

SUMMARY
During development, oligodendrocyte progenitor cells (OPCs) migrate extensively throughout the spinal cord. However, their migration is restricted at transition zones (TZs). At these specialized locations, unique glial cells in both zebrafish and mice play a role in preventing peripheral OPC migration, but the mechanisms of this regulation are not understood. To elucidate the mechanisms that mediate OPC segregation at motor exit point (MEP) TZs, we performed an unbiased small-molecule screen. Using chemical screening and in vivo imaging, we discovered that inhibition of A2a adenosine receptors (ARs) causes ectopic OPC migration out of the spinal cord. We provide in vivo evidence that neuromodulation, partially mediated by adenosine, influences OPC migration specifically at the MEP TZ. This work opens exciting possibilities for understanding how OPCs reach their final destinations during development and identifies mechanisms that could promote their migration in disease.
Graphical Abstract
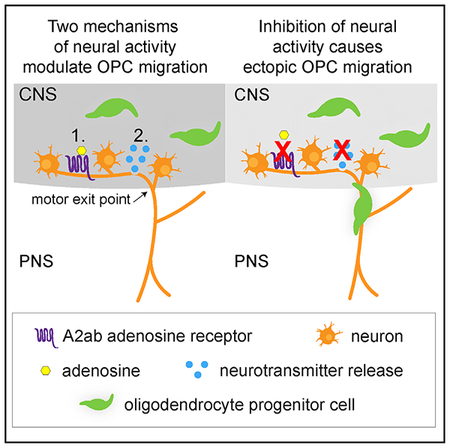
In Brief
Fontenas et al. show that, during development, at least two distinct mediators of neural activity, adenosine signaling via adenosine receptors and vesicular neurotransmitter release, regulate oligodendrocyte progenitor cell (OPC) migration at the motor exit point transition zone. Loss of both mechanisms results in ectopic OPCs in the peripheral nervous system.
INTRODUCTION
Oligodendrocyte progenitor cells (OPC) are migratory, proliferative cells with multiple functions in CNS development and disease (Bergles and Richardson, 2015; Emery and Lu, 2015; Zuchero and Barres, 2015). These progenitors differentiate into oligodendrocytes (OLs), which ensheath axons in an insulating layer of myelin that is essential for saltatory conduction (Simons and Nave, 2015). During gliogenesis, OPCs are specified from precursor domains within the brain and spinal cord, but they migrate extensively to become distributed throughout the entire CNS (Miller, 2002; Rowitch, 2004). In the spinal cord, most OPCs are specified from ventral motor neuron progenitor (pMN) precursors that also give rise to motor neurons during neurogenesis (Richardson et al., 2000). These motor neurons extend axons ventrally toward the motor exit point (MEP) transition zone (TZ) and cross into the peripheral nervous system (PNS), where they ultimately innervate targets including skeletal muscle (Bonanomi and Pfaff, 2010; Lewis and Eisen, 2003). As OPCs disperse from the pMN domain to populate the spinal cord, a subset transiently extends membrane processes through the MEP TZ into the PNS (Fraher and Kaar, 1984; Smith et al., 2014). However, under normal physiological conditions, these processes retract back into the spinal cord, and OPC bodies are restricted from migrating onto peripheral nerves.
The mechanisms allowing selective migration of motor axons and other glial populations, but not OPCs, through the MEP TZ are not understood. Recently, work from our lab and others has shown that OPCs are capable of migrating into the PNS in zebrafish and mouse mutants with PNS defects, and peripheral OPCs have even been described in human peripheral neuropathy patients (Coulpier et al., 2010; Fröb et al., 2012; Kucenas et al., 2009; Smith et al., 2014). On the basis of these studies, we hypothesize that active regulation of OPC migration restricts them to the CNS.
Although the molecular mechanisms that mediate OPC restriction to the CNS are unknown, previous work from our lab identified a population of CNS-derived peripheral glial cells called MEP glia, which are essential for preventing OPC migration onto spinal motor nerves (Smith et al., 2014). A similar population known as boundary cap (BC) cells, located at TZs, exists in mice and is hypothesized to regulate OPC migration at these positions as well (Coulpier et al., 2010; Fröb et al., 2012).
In order to identify additional mechanisms regulating OPC migration at the MEP TZ, we performed an unbiased chemical screen that identified ten small molecules that resulted in peripherally located OPCs. We focused further testing on one of these compounds, an adenosine receptor (AR) antagonist, because adenosine is a well-known modulator of neuronal activity, and neuronal activity has been implicated in regulating OPC differentiation, proliferation, and migration (Chen et al., 2018; Etxeberria et al., 2016; Gibson et al., 2014; Hines et al., 2015; Káradóttir and Attwell, 2007; Mensch et al., 2015; Mitew et al., 2018; Nagy et al., 2017; Stevens et al., 2002). Therefore, we hypothesized that adenosinergic regulation of neuronal activity may be involved in restricting OPC migration at the MEP TZ. Here, we report that adenosine signaling through A2a ARs functions to regulate OPC migration at MEP TZs during development.
RESULTS
Identification of Mechanisms that Induce Ectopic OPC Migration through the MEP TZ
Glial cells that establish a barrier to OPC migration across the MEP TZ exist in mice and fish (Coulpier et al., 2010; Fröb et al., 2012; Smith et al., 2014). In zebrafish, during development, ventral OPCs within the spinal cord extend membrane processes through the MEP TZ and contact MEP glia in the PNS (Figure 1A). When this occurs, the OPC is immediately repelled and retracts its process. When MEP glia are absent, OPCs freely migrate through the MEP TZ and onto peripheral motor nerves (Figure 1A) (Morris et al., 2017; Smith et al., 2014). However, the signals restricting OPC migration at the MEP are unknown. Therefore, to identify molecular mechanisms regulating OPC migration at the MEP TZ, we conducted an unbiased screen of small molecules to identify signaling cascades involved in OPC segregation to the CNS.
Figure 1. Small-Molecule Screen Identifies Compounds that Disrupt OPC Migration at the MEP TZ.

(A) Schematic of a transverse view of the MEP TZ. MEP glia (MEPg; green) prevent OPC (yellow) exit from the spinal cord (gray). OPC processes (arrow) contact MEP glia but are repelled. Without MEP glia, OPCs migrate onto peripheral nerves.
(B) Low-magnification images showing lateral views of negative (top) and positive (bottom) controls at 3 dpf in olig2:dsred larvae. Peripheral olig2+ cells are denoted with yellow arrowheads.
(C) Images of control and validated hits in 3 dpf olig2:dsred larvae showing ectopic OPCs (arrowhead) along spinal motor nerves.(D) Mean ± SEM of peripheral OPC count for larvae injected with injection buffer (control) or TeNT mRNA at 3 dpf. p = 0.0013; n = 14 control, n = 23 TeNT.
Scale bars, 100 μM in (B) and 20 μM in (C).
To conduct our screen, we treated 24 h post-fertilization (hpf) olig2:dsred embryos with compounds from the Library of Pharmacologically Active Compounds (LOPAC1280). In our primary screen, embryos were treated with 10 μM of a compound from the library, 1% DMSO as a negative control, or 4 μM AG1478 as a positive control (Figure 1B). The positive control AG1478 inhibits Erbb3 signaling and phenocopies the erbb3b mutation (Lyons et al., 2005). Previously, we demonstrated that erbb3b mutants have peripherally located OPCs because they lack MEP glia (Morris et al., 2017; Smith et al., 2014). A pilot experiment confirmed that treatment with 4 μM AG1478 from 24 hpf to 3 days post-fertilization (dpf) caused robust peripheral OPC migration, whereas 1% DMSO did not (Figure 1B). For the rest of our screen, we analyzed larvae at 3 dpf for the presence of olig2+ cell bodies in the periphery (Figure 1C). We identified 197 “hits” in our primary screen, and these compounds were retested in a secondary screen, and any that resulted in peripheral olig2+ cells was considered a validated hit (Figure 1C). This protocol resulted in 10 total hits, many of which are involved in neurotransmission and/or modulation of neuronal activity (Table 1).
Table 1.
Validated Hits from Screen Using LOPAC1280
| Drug Name | Description |
|---|---|
| CGS-15943 | highly potent, non-selective adenosine receptor antagonist |
| 1-Phenyl-3-(thiazolyl)-2-thiourea | dopamine β-hydroxylase inhibitor |
| Acetylthiocholine chloride | nicotinic acetylcholine receptor agonist |
| Brefeldin A from Penicillium brefeldianum | causes collapse of Golgi apparatus, blocking exocytosis |
| N-phenylanthranilic acid | Cl− channel blocker |
| Salmeterol xinafoate | selective β2 adrenergic receptor agonist |
| I-OMe-tyrphostin AG 538 | insulin growth factor I (IGF-1) receptor inhibitor |
| L-canavanine sulfate | selective inhibitor of inducible nitric oxide synthase (iNOS) |
| Clofibrate | peroxisome proliferator-activated receptor-α (PPARα) agonist |
| Wortmannin from Penicillium funiculosum | selective phosphatidylinositol 3-kinase (PI3-K) inhibitor |
Because several of the compounds from our screen are neuromodulators, we next tested whether a non-pharmacological method of inhibiting neuronal activity would result in peripheral OPC migration. To do this, we inhibited all vesicular release using tetanus toxin light chain (TeNT) (Yu et al., 2004). Previous studies demonstrate that injections of TeNT mRNA effectively inhibit vesicular release and lead to significant paralysis of zebrafish larvae (Fontenas et al., 2016; Mensch et al., 2015). When we injected olig2:dsred embryos at the one-cell stage with 1 to 2 nL of 200 ng/μL TeNT mRNA, we confirmed that neuronal activity was indeed inhibited, as larvae were paralyzed at 2 dpf (n = 54) and 3 dpf (n = 52) (Figures S1A and S1B; 2 dpf, p < 0.0001; 3 dpf, p < 0.0001). Consistent with our hypothesis, silencing neurons using TeNT resulted in significant numbers of OPC bodies in the periphery (Figure 1D; n = 14 larvae [control], n = 23 larvae [TeNT]; p = 0.0013). From these results, we hypothesize that neural activity influences OPC migration at MEP TZs.
A2a ARs Influence OPC Migration at the MEP TZ
One compound we identified, CGS-15943, is a highly potent, non-selective AR antagonist (Ongini et al., 1999). Adenosine is a well-known modulator of neuronal activity and can either increase or decrease neuronal firing by binding to different ARs (Sebastião and Ribeiro, 2015). Because adenosine can modulate neuronal activity and purinergic signaling has been implicated in OL differentiation and migration (Agresti et al., 2005a, 2005b; Coppi et al., 2013, 2015; Dennis et al., 2012; Othman et al., 2003; Stevens et al., 2002), we focused our experiments on this hit from our screen.
We first confirmed that CGS-15943 caused peripheral OPC migration through the MEP TZ by performing in vivo, time-lapse imaging. In order to distinguish OPCs from motor neurons, we used embryos expressing olig2:dsred and sox10:eos, where OPCs are Eos+ and DsRed+ (yellow). We treated these embryos with 10 μM of CGS-15943 from 24 hpf to 3 dpf and imaged three larvae (ten nerves total) from 55 hpf to 3 dpf. In these time-lapse movies, we observed sox10+/olig2+ cells with highly dynamic membrane processes at the MEP TZ (Figure 2A). In nine of ten nerves (90%) imaged, OPCs extended highly dynamic membrane processes through the MEP TZ. In six of ten nerves (60%), OPC bodies squeezed through this opening and migrated onto peripheral spinal motor nerves during imaging (Figure 2A; Video S1). This migration most often occurred between 60 and 72 hpf, although we also observed OPCs already present on the nerve before 60 hpf (three of ten nerves [30%]). Interestingly, we never observed any olig2+/sox10− motor neurons in the periphery in these time-lapse movies (zero of ten nerves [0%]), indicating that adenosine signaling is involved in restricting OPCs, but not motor neurons, to the spinal cord. This is consistent with evidence from our lab and others demonstrating that distinct mechanisms regulate segregation of glia and neurons at the MEP TZ (Fröb et al., 2012; Kucenas et al., 2009).
