelial cells, isolated as described previously 2, were separated by SDS-PAGE (Bio-Rad, Hercules, CA), transferred to PVDF membranes, and blotted using antibodies to phosphorylated myosin light chain (pMLC) (Cell Signaling Technology, Beverly, MA) and β-actin (Sigma-Aldrich) followed by peroxidase-conjugated secondary antibodies (Cell Signaling Technology), and enhanced chemiluminescent detection. Signal intensity was assessed using ImageJ.
Immunostaining.
Caco-2BBe monolayers grown on 5 cm2 Transwell supports were fixed in 2% paraformaldehyde and immunostained as described previously. Snap-frozen tissues were stored at −80°C, after which 5 μm frozen sections were cut and fixed in 1% paraformaldehyde. EdU was detected using the Alexa Fluor 488 click chemistry detection kit (Invitrogen). Immunostaining used affinity-purified rabbit anti-human MLCK1 (2 μg/ml),7 monoclonal mouse anti-MLCK clone K36 (Sigma-Aldrich, 1 μg/ml)7, affinity-purified rabbit anti-pMLC (Cell Signaling 3671, 1 μg/ml), monoclonal mouse anti-occludin clone OC-3F10 (Invitrogen, 1 μg/ml), monoclonal mouse anti-E-cadherin clone M168 (Abcam, 2 μg/ml), or monoclonal rat anti-CD3 clone CD3–12 (Abcam, 2 μg/ml). Primary antibodies were followed by Alexa Fluor 488- or Alexa Fluor 594-conjugated affinity-purified secondary antibodies (Invitrogen) along with Alexa Fluor 594-conjugated phalloidin (Invitrogen) and Hoechst 33342 (Invitrogen). Stained sections were mounted in Prolong gold (Invitrogen).
Microscopy.
Specimens were imaged using an epifluorescence microscope (DM4000; Leica) equipped with DAPI, Endow GFP, and Texas red zero-pixel shift filter sets (Chroma Technology Corp.), a 63× 1.32 NA oil immersion objective, and a camera (CoolSNAP HQ; Roper Industries) controlled by MetaMorph 7. Z stacks were collected at 0.2 μm intervals and deconvolved using AutoDeblur (version X1; Media Cybernetics) for 10 iterations. Line scans (100 pixels wide) were performed using MetaMorph 7. For formalin-fixed, paraffin-embedded specimens stained as part of tissues microarrays, tiled images were collected on a DM4000 microscope equipped with Chroma filter sets (as above), a 20× HC PLAN APO NA 0.7 objective, CoolSNAP HQ2 grayscale CCD (for fluorescence) and Jenoptik ProgRes C10 Plus color CCD (for transmitted light) cameras, and a motorized xyz-stage (Ludl) controlled with custom MetaMorph 7 journals. Images were stitched using MetaMorph 7. Single images of H&E-stained slides were acquired using a MicroPublisher 5.0 CCD camera (Q Imaging) on a Leica DMLB microscope equipped with 10× NA0.25 and 40× NA0.65 HC FL PLAN objectives.
Image analysis.
For analysis of MLCK1 or MLCK2 localization in cultured monolayers (Figs. 1d, 1e, 2g, 3b), the PAMR was defined using labeled phalloidin. ImageJ was used to generate a region of interest around the entire cell (just outside the PAMR) and a separate intracellular region of interest (just inside the PAMR). Total intensity of the area between the two regions was divided by total intensity of the larger region of interest to determine the fraction of MLCK at the PAMR. At least three independent monolayers (Transwells) were analyzed for each condition, with 3 – 5 fields measured in each monolayer. Each point represents the mean values from cells in one microscope field; n represents the number of fields analyzed.
For analysis of MLCK1 localization in intestinal tissues (Figs. 4e, 4j), phalloidin staining was used to define a region of interest at the PAMR as well as a larger region of interest encompassing the entire cell. Each region of interest included a segment of 5 to 10 epithelial cells (~100 μm). The fraction of PAMR-associated MLCK was determined from the two regions of interest as for monolayers. Each point represents a mean of 3 – 5 fields measured in a single tissue specimen; n represents the number of independent biopsy fragments or mice analyzed for human or mouse tissues, respectively. MLC phosphorylation was quantified using Metamorph. Counting of EdU-labeled nuclei and CD3+ T cells was performed manually.
In vitro kinase assay.
