

Summary
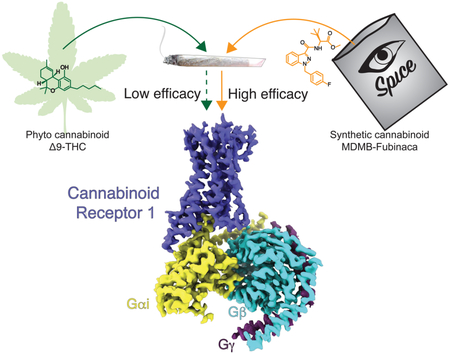
Cannabis elicits its mood-enhancing and analgesic effects through the cannabinoid receptor 1 (CB1), a G protein coupled receptor (GPCR) that signals primarily through the adenylyl cyclase-inhibiting heterotrimeric G protein Gi. Activation of CB1-Gi signaling pathways holds potential for treating a number of neurological disorders, and is thus crucial to understand the mechanism of Gi activation by CB1. Here we present the structure of the CB1-Gi signaling complex bound to the highly potent agonist MDMB-Fubinaca (FUB), a recently emerged illicit synthetic cannabinoid infused in street drugs that have been associated with numerous overdoses and fatalities. The structure illustrates how FUB stabilizes the receptor in an active state to facilitate nucleotide exchange in Gi. The results compose the structural framework to explain CB1 activation by different classes of ligands and provide insights into the G protein coupling and selectivity mechanisms adopted by the receptor.
Keywords: Cannabinoid receptor, GPCR, Cryo-EM, synthetic cannabinoid, Fubinaca, Gi
Graphical Abstract:
Introduction
The cannabinoid receptor 1 (CB1) is the most abundantly expressed G protein coupled receptor (GPCR) in the brain (Marsicano and Lutz, 1999) and the target for Δ9-tetrahydrocannabinol (Δ9-THC), the major psychoactive component of Cannabis which has been used for recreational and therapeutic purposes for millenia. Recently, CB1 has been targeted by designed synthetic cannabinoids which, like their plant-based counterparts, piggyback their pharmacology on a collection of endogenous molecules known as the endocannabinoids. Endocannabinoid signaling has proven to play important roles in memory, mood, sleep, appetite, inflammation and pain sensation (Mackie, 2006), thereby rendering CB1 an attractive target for the development of novel therapeutics towards a variety of conditions.
CB1 elicits its physiological responses by coupling primarily to Gi/o proteins to inhibit adenylate cyclase and cyclic AMP signaling, although coupling with Gs or Gq/11 has also been reported (Glass and Felder, 1997; Lauckner et al., 2005). Activation of CB1 has been shown to have anxiolytic, analgesic, neuroprotective and antinausea effects (Campos et al., 2012; Izzo et al., 2009; Micale et al., 2013), while preclinical data indicate that Δ9-THC and synthetic cannabinoid agonists are effective antinociceptive agents in laboratory animal models of neurodegenerative, neuroinflammatory, and pain-related disease states (Fagan and Campbell, 2014; Guindon and Hohmann, 2009; Pryce and Baker, 2012). However, Δ9-THC and to a much greater extent synthetic cannabinoids can induce side-effects which include dependence, memory impairment, hallucinations, panic attacks, seizures, convulsions and psychoses (Cooper, 2016). Fubinacas, a class of potent synthetic agonists infused in illicit herbal mixes such as “K2” or “Spice”, have been named “zombie drugs” due to their association with users in semicomatose state (Adams et al., 2017). Although side-effects and potential lethality limit their direct therapeutic use, such compounds represent important tools to dissect mechanistic questions regarding the extent of CB1 activation and potency of distinct classes of ligands in order to design drugs with improved pharmaceutical properties.
MDMB-Fubinaca (FUB), a derivatization of the AB-Fubinaca originally developed by Pfizer, has been designated as the deadliest cannabino-mimetic sold to date (Adams et al., 2017). A recent study on 43 synthetic cannabinoids found FUB to have the highest affinity to CB1 in a radioligand binding assay with Ki values of 98 pM (Schoeder et al., 2018). In GTPγS binding assay FUB was found to be 20-fold more potent compared to Δ9-THC (Gamage et al., 2018). Although toxicity of FUB consumption has not been determined directly, Fubinacas have been linked to thousands of hospitalizations and numerous fatalities (Adams et al., 2017; Peace et al., 2017). While synthetic cannabinoids have been shown to have multiple targets, they predominantly signal through CB1. To gain structural insights into the binding and activation of CB1 by FUB, we determined a 3 Å cryo-EM structure of FUB-activated full length CB1 in complex with its down-stream heterotrimeric Gi protein. The structure, complemented by molecular dynamics (MD) and ligand docking calculations, provides a snapshot into the FUB binding properties, its activation of CB1, and the structural basis of G protein coupling. This work sets the framework to integrate a large body of structure-activity relationship (SAR) studies towards understanding cannabinoid receptor activation by different classes of ligands, and also provides insights into the promiscuous coupling of CB1 to both Gs and Gi.
Results and Discussion
Cryo-EM of the CB1-FUB-Gi complex
In our preliminary studies we evaluated 10 synthetic cannabinoids for their ability to activate Gi signaling (Figure 1A). Through GTP turnover and fluoresence-detection size exclusion chromatography (F-SEC) assays (Figure 1B and S1A) we observed a direct correlation between complex stability and ligand ability to induce signaling. In these assays, FUB demonstrated high efficacy and complex stability (Figure 1B).
Figure 1. GTP turnover assay for the CB1 ligands.

(A) Chemical structures of CB1 agonists used in this study, and of PAM ZCZ. (B) GTP turnover assay for the 10 ligands tested. (C) The addition of ZCZ further increases GTP turnover indicating PAM activity. Data are normalized to FUB in (B) and (C).
In addition, we prepared the CB1-FUB-Gi complex in the presence of ZCZ-011 (ZCZ), a positive allosteric modulator (PAM) that was shown to mediate analgesia with no phychoactive effect (Ignatowska-Jankowska et al., 2015). ZCZ further increased the rate of GTP turnover of FUB-bound CB1 confirming positive allosteric modulation (Figure 1C). However, while negative stain EM analysis (Peisley and Skiniotis, 2015) showed an intact CB1-Gi complex, inspection of cryo-EM samples indicated sample dissociation, presumably due to adverse effects during cryo-grid preparation (Noble et al., 2018). To further enhance complex stability, we utilized scFv16, a single-chain variable fragment that we recently employed to obtain the cryo-EM structure of the μ-opioid receptor (μOR)-Gi complex (Koehl et al., 2018). ScFv16 is derived from a monoclonal antibody that was raised against a Rhodopsin:Gi1 complex. It confers GTPγS resistance to receptor-Gi/o complexes, thus enhancing their stability (Maeda et al., 2018). We thus obtained cryo-EM images of the CB1-FUB-Gi-scFv16 complex and used ~177,000 projections to calculate a 3D reconstruction with a global nominal resolution of 3 Å (Figure 2, S1B-C and S2). Local resolution calculations indicate a range of 2.7-3.6 Å in most map regions, with the highest resolution observed at the core of the Gi protein (Figure S2C). This map enabled the building of a model for the fully activated CB1 signaling complex (Figure 2 and S2, Table S1).
Figure 2. Cryo-EM structure of the CB1-Gi complex.

(A) Representative reference-free two-dimensional (2D) cryo-EM average of CB1-FUB-Gi-scFv16 shows high resolution features including the helical pitch of TM α-helices. The labels indicate complex components. The diameter of the circular mask is 18 nm. (B-E) Three-dimensional map (B, D-E) and model (C) obtained from cryo-EM of the CB1-Gi complex. CB1 is colored blue, FUB - orange, Gαi, −β and −γ yellow, cyan and dark magenta, respectively, and scFv16 is colored pink. (F) Snapshot of model vs. map density in the region where the ScFv16 is engaging Gαi and Gβ. The zoomed in region corresponds to the area highlighted by a dashed black box in (E).
The cryo-EM map shows well-defined density for FUB in the orthosteric binding pocket (Figure 3A and S2D) but no observable density to accommodate ZCZ. This might suggest that the PAM engages a flexible site of the receptor, although we cannot exclude the possibility that it may have dissociated from the complex during cryo-EM specimen preparation. The FUB binding pocket is composed of residues in TM2-3 and TM5-7, overlapping with the orthosteric site observed in previously reported crystal structures of active CB1 (Hua et al., 2017) (Figure 3B and Figure 4). Compared to other GPCRs (with the exception of rhodopsin), the FUB pocket is further buried in the TM region and is capped by ECL2, which folds into the pocket with F268ECL2 making direct hydrophobic contacts with the ligand. FUB fits well in the map density where the indazole ring is well-identified, and the p-fluoro-benzyl and tert-butylester moieties fit into the remaining density (Figure S2D). Furthermore, molecular dynamics (MD) simulations of several computationally docked poses converged to nearly identical ensembles that agreed well with the EM density (Figure S2E). The polar ester group often formed water-mediated interactions with residues on TM2 and TM7, and the tert-butyl group remained more proximal to TM2 and TM3.