Figure 2. A2a AR Antagonist Causes OPC Migration through the MEP TZ.

(A) Frames captured from a 15 h time-lapse movie of a sox10:eos;olig2:dsred larva treated with CGS-15943 from 24 hpf to 3 dpf and imaged from 57 to 72 hpf; 0’ is 58 hpf. The black asterisk marks OPC body. Arrowheads mark OPC leading process.
(B) Dose-response curve of the number of peripheral OPCs for fish treated from 24 hpf to 3 dpf with SCH-58261. Mean ± SEM, n = 10 larvae per dose; **p < 0.01 and ***p < 0.0001.
(C) Percentage of nerves at 3 dpf that have ectopic OPC processes and/or cell bodies. n = 60 DMSO, n = 98 SCH-58261; p < 0.01.
(D) Mean ± SEM of peripheral OPCs in olig2:dsred larvae treated with 10 μM SCH-58261 alone or in combination with 2.5 μM CGS-21680 or 5 μM adenosine from 36 hpf to 3 dpf. n = 30 fish per treatment; *p < 0.05 and **p < 0.01.
(E) Peripheral OPC counts for larvae treated with 10 μM SCH-58261 during distinct time periods. Green bars indicate mean peripheral OPC counts significantly above DMSO control (p < 0.05). Red bars indicate mean peripheral OPC counts not significantly different than DMSO (p > 0.05). n = 9–12 larvae per condition. Scale bar, 20 μM.
We next sought to determine which AR subtype was involved in preventing peripheral OPC migration. To do this, we treated embryos expressing olig2:dsred from 24 hpf to 3 dpf with varying concentrations of selective antagonists for each of the four AR subtypes: A1 (8-cyclopentyltheophylline [CPT]), A2a (SCH-58261), A2b (MRS-1754), and A3 (MRS-1191), as well as the general AR antagonist CGS-15943 (Jiang et al., 1996; Ongini et al., 1999; Searl and Silinsky, 2012; Wei et al., 2013). We analyzed drug-treated larvae at 3 dpf and quantified the number of peripheral olig2+ OPCs. As expected, CGS-15943 caused significant peripheral OPC migration (Figure S2A; p < 0.01). The selective A2a AR antagonist SCH-58261 also resulted in significant numbers of peripheral olig2+ OPC bodies and membrane processes (Figures 2B and 2C; p < 0.01 for 10 μM SCH-58261 compared with DMSO). In contrast, we did not observe peripheral OPCs following treatment with selective antagonists for any of the other AR subtypes (Figures S2B–S2D). From these data, we conclude that A2a ARs are involved in regulating OPC migration at MEP TZs.
We sought to further confirm the selectivity of SCH-58261 activity to A2a ARs with competitive binding experiments. If SCH-58261 selectively antagonizes the A2a AR, we hypothesized that co-administering an A2a agonist would rescue the peripheral migration phenotype caused by SCH-58261. To test this, we treated 24 hpf olig2:dsred embryos with 10 μM SCH-58261 combined with 5 μM adenosine or 2.5 μM of the A2a selective agonist CGS-21680 and quantified the number of peripheral OPCs at 3 dpf (Jarvis and Williams, 1989). Both adenosine and CGS-21680 significantly decreased the number of peripheral OPCs when co-administered with SCH-58261 (Figure 2D; p <0.05, CGS-21680; p < 0.01, adenosine compared with SCH-58261 alone; n = 30 larvae). These results support the conclusion that SCH-58261 selectively antagonizes A2a ARs to induce ectopic OPC migration. To confirm that adenosine or CGS-21680 treatment alone did not interfere with normal OPC development or migration, we imaged olig2:dsred zebrafish larvae at 3 dpf following treatment with adenosine, CGS-21680, or DMSO from 24 hpf to 3 dpf and quantified dorsal spinal cord olig2+ OPCs in a four-somite region. In these studies, we did not observe any differences among treatments (Figure S2E; p = 0.23; adenosine, n = 4 larvae; CGS-21680, n = 9 larvae; DMSO, n = 11 larvae).
In our initial screen, our treatment protocol spanned from 24 hpf to 3 dpf, encompassing OPC specification, proliferation, and migration (Kirby et al., 2006; Kucenas et al., 2008a; Park et al., 2002). Therefore, we sought to narrow the treatment window. To do this, we treated olig2:dsred embryos with 10 μM SCH-58261 during various time windows and quantified the number of peripheral OPCs at 3 dpf. We found that exposure to SCH-58261 beginning later than 54 hpf or ending earlier than 54 hpf did not result in any peripheral OPC migration (Figures 2E and S2F–S2H; n = 7–12 larvae). At this stage, OPCs are migrating throughout the spinal cord and we previously observed OPCs extending membrane processes into the periphery and contacting MEP glia at this developmental stage (Kirby et al., 2006; Smith et al., 2014). From these experiments, we conclude that adenosine signaling functions when OPCs are migrating.
A2a AR Antagonists Do Not Disrupt Spinal Motor Nerve Development
Previously, we demonstrated that loss of MEP glia results in OPC migration into the periphery (Morris et al., 2017; Smith et al., 2014). We therefore sought to determine whether antagonizing the A2a AR led to ectopic OPC migration by perturbing MEP glial development. MEP glia can be identified by the co-expression of sox10:eos and olig2:dsred, as well as expression of wnt inhibitory factor 1 (wif1) (Smith et al., 2014). To determine if MEP glia were present in larvae treated with the A2a AR antagonist, we quantified the percentage of motor nerves with sox10+/olig2+ MEP glia at the MEP TZ and observed no differences among larvae treated from 36 hpf to 3 dpf with 1.25 μM CGS-15943 (n = 6 larvae), 10 μM SCH-58261 (n = 9 larvae), or DMSO (n = 5 larvae) (Figures 3A and 3B; p = 0.14, ten nerves analyzed per larvae). We also observed wif1+ MEP glia at 54 hpf in larvae treated with 1.25 μM CGS-15943 from 36 to 54 hpf (Figure S3A). From these data, we conclude that A2a AR antagonism does not affect MEP glial development.
Figure 3. A2a AR Antagonism Does Not Affect Spinal Motor Nerve Development.

(A) Motor nerve in a 3 dpf olig2:dsred;sox10:eos larvae treated with SCH-58261 from 36 hpf to 3 dpf. Red asterisks mark sox10+/olig2+ MEP glia at the MEP TZ. Black pound signs denote sox10+/olig2− dorsal root ganglia.
(B) Mean ± SEM of the percentage of nerves per larva at 3 dpf with sox10+/olig2+ MEP glia after treatment from 36 hpf to 3 dpf with DMSO (n = 5), CGS-15943 (n = 6), or SCH-58261 (n = 9). Ten nerves were quantified per larva. p = 0.14.
(C) Fifty-five hours post-fertilization nkx2.2a: megfp;olig2:dsred larvae treated with DMSO or CGS-15943 from 36 to 55 hpf showing PG extension (arrowheads) on the nerve.
(D) Quantification of Eos+ cells per nerve at 3 dpf in DMSO and SCH-58261-treated larvae. Mean ± SEM for DMSO (n = 6 fish) and SCH-58261 (n = 10 fish). Ten nerves per fish. p = 0.39.
(E) Frames captured from a 20 h time-lapse video starting at 36 hpf in sox10:nls-eos embryos treated with DMSO or SCH-58261. The spinal cord is denoted by a dashed line. SCs divide (yellow arrowheads) along the nerves in both treatments.
(F) Quantification of SC proliferation during these time lapses. Mean ± SEM for DMSO (n = 6 fish, 21 nerves) and SCH-58261 (n = 6 fish, 23 nerves). p = 0.35.
(G) Quantification of SC death during these time-lapse movies. Mean ± SEM for DMSO (n = 6 fish, 21 nerves) and SCH-58261 (n = 6 fish, 23 nerves). p = 0.20. Scale bars, 20 μM.
To determine whether glial migration was generally disrupted at the MEP TZ in larvae treated with the A2a AR antagonist, we assayed the migration of perineurial glia (PG). These cells originate in the ventral spinal cord, exit the CNS via the MEP TZ, and migrate along motor nerves to form the perineurium (Clark et al., 2014; Kucenas et al., 2008b). If the A2a AR antagonist non-selectively disrupted cell migration at the MEP TZ, we hypothesized that we would observe altered extension of PG along spinal motor nerves. To investigate this, we measured extension of PG from the spinal cord along motor nerves in 55 hpf nkx2.2a:megfp;olig2:dsred larvae treated from 36 to 55 hpf with 1.25 μm CGS-15943. In these studies, we found no difference in PG extension between DMSO and CGS-15943-treated larvae, indicating that A2a AR signaling does not generally affect glial migration at the MEP TZ (Figure 3C; p = 0.28; DMSO22.27 ± 0.88 μm [mean ± SEM], n = 6 larvae, ten nerves analyzed per larva; CGS-15943 24.94 ± 1.75 μm [mean ± SEM], n = 10 larvae, ten nerves analyzed per larva).
Finally, we assessed Schwann cell (SC) development along spinal motor nerve roots. Using 3 dpf sox10:eos larvae, we quantified the number of Eos+ cells between the MEP TZ and horizontal myoseptum. In this experiment, we observed that the number of Eos+ cells per nerve was not significantly different between animals treated from 36 hpf to 3 dpf with DMSO or SCH-58261 (p = 0.08), and we saw no difference in the number of Eos+ glia when we compared counts between nerves populated with ectopic OPCs and those without (Figure 3D). We also performed time-lapse imaging in sox10:nls-eos larvae starting at 36 hpf and observed that migration, proliferation, and apoptosis were indistinguishable between SCH-58261-treated and DMSO-treated larvae (Figures 3E–3G). Finally, we confirmed that SCs produced MBP+ myelin sheaths at 5 dpf in both DMSO- and SCH-58261-treated larvae (Figures S3B). From these experiments, we conclude that A2a AR antagonism does not cause peripheral OPC migration by disrupting motor nerve development.
The A2ab AR Influences OPC Migration at MEP TZs
Zebrafish have two orthologous A2a ARs, A2aa and A2ab, which are encoded by adora2aa and adora2ab, respectively (Boehmler et al., 2009). We used Clustal Omega to compare the zebrafish A2aa, A2ab, and the human A2A AR protein sequences (Goujon et al., 2010; Sievers et al., 2011). Zebrafish A2aa and A2ab ARs have 55% and 52% identical amino acids to human A2A AR (Figure S4). However, most of this variability is in the C-terminal cytoplasmic tail, which mediates interactions with other proteins and not ligand binding (Keuerleber et al., 2010). The ligand binding domains of zebrafish A2aa and A2ab ARs are 72% (21 of 29) conserved with human A2A AR, and A2aa and A2ab ARs have 76% (22 of 29) conserved amino acids with each other in this domain, so they likely have similar ligand affinities (Figure S4). However, adora2aa and adora2ab have distinct expression patterns, so we hypothesized that they may have unique functions in nervous system development (Boehmler et al., 2009).