Confluent Caco-2BBe monolayers expressing MLCK1 and MLCK2 were used as the source of MLC kinase. After dilution in kinase reaction buffer (20 mM morpholinepropanesulfonic acid, pH 7.4; 2 mM MgCl2; 0.25 mM CaCl2; and 0.2 μM calmodulin), small molecule inhibitor compounds obtained from the NCI or PIK were added to the reactions, as indicated. The reaction was initiated by the addition of γ32P-ATP (ICN, Costa Mesa, CA) and 5 μM recombinant MLC12 and transferring to 30°C. MLC phosphorylation was determined by autoradiography after SDS-PAGE. All experiments were performed at least 3 times with triplicate or greater samples in each experiment.
Gel contraction assay.
Human aortic smooth muscle cells (HASMC, Invitrogen) were maintained in Medium 231 supplemented with SMGS. Collagen lattices were prepared by mixing, on ice, collagen I (2 mg/ml) in 0.1% acetic acid with supplemented Medium 231 and 0.1 N NaOH to neutralize the pH before adding a cell suspension, as described.55,56 After polymerization (30 min at 37°C), gels were detached and covered with supplemented Medium 231, with or without small molecule inhibitor compounds obtained from the NCI as indicated. After 24 hours, lattices were photographed and the percent gel contraction was calculated using ImageJ.
Wound closure assay.
Confluent Caco-2BBe monolayers were wounded with a suction pipette, treated with 20 μM Y27632, 150 μM PIK, or 250 μM NSC55937 (Divertin) during recovery. Each wound was measured immediately after injury and following 72 hrs of recovery. Closure of each wound is shown relative to the initial wound size. As shown previously,57 inhibition of either rho kinase or MLCK limited repair.
Statistical analysis.
Statistical significance (P) was determined by paired t-test, unpaired t test with Welch’s correction, two-tailed Kruskal-Wallis test, ANOVA with Dunn’s multiple comparison test, ANOVA with Bonferroni correction, ANOVA with Tukey’s multiple comparison test, or Gehan-Breslow-Wilcoxon test, as indicated in each figure legend.
Data Availability Statement.
All requests for raw and analyzed data and materials are promptly reviewed by to verify if the request is subject to any intellectual property or confidentiality obligations. Human subjects were de-identified and no further data are available. Any data and materials that can be shared will be released via a Material Transfer Agreement. Crystal structure data are available as dataset ID D_1000232147 and PDB ID 6C6M.
Extended Data
Figure Extended Data 1.
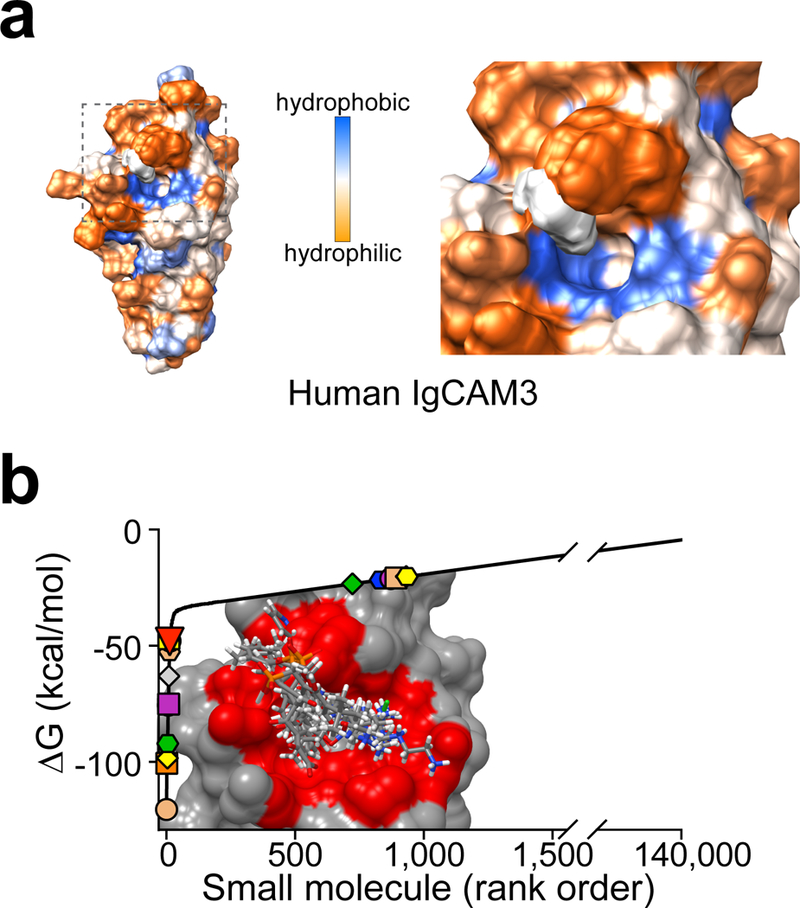
Figure Extended Data 2.