Figure 3. The FUB binding pocket.

(A) Cut-through view of CB1 cryo-EM map with FUB bound in the orthosteric pocket. Density corresponding to FUB is colored orange, and CB1 purple. (B) FUB interactions in the CB1 binding site.
Figure 4. CB1 activation by FUB.

(A) Superposition of the FUB activated complex (blue) with an inverse agonist bound receptor (AM-6358, PDB 5TGZ, grey). (B) FUB and AM-11542 (PDB 5XRA) bound at the CB1 orthosteric pocket make direct contacts with residues F2003.36 and W3566.48. The rotation of F2003.36 to interact with the indazole ring of FUB allows W3566.48 to rotate outwards, with a consequent outward movement of the cytoplasmic end of TM6 that serves to create a cavity for G protein binding. The groups interacting with the ‘toggle twin switch’ of CB1 (indazole ring of FUB and GDH moiety of AM-11542) are marked in orange. The inactive receptor structure is shown in grey (PDB 5TGZ). (C) A comparison of binding pockets of AM-6538 (antagonist, grey, PDB 5TGZ), AM-11542 (agonist, cyan, PDB 5XRA) and FUB (orange). The receptor is presented as cartoon with the active conformation in blue (present cryo-EM structure) and the inactive conformation in gray (PDB 5TGZ).
CB1 activation by FUB
Compared to the inverse agonist (taranabant)- and antagonist (AM-6538)-bound structures of CB1 (Hua et al., 2016; Shao et al., 2016), the ligand binding pocket in FUB-bound CB1 undergoes an extensive structural rearrangement that agrees well with that observed in the agonist-bound CB1 structures (Hua et al., 2017). This structural rearrangement involves the inward displacement of TM1 and TM2, along with the displacement of the N-terminus out of the transmembrane core (Figure 4). Such large differences in the binding pocket between active and inactive states have not been observed in other GPCRs; RMSD values between binding pocket residues in the active and inactive states range between 2.7 Å in the β2-adrenergic receptor (β2AR) and 5.1 Å in the μOR compared to 8.5 Å in CB1. The TM2 movement is accompanied by the repositioning of residues F1702.5, F1742.61, F1772.64 and H1782.65 (superscripts indicate Ballesteros-Weinstein numbering for GPCRs (Ballesteros and Weinstein, 1995)), that turn towards the pocket in the active conformation and contribute to interactions with the agonist (Figure 4A). Notably, F2003.36, which in the inactive receptor state is stabilized by interactions with W3566.48, rotates away to interact with the indazole ring of FUB (Figure 4A-B). The role of the interaction between F2003.36 and W3566.48, also known as the ‘toggle twin switch’, in stabilizing the inactive conformation of CB1 was previously demonstrated by mutation of F3.36A, which resulted in high basal activity in a GTPγS binding assay (McAllister et al., 2004). In our structure, the F2003.36 repositioning allows W3566.48 to rotate inward, resulting in the relaxation of the kink at the highly conserved P3586.50, with a consequent straightening and an outward movement of the cytoplasmic end of TM6 that serves to create a cavity for G protein binding (Figure 4B).
Furthermore, the structural rearrangements to accommodate the agonist involve the displacement of the receptor N-terminus, a region which along the proceeding TM1 helix has limited resolution in our map (~ 4Å). These observations suggest that these elements are relatively mobile, consistent with their dynamics observed in MD simulations (Figure S3A). It is clear however, that the N-terminal displacement is necessary to accommodate the agonist, due to steric clash with the tert-butyl moiety in FUB or the terpenoid scaffold in the previously reported CB1 agonists AM-11542 and AM-841 (Figure 4C). This steric clash is not present in the CB1 structures bound to antagonists or inverse agonists reported to date (Hua et al., 2016; Shao et al., 2016), raising the possibility that the N-terminal displacement may be part of the activation mechanism. Indeed, in all MD simulations performed here, the N-terminus regularly fluctuated between contacting the extracellular surface of the receptor and residing in the bulk solvent (Figure S3A).
The MD simulations further hint at a possible mechanism for ligand entry at the CB1 receptor. In four out of six simulations, TM1 moves outwards to create a gap between TM1 and TM7 (Figure S3A). This small opening is similar to one observed in the lipid bilayer between TM1 and TM7 in an inactive-state structure (Figure S3B) and has been proposed to facilitate ligand entry and dissociation in CB1 and all lipid-activated receptors (Shao et al., 2016). Intriguingly, the simulations also show that the opening of the TM1-TM7 gap coincides with both the binding of a lipid molecule (POPC) and the stabilization of the upward position of the N-terminus relative to the extracellular surface of the receptor (Figure S3C-F).
FUB binding and comparison with other CB1 agonists
Crystal structures of CB1 constructs truncated at N- and C-termini have been previously determined in complex with agonists AM-11542 and AM-841 that possess a THC-like scaffold and are structurally distinct from FUB (Hua et al., 2017). Despite the differences in chemical composition, FUB assumes the same overall C-shape geometry as AM-11542 and AM-841, with overlapping ligand binding pockets (Figure 4B and S4). The p-fluorobenzyl that π-π stacks with W2795.43, and the indazole group of FUB engage in hydrophobic interactions with aromatic residues in TM5 and TM6 (Figure 4A). Notably, the C3 alkyl chain of the AM-derivatives and the p-fluorobenzyl of FUB occupy the same position in a narrow side pocket comprised of residues in TM 3,5,6 and ECL2 (Figure 4B-C). This pocket has been observed to be occupied by the aliphatic chain of CB1 antagonist AM-6538 (Hua et al., 2016) (Figure 4C), and is also present in the sphingosine 1-phosphate receptor (S1P1) where it was shown to be occupied by the aliphatic moiety of a selective antagonist (Hanson et al., 2012). It thus appears that the side pocket is a conserved docking region for aliphatic chains in lipid binding receptors, where it seems to be important for ligand affinity regardless of its ability to activate the receptor. This might explain the similar affinities observed for the AM-derivatives and FUB (indicated Ki values are 0.098 and 0.11 nM for FUB, and AM-11542, respectively) (Schoeder et al., 2018) as also supported by SAR studies showing that the replacement of the FUB p-fluorobenzyl with an alkyl chain yields a ligand with similar affinity (Schoeder et al., 2018) and EC50 (Adams et al., 2017). In agreement with this interpretation, our ligand docking calculations show that the aliphatic moiety of a Fubinaca derivative (5F-MDMB-Pinaca) overlaps with the p-fluorobenzyl group of FUB (Figure S4B).
Δ9-THC has a much greater safety profile compared to synthetic cannabinoids (Fantegrossi et al., 2014). One reason that could attribute to this toxicity difference is that Δ9-THC is a partial agonist whereas synthetic cannabinoids like FUB are full agonists (Atwood et al., 2010). Though structurally similar to Δ9-THC, AM-11542 shows enhanced potency and efficacy for CB1 due to the addition of a 1′,′-gem-dimethylheptyl (GDH) chain at the C3 position. The GDH group in AM-11542 mediates hydrophobic interactions with the ‘toggle twin switch’, which undergoes concerted conformational changes upon agonist-binding and activation (Figure 4B). In FUB instead, strong aromatic interactions with both F2003.36 and W3566.48 are maintained by the indazole ring, thereby explaining the high efficacy of this ligand (Figure 4B). The lack of the toggle switch interaction has been suggested as the explanation of the partial agonist activity observed for Δ9-THC compared to the full agonist activity of the AM compounds. In support of this notion, our docking calculations of Δ9-THC in the CB1 cryo-EM structure yields several poses where the terpenoid ring aligns well with that of AM-11542 in the CB1 crystal structure (PDB 5XRA) (Hua et al., 2017) but the aliphatic moiety of Δ9-THC fluctuates between penetrating the hydrophobic cavity occupied by the p-fluorobenzyl group in FUB and a downward conformation that points towards the toggle switch to activate the receptor (Figure S4C-E). Hence, it appears that the conformational variability of Δ9-THC likely compromises both its affinity and potency for CB1, a characteristic that presumambly makes it safer compared to more rigid and potent synthetic cannabinoids.
In contrast, the high efficacy of FUB is partly due to its structural rigidity in the characteristic C-shape configuration that stereotypically recognizes the CB1 binding site and stabilizes the active receptor conformation (Figure 4 and Figure S4A). FUB analogues demonstrate that intramolecular interactions between the heterocyclic core and the linker of the tert-butyl can significantly stabilize the bound conformation, with a marked loss of potency when the indazole core is replaced with an indole or when the amide is replaced by a ketone (Schoeder et al., 2018). Accordingly, our quantum mechanics (QM) calculations show that the bound conformation in FUB is much lower in energy compared to the alternate conformation with a flipped dihedral angle between the amide and the indazole (Figure S5). Because this intramolecular interaction is absent in less potent analogues, the energy difference between the bound and flipped pose is significantly lower and thus these compounds display a mix of conformations compromising their activity.