To determine if A2aa and/or A2ab ARs were required to prevent peripheral OPC migration, we created genetic mutants for adora2aa and adora2ab. We created germline mutations in adora2aa using transcription activator-like effector nuclease (TALEN) targeted mutagenesis (Boch et al., 2009; Moscou and Bogdanove, 2009). Adora2aact845 is a 7 bp deletion within exon 3, which causes a frameshift and early stop codon, and this results in a severely truncated protein sequence (Figure S5A). The mutated A2aa AR protein lacks trans-membrane domains 6 and 7, which contain conserved ligand binding residues (de Lera Ruiz et al., 2014). It also lacks the entire C-terminal cytoplasmic tail, which mediates interaction with G proteins and protects the receptor from ubiquitination and degradation (Keuerleber et al., 2010). For these reasons, we hypothesize that adora2aact845 is a presumptive null. Similar to A2a−/− mice, adora2aa−/− zebrafish mutants are homozygous viable, produce viable offspring, and do not show any morphological defects compared with wild-type (WT) siblings at all stages investigated (Figure S5B) (Chen et al., 1999).
We also created germline mutations in adora2ab using CRISPR/Cas9 (Hwang et al., 2013). We designed a single guide RNA (sgRNA) targeting adora2ab exon 2, just downstream of the translation start site, and identified three germline frameshift mutations in the F1 generation (Figure 4A). These alleles include adora2abuva6, which has a 52 bp deletion in exon 2; adora2abuva7, which is a 66 bp insertion within exon 2; and adora2abuva9, which has a 13 bp deletion in exon 2 (Figure 4B). Because all three adora2ab mutant alleles lack all conserved ligand binding domains and C-terminal signaling, we hypothesize that they are also presumptive null mutations. Similar to adora2aa−/− mutant larvae, adora2ab−/− mutant larvae do not show any morphological defects compared with WT siblings at all stages investigated (Figure 4C).
Figure 4. A2ab ARs Influence OPC Migra tion at the MEP TZ.

(A) adora2ab WT and germline CRISPR mutant DNA sequences. sgRNA target and PAM site (underlined) are shown on WT sequence. Red letters or dashes indicate insertions or deletions.
(B) Diagram of A2ab protein with locations of early stops caused by adora2abuva6, adora2abuva7, and adora2abuva9.
(C) Bright-field images of WT and adora2ab−/− siblings at 3 and 5 dpf.
(D) Mean ± SEM of peripheral OPCs per fish for olig2:dsred;adora2aa−/− and WT larvae treated with water, DMSO, or 10 μM SCH-58261 from 36 hpf to 3 dpf. n = 12 fish; p < 0.0001 for treatment, p = 0.53 for genotype, p = 0.93 for interaction of treatment and genotype.
(E) Mean ± SEM of peripheral OPCs per fish for olig2:dsred;adora2ab−/−, adora2ab+/−, and WT larvae. p = 0.32; n = 19 adora2ab−/−, n = 26 adora2ab+/−, and n = 16 WT.
(F) Mean ± SEM of dorsally migrated OPCs per somite at 72 hpf. p = 0.43; n = 6 WT, n = 16 adora2ab+/−, n = 9 adora2ab−/−.
(G) Mean ± SEM of peripheral OPCs per fish for olig2:dsred;adora2ab−/−, gpr126−/−, and WT larvae injected with injection buffer (control) or TeNT mRNA. **p = 0.0088 (WT versus WT + TeNT), **p = 0.0053 (adora2ab+/− versus adora2ab+/− + TeNT), and ****p < 0.0001 (adora2ab−/− versus adora2ab−/− + TeNT); p = 0.99 (WT + TeNT versus gpr126−/− + TeNT) and (WT + TeNT versus adora2ab−/− + TeNT). n = 22 WT, n = 22 adora2ab+/−, n = 13 adora2ab−/−, n = 36 WT + TeNT, n = 32 adora2ab+/− + TeNT, n = 20 adora2ab−/− + TeNT, n = 4 gpr126+/+, and n = 5 gpr126−/− + TeNT. Red asterisks highlight significant comparisons relevant for the experiment.
(H) Mean ± SEM of the percentage of nerves with peripheral OPC bodies and/or processes at 3 dpf following TeNT mRNA injections and/or MEP glial ablation. For each WT or TeNT-injected larva, two nerves were analyzed with MEP glia ablated, and two nerves were analyzed with MEP glia intact in the same larva. n = 5 larvae (uninjected), n = 7 larvae (TeNT). **p = 0.004 for TeNT versus WT, p < 0.0001 for MEP glia ablated versus MEP glia present. Scale bar, 0.5 mm.
To determine if adora2aa mediated OPC restriction from the PNS, we quantified the number of peripheral OPCs at 3 dpf in olig2:dsred;adora2aa mutant larvae. In these mutants, we did not observe significant numbers of peripheral OPCs compared with WT and heterozygous siblings (Figure S5C; p = 0.69; n = 7 adora2aa+/+, n = 20 adora2aa+/−, n = 26 adora2aa−/−). To determine if both A2a paralogs in zebrafish were involved in segregating OPCs to the spinal cord, we treated olig2:dsred;adora2aa−/− larvae, which still have functional A2ab ARs, with the A2a AR antagonist SCH-58261 at 36 hpf. At 3 dpf, we did not observe peripherally migrated OPCs in WT or adora2aa−/− larvae treated with DMSO (Figure 4D; n = 12). However, when WT or adora2aa mutant larvae were treated with SCH-58261 from 36 hpf to 3 dpf, we observed peripheral OPCs in both genotypes (Figure 4D; n = 12). Two-way ANOVA analysis showed a significant effect of drug treatment (p < 0.0001) but not for genotype (p = 0.53) or the interaction between treatment and genotype (p = 0.93). In other words, both WT and adora2aa mutant larvae have the same number of peripherally migrated OPCs when treated with the A2a AR antagonist. These results led us to hypothesize that A2ab may be involved in OPC migration at the MEP TZ. Therefore, we quantified peripheral OPCs in 3 dpf olig2:egfp;adora2ab−/− larvae. We did not observe any peripheral OPC migration in WT larvae, but we did observe variable numbers of olig2+ OPCs along peripheral motor nerves in adora2ab+/− and adora2ab−/− larvae (Figure 4E; p = 0.32; n = 16 WT, n = 26 adora2ab+/−, n = 19 adora2ab−/−). However, the phenotype of peripheral OPC migration in adora2ab−/− larvae was not as robust as in larvae treated with the A2a antagonist SCH-58261 and was not significantly different from WT siblings. We discuss this in greater detail later. To rule out a general defect in OPC migration caused by a mutation in adora2ab, we time-lapse-imaged olig2:egfp;adora2ab mutants and their siblings from 55 to 72 hpf and quantified the number of dorsally migrated OPCs in the spinal cord. In these movies, we did not observe any significant difference in OPC activity between adora2ab mutant larvae and siblings (Figure 4F; p = 0.43; n = 6 WT, n = 16 adora2ab+/−, n = 9 adora2ab−/−).
As an independent test of the role of A2ab ARs in OPC restriction to the spinal cord, we performed genetic knockdown of adora2aa and adora2ab using morpholino oligonucleotides (MOs). When we quantified peripherally migrated OPCs in larvae injected with MOs at the one-cell stage, adora2abMO1 morphants (n = 42), but not adora2aaMO1 morphants (n = 65), had significantly more peripheral OPCs at 3 dpf compared with uninjected (n = 42) and phenol red-injected (n = 11) larvae (p < 0.0001) (Figures S6A and S6C). This result is consistent with our finding that adora2aa−/− larvae do not have peripherally migrated OPCs (Figure S5C). Similarly, we observed ectopic peripheral OPCs only in adora2aa−/− larvae injected with adora2ab MO (n = 84) but not in control-injected (n =30) adora2aa−/− larvae (Figure S6B; p < 0.0001). To directly rule out the possibility that the phenotype we see in adora2ab morphants is due to an off-target effect of the MO, we injected WT and adora2ab mutants with 1 ng/nL adora2ab MO at the one-cell stage. We quantified the number of peripheral OPCs and observed that the effect of the MO was lost in adora2ab−/− larvae and that the number of peripheral OPCs was comparable with that in uninjected adora2ab mutant larvae (Figure S6E; p = 0.03 [adora2ab MO-injected adora2ab+/+ versus adora2ab MO-injected adora2ab−/−]; n = 20 adora2ab+/+, n = 13 adora2ab−/−).
In these studies, we discovered a difference between our adora2ab MO-injected and adora2ab−/− embryos. The genetic perturbation of adenosine signaling through A2ab loss of function in our CRISPR mutants could trigger genetic compensation, leading to a subsequent rebalancing of neuronal activity, and mask the role of A2ab ARs in OPC migration at the MEP TZ (El-Brolosy et al., 2018; Rossi et al., 2015). Because of the high homology between the paralogs adora2aa and adora2ab, we hypothesized that A2aa could compensate for A2ab to mediate adenosine signaling in adora2ab−/− larvae. To test this hypothesis, we examined peripheral OPC migration in olig2:dsred; adora2aa;adora2ab double-mutant larvae at 3 dpf. In these larvae, we did not see any significant change in the number of peripherally migrated OPCs (Figure S6D; p = 0.75). In support of this, we performed qRT-PCR at 72 hpf and did not detect up-regulation of adora2aa mRNA expression in adora2ab−/− larvae (data not shown). We conclude from these experiments that the compensatory mechanisms triggered by mutations in adora2ab do not function through adora2aa but likely through other mechanisms regulating neuronal activity.
Because A2a ARs are excitatory G protein-coupled receptors that are known to function in neuromodulation, we hypothesized that a plausible compensatory mechanism at play could involve the enhancement of other mechanisms that drive neural activity. Therefore, we asked whether silencing neurons via inhibition of vesicular release would attenuate these compensatory mechanisms and consequently elicit enhanced OPC migration in adora2ab mutant larvae. To test this hypothesis, we injected olig2:dsred;adora2ab mutant embryos at the one-cell stage with 1–2 nL of 200 ng/μL TeNT mRNA to block vesicular release. Consistent with our hypothesis, we observed a significant increase in the number of peripheral OPCs in adora2ab−/− mutant larvae compared with WT when neuronal activity was inhibited (Figure 4G; n = 22 WT, n = 22 adora2ab+/−, n = 13 adora2ab−/−, n = 36 WT + TeNT, n = 32 adora2ab+/− + TeNT, n = 20 adora2ab−/− + TeNT; p < 0.0001, WT + TeNT versus adora2ab−/− + TeNT; p < 0.0001, adora2ab−/− versus adora2ab−/− + TeNT). As a way to demonstrate that A2ab is specifically enhancing the TeNT phenotype, we injected TeNT mRNA into zebrafish embryos harboring a mutation in G protein coupled receptor 126 (gpr126) at the one-cell stage. In these mutant larvae, we never see ectopic OPCs under normal conditions, and gpr126 does not play a role in neuromodulation. However, we would expect injection of TeNT mRNA to result in the ectopic migration of OPCs. We therefore quantified the number of ectopic OPCs in TeNT-injected gpr126 mutant larvae at 3 dpf and observed that the number of ectopic OPCs is not significantly different from TeNT-injected WT larvae (Figure 4G; p = 0.99, WT + TeNT versus gpr126−/− + TeNT; n = 4 gpr126+/+, n = 5 gpr126−/− + TeNT). Similar to control adora2ab−/− larvae, TeNT-injected adora2ab−/−, gpr126−/−, and WT larvae do not show any morphological defects at 3 or 5 dpf (Figures S1C and S1D). From these data, we hypothesize that the enhanced phenotype in adora2ab mutant larvae injected with TeNT mRNA is due to a synergistic inhibition of neural activity. Therefore, we conclude that neural activity, driven partially by A2a ARs, leads to OPC restriction at the MEP TZ.
