

SUMMARY
Transcriptional circuit architectures in several organisms have been evolutionarily selected to dictate precise given responses. Unlike these cellular systems, HIV is regulated through a complex circuit composed of two successive phases (host and viral), which create a positive feedback loop facilitating viral replication. However, it has long remained unclear whether both phases operate identically and to what extent the host phase influences the entire circuit. Here, we report that, although the host phase is regulated by a checkpoint whereby KAP1 mediates transcription activation, the virus evolved a mini-malist system bypassing KAP1. Given the complex circuit’s architecture, cell-to-cell KAP1 fluctuations impart heterogeneity in the host transcriptional responses, thus affecting the feedback loop. Mathematical modeling of a complete circuit reveals how these oscillations ultimately influence homogeneous reactivation potential of a latent virus. Thus, although HIV drives molecular innovation to fuel robust gene activation, it experiences transcriptional fragility, thereby influencing viral fate and cure efforts.
Graphical Abstract
In Brief
Morton et al. show that HIV has evolved a minimalist but robust transcriptional circuit that bypasses host regulatory checkpoints. However, they demonstrate that the fragility of the circuit in the host phase (which primes HIV for activation) largely affects proviral transcription and fate.
INTRODUCTION
Transcriptional regulatory circuits are essential for controlling several key biological processes, such as development, differentiation, and cell fate responses. As such, transcriptional circuit architecture have been evolutionarily selected to precisely dictate the appropriate cellular responses. In contrast to these highly evolvable circuits, viruses such as HIV type 1, which integrate into the human genome (Hughes and Coffin, 2016; Schröder et al., 2002), initially fall under the control of host circuits. Given that HIV integration is “quasi”-random, the heterogeneous integration landscape may affect transcriptional circuit architecture, leading to variable outcomes and thereby generating profound phenotypic diversity among different infections, here referred to as “proviral fate” (Figure 1A).
Figure 1. Establishing an Experimental-Mathematical Modeling Framework for Understanding a Complete HIV Transcriptional Circuit.

(A) Simplistic scheme of HIV proviral fate after infection and integration into the host cell genome. Latent viruses can be reactivated in response to immune stimulation.
(B) Scheme depicting the latent proviral state and its associated transcriptional circuit (basal) and output.
(C) Scheme depicting the active proviral state and its associated transcriptional circuit (host) and output.
(D) Scheme depicting the super-active proviral state and its associated transcriptional circuit (viral) and output.
(E) Scheme of an incomplete HIV transcriptional circuit.
(F) Scheme of a complete HIV transcriptional circuit.
Over the past decades, one of the most exciting breakthroughs in biomedical research has been the discovery of anti-retroviral therapy (ART), which suppresses active replication to nearly undetectable levels. However, ART fails to cure latent infections, because the targeted proteins are not expressed or are expressed at extremely low levels. Consequently, HIV establishes long-lived latent reservoirs in vivo by persisting as a stable integrated provirus in resting memory CD4+ T lymphocytes and myeloid cells and by remaining undetected by immune surveil-lance mechanisms. Although these constitute a very small population, they do not apparently produce appreciable virus and are considered the largest barrier for HIV eradication from a patient (Chun et al., 1995; Finzi et al., 1999). Although the molecular rules governing proviral latency appear to be pleiotropic, one common feature is the resting state of the infected cell, leading to low, or even undetectable, levels of transcription activity. Thus, HIV latency is a state of non-productive infection due to major transcriptional restrictions (Karn, 2011; Ruelas and Greene, 2013).
Because cessation of therapy leads to viral rebound within weeks, HIV-infected individuals must remain on therapy permanently. Given the secondary effects associated with the long-term regime, pharmacological strategies designed to eradicate the viral latent reservoir represent a critical unmet need. There is enormous enthusiasm for the potential of precision therapies targeting the latent reservoir in clinical settings. Thus, HIV latency has become the center of attention. As such, a large body of research has identified the role of individual host factors and epigenetics on HIV transcription activation or silencing and elucidated host enzymes as targets that could be manipulated using chemical probes to induce latency reversal. Despite several landmark discoveries, we currently lack a complete understanding of the fundamental regulatory principles of the HIV transcriptional circuit and its implications for proviral fate control, including latency.