The tert-butyl position in FUB, which overlaps with the terpenoid scaffold of the AM agonists, greatly diversifies in various derivatives of the Fubinaca family (Figure 1A). In the CB1-FUB-Gi-scFv16 structure, the ester moiety of FUB is in position to form polar interactions with H1782.65 (Figure S4F). In addition, molecular mechanics calculations using JAWS (Michel et al., 2009) consistently showed the presence of a strongly bound (>3.5 kcal/mol) water molecule in between the amide linker, H1782.65, and S3837.39 (Figure S4F-H). MD simulations further support the presence of a polar network involving the ligand’s ester moiety, H1782.65, and S3837.39, but the precise arrangement and behavior of waters tends to differ slightly from the JAWS-predicted network. Both ligand docking and comparison to an agonist bound-crystal structure (PDB 5XRA) reveal that AM analogues and Δ9-THC have a hydrogen bonding group able to interact with this water molecule and/or S3837.39. In agreement with these findings, mutagenesis of S3837.39 decreases agonist binding (Hua et al., 2017; Kapur et al., 2007), while SAR data with the hydrogen bonding moiety removed (-OH in terpenoid or cannabidiol scaffolds and amino-group in the FUB linker) reveal lower compound potency (Bow and Rimoldi, 2016).
Propagation of agonist-stabilized structural changes
Excluding differences in regions that are stabilized by Gi binding to the receptor, the structures of FUB-Gi-bound and agonist-bound CB1 are remarkably similar (RMSD of Cα is 1.2) (Figure S6) despite the fact that the modified receptor construct used for determining the agonist-bound structure cannot signal (Hua et al., 2017). This finding is in contrast to the β2AR and μOR (Rosenbaum et al., 2011); (Nygaard et al., 2013) where agonist-binding alone, in the absence of G protein, cannot stabilize the fully extended active-state conformation of TM6. The higher propensity of CB1 to transition to an active conformation likely explains its inherently high basal activity (Seifert and Wenzel-Seifert, 2002), a feature that has been shown to be important for modulating neuronal development (Njoo et al., 2015).
A striking difference between CB1 as well as a number of lipid-activated receptors with most other family A GPCRs is the absence of the conserved P5.50, whose insertion creates a bulge in TM5 and local unwinding between residues 5.45 and 5.48 to relieve geometric constraints and orient ligand-interacting residues to the binding pocket (Sansuk et al., 2011) (Figure 5A). In the β2AR and μOR, P5.50 is involved in packing interactions with I3.40 (found in 42% of Class A GPCRs) and F6.44 (found in 82% of Class A GPCRs), which rearrange upon receptor activation (Figure 5A-B) (Huang et al., 2015). Although in activated CB1 we observe relatively small rearrangements in the homologous corresponding residues L5.50, V3.40, and L6.44, the discussed structural changes in TM6 drive L6.44 to move away from V3.40 and L5.50 (Figure 5A). Crucially, the lack of unwinding in CB1 due to the absence of a proline at position 5.50, makes TM5 a more rigid helix connecting the binding pocket to the G protein coupling domain. While this highly conserved P5.50 has been proposed to play a key role in the activation of many family A receptors (Deupi, 2014), the L5.50P mutation in the CB2 receptor disrupts signaling, indicating that a non-kinked TM5 is a prerequisite for cannabinoid receptor function (Zhou and Song, 2002).
Figure 5. Structural changes in CB1 on nucleotide-free Gi binding.

(A) Structural rearrangement of P-I-F motif in CB1, μOR (inactive- PDB 4DKL, yellow; PDB 6DDE, magenta) and β2AR (inactive-PDB 2RH1, grey; active-PDB 3SN6, green) upon activation. (B) The local unwinding of TM5 due to P5.50 in active and inactive β2AR (green) and μOR (magenta) is not seen in CB1.
Structure of the CB1-Gi interface
Globally, the structure of the CB1-Gi complex reveals a similar mode of interaction when compared to other Gi bound receptors. However, the N-terminus of α5 in the CB1-Gi complex deviates from that of the G-protein in the μOR-Gi and β2AR-Gs complexes, resulting in a different relative orientation of the G protein. When aligned on the receptors, the Gi in complex with CB1 is rotated along the membrane by 18° when compared to μOR-Gi (Figure 6A). The difference is attributed to the more extensive interactions between the N-terminus of the α5 helix and the extended TM5 of CB1 (Figure 6B and 6C), resulting in a relative configuration that is similar to a recently reported cryo-EM structure of a Rho-Gi complex (Kang et al., 2018). Although TM5 is also extended in the β2AR-Gs complex, it does not form strong interactions with the N-terminus of the α5 helix due to the difference in G protein orientation (Figure 6D). Thus, the relative orientational differences between G protein and its respective receptors, as exemplified in the structures of CB1, μOR and β2AR complexes, is underlined by a change of interaction profiles. Gi interactions with CB1 are primarily between the α5 helix of Gi and ICL2, TM5, TM6 and H8 of CB1 (Figure 6B, E), while only a weak hydrophobic contact is observed between the β2-β3 loop of Gi and ICL. In contrast, more extensive interactions are maintained between the ICL2 of the μOR and the αN-β1 loop of Gi, and between ICL2 of the β2AR and the αN-β1 loop of Gs (Figure 6C, D, F, G). A complete list of contacts is shown in Table S2.
Figure 6. Relative orientation of CB1 and Gi and Role of ICL2 in coupling selectivity.

(A) Comparison of the relative orientation of Gi bound to CB1 (blue), μOR (PDB 6DDE, magenta), and β2AR (PDB 3SN6, green) when aligned on the receptor. A magnified view is provided of the position of the α5 helices in CB1-Gαi (yellow), μOR-Gαi (wheat) and β2AR-Gαs (orange). (B, C, D) Residues in the TM5-TM6 helices of CB1 (B), μOR (C) and β2AR (D) interacting with the α5 helix of Gαi (bound to CB1, yellow and bound to μOR, wheat) and Gαs (orange). (E, F, G) Interactions between ICL2 of CB1 (blue) and Gαi (yellow), μOR (magenta) and Gαi (wheat) and β2AR (green) and Gαs (orange).
The C-terminus of the α5 helix of Gi is in a similar position when bound to CB1 or μOR (Figure 6A), while the Gi complexes overlay well with the structures of rhodopsin, adenosine receptor, A1 (A1A) (Draper-Joyce et al., 2018) and the serotonin receptor - 5HT1B (García-Nafría et al., 2018) in complex with Gi. On the other hand, the C-terminus of the α5 helix of Gs coupled to receptors, e.g. the β2AR, is displaced by ~6.5 Å, requiring a larger outward movement of TM6 compared to Gi coupled receptors. Accordingly, TM6 repositions by 12 Å and 11.3 Å in CB1-Gi and μOR-Gi, respectively, compared to ~14 Å in β2AR-Gs (Figure 6A), while an even larger outward displacement of TM6 is observed in the family B receptors in complex with Gs. Given these findings, we postulate that the ability to accommodate the C-terminus of Gαs is one of the determinants of Gs coupling specificity. For CB1 to couple to Gs, its TM6 would have to be able to move outward to a greater extent than that found in the CB1-Gi complex. In the β2AR, the larger displacement of TM6 can be attributed to G6.38 and G6.42. Although there are no glycine residues at similar positions of CB1, G3576.49 in the conserved C6.47W6.48X6.49P6.50 motif may add extra flexibility to TM6. It is also worth noting that the homologous amino acid at position 6.49 in CB2 is phenylalanine and in μOR is threonine, and both of these receptors couple very poorly to Gs (Connor and Christie, 1999; Mnpotra et al., 2014). Conformational memory calculations showed that the flexibility of TM6 around positions 6.49 and 6.50 was significantly greater for CB1 compared to CB2, but the flexibility was greater for CB2 with the F6.49G mutation compared to CB1 with the G6.49F mutation (Barnett-Norris et al., 2002).
Role of CB1 ICL2 in coupling Gi/s coupling promiscuity
CB1 signals primarily through the Gi/Go family of G proteins but several studies indicate that the receptor can also couple to Gs (Felder and Glass, 1998; Glass and Felder, 1997). Multiple GPCRs have been shown to couple to different G protein subtypes, such as β2AR that couples to Gs and Gi, while some receptors couple almost exclusively to one G protein subtype, e.g. the μOR (Connor and Christie, 1999), which couples predominantly to the Gi/Go family. While several studies have provided evidence that the μOR may couple to Gs in GTPγS binding and cAMP accumulation assays (Chakrabarti et al., 2010; Szücs et al., 2004), the coupling is very weak, requiring much higher concentrations of agonist than is needed for Gi activation. Earlier work has shown that Gs- and Gi-coupled receptors appear to have different amino acid preferences at the ICL2 position, with L222ICL2, in particular, reported to determine Gs protein coupling (Chen et al., 2010). A L222ICL2F mutation in CB1 increased basal signaling through Gs, whereas a L222ICL2A/L222ICL2P mutations led to a loss of Gs coupling but retained coupling to Gi. In the CB1-Gi structure, the ICL2 of the receptor interacts primarily with the α5 helix of Gi, but not with the αN-β1 loop as observed in the μOR-Gi and β2AR-Gs complexes.