We next sought to demonstrate that OPC migration at MEP TZs was regulated by multiple mechanisms involving both neural modulation and cellular restriction. On the basis of our previous finding that laser ablation of MEP glia results in ectopic migration of OPCs into the periphery (Smith et al., 2014), we performed MEP glial ablation at 55 hpf in sox10:eos larvae that were injected with 1–2 nL of 200 ng/μL TeNT mRNA at the one-cell stage. In these larvae, we observed a significant increase in the percentage of peripheral nerves with OPC bodies and/or processes when MEP glia and vesicular release were ablated in comparison with MEP glial ablation alone or inhibition of neuronal activity alone (Figure 4H; p = 0.004, TeNT versus WT; p < 0.0001, MEP glia ablated versus MEP glia present; n = 5 larvae [uninjected], n = 7 larvae [TeNT]). Together, these data indicate that there are multiple, independent mechanisms regulating OPC migration at MEP TZs.
Neuronal Activity Influences Peripheral OPC Migration
Adenosine signaling through A2a ARs is a neuromodulatory pathway that acts to increase neurotransmitter release (Ciruela et al., 2006; Golder et al., 2008; Rebola et al., 2008). To confirm that adenosine signaling through A2a ARs regulates peripheral OPC migration by modifying neuronal activity in the spinal cord, we assessed the expression of the A2a AR.
In these studies, we observed A2a antibody expression at 60 and 72 hpf on peripheral motor and sensory axons, as well as on neuronal cell bodies and axons in the spinal cord, including olig2+ motor neurons (Figures 5A and S7A). As another method to detect expression, we used in situ hybridization to observe adora2ab mRNA expression. Consistent with our results using the A2a antibody, we observed adora2ab expression in spinal cord neurons, some of which were located in the ventral spinal cord, where motor neurons reside (Figure 5B). We did not detect any A2a AR or adora2ab expression on OPCs or any other glial cells at these stages, which is consistent with published RNA sequencing (RNA-seq) datasets that do not show adora2a expression in mouse OPCs (Zhang et al., 2014). When we used a fluorescently tagged SCH-58261 (SCH-red), we observed SCH-red binding to olig2+ spinal cord motor neurons at 25 hpf (Figure 5C). This expression pattern matches the previously published expression of adora2ab mRNA in spinal cord neurons and further supports the selectivity of SCH-58261 for zebrafish A2a ARs on neurons (Boehmler et al., 2009).
Figure 5. A2a AR Inhibition Reduces Neural Activity of Spinal Cord Neurons.

(A) Lateral views of a 60 hpf WT olig2:dsred;sox10:eos larva stained with an A2a AR antibody. Top: spinal cord (CNS) with A2a expression in a subset of olig2+ motor neurons (asterisks). Bottom (PNS): peripheral spinal nerves with A2a expression in motor (closed arrowheads) and sensory (open arrowheads) axons.
(B) adora2ab mRNA expression at 72 hpf in the spinal cord of a WT larva. Yellow asterisks denote motor neurons.
(C) Twenty-five hours post-fertilization olig2:egfp embryo labeled with SCH-red fluorescent antagonist. Arrowheads mark spinal cord olig2+/SCH-red+ motor neurons.
(D) Mean ± SEM of the fold change in ΔF/F0 over time per individual firing event for larvae treated with DMSO (n = 8 neurons from eight larvae) and SCH-58261 (n = 11 neurons from ten larvae); p = 0.01. Scale bars, 20 μM.
Extracellular adenosine binding to A2a ARs on neurons increases firing, and in the spinal cord, A2a ARs increase motor neuron firing (Golder et al., 2008; Patel et al., 2001; Sebastião and Ribeiro, 2015). Therefore, we hypothesized that inhibiting A2a ARs decreased neuronal activity, which resulted in peripheral OPC migration. To test this hypothesis, we first tested whether inhibiting A2a ARs decreased neuronal activity in vivo. Using the calcium indicator GCaMP5 to detect neuronal firing, we quantified calcium transients as a measure of neuronal activity in 72 hpf neurod:gal4;UAS:GCaMP5 larvae treated with SCH-58261 or DMSO. Because spontaneous neuronal activity in the spinal cord was not robust in 72 hpf larvae, we induced neuronal firing by treating the larvae with the potassium channel blocker 4-aminopyridine 20 min prior to imaging (Ellis et al., 2012). We then measured the change in fluorescence intensity over time in ventral spinal cord neurons and quantified fluorescence spikes compared with baseline. Treatment with the A2a antagonist SCH-58261 significantly reduced neuronal firing compared with control, such that ΔF/F0 values rarely rose above baseline (Figures 5D and S7B). We conclude from this experiment that, consistent with the effects of A2a antagonists in other species, antagonizing A2a receptors with SCH-58261 decreases neuronal activity in zebrafish.
Because developing zebrafish have a combination of electrically and chemically coupled neurons, we also tested the effect of inhibiting activity of electrically coupled neurons with carbenoxolone, a gap junction blocker (Rozental et al., 2001; Saint-Amant and Drapeau, 2001; Solomon et al., 2003; Weber and Ross, 2003). We treated olig2:dsred embryos from 30 hpf to 3 dpf and quantified peripheral OPCs at 3 dpf. Consistent with our hypothesis, blocking gap junctions also resulted in significant numbers of peripherally migrated OPCs (Figure S7C; p <0.001, 10 μM carbenoxolone; p < 0.0001, 20 μM carbenoxolone; n = 5–10 larvae per dose) (Saint-Amant and Drapeau, 2001).
Peripherally Migrated OPCs Rescue Myelin Deficits in Peripheral Myelin Mutants
Our data demonstrate that A2a AR inhibition results in OPC migration onto peripheral spinal motor nerves without otherwise disturbing overall nerve development. This led us to hypothesize that OPCs could be recruited onto peripheral nerves in order to myelinate them. Previous studies from our lab demonstrate that peripheral OPCs are capable of initiating myelination of peripheral nerves (Kucenas et al., 2009; Morris et al., 2017; Smith et al., 2014). However, in these studies, OPCs populated the nerve in the context of genetic mutations resulting in the death or absence of both SCs and MEP glia. Therefore, we wanted to determine if it was possible to recruit OPCs to myelinate peripheral motor nerves in a model more closely resembling human peripheral neuropathies. To do this, we used gpr126 mutant larvae, a model in which SCs ensheath, but fail to myelinate, peripheral nerves (Monk et al., 2009). Importantly, OLs are present in normal numbers in these mutants, and central myelination is indistinguishable from WT larvae (Monk et al., 2009).
We first tested whether untreated gpr126 mutant larvae had functional MEP glia. MEP glia along the spinal motor root can be identified by expression of wif1 or by fate mapping with photoconversion of the sox10:eos transgenic line (Smith et al., 2014). Using these tools, we found that MEP glia were present on spinal motor roots in gpr126 mutant larvae at 72 hpf (Figures 6A and 6B). When we quantified peripheral OPC migration in olig2:dsred;gpr126−/− larvae at 3 dpf, we did not observe any peripheral OPCs in mutants treated with DMSO (Figure 6C; n = 8). These data demonstrate that MEP glia are present and functioning properly and that the absence of SC myelin on peripheral nerves is not sufficient to elicit peripheral OPC migration.
Figure 6. Peripheral OPCs Myelinate Spinal Motor Axons in gpr126 Mutant Larvae.

(A) wif1 mRNA expression in WT and gpr126−/− larvae at 3 dpf shows MEP glia (arrowheads). SC, spinal cord; N, notochord.
(B) Live images of photoconverted sox10:eos larvae at 3 dpf show Eos+ MEP glia (arrowheads) in both WT and gpr126−/− larvae.
(C) Mean ± SEM of peripheral OPCs in 3 dpf gpr126−/− larvae treated with DMSO or SCH-58261 from 36 hpf to 3 dpf. n = 8 DMSO, n = 6 SCH-58261. **p = 0.009.
(D) Frames from a 15 h time-lapse movie of a olig2:dsred;nkx2.2a:megfp;gpr126−/− larva treated with SCH-58261 from 36 hpf to 3 dpf; 0’ is 57 hpf. Arrowheads mark a nkx2.2a+/olig2+ OPC ensheathing motor axons.
(E) Images of MBP antibody (arrowheads) on peripheral nerves of 4 dpf olig2:dsred;gpr126−/− larvae treated with DMSO or SCH-58261 from 36 hpf to 3 dpf. Asterisk marks peripheral OPC.(F) Live images of 4 dpf WT and gpr126−/−;olig2:dsred;mbp:egfp larvae treated with DMSO or SCH-58261 from 36 hpf to 3 dpf. Asterisk marks olig2+ peripheral OPC with mbp+ membrane sheaths (arrowheads) around peripheral motor axons. Scale bars, 20 μm.
Therefore, we tested whether treatment with the A2a AR antagonist would result in peripherally migrated OPCs. When we treated gpr126 mutant larvae with 10 μM of the A2a AR antagonist SCH-58261 from 36 hpf to 3 dpf, we observed a significant number of olig2+ OPCs in the periphery compared with DMSO-treated controls (Figure 6C; p = 0.009; n = 6). We then performed in vivo, time-lapse imaging on nkx2.2a:megfp;olig2:dsred;gpr126−/− larvae in order to visualize whether these peripheral OPCs initiated myelination of peripheral motor axons (Kucenas et al., 2008a). In larvae treated with SCH-58261 from 36 hpf to 3 dpf, we observed nkx2.2a+/olig2+ OPCs initiate ensheathment of peripheral spinal motor axons (Figure 6D; Video S2). These time-lapse data rule out the possibility that SCH-58261 treatment stimulates SCs to myelinate the nerve.
To examine whether these membrane sheaths were in fact myelin, we treated gpr126 mutant larvae from 36 hpf to 3 dpf with SCH-58261 or DMSO, fixed them at 4 dpf, and labeled with an antibody specific to myelin basic protein (MBP) (Kucenas et al., 2009). In mutant larvae treated with DMSO, we observed MBP only on a short segment of the proximal nerve root(Figure6E), which is consistent with our previous data showing that differentiated MEP glia express MBP in this location (Smith et al., 2014). In contrast, in mutant larvae treated with the A2a AR antagonist, MBP labeling was present further distally along the nerve (Figure 6E). As further confirmation that peripheral OPCs myelinate spinal motor nerve roots following treatment with SCH-58261, we imaged mbp:egfp;olig2:dsred;gpr126−/− larvae from 72 to 86 hpf (n = 3) (Figure 6F). In these time-lapses, we observed olig2+ OPCs ensheathing peripheral nerves with mbp+ membrane tubes (Video S3). Both the MBP antibody and mbp:egfp labeling were closely associated with an olig2+ cell body, providing further evidence that peripherally migrated OPCs, and not SCs, initiate myelination in gpr126 mutant larvae treated with SCH-58261 (Figures 6D and 6E). From these experiments, we conclude that OPCs can be recruited onto peripheral motor nerves and initiate myelination, even in the presence of MEP glia and SCs.