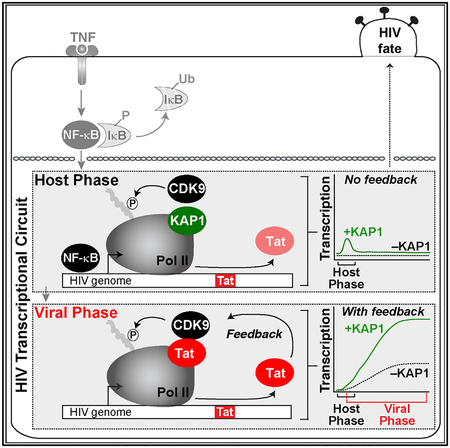
The HIV transcriptional circuit is regulated at different levels. First, during normal cell homeostasis, “basal” steady-state transcription maintains a low level of non-productive RNA synthesis, leading to short, immature transcripts (Figure 1B). In this state, the viral activator Tat is not expressed, and thus, HIV does not replicate (latent state). In the “host” phase, when cells are exposed to immune stimulation, transcription factors such as NF-κB and NFAT are activated, leading to an initial low-level “boost” in proviral transcription. In proviruses lacking Tat, this phase shows a unimodal pattern of activation that is quickly turned off, leading to a small amount of viral products (Figure 1C). During productive infections with wild-type proviruses, the initial transcriptional boost is critical because it enables Tat synthesis before the host phase turns off. In this case, the host phase is rapidly followed by a “viral” phase in which Tat amplifies transcription by more than 100-fold, promoting a positive transcriptional feedback loop and robust viral replication (Karn, 2011) (Figure 1D).
In the resting scenario, most of the cellular activators are found in a latent state, but they become activated when the infected immune cells encounter a stimulus from the microenvironment. For example, the pro-inflammatory cytokine tumor necrosis factor (TNF) activates the canonical NF-κB pathway, where it trans-locates from the cytoplasm into the nucleus and recognizes its binding element at the viral promoter, driving proviral transcription (Nabel and Baltimore, 1987). Similarly, the CD40 ligand and lymphotoxin induce proviral transcription upon receptor activation signaling through non-canonical NF-κB (Pache et al., 2015). T cell stimulation functions broadly through multiple signaling pathways, including several master cellular activators (Kinoshita et al., 1997; Nabel and Baltimore, 1987).
The cellular activators and Tat promote transcription in the host and viral phases, respectively, through a complex layer of host factors, including general transcription factors, RNA polymerase II (Pol II), and co-activators and co-repressors. One key factor is the positive transcription elongation factor b (PTEFb), which is composed of a cyclin T subunit and the catalytic CDK9 subunit (hereafter referred as CDK9). Both cellular activators and Tat use CDK9 to facilitate the transcription elongation program, a critical step in the viral life cycle (Ott et al., 2011; Bacon and D’Orso, 2018).
Despite the relevance of CDK9, it has long remained unclear whether both host and viral programs operate through identical mechanisms and how their malfunction affects proviral latency. Recent studies have suggested a role for the transcriptional regulator KAP1 (TRIM28, TIF1β) in proviral transcription through CDK9 recruitment to the promoter as part of the 7SK complex, in which the kinase remains in a primed state (D’Orso, 2016). In this context, the 7SK complex (composed of 7SK RNA and kinase inhibitor HEXIM) not only inactivates the kinase, but more important, it has a positive role in delivering the kinase for on site activation at the promoter (McNamara et al., 2016a, 2016b). These recent discoveries provide an unprecedented function for KAP1, which has been previously implicated in transcriptional repression through epigenetic silencing of genes and retroele ments in progenitor and non-committed cells as well as repression of viruses in embryonic stem cells (ESCs) (Rowe et al., 2010; Wolf and Goff, 2007).