Notably, L222ICL2 is only weakly interacting with L194 in the β2-β3 loop of Gi (Figure 6E). The homologous F139ICL2 in the β2AR is buried in a hydrophobic pocket formed by the α5 helix, the αN-β2 loop and the β2-β3 loop of Gs (Figure 6G), whereas the corresponding smaller residue V173ICL2 in the μOR-Gi complex is surrounded by a similar hydrophobic pocket formed by I343, F336 and T340 of Gi (Figure 6B). In contrast, L222ICL2 of CB1 points towards, but does not engage the Gi hydrophobic pocket (Figure 6E). Although alanine substitution of F139ICL2 in the β2AR and other Gs coupled receptors impairs their ability to activate G proteins (Moro et al., 1993), the L222ICL2A mutation in CB1 does not alter Gi coupling, suggesting that interactions between ICL2 and the hydrophobic pocket are less important for Gi coupling of this receptor. On the other hand, the bulkier phenylalanine in the CB1 L222ICL2F mutation would be expected to engage the hydrophobic pocket in Gs, which has been shown to be required for promoting GDP release.
Structural changes in Gi upon binding CB1
Upon coupling to CB1, the α5 helix of Gi undergoes a 6 Å translation and a 60° rotation to engage the core of the receptor (Figure 7A, B). This is in good agreement with other complex structures where the translation of α5 has been shown to influence the position of the β6-α5 loop containing the conserved TCAT motif that directly contacts the guanosine base of GDP. The TCAT motif in Gi bound to CB1 has moved ~2.0 Å closer to the nucleotide-binding site compared to the recently published μOR-Gi structure (Figure 7C). Movement of the α5 helix disrupts interactions between its N-terminus and the α1 helix, leading to displacement of the P-loop (Figure 7B) and the destabilization of its contacts GDP, with eventual release of the nucleotide. The P loop in the CB1-Gi complex deviates from μOR-Gi by 3.5 Å (at E43) but overlays well with the structures of GDP-bound Gαi subunit (Figure 7C). Interestingly, the observed P loop conformation in the CB1-Gi complex may accommodate nucleotide binding, in contrast to the μOR-Gi complex where this is precluded due to potential steric clashes (Figure 7C). Thus, while both μOR-Gi and CB1-Gi complexes are nucleotide-free, the CB1-Gi complex may represent a conformation that is poised for GTP binding and activation (Figure 7C).
Figure 7. Structural changes in Gi on CB1 binding.

(A) Comparison of GDP-bound Gαi (PDB 1GP2, green) and nucleotide-free Gαi from the CB1-Gi complex (yellow). The structures are aligned on the β-subunit (CB1-Gi, cyan and GDP-bound Gαi, grey). GDP is shown as sticks. The alpha helical domain (AHD) seen in the GDP-bound structure is not resolved in the nucleotide-free Gαi bound to CB1. (B) The α5 helix of Gαi moves upward by 6 Å and rotates ~ 60° to engage the receptor core. The TCAT motif that coordinates the guanosine base of GDP in the GDP-bound structure (green) has shifted upwards in the nucleotide-free Gαi bound to μOR (wheat). In CB1-bound Gαi (yellow), the TCAT motif is in a similar position as that seen in A2A-bound mini-Gαs (with GDP) (PDB 5G53, purple). (C) The conformation of the P loop in CB1-bound Gi (yellow) will allow nucleotide binding as seen when overlayed with GDP-bound Gi (1GP2, smudge). However in the nucleotide-free G protein bound to μOR (μOR-Gi, sand), there is a clash of the P loop with the GDP.
Conclusion
Delineating the structural basis for ligand efficacy and G protein recruitment at CB1 will aid in the design of drugs with high specificity and optimal therapeutic effects. Here we report a cryo-EM structure of CB1-Gi complex revealing the binding mode of the highly potent synthetic cannabinoid FUB and the molecular characteristics of Gi protein coupling and activation. FUB binding stabilizes the receptor in an active conformation through interference with the CB1 ‘toggle twin switch’ residues F2003.36 and W3566.48. The W3566.48 repositioning results in the relaxation of the kink at P3586.50, thereby allowing the cytoplasmic part of TM6 to straighten and open up to accommodate the C-terminal α5 helix of Gi. The rigid C-shape geometry of FUB along with the strong aromatic interactions of its indazole ring with CB1 residues F2003.36 and W3566.48 might distinguish this full agonist from partial agonists like Δ9-THC, which has a better safety profile.
Comparison of the CB1-Gi complex with the previously determined nucleotide-free structures of μOR-Gi, A1A-Gi, 5HT1B-Go, rhodopsin-Gi and β2AR-Gs reveal largely similar overall interaction profiles for receptor-G protein binding. However, compared to the previously determined complex structures, the relative orientation of the CB1 and Gi is different, with a weaker interaction between the Ras domain of Gi and ICL2 of CB1. Determining G protein coupling specificity has been a major unanswered question in the field of GPCR biology. Although, the CB1-Gi complex provides insights into the promiscuous coupling of CB1 relative to CB2, it does not provide a universal molecular basis of G protein coupling specificity across all receptors. It is entirely possible that coupling specificity is determined at the initial stages of complex formation wherein a GDP-bound G protein engages the agonist-activated receptor. However, due to their highly dynamic and transient nature, such pre-equilibrium complexes are currently challenging for structure determination.
STAR Methods
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for reasorces and reagents should be directed to and will be fulfilled by the Lead Contact, Georgios Skiniotis (yiorgo@stanford.com).
METHOD DETAILS
Purification of CB1
Human full-length CB1 with N-terminal FLAG and C-terminal hexahistadine tag was expressed in Spodoptera frugiperda Sf9 insect cells using the baculovirus method (Expression Systems). The same construct with an eGFP (CB1-eGFP) at the C-terminus was used for small-scale coupling and FSEC studies. The receptor was extracted from insect cell membranes with 1% lauryl maltose neopentyl glycol (L-MNG) and purified by nickel-chelating sepharose chromatography. The Ni-NTA pure eluate was applied to a Ml anti-FLAG immunoaffinity resin and washed with progressively decreasing concentration of inverse agonist, SR and increasing concentration of agonist FUB. The receptor was eluted in a buffer consisting of 20 mM HEPES pH 7.5, 150 mM NaCl, 0.05% L-MNG, 0.005% cholesterol hemisuccinate (CHS), 2 μM FUB, FLAG peptide and 5 mM EDTA. The final step of purification was size exclusion chromatography on Superdex 200 10/300 gel filtration column (GE) in 20 mM HEPES pH 7.5, 150 mM NaCl, 0.02% L-MNG, 0.002% CHS, and 2 μM FUB. Finally, agonist-bound CB1 was concentrated to ~500 μM and flash frozen and stored in −80 °C.
Expression and purification of Gi heterotrimer
Heterotrimeric Gi was expressed and purified as previously described (Dror et al, 2015). Briefly, Trichuplusia ni Hi5 insect cells were infected with two viruses, one encoding the wild-type human Gαi subunit and another encoding the wild-type human β1γ2 subunits with an histidine tag inserted at the amino terminus of the β subunit. Cells expressing the heterotrimetric G-protein were harvested 48 hours post infection. After cells were lysed in hypotonic buffer, heterotrimeric Giβ1γ2 was extracted in a buffer containing 1% sodium cholate and 0.05% n-dodecyl-β-D-maltoside (DDM, Anatrace). The heterotrimer containing soluble fraction was purified using Ni-NTA chromatography, and the detergent was exchanged from cholate/DDM to DDM on column. Human rhinovirus 3C protease (3C protease) was added and the histidine tag was cleaved on-column overnight at 4 °C. The flow through was collected and was further purified by size exclusion chromatography on Superdex 200 10/300 gel filtration column (GE) into 20 mM HEPES pH 7.5, 100 mM NaCl, 0.02% DDM, 100 μM TCEP, 10 μM GDP, and concentrated to ~20 mg/mL for further complexing with the CB1.
Fluorescence - size exclusion chromatography
Purified CB1-eGFP (1.25 M excess) was incubated with Gi (that has been incubated with 1 % L-MNG for 1 hour), in the presence of the tested ligands at room temperature for 1 hour, after which apyrase was added and further incubated on ice for 1 hour. The sample was applied to a Superdex 200 10/300 column (equilibrated in 20mM HEPES pH 7.5, 100mM NaCl, 0.01% L-MNG/0.001% CHS) in-line with a Jasco FP 2020 Plus fluorescence detector set to an excitation of 480 nm and an emission of 512 nm. Complex formation and complex stability was monitored for each ligand by analysing the extent of free receptor and complex peaks.