DISCUSSION
At MEP TZs, motor axons originating within the spinal cord cross through the glia limitans and transition into the PNS (Bonanomi and Pfaff, 2010; Siebel et al., 2014). OPCs, in contrast, originate within the CNS but do not cross into the periphery, despite being highly migratory and extending membrane processes into the TZ (Fraher and Kaar, 1984; Smith et al., 2014). The mechanisms responsible for allowing axons and other glial populations to cross through the MEP TZ while preventing OPCs are not fully understood. Using selective inhibitors for AR subtypes combined with genetic manipulation, we show that neuronal activity, at least partially mediated by A2a ARs, is involved in regulating OPC migration at the MEP TZ.
Adenosine and Activity-Dependent Regulation of OPCs
Adenosine is a ubiquitous extracellular signaling ligand in the CNS and PNS (Abbracchio et al., 2009; Welsh and Kucenas, 2018). It can be released directly from cells into the extracellular space through nucleoside transporters and can also be derived from hydrolysis of extracellular ATP, which is released from synaptic vesicles by most, if not all, neurons (Burnstock, 1972; Fields and Burnstock, 2006). In the spinal cord, adenosine modulates the activity of motor neurons (Golder et al., 2008; Patel et al., 2001). In our studies, we demonstrate that motor neurons and interneurons within the spinal cord express A2ab receptors and that inhibiting A2ab decreases neuronal activity. These results are consistent with data from many other model systems showing that neuronal A2a receptors act to promote activity of spinal cord motor neurons as well as other kinds of CNS neurons (Sebastião and Ribeiro, 2015).
The data we present fit well with growing evidence from multiple labs that neuronal activity regulates OPC and OL behaviors, including migration, differentiation, and myelination (Almeida and Lyons, 2017; Czopka, 2016; Gallo et al., 2008). Electrophysiological and in vivo time-lapse imaging studies demonstrate that OPCs detect changes in neuronal activity or a variety of directly applied neurotransmitters (Baraban et al., 2018; Lin and Bergles, 2004). Studies in mice and zebrafish also demonstrate that increased neuronal activity promotes OPC differentiation and myelination, whereas decreased activity inhibits differentiation and leads to hypomyelination (Gibson et al., 2014; Hines et al., 2015; Makinodan et al., 2012; Mensch et al., 2015; Mitew et al., 2018; Stevens et al., 2002). In addition, decreased neuronal activity results in altered OPC distribution and migration in mouse brains (Mangin et al., 2012; Tong et al., 2009). We hypothesize that because neuronal activity leads to OPC differentiation, decreased activity in our experiments may affect OPC differentiation, which could cause them to maintain their highly migratory state. Our findings that perturbing neural activity with SCH-58261, genetic disruption of A2ab ARs or TeNT injection leads to ectopic OPC migration, are consistent with these studies and the hypothesis that neuronal activity regulates OPC migration and differentiation.
Therefore, we propose a model in which A2a AR regulation of neuronal activity participates in preventing peripheral migration of OPCs. A2a ARs are excitatory Gs protein-coupled receptors, which upon activation by adenosine, activate adenylyl cyclase that in turn causes increased intracellular cyclic AMP levels and activation of PKA (Borea et al., 2018). Through this cascade, A2a AR activation not only modulates neuronal activity, both pre- and post-synaptically, by increasing neurotransmitter release and cellular excitability respectively, but also modulates neuronal activity non-synaptically by depolarizing neurons through the regulation of ion channels such as calcium and potassium channels (Ribeiro and Sebastião, 2010). For these reasons, we believe that inhibiting vesicular release by tetanus toxin and the blocking of A2a AR signaling are distinct mechanisms that independently result in decreased neuronal activity, and when disrupted together, lead to a synergistic inhibition of neuronal activity and an enhanced phenotype of peripheral OPCs.
Multiple Mechanisms Mediate OPC Segregation at the MEP TZ
In a previous study, we showed that MEP glia restrict OPCs to the CNS by contact-mediated repulsion at MEP TZs (Smith et al., 2014). In the present article, we demonstrate that adenosine signaling, mediated at least partially by A2ab ARs, is another mechanism regulating OPC restriction at the MEP TZ, independent of MEP glial function. We provide evidence that unbalancing neuronal activity, either by antagonizing adenosine signaling or by blocking vesicular release or gap junctions, leads to ectopic peripherally migrated OPCs. Our current data show that affecting several of these mechanisms results in a cumulative phenotype and leads to more OPCs migrating onto peripheral nerves, suggesting that multiple independent mechanisms regulate OPC migration at the MEP TZ.
This supports our hypothesis that a large range of mechanisms could be compensating for the disrupted adenosine signaling in adora2ab mutant larvae and balance neuronal activity. We sought to identify the compensatory mechanisms masking the function of A2ab in OPC restriction in adora2ab mutant larvae and hypothesized that adora2aa was compensating for adora2ab. Surprisingly, there was no genetic compensation of adora2ab by its paralog adora2aa. Other mechanisms, including other ARs, could be compensating for the lack of A2ab. Eight other ARs, encoded by eight distinct genes, including the newly identified adora2c (Wakisaka et al., 2017), could individually or synergistically compensate for A2ab. Because adenosine is a neuromodulator and because genetic compensation is not limited to genes from a same family (Rossi et al., 2015), we also cannot rule out the possibility that other neuromodulators and neurotransmitters could compensate for the loss of A2ab ARs and balance neuronal activity. Glutamate signaling is an attractive candidate, as OPCs express receptors for this neuro-transmitter (Lin and Bergles, 2004) and motor neurons can release glutamate in the spinal cord (Nishimaru et al., 2005). Synapses between OPCs and neurons have been observed using electron microscopy, and vesicular release of glutamate from neurons activates AMPA and/or NMDA receptors on OPCs (Bergles et al., 2000). We also acknowledge the possibility that OPCs could respond to other factors released from neurons in an activity-dependent manner, such as ATP (Fields and Stevens, 2000).
Potential for Peripheral OPCs to Myelinate Peripheral Nerves
Our findings also offer intriguing possibilities for the treatment of peripheral neuropathies. We and others have shown that OPCs are capable of myelinating peripheral axons (Coulpier et al., 2010; Kucenas et al., 2009; Morris et al., 2017; Smith et al., 2014). However, these previous studies were in the context of genetic mutations that result in the loss of all peripheral myelinating glia. In our studies using gpr126 mutant larvae, SCs are present on nerves, but they fail to make myelin, which is a feature of some hypomyelinating peripheral myelinopathies (Monk et al., 2011). Our data as well as previous studies in mice and zebrafish demonstrate no peripheral OPC migration in mutants with peripheral hypomyelination, and it is unclear whether OPCs would naturally migrate onto peripheral nerves in human neuropathy patients (Coulpier et al., 2010; Monk et al., 2009, 2011). Because of this, we are excited by the possibility of inducing OPCs to migrate onto peripheral nerves that lack myelin. Once in the periphery, our studies demonstrate that OPCs are competent to initiate myelination without continued drug treatment. In combination with previous studies, this is strong evidence for the potential for OPCs to myelinate peripheral nerves in disease.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Sarah Kucenas (sk4ub@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fish Husbandry
All animal studies were approved by the University of Virginia Institutional Animal Care and Use Committee. Zebrafish strains used in this study were: AB*, Tg(sox10(4.9):eos)w9, Tg(sox10(4.9):nls-eos)w18 (Prendergast et al., 2012), Tg(olig2:egfp)vu12, Tg(olig2:dsred)vu19 (Shin et al., 2003), Tg(nkx2.2a:megfp)vu17 (Kucenas et al., 2008b), Tg(mbp:egfp-CAAX) (Almeida et al., 2011), adora2aact845, adora2abuva6, adora2abuva7, adora2abuva9, Tg(neuroD:gal4)uva22 and gpr126st49 (Monk et al., 2009). Table S1 denotes abbreviations used for each strain and summarizes what each transgene labels. Embryos were raised at 28.5°C in egg water and staged by hours or days post fertilization (hpf and dpf, respectively). Embryos of either sex were used for all experiments (Kimmel et al., 1995). Phenylthiourea (PTU) (0.004%) in egg water was used to reduce pigmentation for imaging. Stable, germline transgenic lines were used in all experiments.
METHOD DETAILS
Chemical treatments
The LOPAC®1280 library (Sigma Cat. No. LO1280) was used for the chemical screen. 384 well masterplates, a gift from Drs. John Lazo and Elizabeth Sharlow, were replated and diluted with DMSO to create 1 mM 96-well stock plates. At 24 hours post fertilization (hpf), olig2:dsred embryos were dechorionated using 0.2% pronase. Embryos remained in pronase for 2 minutes at 25°C and were washed 4 times with egg water at 25°C. One embryo was placed in each well of a 96 well plate with 198 μL of water. 2 μL of compound from the 96 well stock plate was added to the water for a final concentration of 10 μM compound and 1% DMSO. For the initial screen, we tested each drug once in 1 well of a 96 well plate. Each row of the plate also contained 1 positive and 1 negative control well. 4 μM AG1478 was used as a positive control and 1% DMSO was used as a negative control for each plate. Plates were covered with a low evaporation lid and placed in a 28.5°C incubator until analysis at 3 dpf. Plates were analyzed on a Zeiss AxioObserver inverted microscope equipped with epifluorescence using a 10× objective (NA = 0.3). At 3 dpf, the presence or absence of peripheral olig2+ cells was scored for each larva. All positive hits from the primary screen were retested on 3 embryos in a secondary screen in which LOPAC®1280 compounds, AG1478, and 1% DMSO treatments were randomized across the plate and all experimenters were blinded to the identity of compounds or controls until after analysis. Any experiment in the secondary screen in which fewer than 8 positive control embryos or greater than one DMSO treated embryo had ectopic OPCs was excluded and later repeated. For all subsequent drug experiments, new chemical stocks were obtained from suppliers, and embryos were manually dechorionated using forceps. 1% DMSO was used as a negative control for all drug treatment experiments, except carbenoxolone and MK-801, which were dissolved in distilled water. Using the same microscope and objective as in the original screen, we quantified peripheral olig2+ cells for whole larvae, excluding the first (anterior) 2 somites which are obscured by the yolk, and the last (posterior) 4 somites in the tail, which develop more slowly than the rest of the trunk.
In vivo imaging and photoconversion
Embryos were anesthetized with 0.01% 3-aminobenzoic acid ester (Tricaine), immersed in 0.8% low-melting point agarose and mounted laterally in glass-bottomed 35 mm Petri dishes (Electron Microscopy Sciences). After mounting, the Petri dish was filled with egg water containing PTU and Tricaine. For some experiments, chemical compounds were also dissolved in the water. A 25× multi-immersion objective (NA = 0.8), 40× oil objective (NA = 1.4) and a 40× water objective (NA = 1.1) mounted on a motorized Zeiss AxioObserver ZI microscope equipped with a Quorum WaveFX-XI spinning disc confocal system (Quorum Technologies Inc.) were used to capture images. Image processing was performed with MetaMorph and Photoshop to enhance brightness and contrast of images. The Fiji plugin MTrackJ was used to annotate time-lapse movies (Meijering et al., 2012).
We used fate mapping with photoconversion of the sox10:eos transgenic line to identify MEP glia in some experiments. The nascent Eos protein exists in a green fluorescent state, but when exposed to ultraviolet (UV) light, it permanently shifts to a red fluorescent state. When exposed to UV light at 48 hpf, neural crest-derived cells are photoconverted to red fluorescence. MEP glia, which are not neural crest-derived and begin expressing sox10:eos after 48 hpf, are not photoconverted and can be identified as GFP+ cells on the nerve root (Smith et al., 2014). For photoconversion, the entire trunk of sox10:eos larvae was exposed to 30 s of UV light through a DAPI filter using a 20× objective (NA = 0.8).