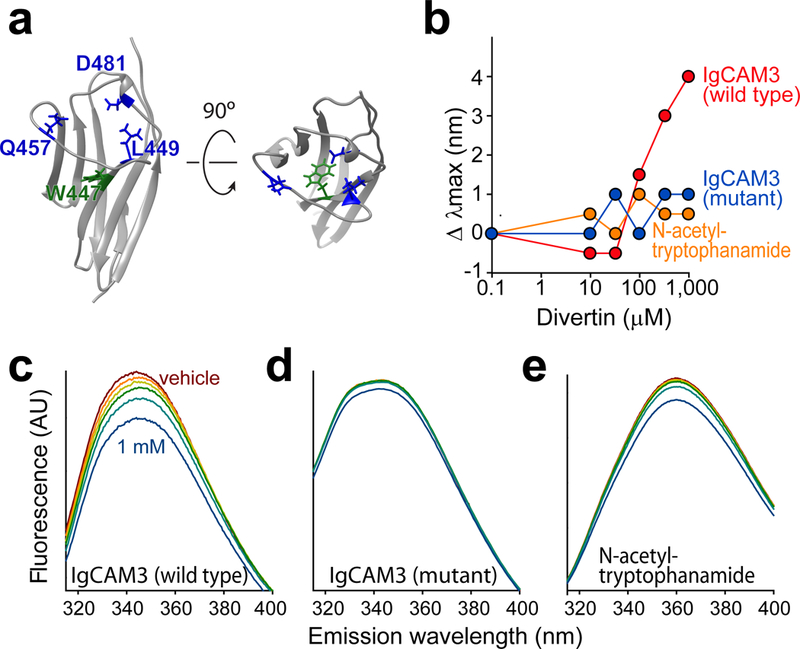
Figure Extended Data 3.
Supplementary Material
Acknowledgements.
Supported by the National Institutes of Health R01DK61931 (JRT), R01DK068271 (JRT), R24DK099803 (JRT), R01GM081030 (LWM), R01AG048793 (SCM), P30CA014599 (The University of Chicago Comprehensive Cancer Center), P30DK034854 (the Harvard Digestive Disease Center), and T32HL007237 (WVG, AMM), the Broad Medical Research Foundation (IBD-022), the Department of Defense W81XWH-09–1-0341 (JRT), a Catalyst Award from the Chicago Biomedical Consortium (JRT, LWM), the National Natural Science Foundation of China 81470804 (WH) and 31401229 (WH). The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02–05CH11231.
Footnotes
Competing interests statement. The authors have no competing interests.
References
- 1.Odenwald MA & Turner JR The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol 14, 9–21 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clayburgh DR, et al. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Invest. 115, 2702–2715 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turner JR Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9, 799–809 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Lazar V & Garcia JG A single human myosin light chain kinase gene (MLCK; MYLK). Genomics 57, 256–267 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Blue EK, et al. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am. J. Physiol. - Cell Physiol. 282, C451–460 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamm KE & Stull JT Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 276, 4527–4530 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Clayburgh DR, et al. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J. Biol. Chem. 279, 55506–55513 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birukov KG, et al. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src). J. Biol. Chem. 276, 8567–8573 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Wainwright MS, et al. Protein kinase involved in lung injury susceptibility: evidence from enzyme isoform genetic knockout and in vivo inhibitor treatment. Proc. Natl. Acad. Sci. U.S.A. 100, 6233–6238 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su L, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterol. 145, 407–415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Somlyo AV, et al. Myosin light chain kinase knockout. J. Muscle Res. Cell Motil. 25, 241–242 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Zolotarevsky Y, et al. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterol. 123, 163–172 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Wang F, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterol. 131, 1153–1163 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Powrie F, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Williams AF & Barclay AN The immunoglobulin superfamily--domains for cell surface recognition. Annu. Rev. Immunol. 6, 381–405 (1988). [DOI] [PubMed] [Google Scholar]
- 16.Holden HM, Ito M, Hartshorne DJ & Rayment I X-ray structure determination of telokin, the C-terminal domain of myosin light chain kinase, at 2.8 A resolution. J. Mol. Biol. 227, 840–851 (1992). [DOI] [PubMed] [Google Scholar]
- 17.Turner JR, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 273, C1378–1385 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Owens SE, Graham WV, Siccardi D, Turner JR & Mrsny RJ A strategy to identify stable membrane-permeant peptide inhibitors of myosin light chain kinase. Pharm. Res. 22, 703–709 (2005). [DOI] [PubMed] [Google Scholar]
- 19.He WQ, et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterol. 135, 610–620 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marchiando AM, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 189, 111–126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham WV, et al. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J. Biol. Chem. 281, 26205–26215 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Developmental Therapeutics Program N.C.I Datawarehouse Index Results. (2004).