Purification of scFv16
scFv16 was purified as previously described (Koehl et al., 2018). Briefly, hexahistidine-tagged scFv was expressed in secreted form from Trichuplusia ni Hi5 insect cells using the baculoviral method, and purified by Ni-NTA chromatography. After balancing the pH and quenching chelating agents, the supernatant from baculoviral infected cells was loaded onto Ni-NTA resin. The protein was eluted in 20 mM HEPES pH 7.5, 500 mM NaCl, and 250 mM imidazole and incubated with 3C protease to cleave the carboxy-terminal hexahistidine tag. Following dialysis into a buffer consisting of 20mM HEPES pH 7.5 and 100 mM NaCl, cleaved scFv16 was further purified by reloading over Ni-NTA resin. The flowthrough was collected and applied over a Superdex 200 16/60 column. scFv16 fractions were pooled, concentrated, and flash frozen.
CB1-Gi complex formation and purification
Purified Gi1 heterotrimer in DDM was incubated with 1% L-MNG for 1 hour at 4 °C to exchange the detergent and simultaneously, FUB-bound CB1 was incubated with ZCZ at 24 °C. The FUB- and ZCZ-bound CB1 was incubated with a 1.25 molar excess of detergent exchanged Gi heterotrimer. The reaction tube was incubated at 24 °C for 3 hours and was followed by the addition of apyrase, to stabilise a nucleotide-free complex (Westfield et al., 2011), for an additional 1.5 hour at 4 °C. A 4-fold volume of 20 mM HEPES pH 7.5, 100 mM NaCl, 0.8% L-MNG/0.08% CHS, 0.27% GDN/0.027% CHS, 1 mM MgCl2, 10 μM FUB, 1 μM ZCZ and 2 mM CaCl2 was added to the complexing reaction and purified by M1 anti-FLAG affinity chromatography to remove excess G protein. The complex was eluted in 20mM HEPES pH 7.5, 100mM NaCl, 0.01% L-MNG/0.001% CHS, 0.0033% GDN/0.00033% CHS 10 μM FUB, 1 μM ZCZ, 5 mM EDTA, and FLAG peptide. The eluted complex was supplemented with 100 μM TCEP to provide a reducing environment. A 2 molar excess of scFv16 was added to the preparation and incubated overnight at 4 °C. The CB1-Gi-scFv16 complex was purified by size exclusion chromatography on a Superdex 200 10/300 Increase column in 20mM HEPES pH 7.5, 100mM NaCl, 10 μM FUB, 1 μM ZCZ, 0.00075% L-MNG/0.000075% CHS and 0.00025% GDN/0.000025% CHS. Peak fractions were concentrated to ~16 mg/mL for electron microscopy studies.
Cryo-EM data acquisition
For cryo-EM 3.5 μL of purified CB1-Gi complex at 5 mg/ml concentration were applied on glow-discharged holey carbon gold grids (Quantifoil R1.2/1.3, 200 mesh). The grids were blotted using a Vitrobot Mark IV (FEI) with 1 s blotting time at 100% humidity and plunge-frozen in liquid ethane. A total of 2,759 movies were recorded on a Titan Krios electron microscope (Thermo Fisher Scientific - FEI) operating at 300 kV at a calibrated magnification of x29,000 and corresponding to a magnified pixel size of 0.86 Å. Micrographs were recorded using a K2 Summit direct electron camera (Gatan Inc.) with a dose rate of ~5.0 electrons/Å2/s and defocus values ranging from −0.7μm to −2.5 μm. The total exposure time was 10.0 s and intermediate frames were recorded in 0.2 s intervals resulting in an accumulated dose of ~58 electrons per Å2 and a total of 50 frames per micrograph. Automatic data acquisition was done using SerialEM (Mastronarde, 2005).
Image processing and 3D reconstructions
Micrographs were subjected to beam-induced motion correction using MotionCor2 (Zheng et al., 2017). CTF parameters for each micrograph were determined by CTFFIND4 (Rohou and Grigorieff, 2015). An initial set of 1,178,914 particle projections were extracted using semi-automated procedures and subjected to reference-free two-dimensional classification in RELION 2.1.0 (Fernandez-Leiro and Scheres, 2017). From this step, 562,312 particle projections were selected for further processing. The map of μ-opioid receptor (EMDB- 7868) low passed filtered to 60 Å was used as an initial reference model for maximum-likelihood-based three-dimensional classifications. Conformationally homogeneous groups accounting for 177,787 particles, forming class averages with well resolved features for all subunits, were subjected to 3D masked refinement in Frealign (CisTEM (Grant et al., 2018)) followed by map sharpening applying temperature-factors of −90 Å2 and −60 Å2 for the low- and high- resolution ends of the amplitude spectrum, respectively. The final map has an indicated global nominal resolution of 3.0 Å (Figure S1-2). Reported resolution is based on the gold-standard Fourier shell correlation (FSC) using the 0.143 criterion and is in agreement with both Relion 2.1.0 and M-triage as implemented in Phenix (Afonine et al., 2018). Local resolution was determined using B-soft (Heymann, 2018) with half map reconstructions as input maps (Figure S2C).
Model building and refinement
The initial template of CB1 was derived from the crystal structure of agonist-bound CB1 (PDB 5XRA). The μ-opioid receptor coordinates (PDB 6DDE) were used as initial models for the Gi and scFv16. Agonist coordinates and geometry restrains were generated using phenix.elbow (Moriarty et al., 2009). Models were docked into the EM density map using UCSF Chimera (Pettersen et al., 2004), followed by iterative manual building in Coot (Emsley and Cowtan, 2004). The final model was subjected to global refinement and minimization in real space using phenix.real_space_refine in Phenix (Adams et al., 2011). Residues in regions of weak density were stubbed to their Cβ position, while preserving sequence information (Stubbed CB1 residues shown in Table S2). Molprobity (Williams et al., 2018) was used to evaluate model geometry. FSC curves were calculated between the resulting model and the half map used for refinement as well as between the resulting model and the other half map for cross-validation (Figure S2). The final refinement parameters are provided in Table S1.
GTP turnover assay
Analysis of GTP turnover was performed by using a modified protocol of the GTPase-Glo™ assay (Promega) described previously (Gregorio et al., 2017). This assay detects the amount of GTP remaining after GTP hydrolysis, which is enhanced upon activation of the G protein by the ligand-bound receptor. After the GTPase reaction, addition of GTPase-Glo-reagent converts the remaining GTP to ATP that is converted to a luminescent signal by the detection reagent. CB1 was incubated with and without ligands for 30 minutes at room temperature. The reaction was started by mixing the unliganded or liganded-CB1 and Gi in an assay buffer containing 20 mM HEPES, pH 7.5, 50 mM NaCl, 0.01% L-MNG, 100 μM TCEP, 10 mM μM GDP and 5 μM GTP. After incubation for 60 minutes (agonists assay) and 30 minutes (for PAM assay), reconstituted GTPase-Glo-reagent was added to the sample and incubated for 30 min at room temperature. Luminescence was measured after the addition of detection reagent and incubation for 10 min at room temperature using a SpectraMax Paradigm plate reader.
Docking and pose refinement
To select ligand poses for simulation, we performed docking with Glide SP (Schrödinger) against an earlier refinement of the cryo-EM structure (model available upon request). We used the enhanced sampling option to improve conformer generation (set to four times the usual amount of sampling) and requested at most 100 output poses. Subsequent poses were aligned within the electron density map, and two distinct poses that fit within the density were selected for simulation, along with a pose obtained through manual modeling whose orientation also differed from the two Glide-generated poses. In order to compare docking poses for other CB1 agonists, Glide XP docking was performed with CP55,940, Δ9-THC, and the fluoroalkyl analogue of FUB.
System setup for MD simulations
Prior to running MD simulations of CB1, we performed several steps of refinement and modeling of the cryo-EM structure. The CB1 model was treated in isolation after removing the Gi atoms form the cryo-EM structure. The Advanced Homology Modeling tool in Maestro (Schrödinger) was used to remodel ECL2 to match the conformation observed in 5XRA. Prime (Schrödinger) was used to insert missing side chains, hydrogens and cap the termini of the protein while D163 and D213 were manually protonated, in accordance with evidence that these residues become protonated upon GPCR activation (Ghanouni et al., 2000; Ranganathan et al., 2014). For each of the final candidate ligand poses, the ligand was added to the prepared protein resulting in three unique protein-ligand complexes. Each prepared protein-ligand complex was inserted into a pre-equilibrated palmitoyl oleoylphosphatidylcholine (POPC) bilayer using Dabble (Betz, 2017). The final system dimensions were 74.1 × 76.8 × 94.3 Å, including 121 lipids, ~10,600 water molecules, 13 sodium ions and 29 chloride ions.