Cell ablation
Single cell ablations were performed with a nitrogen-pulsed MicroPoint laser using a coumarin dye (435nm) with a 63× (MEP glia ablation) water immersion objectives. After pre ablation images were acquired, a region of interest (ROI) was created based off the merged-color image around MEP glia to selectively laser ablate single cells.
Calcium imaging
We injected the calcium indicator UAS:GCaMP5 DNA construct into stable transgenic zebrafish embryos neuroD:gal4. The combination of UAS:GCaMP5 and neuroD:gal4 leads to mosaic expression of GcAMP5 in CNS neurons. Prior to imaging, larvae expressing GCaMP5 in the ventral spinal cord were treated with 800 μM 4-aminopyridine (4-AP) (Sigma A78403) diluted in egg water for 15 minutes to induce neuronal activity (Ellis et al., 2012), then immediately paralyzed using the neuromuscular junction (NMJ) blocking nicotinic receptor antagonist pancuronium bromide (Sigma P1918) for 10 minutes, which was dissolved in egg water (0.3 mg/mL) (Baraban et al., 2018). Larvae were then embedded in 0.8% low melting point agarose for imaging. To record Ca2+ dynamics, individual neurons of the ventral spinal cord were imaged every 250 ms for 2 minutes, using an exposure time of 100 ms on single z-planes. Images were captured with a 63× water immersion objective (na = 1.2) mounted on a motorized Zeiss AxioObserver ZI microscope equipped with a Quorum Wave FX-XI spinning disc confocal system (Quorum Technologies Inc.). Single-plane time-lapses were processed in MetaMorph with no intensity averaging.
Immunohistochemistry and fluorescent antagonist treatment
Embryos were fixed and stained using the procedure previously described (Smith et al., 2014). Antibodies used were: rabbit anti-A2a (1:100 GeneTex (Andersson et al., 2012)), rabbit anti-Sox10 (1:5000 [Binari et al., 2013]), rabbit anti-MBP (1:250 [Kucenas et al., 2009]), and Alexa 647 goat anti-rabbit (1:600) (ThermoFisher). Fluorescent SCH-58261 (SCH-red) was purchased from CisBio. 25 hpf embryos were immersed in 7.14 μM SCH-red in 30% DMSO for 30 minutes, then fixed in 4% PFA at 25°C for 3 hours. Embryos were mounted in glass-bottomed Petri dishes for imaging as described above.
Morpholino and mRNA injections
Antisense morpholino oligonucleotides (MO) were purchased from Gene Tools. adora2aaMO1 (CATTGTTCAGCATGGTGAGG TCGCT) (Haas et al., 2013) is complementary to the region spanning the translation start codon of adora2aa mRNA, and adora2abMO1 (GTGCTATCAACCAGTGTGAAAGGAT) is complementary to the region immediately 5’ to the start codon of adora2ab mRNA. Embryos were injected with 2 to 3 nL of injection solution (distilled water, 4 mg/ml phenol red, diluted MO) at the 1-cell stage. Any embryos damaged during the injection procedure were removed, and the rest were incubated at 28.5°C. Data presented for each MO are combined from at least 3 independent experiments. Uninjected controls and phenol red control injections were performed at least twice for each dataset.
For Tetanus Toxin experiments, TeNT cDNA was prepared from plasmid pGEMTEZ-TeTxLC (a gift from Marcel Tawk) as previously described (Fontenas et al., 2016). TeNT mRNA was prepared using the mMessage mMachine SP6 in vitro transcription kit (ThermoFisher). Injections of mRNA encoding Tetanus Toxin Light Chain (TeNT) were performed at the one-cell stage. A 2 nL volume of 200 ng/μl mRNA in DEPC-treated distilled water was used. All embryos were manually dechorionated at 2 dpf and evaluated for paralysis at that time. The startle reflex was also analyzed at 3 dpf as further evidence that TeNT expression caused inhibition of neuronal firing. For calcium imaging experiments, 2nL of 100 ng/μL UAS-GCaMP5 DNA construct diluted in injection buffer were injected into neuroD:gal4 embryos at the one cell stage.
Creation of adora2ab mutant line
sgRNA targeting adora2ab was designed using CHOPCHOP (http://chopchop.cbu.uib.no/) and the protocol from https://www.schierlab.fas.harvard.edu/resources/ (Gagnon et al., 2014). We annealed the 5’ gene-specific oligo and the 3’ constant oligo using the PCR protocol described in Nakayama et al. (2014). We transcribed sgRNA using Ambion Megascript T7 kit and injected 2 nL of 200–400 ng/μl sgRNA with 500 ng/μl Cas9 protein (PNA Bio) dissolved in nuclease-free water into cells of olig2:dsred or olig2:egfp embryos at the 1 cell stage. To identify founders, injected embryos were raised to adulthood and mated, then genomic DNA was isolated from a pool of 8 embryos from each potential founder. PCR was performed using the primers CAACTATGTGTGTCCCT GAGGA and ATGAAGAGGCATCCATGAAAAT were to amplify a 279 base pair region of genomic adora2ab, and Sanger sequencing with the primer CAACTATGTGTGTCCCTGAGGA was used to identify CRISPR-induced indels. Sequence trace files were analyzed with ApE or SnapGene. To identify alleles in F1 animals, we cloned single mutated DNA fragments using TOPO cloning. To analyze the CRISPR induced-mutations, DNA from adult fin clips was extracted and amplified by PCR using the adora2ab forward (5’-CAACTATGTGTGTCCCTGAGGA-3’) and reverse (5’-ATGAAGAGGCATCCATGAAAAT-3’) primers. PCR was done with GoTaq green master mix (Promega) and conditions were as follows: 95°C for 3 min, followed by 35 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 1 min. Final extension was at 72°C for 20 min. PCR products from individual embryos were TOPO TA cloned into pCR8/GW or pCR4 vectors (Invitrogen). For each embryo, clones were sequenced using the M13 forward or M13 reverse primer. Sequences were aligned to WT genomic DNA using BLAST global alignment tool. All CRISPR data presented in this manuscript was obtained using stable, germline mutations identified in the F1 generation.
Creation of adora2aa mutant line
A TALEN targeting zebrafish adora2aa was designed using the ZiFIT Targeter (https://omictools.com/zifit-tool) (Sander et al., 2011) and constructed as previously described (Chen et al., 2013). TALEN mRNA was synthesized from purified linear DNA using the mMessage mMachine T7 Ultra kit (Ambion), 50–100pg of TALEN mRNA dissolved in nuclease-free water was injected into zebrafish embryos at the 1-cell stage. To identify founders, injected embryos were raised to adulthood and mated, then genomic DNA was isolated from a pool of 1 to 6 embryos from each potential founder. PCR was performed using the primers GTTTGTTCGAGGA GGTGGTG and ACCACTGAGTTTGCGTGTGA, and indels were identified by analyzing PCR product sizes using Peak Scanner (Applied Biosystems). To confirm indel sequences, genomic DNA from single embryos was amplified and subcloned using the Strataclone PCR Cloning Kit (Stratagene), and DNA sequences were then analyzed with SeqBuilder and SeqMan (DNAStar Lasergene).
Genotyping
Genomic DNA was extracted using HotSHOT (hot sodium hydroxide and tris) (Truett et al., 2000). The primers used for genotyping gpr126st49 have previously been published (Monk et al., 2009). For adora2abuva6, adora2abuva7 and adora2abuva9, PCR was performed using the primers CAACTATGTGTGTCCCTGAGGA and ATGAAGAGGCATCCATGAAAAT. For adora2abuva6 and adora2abuva7 the mutant alleles were identified using agarose gel electrophoresis. For adora2abuva9, the WT PCR product was digested with Tsp45i. The primers AAGCCATCCCATGTGAACTC and TCACATTCAGGGCAGAACAG were used to amplify a 151 bp product within adora2aa. Because the mutation is a 7 bp deletion and is difficult to resolve on an agarose gel, we performed Sanger sequencing using the primer TCACATTCAGGGCAGAACAG to identify WT, heterozygous, and homozygous mutants.
In situ hybridization and sectioning
Larvae were fixed in 4% PFA at 4°C overnight and stored in 100% methanol at 20°C and processed for in situ RNA hybridization. Plasmids were linearized with appropriate restriction enzymes and cRNA preparation was carried out using Roche DIG-labeling reagents and RNA polymerases (NEB). We used previously published probes for wif1 and adora2ab (Boehmler et al., 2009; Smith et al., 2014). After in situ hybridization, embryos were embedded in 1.5% agarose/30% sucrose and frozen in 2-methylbutane chilled by immersion in liquid nitrogen. We collected 20 μm transverse sections on microscope slides using a cryostat microtome and covered with 75% glycerol. For adora2ab, in situ hybridization was performed on 20 μm tissue sections. Images were obtained using a Zeiss AxioObserver inverted microscope using a 40× oil immersion objective. All images were imported into Adobe Photoshop. Adjustments were limited to levels, contrast, and cropping.
QUANTIFICATION AND STATISTICAL ANALYSIS
GCaMP5 analysis
Fluorescence intensity measurements were extracted from ROIs using ImageJ and imported to Excel. ΔF/F0 were measured by applying the following formula ΔF/F0 = (Ft-F0)/(F0-Fbackground), where Ft is the fluorescence intensity in the ROI in which the calcium transient was observed at time t, F0 is the average fluorescence intensity of first three frames of the timelapse in the same ROI, and Fbackground is the fluorescence intensity of the background at time t (Baraban et al., 2018). For each individual neuron, the average baseline fluorescence was determined by averaging the ΔF/F0 of all 500 time points. For fold change quantification, all ΔF/F0 values one standard deviation or more above the baseline were considered firing events, and their average was compared to the baseline.