- 23.Su L, et al. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterol. 136, 551–563 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berg DJ, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Invest. 98, 1010–1020 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhn R, Lohler J, Rennick D, Rajewsky K & Muller W Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Odenwald MA & Turner JR Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol 11, 1075–1083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buhner S, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut 55, 342–347 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prager M, et al. Myosin IXb variants and their pivotal role in maintaining the intestinal barrier: a study in Crohn’s disease. Scand. J. Gastroenterol. 49, 1191–1200 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Wapenaar MC, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut 57, 463–467 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Wyatt J, Vogelsang H, Hubl W, Waldhoer T & Lochs H Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 341, 1437–1439 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Arrieta MC, Madsen K, Doyle J & Meddings J Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut 58, 41–48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vetrano S, et al. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterol. 135, 173–184 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Lin HB, Cadete VJ, Sawicka J, Wozniak M & Sawicki G Effect of the myosin light chain kinase inhibitor ML-7 on the proteome of hearts subjected to ischemia-reperfusion injury. Journal of proteomics 75, 5386–5395 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Clayburgh DR, Musch MW, Leitges M, Fu YX & Turner JR Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J. Clin. Invest. 116, 2682–2694 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pearson AD, Eastham EJ, Laker MF, Craft AW & Nelson R Intestinal permeability in children with Crohn’s disease and coeliac disease. Br Med J (Clin Res Ed) 285, 20–21 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gruber R, et al. Diverse regulation of claudin-1 and claudin-4 in atopic dermatitis. Am. J. Pathol. 185, 2777–2789 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Mirzapoiazova T, et al. Non-muscle myosin light chain kinase isoform is a viable molecular target in acute inflammatory lung injury. Am. J. Respir. Cell Mol. Biol. 44, 40–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, et al. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat. Immunol. 9, 880–886 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown GR, et al. Tumor necrosis factor inhibitor ameliorates murine intestinal graft-versus-host disease. Gastroenterol. 116, 593–601 (1999). [DOI] [PubMed] [Google Scholar]
- 40.Bennett J, et al. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J. Neuroimmunol. 229, 180–191 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Kebir H, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moravcevic K, Oxley CL & Lemmon MA Conditional peripheral membrane proteins: facing up to limited specificity. Structure 20, 15–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sagona AP, et al. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat. Cell Biol. 12, 362–371 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Webb BA, et al. A Histidine Cluster in the Cytoplasmic Domain of the Na-H Exchanger NHE1 Confers pH-sensitive Phospholipid Binding and Regulates Transporter Activity. J. Biol. Chem. 291, 24096–24104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buschmann MM, et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol. Biol. Cell 24, 3056–3068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keegan RM & Winn MD Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Crystallogr D Biol Crystallogr 63, 447–457 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bricogne G, et al. BUSTER version 2.11.7. (Cambridge, United Kingdom: Global Phasing Ltd., 2017; ). [Google Scholar]
- 49.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Irwin JJ & Shoichet BK ZINC--a free database of commercially available compounds for virtual screening. J Chem Inf Model 45, 177–182 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berglund JJ, Riegler M, Zolotarevsky Y, Wenzl E & Turner JR Regulation of human jejunal transmucosal resistance and MLC phosphorylation by Na(+)-glucose cotransport. Am. J. Physiol. - Gastrointest. Liver Physiol. 281, G1487–1493 (2001). [DOI] [PubMed] [Google Scholar]
- 52.Turner JR & Black ED NHE3-dependent cytoplasmic alkalinization is triggered by Na(+)-glucose cotransport in intestinal epithelia. Am. J. Physiol. - Cell Physiol. 281, C1533–1541 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Wang F, et al. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 166, 409–419 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salic A & Mitchison TJ A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. U.S.A. 105, 2415–2420 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Badri KR, et al. Blood pressure homeostasis is maintained by a P311-TGF-beta axis. J. Clin. Invest. 123, 4502–4512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oishi K, et al. Agonist-induced isometric contraction of smooth muscle cell-populated collagen gel fiber. Am. J. Physiol. - Cell Physiol. 279, C1432–1442 (2000). [DOI] [PubMed] [Google Scholar]
- 57.Russo JM, et al. Distinct temporal-spatial roles for rho kinase and myosin light chain kinase in epithelial purse-string wound closure. Gastroenterol. 128, 987–1001 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All requests for raw and analyzed data and materials are promptly reviewed by to verify if the request is subject to any intellectual property or confidentiality obligations. Human subjects were de-identified and no further data are available. Any data and materials that can be shared will be released via a Material Transfer Agreement. Crystal structure data are available as dataset ID D_1000232147 and PDB ID 6C6M.