MD simulation force field parameters
The CHARMM36m parameters were used to model protein molecules, CHARMM36 parameters for lipids and salt and the CHARMM36 TIP3P model for water (Huang et al., 2017; Klauda et al., 2010). Parameters for the ligand were generated using the ParamChem Webserver and CGenFF parameterset (Vanommeslaeghe and MacKerell, 2012; Vanommeslaeghe et al., 2012)
MD simulation protocol
MD simulations were performed on GPUs with the CUDA enabled version of PMEMD in AMBER16 (Case et al., 2008; Salomon-Ferrer et al., 2013). Each simulation underwent a similar equilibration procedure. Following an initial minimization, each system underwent a heating using the Langevin thermostat from 0K to 100K in the NVT ensemble over 12.5 picoseconds (ps) with 10 kcal mol−1 Å−2 harmonic restraints on all non-hydrogen atoms in the protein, ligand and lipid. The heated then continued in the NPT ensemble with semi-isotropic coupling for 125 ps and a pressure of 1 bar to a final temperature of 310K with 5.0 kcal mol−1 Å−2 harmonic restraints. Further equilibration was then carried out at 310K with harmonic restraints applied to the protein starting at 5.0 kcal mol−1 Å−2 and reduced in a stepwise fashion every 2 nanoseconds (ns) for 10 ns, followed by 0.1 kcal mol−1 Å−2 restraints for 20 ns for a total of 30 ns of equilibration. Production simulations were run at 310K and 1 bar in the NPT ensemble using the Langevin thermostat and Monte Carlo barostat. Throughout the final stages of equilibration and production, 5.0 kcal mol−1 Å−2 harmonic restraints were placed on all residues of CB1 that were within 5Å of Gi in the CB1- Gi complex to ensure that the receptor remained in the active state in the absence of the G-protein.
Each simulation used periodic boundaries and employed a time step of 4.0 fs using hydrogen mass repartitioning (Hopkins et al., 2015). All bond lengths to hydrogens were constrained by SHAKE (Ryckaert et al., 1977). Short range electrostatic and van der Waals interactions were cut off at 9.0 Å, while long range electrostatic interactions were computed using the particle mesh Ewald method. The FFT grid size was chosen such that the width of a single grid cell was approximately 1 Å. For each of the three simulated poses, we performed three independent simulations, each of 2.0 μs in length.
Snapshots from each trajectory were saved every 200 ps during the production phase of each simulation and visualized using VMD (Humphrey et al., 1996). Analysis was carried out using a combination of VMD and locally developed analysis tools.
Quantum Chemical Calculations
Quantum chemical dihedral scans were prepared using the GAUSSIAN software (Frisch et al., 2009) for each of the indazole-amide derivatives. All calculations were performed with the ωB97-xd functional (Chai and Head-Gordon, 2008) and the 6-311++(2d,2p) basis set. The annotated dihedral was scanned in increments of 15 degrees with other degrees of freedom optimized. All compounds had minima at ~0 and ~180 degrees, the relative energies of which are given in Figure S5.
Molecular Mechanics JAWS Simulations
JAWS simulations (Michel et al., 2009) were prepared from the cryo-EM structure of the CB1 receptor with the MCPRO software package (Jorgensen and Tirado-Rives, 2005). A 15Å sphere around FUB was solvated with theta waters to be sampled in the simulation and treated as flexible. The protein was simulated with the OPLS-AA/M force field (Robertson et al., 2015), the ligand used the OPLS-AA/CM1A force field (Udier-Blagović et al., 2004) and the TIP4P model was used for the water (Jorgensen et al., 1996). Simulations used 5 million Monte Carlo steps for solvent equilibration, 10 million Monte Carlo steps in identifying hydration sites, and 50 million Monte Carlo steps in the production phase. Free energies of binding were then calculated from the percentage of the simulation where a given water molecule was predicted to exist, with water molecules sufficiently energetically favourable to always be existent given an energy of > 3.5 kcal/mol.
Figure preparation
Figures were created using PyMol (http://pymol.org/), and the UCSF Chimera X package (Goddard et al., 2018).
DATA AVAILABILITY
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-0339 and model coordinates have been deposited in the Protein Data Bank (PDB) under accession number 6N4B.
Supplementary Material
Figure S1. Complex formation and cryo-EM data processing. Related to Figures 1 and 2. (A) Fluorescence-detection size exclusion chromatography (FSEC) traces showing complex peak and free receptor peak with the different ligands. (B) Representative reference-free 2D cryo-EM averages of CB1-Gi. The diameter of the circular mask is 18 nm. (C) Cryo-EM data processing flow chart of CB1, including particle selection, classifications and density map reconstruction. Details are provided in the methods section.
Figure S2. Map/model quality and local resolution. Related to Figures 2 and 3. (A) Gold standard Fourier shell correlation (FSC) curves of two individual half maps indicating an average resolution of 3 Å at 0.143 FSC. (B) FSC curves of the model vs. the final EM map (blue) and the two independent half maps (gold and green). FSC plots were calculated with Phenix_M-triage. (C) Density map colored by local resolution indicating a range of 2.7-3.6 Å resolution in most map regions. Highest resolution is observed at the core of the Gi-protein complex with indicated local resolution better than 2.8 Å for most residues. Color scheme for resolution values is presented in the bottom. (D) FUB in density. Density is contoured at σ=3.0 around the ligand. (E) Ensemble of FUB poses observed in MD simulations sampled every 150 ns fitted in the cryo-EM map density. (F) CB1 helixes 1-8 in density.
Figure S3. Ligand binding pocket and entry pathway. Related to Figure 4. (A) A snapshot from MD simulation (yellow) with the N-terminus in the most outward position and TM1-TM7 farthest apart compared to the CB1-Gi structure (blue). (B-C) The presence and absence of lipid access channel between TM1 and TM7 in taranabant- and FUB-bound structures respectively. (D) Surface representation of a snapshot from MD simulation showing the ligand access channel between TM1 and TM7. (E) A graph showing the CB1 N-terminus (in black) outward movement as measured by the distance observed between the N-terminus (Cα F108N-ter) and ICL2 (Cα P269ICL2). This N-terminus movement coincides with the increase in TM1-TM7 distance (shown in red) distance as measured by (Cα Q1161.32) and ICL2 (Cα T3777.32). The yellow background colours indicate the presence of lipid binding during the simulation. (F) An example of lipid binding the TM1-TM7 gap during MD simulation and the lipid molecule overlaid on the taranabant-bound structure.
Figure S4. Interaction of agonists with the CB1. Related to Figure 4. (A) FUB assumes a C-shape configuration that occupies most of the binding pocket volume. (B) Docking of FUB analogue 5F-MDMB-PINACA (PIN) with a fluoro-alkyl moiety to the CB1 binding pocket, with alkyl chain overlapping with the p-fluorobenzyl group in FUB. (C) Docking results of one pose of Δ9-THC (green) superimposed with the structure of FUB (orange) and the crystal structure of AM-11542 (cyan, PDB 5XRA) indicating that the alkyl chain in Δ9-THC overlaps with the p-Fluorobenzyl group and the alkyl chains in FUB and AM-11542, respectively. (D) The docking pose from C (light green) superimposed with an alternative docked pose for Δ9-THC (dark green), indicating that the alkyl chain in Δ9-THC fluctuates between two main poses. (E) Chemical structures of FUB, AM-11542 and Δ9-THC, with suggested mechanism for Δ9-THC partial agonism as implemented from the docking experiments. Moieties that interfere with toggle switch activation are highlighted in orange. (F-H) JAWS calculation demonstrating a tightly bound water molecule mediating interactions between FUB (F), H1782.65, and S3837.39. Both AM-11542 (PDB 5XRA, G) and CP-55,940 (docked, H) have hydrogen bonding groups in this region. Chemical structures are showed on bottom, hydrogen bonding group highlighted in light yellow.
Figure S5. QM calculations. Related to Figure 4. (A) Diagram of the dihedral being flipped in the full FUB molecule. (B) Comparison of the relative QM energies for the cis/trans flip in derivatives of FUB (orange) and other analogues with the same scaffold. The compounds are: Orange (FUB) with both an indazole and an amide (X=N, Y=NH); cyan with an indazole but no amide (X=N, Y=CH2); green an amide and an indole (X=CH, Y=NH2); and yellow with an indole and no amide (X=CH, Y=CH2). Positions X and Y are denoted with red circles.
Figure S6. Structural changes in CB1 on nucleotide-free Gi binding. Related to Figure 5. (A) Comparison of R3.50 and the NPXXY motif conformations in agonist-bound (PDB 5XRA, cyan) and Gi-bound CB1 (blue). (B) The TM6 outward movement on agonist binding (cyan) is further enhanced upon Gi binding (blue) in comparison to antagonist-bound CB1 (PDB 5U09, orange).
Figure S7. Coupling selectivity. Related to Figure 6. Comparison of the position of residues at 6.33 and 6.34 in CB1-Gi (blue and yellow) and β2AR-Gs (green and orange) structures.
Highlights.