Statistical analysis
GraphPad Prism was used for all statistical analyses. Unpaired Student’s t test or, for multiple comparisons, 1-way ANOVA followed by Bonferonni post test, were used for quantifications of OPCs, MEP glia, Eos+ peripheral cells. 2-way ANOVA followed by Tukey’s multiple comparison test was used for quantification of peripheral OPCs in adora2ab mutant larvae injected with TeNT, and for MEP glial ablation combined with TeNT. Chi-square analysis was used for quantification of paralysis and startle reflex, and survival test was used for analyzing mutant survival and hatching. A p value less than 0.05 was considered statistically significant. For dose response experiments, 10 to 12 embryos were treated per dose, and n is reported as the range of larvae analyzed per dose.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-A2a | GeneTex | Cat#GTX23461; RRID: AB_367170 |
| Rabbit anti-Sox10 | Binari et al., 2013 | N/A |
| Rabbit anti-MBP | Kucenas et al., 2009 | N/A |
| Alexa Fluor 647 goat anti-rabbit IgG(H+L) | ThermoFisher | Cat#A-21244; RRID: AB_2535812 |
| Sheep Anti-Digoxigenin-AP, Fab fragments | Sigma | Cat#11093274910; RRID: AB_514497 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fluorescent SCH-58261 | CisBio | Cat#L0058RED |
| Cas9 protein | PNA Bio | Cat#CP01 |
| 4-aminopyridine | Sigma | Cat#A78403; CAS number 504-24-5 |
| Pancuronium bromide | Sigma | Cat#P1918; CAS number 15500-66-0 |
| 3-aminobenzoic acid ester (Tricaine) | Syndel | N/A |
| LOPAC®1280 | Sigma | Cat#LO1280 |
| 1-Phenyl-2-thiourea (PTU) | Sigma | Cat#P7629; CAS number 103-85-5 |
| SCH-58261 | Sigma | Cat#S4568; CAS number160098-96-4 |
| AG1478 | Sigma | Cat#658548; CAS number 175178-82-2 |
| CGS-15943 | Sigma | Cat#C199; CAS number 104615-18-1 |
| MRS-1754 | Sigma | Cat#M6316 |
| 8-cyclopentyltheophylline (CPT) | Sigma | Cat#C102; CAS number 35873-49-5 |
| MRS-1191 | Sigma | Cat#M227; CAS number 185222-90-6 |
| CGS-21680 | Sigma | Cat#119137 CAS number 124182-57-6 |
| Dimethyl sulfoxide (DMSO) | Sigma | Cat#D2650; CAS number 67-68-5 |
| Carbenoxolone | Sigma | Cat#C4790; CAS number 7421-40-1 |
| DIG RNA labeling mix | Sigma | Cat#11 277 073 910 |
| Critical Commercial Assays | ||
| mMessage mMachine SP6 in vitro transcription kit | ThermoFisher | Cat#AM1340 |
| Megascript T7 kit | Thermofisher | Cat#AM1334 |
| mMessage mMachine T7 Ultra kit | Thermofisher | Cat#AM1345 |
| Strataclone PCR Cloning Kit | Stratagene | Cat# 240205 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish: Tg(sox10(4.9):eos)w9 | Prendergast et al., 2012 | ZDB-ALT-110721–1 |
| Zebrafish: Tg(sox10(4.9):nls-eos)w18 | Prendergast et al., 2012 | ZDB-ALT-110721–2 |
| Zebrafish: Tg(olig2:egfp)vu12 | Shin et al., 2003 | ZDB-ALT-041129–8 |
| Zebrafish: Tg(olig2:dsred)vu19 | Shin et al., 2003 | ZDB-ALT-080321–2 |
| Zebrafish: Tg(nkx2.2a:megfp)vu17 | Kucenas et al., 2008b | ZDB-ALT-080321–1 |
| Zebrafish: Tg(mbp:egfp-caax)ue2 | Almeida et al., 2011 | ZDB-ALT-120103–2 |
| Zebrafish: adora2aact845 | This paper | N/A |
| Zebrafish: adora2abuva6 | This paper | N/A |
| Zebrafish: adora2abuva7 | This paper | N/A |
| Zebrafish: adora2abuva9 | This paper | N/A |
| Zebrafish: gpr126st49 | Monk et al., 2009 | ZDB-ALT-061207–4 |
| Zebrafish: Tg(neuroD:gal4)uva22 | This paper | N/A |
| Oligonucleotides | ||
| adora2aaMO1 (CATTGTTCAGCATGGTGAGGTCGCT) | GeneTools | N/A |
| adora2abMO1 (GTGCTATCAACCAGTGTGAAAGGAT) | GeneTools | N/A |
| Adora2aa FWD AAGCCATCCCATGTGAACTC | IDT | N/A |
| Adora2aa REV TCACATTCAGGGCAGAACAG | IDT | N/A |
| Adora2ab FWD CAACTATGTGTGTCCCTGAGGA | IDT | N/A |
| Adora2ab REV ATGAAGAGGCATCCATGAAAAT | IDT | N/A |
| Gpr126 FWD TAGAGATTGCACATTTGGATTA | Monk et al., 2009 | N/A |
| Gpr126 REV ATCGGAACACACCAAACAGG | Monk et al., 2009 | N/A |
| Recombinant DNA | ||
| pGEMTEZ-TeTxLC | Addgene | Cat#32640 |
| pCR8/GW | Invitrogen | Cat#K250020 |
| pCR4 | Invitrogen | Cat#450030 |
| pDestTol2-pA2-UAS-GCaMP5 | This paper | N/A |
| Software and Algorithms | ||
| MetaMorph | Molecular Devices | moleculardevices.com |
| ImageJ | NIH | ImageJ |
| Adobe Photoshop CC 2017 | Adobe | Adobe |
| Adobe Illustrator CC 2017 | Adobe | Adobe |
| Microsoft Excel | Microsoft | Microsoft |
| Prism 6 | GraphPad | graphpad.com |
Highlights.
A2ab adenosine receptor regulates oligodendrocyte migration at transition zones (TZs)
Vesicular-mediated release of neurotransmitter compensates in A2ab mutants
Inhibition of both processes cooperatively results in enhanced OPC migration at TZs
Ectopic OPCs can rescue the lack of myelin in peripheral hypomyelination mutants
ACKNOWLEDGMENTS
We would like to thank Drs. John Lazo and Elizabeth Sharlow for the LOPAC1280 library and for advice on designing the chemical screen. We thank Dr. Marcel Tawk for providing the pGEMTEZ-TeTxLC plasmid and Dr. Cody Smith for the UAS:GCaMP5 plasmid. We are extremely grateful to Lori Tocke for care and maintenance of the zebrafish facility and to all the members of the Kucenas lab and Dr. Dave Parichy for helpful comments and advice. This work was funded by the NIH through grants NS072212 and NS092070 (S.K.), NS070911 (D.A.P.), and NS101665 (D.A.P.) and by The Hartwell Foundation (S.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.013.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abbracchio MP, Burnstock G, Verkhratsky A, and Zimmermann H (2009). Purinergic signalling in the nervous system: an overview. Trends Neurosci 32, 19–29. [DOI] [PubMed] [Google Scholar]
- Agresti C, Meomartini ME, Amadio S, Ambrosini E, Serafini B, Fran-chini L, Volonté C, Aloisi F, and Visentin S (2005a). Metabotropic P2 receptor activation regulates oligodendrocyte progenitor migration and development. Glia 50, 132–144. [DOI] [PubMed] [Google Scholar]
- Agresti C, Meomartini ME, Amadio S, Ambrosini E, Volonté C, Aloisi F, and Visentin S (2005b). ATP regulates oligodendrocyte progenitor migration, proliferation, and differentiation: involvement of metabotropic P2 receptors. Brain Res. Brain Res. Rev 48, 157–165. [DOI] [PubMed] [Google Scholar]
- Almeida RG, and Lyons DA (2017). On myelinated axon plasticity and neuronal circuit formation and function. J. Neurosci 37, 10023–10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RG, Czopka T, Ffrench-Constant C, and Lyons DA (2011). Individual axons regulate the myelinating potential of single oligodendrocytes in vivo. Development 138, 4443–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, German MS, and Stainier DY (2012). Adenosine signaling promotes regeneration of pancreatic b cells in vivo. Cell Metab 15, 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban M, Koudelka S, and Lyons DA (2018). Ca2+ activity signatures of myelin sheath formation and growth in vivo. Nat. Neurosci 21, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, and Richardson WD (2015). Oligodendrocyte development and plasticity. Cold Spring Harb. Perspect. Biol 8, a020453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, and Jahr CE (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191. [DOI] [PubMed] [Google Scholar]
- Binari LA, Lewis GM, and Kucenas S (2013). Perineurial glia require Notch signaling during motor nerve development but not regeneration. J. Neurosci 33, 4241–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, and Bonas U (2009). Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Boehmler W, Petko J, Woll M, Frey C, Thisse B, Thisse C, Canfield VA, and Levenson R (2009). Identification of zebrafish A2 adenosine receptors and expression in developing embryos. Gene Expr. Patterns 9, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanomi D, and Pfaff SL (2010). Motor axon pathfinding. Cold Spring Harb. Perspect. Biol 2, a001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borea PA, Gessi S, Merighi S, Vincenzi F, and Varani K (2018). Pharmacology of adenosine receptors: the state of the art. Physiol. Rev 98, 1591–1625. [DOI] [PubMed] [Google Scholar]
- Burnstock G (1972). Purinergic nerves. Pharmacol. Rev 24, 509–581. [PubMed] [Google Scholar]
- Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, and Schwarzschild MA (1999). A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J. Neurosci 19, 9192–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Oikonomou G, Chiu CN, Niles BJ, Liu J, Lee DA, Antoshechkin I, and Prober DA (2013). A large-scale in vivo analysis reveals that TALENs are significantly more mutagenic than ZFNs generated using context-dependent assembly. Nucleic Acids Res 41, 2769–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TJ, Kula B, Nagy B, Barzan R, Gall A, Ehrlich I, and Kukley M (2018). In vivo regulation of oligodendrocyte precursor cell proliferation and differentiation by the AMPA-receptor subunit GluA2. Cell Rep 25, 852–861.e7. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, et al. (2006). Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1℃A2A receptor heteromers. J. Neurosci 26, 2080–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JK, O’keefe A, Mastracci TL, Sussel L, Matise MP, and Kucenas S (2014). Mammalian Nkx2.2+ perineurial glia are essential for motor nerve development. Dev. Dyn 243, 1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppi E, Maraula G, Fumagalli M, Failli P, Cellai L, Bonfanti E, Mazzoni L, Coppini R, Abbracchio MP, Pedata F, and Pugliese AM (2013). UDP-glucose enhances outward K(+) currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia 61, 1155–1171. [DOI] [PubMed] [Google Scholar]
- Coppi E, Cellai L, Maraula G, Dettori I, Melani A, Pugliese AM, and Pedata F (2015). Role of adenosine in oligodendrocyte precursor maturation. Front. Cell. Neurosci 9, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulpier F, Decker L, Funalot B, Vallat JM, Garcia-Bragado F, Charnay P, and Topilko P (2010). CNS/PNS boundary transgression by central glia in the absence of Schwann cells or Krox20/Egr2 function. J. Neurosci 30, 5958–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czopka T (2016). Insights into mechanisms of central nervous system myeli-nation using zebrafish. Glia 64, 333–349. [DOI] [PubMed] [Google Scholar]
- de Lera Ruiz M, Lim Y-HH, and Zheng J (2014). Adenosine A2A receptor as a drug discovery target. J. Med. Chem 57, 3623–3650. [DOI] [PubMed] [Google Scholar]
- Dennis J, Morgan MK, Graf MR, and Fuss B (2012). P2Y12 receptor expression is a critical determinant of functional responsiveness to ATX’s MORFO domain. Purinergic Signal 8, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy M, Rossi A, Kontarakis Z, Kuenne C, Guenther S, Fukuda N, Takacs C, Lai S-L, Fukuda R, Gerri C, et al. (2018). Genetic compensation is triggered by mutant mRNA degradation. bioRxiv 10.1101/328153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis LD, Seibert J, and Soanes KH (2012). Distinct models of induced hyperactivity in zebrafish larvae. Brain Res 1449, 46–59. [DOI] [PubMed] [Google Scholar]
- Emery B, and Lu RQ (2015). Transcriptional and epigenetic regulation of oligodendrocyte development and myelination in the central nervous system. Cold Spring Harb. Perspect. Biol 7, a020461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria A, Hokanson KC, Dao DQ, Mayoral SR, Mei F, Redmond SA, Ullian EM, and Chan JR (2016). Dynamic modulation of myelination in response to visual stimuli alters optic nerve conduction velocity. J. Neurosci 36, 6937–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, and Burnstock G (2006). Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci 7, 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, and Stevens B (2000). ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci 23, 625–633. [DOI] [PubMed] [Google Scholar]
- Fontenas L, De Santis F, Di Donato V, Degerny C, Chambraud B, Del Bene F, and Tawk M (2016). Neuronal Ndrg4 is essential for nodes of Ranvier organization in zebrafish. PLoS Genet 12, e1006459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraher JP, and Kaar GF (1984). The transitional node of Ranvier at the junction of the central and peripheral nervous systems: an ultrastructural study of its development and mature form. J. Anat 139, 215–238. [PMC free article] [PubMed] [Google Scholar]