Here we found that KAP1 is expressed in primary resting memory CD4+ T cells and is recruited to the proviral genome in a latency model in primary cells, thus providing biological relevance for the pathogenic mechanism described. To our surprise, we also report the unexpected findings that the different phases of the HIV circuit have different functional requirements. Although KAP1 is critical for activation of the host phase, HIV evolved a minimalist system whereby Tat represents a switch to a “higher gear,” bypassing KAP1 to activate transcription. Although KAP1 recruits CDK9 to the promoter to facilitate activation by cellular activators in response to cytokine stimulation, Tat subsequently functions in a KAP1-independent manner, directly recruiting the kinase to sustain transcription elongation. Given that the host phase has a strict requirement for KAP1, its loss affects the positive feedback loop, thus reducing the magnitude of reactivation of a latent virus.
Previous studies have created mathematical models that incompletely interrogate the HIV transcriptional circuit (“basal-viral”) (Weinberger et al., 2005) (Figure 1E). Thus, the roles of host cell factors and immune cell stimulation on the host phase and its effect on the positive feedback loop have not been previously probed. Given that the virus strictly relies on the immune cells’ activation status, we rationalized that generating a model that can recapitulate the complete HIV program can not only provide critical insights into HIV biology but also pave the groundwork for more efficient interventions in the clinical setting. We thus created a mathematical model that recapitulates the complete HIV transcriptional circuit (“basal-host-viral”). This model predicts that fluctuations of KAP1 levels in patient’s cells could affect the host phase and, as a consequence, the magnitude of the Tat feedback (thereby dampening latency reversal potential). We tested this model experimentally and observed how KAP1 oscillations impart heterogeneity in the transcriptional responses thereby influencing the reactivation potential of a latent virus. Our findings provide a mechanistic explanation for the importance of the host phase to ensure the virus is readily and robustly activated during infection to complete the pathogenic cycle.
RESULTS
Establishing an Experimental-Mathematical Modeling Framework for Interrogating a Complete HIV Transcriptional Circuit
HIV infection of immune cells can lead to active and latent infections as a potential consequence of transcriptionally active and silent states, respectively (Figure 1A). Figures 1B–1D illustrate the progression of molecular events leading to activation of the HIV circuit from basal transcription, to activation of the host phase by cellular activators (NF-κB) in response to immune stimulation, and ultimately to activation of the viral phase by Tat. The key feature of this system is that activation of the host phase during productive infection leads to Tat synthesis, which induces a positive feedback loop leading to robust viral replication. The activators of both phases function by recruiting CDK9 to the proviral genome (directly or indirectly) to induce Pol II transcription elongation.
Because the HIV circuit operates through the combined, sequential activity of the host and viral phases, it has been challenging to uncouple the precise contributions of each phase in the program as a whole. To overcome this challenge, here we establish an integrated experimental and mathematical modeling framework for precisely interrogating a complete HIV transcriptional circuit. Experimentally, we use primary and transformed cell-based models of latency containing integrated HIV in which Tat can be either wild-type (Tat+) or defective (Tat−), which are regulated by the complete circuit or its minimalist version (host phase only), respectively. Directly comparing the transcriptional profiles of both proviruses in response to stimulation upon host factor depletion enables us to infer their contributions to the phases of the HIV circuit.
To expand the establishment of experimental approaches, a mathematical model was created that recapitulates the complete HIV circuit architecture (Figure 1F). Although previous studies have modeled HIV activation by Tat using a simple circuit composed of the “basal-viral” phases (Weinberger et al., 2005) (Figure 1E), using detailed kinetic parameters from measurements after Reddy and Yin (1999), those models do not enable one to examine the contribution of the host phase to the viral phase, the magnitude of the feedback loop, or the reactivation potential of a latent virus. Given this large caveat, it has been impossible to predict and test the contributions of host factors to the HIV transcriptional program as a whole. Our integrated approach establishes a framework to interrogate the complete HIV circuit and its implications in the context of proviral latency and reactivation.