3-Å cryo-EM structure of the CB1-Gi complex bound to potent agonist MDMB-Fubinaca
MDMB-Fubinaca locks ‘toggle switch’ residues F2003.36/W3566.48 in active conformation
Quantum mechanics calculations reveal the mechanism for the high affinity of Fubinaca
Molecular dynamic simulations reveal a path for ligand entry between TM1 and TM7
Looking at how a toxic, synthetic ligand locks cannabinoid receptor 1 into a signaling conformation points to ways to understand and modulate receptor activity
Acknowledgements
Kaavya Krishna Kumar was supported by the CJ Martin NHMRC Fellowship. The work is supported by NIH grant R37DA036246 (B.K.K. and G.S.) and NIH grant R01GM083118 (B.K.K.). B.K.K. is a Chan Zuckerberg Biohub investigator.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Lead contact: Georgios Skiniotis
References
- Adams AJ, Banister SD, Irizarry L, Trecki J, Schwartz M, and Gerona R (2017). “Zombie” Outbreak Caused by the Synthetic Cannabinoid AMB-FUBINACA in New York. N. Engl. J. Med 376, 235–242. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung L-W, Jain S, Kapral GJ, Grosse-Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Klaholz BP, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, Adams PD, and Urzhumtsev A (2018). New tools for the analysis and validation of Cryo-EM maps and atomic models. 1–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood BK, Huffman J, Straiker A, and Mackie K (2010). JWH018, a common constituent of “Spice” herbal blends, is a potent and efficacious cannabinoid CB receptor agonist. Br. J. Pharmacol 160, 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, and Weinstein H (1995). Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In Methods in Neurosciences, (Elsevier), pp. 366–428. [Google Scholar]
- Barnett-Norris J, Hurst DP, Buehner K, Ballesteros JA, Guarnieri F, and Reggio PH (2002). Agonist alkyl tail interaction with cannabinoid CB1 receptor V6.43/I6.46 groove induces a helix 6 active conformation. International Journal of Quantum Chemistry 88, 76–86. [Google Scholar]
- Betz RM (2017). Dabble. [Google Scholar]
- Bow EW, and Rimoldi JM (2016). The Structure-Function Relationships of Classical Cannabinoids: CB1/CB2 Modulation. Perspect Medicin Chem 8, 17–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AC, Moreira FA, Gomes FV, Del Bel EA, and Guimarães FS (2012). Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos. Trans. R. Soc. Lond., B, Biol. Sci 367, 3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, and Simmerling CL (2008). Amber 10. [Google Scholar]
- Chai J-D, and Head-Gordon M (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Liu N-J, and Gintzler AR (2010). Formation of mu-/kappa-opioid receptor heterodimer is sex-dependent and mediates female-specific opioid analgesia. Proc. Natl. Acad. Sci. U.S.a 107, 20115–20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XP, Yang W, Fan Y, Luo JS, Hong K, Wang Z, Yan JF, Chen X, Lu JX, Benovic JL, et al. (2010). Structural determinants in the second intracellular loop of the human cannabinoid CB1 receptor mediate selective coupling to G(s) and G(i). Br. J. Pharmacol 161, 1817–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M, and Christie MD (1999). Opioid receptor signalling mechanisms. Clin. Exp. Pharmacol. Physiol 26, 493–499. [DOI] [PubMed] [Google Scholar]
- Cooper ZD (2016). Adverse Effects of Synthetic Cannabinoids: Management of Acute Toxicity and Withdrawal. Curr Psychiatry Rep 18, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupi X (2014). Relevance of rhodopsin studies for GPCR activation. Biochim. Biophys. Acta 1837, 674–682. [DOI] [PubMed] [Google Scholar]
- Draper-Joyce CJ, Khoshouei M, Thal DM, Liang YL, Nguyen ATN, Furness SGB, Venugopal H, Baltos JA, Plitzko JM, Danev R, et al. (2018). Structure of the adenosine-bound human adenosine A1receptor-Gicomplex. Nature 558, 559–565. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fagan SG, and Campbell VA (2014). The influence of cannabinoids on generic traits of neurodegeneration. Br. J. Pharmacol 171, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantegrossi WE, Moran JH, Radominska-Pandya A, and Prather PL (2014). Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to Δ(9)-THC: mechanism underlying greater toxicity? Life Sci. 97, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, and Glass M (1998). Cannabinoid receptors and their endogenous agonists. Annu. Rev. Pharmacol. Toxicol 38, 179–200. [DOI] [PubMed] [Google Scholar]
- Fernandez-Leiro R, and Scheres SHW (2017). A pipeline approach to single-particle processing in RELION. Acta Crystallogr D Struct Biol 73, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, and Petersson GA (2009). Gaussian 09 Revision D. 01, 2009 Gaussian Inc; Wallingford CT. [Google Scholar]
- Gamage TF, Farquhar CE, Lefever TW, Marusich JA, Kevin RC, McGregor IS, Wiley JL, and Thomas BF (2018). Molecular and Behavioral Pharmacological Characterization of Abused Synthetic Cannabinoids MMB- and MDMB-FUBINACA, MN-18, NNEI, CUMYL-PICA, and 5-Fluoro-CUMYL-PICA. J. Pharmacol. Exp. Ther 365, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Nafría J, Nehmé R, Edwards PC, and Tate CG (2018). Cryo-EM structure of the serotonin 5-HT1Breceptor coupled to heterotrimeric Go. Nature 558, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanouni P, Schambye H, Seifert R, Lee TW, Rasmussen SG, Gether U, and Kobilka BK (2000). The effect of pH on beta(2) adrenoceptor function. Evidence for protonation-dependent activation. J. Biol. Chem 275, 3121–3127. [DOI] [PubMed] [Google Scholar]
- Glass M, and Felder CC (1997). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J. Neurosci 17, 5327–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, and Grigorieff N (2018). cisTEM, user-friendly software for single-particle image processing. Elife 7, e14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, et al. (2017). Single-molecule analysis of ligand efficacy in β2AR-G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, and Hohmann AG (2009). The endocannabinoid system and pain. CNS Neurol Disord Drug Targets 8, 403–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. (2012). Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JB (2018). Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins CW, Le Grand S, Walker RC, and Roitberg AE (2015). Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J Chem Theory Comput 11, 1864–1874. [DOI] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho J-H, et al. (2017). Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547, 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, et al. (2016). Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 167, 750–762.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmüller H, and MacKerell AD Jr (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nature Methods 2016 14:1 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, et al. (2015). Structural insights into μ-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: Visual molecular dynamics. Journal of Molecular Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Ignatowska-Jankowska BM, Baillie GL, Kinsey S, Crowe M, Ghosh S, Owens RA, Damaj IM, Poklis J, Wiley JL, Zanda M, et al. (2015). A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology 40, 2948–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Borrelli F, Capasso R, Di Marzo V, and Mechoulam R (2009). Non-psychotropic plant cannabinoids: new therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci 30, 515–527. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, and Tirado-Rives J (2005). Molecular modeling of organic and biomolecular systems using BOSS and MCPRO. J Comput Chem 26, 1689–1700. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, and Tirado-Rives J (1996). Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 118, 11225–11236. [Google Scholar]
- Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, et al. (2018). Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Hurst DP, Fleischer D, Whitnell R, Thakur GA, Makriyannis A, Reggio PH, and Abood ME (2007). Mutation studies of Ser7.39 and Ser2.60 in the human CB1 cannabinoid receptor: evidence for a serine-induced bend in CB1 transmembrane helix 7. Mol. Pharmacol 71, 1512–1524. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, and Pastor RW (2010). Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, Latorraca NR, Hilger D, Dawson R, Matile H, et al. (2018). Structure of the μ-opioid receptor-Gi protein complex. Nature 558, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, and Mackie K (2005). The cannabinoid agonist WIN55,212–2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. U.S.a 102, 19144–19149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K (2006). Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol 46, 101–122. [DOI] [PubMed] [Google Scholar]
- Maeda S, Koehl A, Matile H, Hu H, Hilger D, Schertler GFX, Manglik A, Skiniotis G, Dawson RJP, and Kobilka BK (2018). Development of an antibody fragment that stabilizes GPCR/G-protein complexes. Nat Commun 9, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, and Lutz B (1999). Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur. J. Neurosci 11, 4213–4225. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, and Abood ME (2004). Structural mimicry in class A G protein-coupled receptor rotamer toggle switches: the importance of the F3.36(201)/W6.48(357) interaction in cannabinoid CB1 receptor activation. J. Biol. Chem 279, 48024–48037. [DOI] [PubMed] [Google Scholar]
- Micale V, Di Marzo V, Sulcova A, Wotjak CT, and Drago F (2013). Endocannabinoid system and mood disorders: priming a target for new therapies. Pharmacol. Ther 138, 18–37. [DOI] [PubMed] [Google Scholar]