- Fröb F, Bremer M, Finzsch M, Kichko T, Reeh P, Tamm ER, Charnay P, and Wegner M (2012). Establishment of myelinating Schwann cells and barrier integrity between central and peripheral nervous systems depend on Sox10. Glia 60, 806–819. [DOI] [PubMed] [Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, and Schier AF (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 9, e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Mangin JM, Kukley M, and Dietrich D (2008). Synapses on NG2-expressing progenitors in the brain: multiple functions? J. Physiol 586, 3767–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golder FJ, Ranganathan L, Satriotomo I, Hoffman M, Lovett-Barr MR, Watters JJ, Baker-Herman TL, and Mitchell GS (2008). Spinal adenosine A2a receptor activation elicits long-lasting phrenic motor facilitation.J. Neurosci 28, 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, and Lopez R (2010). A new bioinformatics analysis tools framework at EMBLEBI. Nucleic Acids Res 38, W695–W699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J, Frese KS, Park YJ, Keller A, Vogel B, Lindroth AM, Weichenhan D, Franke J, Fischer S, Bauer A, et al. (2013). Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med 5, 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK, and Appel B (2015). Neuronal activity biases axon selection for myelination in vivo. Nat. Neurosci 18, 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJR, and Joung JK (2013). Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis MF, and Williams M (1989). Direct autoradiographic localization of adenosine A2 receptors in the rat brain using the A2-selective agonist, [3H] CGS 21680. Eur. J. Pharmacol 168, 243–246. [DOI] [PubMed] [Google Scholar]
- Jiang JL, van Rhee AM, Melman N, Ji XD, and Jacobson KA (1996). 6-phenyl-1,4-dihydropyridine derivatives as potent and selective A3 adeno-sine receptor antagonists. J. Med. Chem 39, 4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, and Attwell D (2007). Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience 145, 1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keuerleber S, Gsandtner I, and Freissmuth M (2010). From cradle to twilight: the carboxyl terminus directs the fate of the A2A-adenosine receptor. Biochim. Biophys. Acta 1808, 1350–1357. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev. Dyn 203, 253–310. [DOI] [PubMed] [Google Scholar]
- Kirby BB, Takada N, Latimer AJ, Shin J, Carney TJ, Kelsh RN, and Appel B (2006). In vivo time-lapse imaging shows dynamic oligodendrocyte progenitor behavior during zebrafish development. Nat. Neurosci 9, 1506–1511. [DOI] [PubMed] [Google Scholar]
- Kucenas S, Snell H, and Appel B (2008a). nkx2.2a promotes specification and differentiation of a myelinating subset of oligodendrocyte lineage cells in zebrafish. Neuron Glia Biol 4, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucenas S, Takada N, Park HC, Woodruff E, Broadie K, and Appel B (2008b). CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat. Neurosci 11, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucenas S, Wang WD, Knapik EW, and Appel B (2009). A selective glial barrier at motor axon exit points prevents oligodendrocyte migration from the spinal cord. J. Neurosci 29, 15187–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KE, and Eisen JS (2003). From cells to circuits: development of the zebrafish spinal cord. Prog. Neurobiol 69, 419–449. [DOI] [PubMed] [Google Scholar]
- Lin S-CC, and Bergles DE (2004). Synaptic signaling between neurons and glia. Glia 47, 290–298. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Pogoda HM, Voas MG, Woods IG, Diamond B, Nix R, Arana N, Jacobs J, and Talbot WS (2005). erbb3 and erbb2 are essential for Schwann cell migration and myelination in zebrafish. Curr. Biol 15, 513–524. [DOI] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, and Corfas G (2012). A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangin J-MM, Li P, Scafidi J, and Gallo V (2012). Experience-dependent regulation of NG2 progenitors in the developing barrel cortex. Nat. Neurosci 15, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, and Smal I (2012). Methods for cell and particle tracking. Methods Enzymol 504, 183–200. [DOI] [PubMed] [Google Scholar]
- Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A, and Lyons DA (2015). Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat. Neurosci 18, 628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH (2002). Regulation of oligodendrocyte development in the vertebrate CNS. Prog. Neurobiol 67, 451–467. [DOI] [PubMed] [Google Scholar]
- Mitew S, Gobius I, Fenlon LR, McDougall SJ, Hawkes D, Xing YL, Bujalka H, Gundlach AL, Richards LJ, Kilpatrick TJ, et al. (2018). Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun 9, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk KR, Naylor SG, Glenn TD, Mercurio S, Perlin JR, Dominguez C, Moens CB, and Talbot WS (2009). A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science 325, 1402–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk KR, Oshima K, Jörs S, Heller S, and Talbot WS (2011). Gpr126 is essential for peripheral nerve development and myelination in mammals. Development 138, 2673–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AD, Lewis GM, and Kucenas S (2017). Perineurial glial plasticity and the role of TGF-β in the development of the blood-nerve barrier. J. Neurosci 37, 4790–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou MJ, and Bogdanove AJ (2009). A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501. [DOI] [PubMed] [Google Scholar]
- Nagy B, Hovhannisyan A, Barzan R, Chen TJ, and Kukley M (2017). Different patterns of neuronal activity trigger distinct responses of oligodendrocyte precursor cells in the corpus callosum. PLoS Biol 15, e2001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Blitz IL, Fish MB, Odeleye AO, Manohar S, Cho KW, and Grainger RM (2014). Cas9-based genome editing in Xenopus tropicalis. Methods Enzymol 546, 355–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimaru H, Restrepo CE, Ryge J, Yanagawa Y, and Kiehn O (2005). Mammalian motor neurons corelease glutamate and acetylcholine at central synapses. Proc. Natl. Acad. Sci. USA 102, 5245–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongini E, Dionisotti S, Gessi S, Irenius E, and Fredholm BB (1999). Comparison of CGS 15943, ZM 241385 and SCH 58261 as antagonists at human adenosine receptors. Naunyn Schmiedebergs Arch. Pharmacol 359, 7–10. [DOI] [PubMed] [Google Scholar]
- Othman T, Yan H, and Rivkees SA (2003). Oligodendrocytes express functional A1 adenosine receptors that stimulate cellular migration. Glia 44, 166–172. [DOI] [PubMed] [Google Scholar]
- Park H-C, Mehta A, Richardson JS, and Appel B (2002). olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev. Biol 248, 356–368. [DOI] [PubMed] [Google Scholar]
- Patel MK, Pinnock RD, and Lee K (2001). Adenosine exerts multiple effects in dorsal horn neurones of the adult rat spinal cord. Brain Res 920, 19–26. [DOI] [PubMed] [Google Scholar]
- Prendergast A, Linbo TH, Swarts T, Ungos JM, McGraw HF, Krispin S, Weinstein BM, and Raible DW (2012). The metalloproteinase inhibitor Reck is essential for zebrafish DRG development. Development 139, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, and Mulle C (2008). Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 57, 121–134. [DOI] [PubMed] [Google Scholar]
- Ribeiro JA, and Sebastião AM (2010). Modulation and metamodulation of synapses by adenosine. Acta Physiol. (Oxf.) 199, 161–169. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Smith HK, Sun T, Pringle NP, Hall A, and Woodruff R (2000). Oligodendrocyte lineage and the motor neuron connection. Glia 29, 136–142. [DOI] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M, and Stain-ier DYR (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–233. [DOI] [PubMed] [Google Scholar]
- Rowitch DH (2004). Glial specification in the vertebrate neural tube. Nat. Rev. Neurosci 5, 409–419. [DOI] [PubMed] [Google Scholar]
- Rozental R, Srinivas M, and Spray DC (2001). How to close a gap junction channel. Efficacies and potencies of uncoupling agents. Methods Mol. Biol 154, 447–476. [DOI] [PubMed] [Google Scholar]
- Saint-Amant L, and Drapeau P (2001). Synchronization of an embryonic network of identified spinal interneurons solely by electrical coupling. Neuron 31, 1035–1046. [DOI] [PubMed] [Google Scholar]
- Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, and Yeh J-RJR (2011). Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol 29, 697–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searl TJ, and Silinsky EM (2012). Evidence for constitutively-active aden-osine receptors at mammalian motor nerve endings. Eur. J. Pharmacol 685, 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastião AM, and Ribeiro JA (2015). Neuromodulation and metamodulation by adenosine: Impact and subtleties upon synaptic plasticity regulation. Brain Res 1621, 102–113. [DOI] [PubMed] [Google Scholar]
- Shin J, Park H-CC, Topczewska JM, Mawdsley DJ, and Appel B (2003). Neural cell fate analysis in zebrafish using olig2 BAC transgenics. Methods Cell Sci 25, 7–14. [DOI] [PubMed] [Google Scholar]
- Siebel AM, Vianna MR, and Bonan CD (2014). Pharmacological and toxicological effects of lithium in zebrafish. ACS Chem. Neurosci 5, 468–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, and Nave KA (2015). Oligodendrocytes: myelination and axonal support. Cold Spring Harb. Perspect. Biol 8, a020479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Morris AD, Welsh TG, and Kucenas S (2014). Contact-mediated inhibition between oligodendrocyte progenitor cells and motor exit point glia establishes the spinal cord transition zone. PLoS Biol 12, e1001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon IC, Chon KH, and Rodriguez MN (2003). Blockade of brain stem gap junctions increases phrenic burst frequency and reduces phrenic burst synchronization in adult rat. J. Neurophysiol 89, 135–149. [DOI] [PubMed] [Google Scholar]
- Stevens B, Porta S, Haak LL, Gallo V, and Fields RD (2002). Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron 36, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XP, Li XY, Zhou B, Shen W, Zhang ZJ, Xu TL, and Duan S (2009). Ca(2+) signaling evoked by activation of Na(+) channels and Na(+)/Ca(2+) exchangers is required for GABA-induced NG2 cell migration. J. Cell Biol 186, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, and Warman ML (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29, 52–54. [DOI] [PubMed] [Google Scholar]
- Wakisaka N, Miyasaka N, Koide T, Masuda M, Hiraki-Kajiyama T, and Yoshihara Y (2017). An adenosine receptor for olfaction in fish. Curr. Biol 27, 1437–1447.e4. [DOI] [PubMed] [Google Scholar]
- Weber SA, and Ross LS (2003). Gap junctional coupling in the olfactory organ of zebrafish embryos. Brain Res. Dev. Brain Res 143, 25–31. [DOI] [PubMed] [Google Scholar]
- Wei W, Du C, Lv J, Zhao G, Li Z, Wu Z, Haskó G, and Xie X (2013). Blocking A2B adenosine receptor alleviates pathogenesis of experimental autoimmune encephalomyelitis via inhibition of IL-6 production and Th17 differentiation. J. Immunol 190, 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh TG, and Kucenas S (2018). Purinergic signaling in oligodendrocyte development and function. J. Neurochem 145, 6–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RC, Power J, Barnea G, O’Donnell S, Brown HEV, Osborne J, Axel R, and Gogos JA (2004). Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron 42, 553–566. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchero JB, and Barres BA (2015). Glia in mammalian development and disease. Development 142, 3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.