Defining Host Cell Factor Contributions to the Transcriptional Circuit of a Latent Virus
KAP1 has been previously shown to play an important role in epigenetic silencing of retroelements. As part of this mechanism, KAP1 appears to be recruited to gene promoters through interaction with a family of KRAB-domain zinc finger proteins (KRAB-ZnF). Then, KAP1 recruits chromatin-modifying enzymes that promote epigenetic silencing (Iyengar and Farnham, 2011; Macfarlan et al., 2011) (Figure 2A).
Figure 2. Loss of KAP1 Does Not Reactivate HIV from Cell-Based Models of Latency.

(A) Simplified current model of KAP1-mediated transcriptional silencing based on previous studies.
(B) Overview of the protocol used to transduce and analyze the cell-based models of HIV latency.
(C) Western blots of the indicated cell-based models.
(D) Flow cytometry analysis of the HIV Tat+ cell-based models from (C). The number of GFP+ cells from three independent runs is indicated.
(E) Western blots of the indicated cell-based models.
(F) Flow cytometry analysis of the HIV Tat+ cell-based models from (E). The number of GFP+ cells from three independent runs is indicated.
To test the hypothesis that KAP1 is recruited to the HIV genome to induce proviral latency through epigenetic silencing of the host phase of the circuit, we transduced the Jurkat cell-based models of latency E4 and 10.6, which contain Tat (HIV Tat+) and a GFP marker for ease of measurement (Jordan et al., 2003; Pearson et al., 2008), with self-inactivating lentiviruses (pLVTHM) expressing non-targeting (NT) or KAP1 short hairpin RNAs (shRNAs) (Figure 2B). As a positive control for our experiments, we used a shRNA targeting the NELF-E subunit of the negative elongation factor complex (NELF), which has been shown to relieve Pol II pausing at the HIV promoter to spontaneously induce latency reversal (Jadlowsky et al., 2014). Given that pLVTHM co-expresses the shRNA and a fluorescent marker (mCherry), we used fluorescence-activated cell sorting (FACS) to separate efficiently transduced cells from untransduced cells to assess knockdown (KD) efficiency in the population by western blot and latent HIV reactivation by flow cytometry by measuring GFP+ cells (Figures 2C–2F).
Despite the remarkably efficient KD of KAP1 in the E4 clone (>90% KD compared with shNT), we did not observe reactivation of latent HIV, as revealed by the similar levels of GFP+ cells in shNT and shKAP1 cell lines (Figures 2C and 2D). However, as expected, loss of NELF resulted in a slight increase in the percentage of GFP+ cells (~2.8-fold increase over NT; Figure 2D), consistent with previous studies (Jadlowsky et al., 2014).
Although the previous analysis was performed in a bulk population, we sought to determine whether these results could be reproduced at a single-cell level. To that end, we sorted individual cells, generated clonal cell lines and examined using FACS their latency-reversal potential. Consistent with the results obtained at the population level, individual shKAP1 clones did not show latent HIV reactivation compared with shNT clones (data not shown).
To test if the results obtained in E4 were cell model independent and thus generalizable, we recreated a collection of cell lines on the 10.6 model. Consistent with the data obtained in E4, we did not observe significant changes in the percentage of GFP+ cells between all cell lines (Figures 2E and 2F). Again, efficient NELF KD (shNELF) led to increase in GFP+ cells compared with parental and shNT cell lines (Figure 2F), indicating reactivation of latent HIV, with slightly higher reactivation levels in 10.6 cells compared with those in E4 cells (4.7-fold versus ~2.8-fold, respectively), probably due to intrinsic differences in the two systems (Figures 2D and 2F).