- Michel J, Tirado-Rives J, and Jorgensen WL (2009). Prediction of the water content in protein binding sites. J Phys Chem B 113, 13337–13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnpotra JS, Qiao Z, Cai J, Lynch DL, Grossfield A, Leioatts N, Hurst DP, Pitman MC, Song ZH, and Reggio PH (2014). Structural basis of G protein-coupled receptor-Giprotein interaction: Formation of the cannabinoid CB2receptor-Giprotein complex. J. Biol. Chem 289, 20259–20272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty NW, Grosse-Kunstleve RW, and Adams PD (2009). electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro O, Lameh J, Högger P, and Sadée W (1993). Hydrophobic amino acid in the i2 loop plays a key role in receptor-G protein coupling. J. Biol. Chem 268, 22273–22276. [PubMed] [Google Scholar]
- Njoo C, Agarwal N, Lutz B, and Kuner R (2015). The Cannabinoid Receptor CB1 Interacts with the WAVE1 Complex and Plays a Role in Actin Dynamics and Structural Plasticity in Neurons. PLoS Biol. 13, e1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble AJ, Dandey VP, Wei H, Brasch J, Chase J, Acharya P, Tan YZ, Zhang Z, Kim LY, Scapin G, et al. (2018). Routine single particle CryoEM sample and grid characterization by tomography. Elife 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. (2013). The dynamic process of β(2)-adrenergic receptor activation. Cell 152, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peace MR, Krakowiak RI, Wolf CE, Poklis A, and Poklis JL (2017). Identification of MDMB-FUBINACA in commercially available e-liquid formulations sold for use in electronic cigarettes. Forensic Science International 271, 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, and Skiniotis G (2015). 2D Projection Analysis of GPCR Complexes by Negative Stain Electron Microscopy In G Protein-Coupled Receptors in Drug Discovery, (New York, NY: Springer New York; ), pp. 29–38. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pryce G, and Baker D (2012). Potential control of multiple sclerosis by cannabis and the endocannabinoid system. CNS Neurol Disord Drug Targets 11, 624–641. [DOI] [PubMed] [Google Scholar]
- Ranganathan A, Dror RO, and Carlsson J (2014). Insights into the role of Asp79(2.50) in β2 adrenergic receptor activation from molecular dynamics simulations. Biochemistry 53, 7283–7296. [DOI] [PubMed] [Google Scholar]
- Robertson MJ, Tirado-Rives J, and Jorgensen WL (2015). Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J Chem Theory Comput 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi H-J, Devree BT, Sunahara RK, et al. (2011). Structure and function of an irreversible agonist-β(2) adrenoceptor complex. Nature 469, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J-P, Ciccotti G, and Berendsen HJC (1977). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics 23, 327–341. [Google Scholar]
- Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, and Walker RC (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- Sansuk K, Deupi X, Torrecillas IR, Jongejan A, Nijmeijer S, Bakker RA, Pardo L, and Leurs R (2011). A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol. Pharmacol 79, 262–269. [DOI] [PubMed] [Google Scholar]
- Schoeder CT, Hess C, Madea B, Meiler J, and Müller CE (2018). Pharmacological evaluation of new constituents of “Spice”: synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicology 191, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, and Wenzel-Seifert K (2002). Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch. Pharmacol 366, 381–416. [DOI] [PubMed] [Google Scholar]
- Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, and Rosenbaum DM (2016). High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 540, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szücs M, Boda K, and Gintzler AR (2004). Dual effects of DAMGO [D-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin and CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) on adenylyl cyclase activity: implications for mu-opioid receptor Gs coupling. J. Pharmacol. Exp. Ther 310, 256–262. [DOI] [PubMed] [Google Scholar]
- Udier-Blagović M, Morales De Tirado P, Pearlman SA, and Jorgensen WL (2004). Accuracy of free energies of hydration using CM1 and CM3 atomic charges. J Comput Chem 25, 1322–1332. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, and MacKerell AD (2012). Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Raman EP, and MacKerell AD (2012). Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfield GH, Rasmussen SGF, Su M, Dutta S, Devree BT, Chung KY, Calinski D, Velez-Ruiz G, Oleskie AN, Pardon E, et al. (2011). Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci. U.S.a 108, 16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 2016 14:1 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, and Song ZH (2002). CB1 cannabinoid receptor-mediated tyrosine phosphorylation of focal adhesion kinase-related non-kinase. FEBS Lett. 525, 164–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Complex formation and cryo-EM data processing. Related to Figures 1 and 2. (A) Fluorescence-detection size exclusion chromatography (FSEC) traces showing complex peak and free receptor peak with the different ligands. (B) Representative reference-free 2D cryo-EM averages of CB1-Gi. The diameter of the circular mask is 18 nm. (C) Cryo-EM data processing flow chart of CB1, including particle selection, classifications and density map reconstruction. Details are provided in the methods section.
Figure S2. Map/model quality and local resolution. Related to Figures 2 and 3. (A) Gold standard Fourier shell correlation (FSC) curves of two individual half maps indicating an average resolution of 3 Å at 0.143 FSC. (B) FSC curves of the model vs. the final EM map (blue) and the two independent half maps (gold and green). FSC plots were calculated with Phenix_M-triage. (C) Density map colored by local resolution indicating a range of 2.7-3.6 Å resolution in most map regions. Highest resolution is observed at the core of the Gi-protein complex with indicated local resolution better than 2.8 Å for most residues. Color scheme for resolution values is presented in the bottom. (D) FUB in density. Density is contoured at σ=3.0 around the ligand. (E) Ensemble of FUB poses observed in MD simulations sampled every 150 ns fitted in the cryo-EM map density. (F) CB1 helixes 1-8 in density.
Figure S3. Ligand binding pocket and entry pathway. Related to Figure 4. (A) A snapshot from MD simulation (yellow) with the N-terminus in the most outward position and TM1-TM7 farthest apart compared to the CB1-Gi structure (blue). (B-C) The presence and absence of lipid access channel between TM1 and TM7 in taranabant- and FUB-bound structures respectively. (D) Surface representation of a snapshot from MD simulation showing the ligand access channel between TM1 and TM7. (E) A graph showing the CB1 N-terminus (in black) outward movement as measured by the distance observed between the N-terminus (Cα F108N-ter) and ICL2 (Cα P269ICL2). This N-terminus movement coincides with the increase in TM1-TM7 distance (shown in red) distance as measured by (Cα Q1161.32) and ICL2 (Cα T3777.32). The yellow background colours indicate the presence of lipid binding during the simulation. (F) An example of lipid binding the TM1-TM7 gap during MD simulation and the lipid molecule overlaid on the taranabant-bound structure.
Figure S4. Interaction of agonists with the CB1. Related to Figure 4. (A) FUB assumes a C-shape configuration that occupies most of the binding pocket volume. (B) Docking of FUB analogue 5F-MDMB-PINACA (PIN) with a fluoro-alkyl moiety to the CB1 binding pocket, with alkyl chain overlapping with the p-fluorobenzyl group in FUB. (C) Docking results of one pose of Δ9-THC (green) superimposed with the structure of FUB (orange) and the crystal structure of AM-11542 (cyan, PDB 5XRA) indicating that the alkyl chain in Δ9-THC overlaps with the p-Fluorobenzyl group and the alkyl chains in FUB and AM-11542, respectively. (D) The docking pose from C (light green) superimposed with an alternative docked pose for Δ9-THC (dark green), indicating that the alkyl chain in Δ9-THC fluctuates between two main poses. (E) Chemical structures of FUB, AM-11542 and Δ9-THC, with suggested mechanism for Δ9-THC partial agonism as implemented from the docking experiments. Moieties that interfere with toggle switch activation are highlighted in orange. (F-H) JAWS calculation demonstrating a tightly bound water molecule mediating interactions between FUB (F), H1782.65, and S3837.39. Both AM-11542 (PDB 5XRA, G) and CP-55,940 (docked, H) have hydrogen bonding groups in this region. Chemical structures are showed on bottom, hydrogen bonding group highlighted in light yellow.
Figure S5. QM calculations. Related to Figure 4. (A) Diagram of the dihedral being flipped in the full FUB molecule. (B) Comparison of the relative QM energies for the cis/trans flip in derivatives of FUB (orange) and other analogues with the same scaffold. The compounds are: Orange (FUB) with both an indazole and an amide (X=N, Y=NH); cyan with an indazole but no amide (X=N, Y=CH2); green an amide and an indole (X=CH, Y=NH2); and yellow with an indole and no amide (X=CH, Y=CH2). Positions X and Y are denoted with red circles.
Figure S6. Structural changes in CB1 on nucleotide-free Gi binding. Related to Figure 5. (A) Comparison of R3.50 and the NPXXY motif conformations in agonist-bound (PDB 5XRA, cyan) and Gi-bound CB1 (blue). (B) The TM6 outward movement on agonist binding (cyan) is further enhanced upon Gi binding (blue) in comparison to antagonist-bound CB1 (PDB 5U09, orange).
Figure S7. Coupling selectivity. Related to Figure 6. Comparison of the position of residues at 6.33 and 6.34 in CB1-Gi (blue and yellow) and β2AR-Gs (green and orange) structures.
Data Availability Statement
The cryo-EM density map has been deposited in the Electron Microscopy Data Bank (EMDB) under accession code EMD-0339 and model coordinates have been deposited in the Protein Data Bank (PDB) under accession number 6N4B.