Given that HIV integrates semi-randomly and can be found in sites with different chromatin accessibility, we asked whether KAP1 contributes to epigenetic silencing in models in which the HIV long terminal repeat (LTR) is relatively inaccessible. To test this, we silenced KAP1 expression in cell-based models showing lower promoter chromatin accessibility (6.3, 8.4, and 9.2) compared with the previous models (E4 and 10.6). Despite the efficient (>95%) loss of KAP1 in every model examined, as revealed by western blot (Figure S1A), we observed no signifi-cant changes in the percentage of GFP+ cells in response to KAP1 KD (Figure S1B), strongly indicating that KAP1 does not contribute to proviral latency maintenance, at least in these permissive CD4+ T cell-based models.
The expression of GFP encoded by the proviruses in these models requires Tat activity and high levels of transcription. Thus, it remains possible that loss of KAP1 could promote some degree of latency reversal but at levels below the GFP detection threshold. To test this possibility, we developed a real-time qPCR assay on the basis of methods that allow us to accurately and efficiently purify and quantitate short (~17–200 nt), promoter-proximal transcripts (indicative of transcription initiation) and long, promoter-distal transcripts (indicative of transcription elongation), irrespective of the amount of total RNA inputted in the reaction mixture, in the absence and presence of TNF stimulation (Figures S1C–S1F).
Additionally, to provide further evidence that the amplicons were correctly amplifying the initiating and elongating transcripts, we pre-treated E4 cells for 30 min with the potent inhibitor of transcription initiation triptolide (TRP), which blocks the ATPase activity of the TFIIH helicase, and the inhibitor of transcription elongation flavopiridol (FP), which blocks P-TEFb. We observed that TRP, expectedly, blocks TNF-mediated induction of both short and long transcripts, whereas FP only interferes with the synthesis of long transcripts, consistent with an initiation and elongation block, respectively (Figures S1G and S1H).
Using this robust, quantitative method, we detected that KAP1 loss does not promote reactivation of latent HIV in the absence of immune stimulation, consistent with the FACS data. KAP1 KD shows a slight (<1.5-fold), non-significant decrease in the levels of both classes of transcripts in several HIV Tat+ cell models (Figure S1I), indicating that KAP1 major’s role is not to promote transcription control under basal conditions but to allow transcriptional responses to immune stimulation.
KAP1 Is Expressed and Recruited along with the Host Transcription Elongation Complex to the HIV Genome in Primary Resting CD4+ T Cells
The above data showed that KAP1 does not mediate HIV epigenetic silencing in CD4+ T cells. However, our initial experiments were performed in transformed cell-based models and in the absence of immune stimulation, which is key for robust activation of the HIV circuit. Given that this system may not completely recapitulate the establishment and maintenance of latency in CD4+ T cells from patients, we thus wanted to test whether (1) KAP1 expression changes in response to T cell state alterations (active versus resting) and HIV infection, (2) KAP1 is recruited to the proviral genome in the active and resting states, and (3) KAP1 could facilitate HIV transcription activation in a more biologically relevant setting (primary CD4+ T cells) in response to immune stimulation.
To address the first point, we used the primary model in central memory CD4+ T cells (TCM) (Bosque and Planelles, 2009; Martins et al., 2016). Briefly, naive CD4+ T cells from healthy donors are activated and induced to differentiate into central memory, infected with replication-competent virus (HIV-1NL4.3) or mock infected (uninfected), and active infections (p24-positive and CD4-negative) are excluded through magnetic sorting to enrich in latent and uninfected states (Figures 3A and 3B). These cells are then maintained in the active T cell state or allowed to transition into a memory resting state in the presence of ART to better mimic viral suppression in patients and thus create four different experimental groups: “uninfected (activated),” “productively infected (activated),” “uninfected (resting),” and “latently infected (resting).” Of note, the latently infected population is a mixture of uninfected and latently infected cells, which are indistinguishable phenotypically.
Figure 3. KAP1 Recruitment to the HIV Genome during Productive and Latent Infections in Primary Cells Is Required for Provirus Activation.

(A) Experimental outline through which naive cells were used to generate TCM infected or not with replication-competent HIV (HIV-1NL4.3) and either activated only or activated and then allowed to transition into a resting state.
(B) FACS plots of cells from (A).
(C) Quantitation of HIV RNAs (+7232 amplicon) normalized to ACTB by real-time qPCR (mean ± SEM; n = 3).
(D) Western blots of cells from (A).
(E) Top: HIV proviral scheme. The arrow denotes the transcription start site (TSS) position. Bottom: ChIP assays were performed with protein extracts from cells from (A) and the indicated antibodies followed by qPCR assays with a series of amplicons mapping throughout the provirus to monitor factor interactions with the HIV genome. Values represent the percentage of input DNA immunoprecipitated (IP DNA) and are the averages of three independent experiments (mean ± SEM; n = 3). Note that “uninfected” refers to both activated (A) and resting (R) states.
(F) Experimental outline through which naive cells were used to generate TCM that were then infected with pseudotyped viruses pNL4.3-deltaEnv-nLuc-2ANef and used for KO of host factors.
(G) FACS plots in control TCM (not nucleofected) and TCM nucleofected with Cas9-gRNA complexes for a non-targeting control (NT) and targeting CXCR4.
(H) Western blots of the indicated cells.
(I) Luciferase assay of TCM containing KO of host factors and treated with PMA or vehicle (DMSO). Luciferase is expressed as relative luciferase units (RLU).
(J) p24 staining of TCM containing KO of host factors and treated with DMSO and PMA (−/+ PMA, respectively).
The four cell states were then collected for subsequent analysis of viral gene expression by real-time qPCR, KAP1 expression by western blot, and factor occupancy at the proviral genome by chromatin immunoprecipitation (ChIP) assay (Figures 3C–3E). Because of the low levels of reactivation in the presence of T cell activation in this model (1%–3%), as judged by p24 levels on FACS, we collected productive infection data to circumvent the low levels of latency reversal. FACS analysis confirmed the expectation that uninfected cells displayed high levels of CD4 with no p24 staining, while infected cells showed a reduction of CD4 levels (Figure 3B). Consistently, quantitation of HIV transcripts indicated that the productively infected state had ~25-fold higher transcript levels than the latent state and that, as expected, no transcripts were detected in the uninfected states, whether resting or activated (Figure 3C). Given that a low level of HIV RNA molecules was detected in the latent state, we cannot exclude the possibility that this reservoir is made up of a combination of inactive and low-level transcribed proviruses.
To determine whether KAP1 is expressed in the four cell states, we performed western blot and observed that KAP1 is detected at similar levels in both infected and uninfected cells with no, or little, effect of the cell state (Figure 3D). The fact that KAP1 is expressed in both activated and resting primary TCM prompted us to determine whether KAP1 is recruited to the HIV proviral genome during productive and latent infections. Given that in transformed cell-based models, we observed KAP1 recruitment to the proviral 5′-LTR (McNamara et al., 2016b), we predicted that latently infected cells would contain KAP1 at the LTR as well. To test this possibility, we performed ChIP assays in the four cell groups and observed that KAP1 along with Pol II and the elongation complex (CDK9) are bound to the LTR both in productively and latently infected cells (Figure 3E). In addition, we detected higher KAP1 levels bound to the proviral 5′-LTR and within the genome in productively infected cells, compared with latently infected cells, probably because of higher levels of transcription during productive infection (as observed with Pol II and CDK9), consistent with HIV expression data (Figure 3C).
Taken together, we report that KAP1 is expressed in primary T cells irrespective of cell state, KAP1 and the transcription elongation complex is recruited to the proviral genome in the primary TCM model, and the levels of elongation complex recruitment mirror the proviral fate state, with higher levels in the active compared to the latent state.
CRISPR-Cas9 Reveals a Critical Role for KAP1 in Reactivation of Latent HIV in Primary Cells
Given that KAP1 is expressed and recruited to the proviral genome in the primary TCM model, we wanted to examine whether KAP1 could drive HIV transcription of latent proviruses in response to immune stimulation. To test this, we infected TCM cells with replication-incompetent luciferase-tagged virus, and active infections were isolated using a magnetic sorting kit to enrich in latent infections (Figure 3F). After expansion of the remaining cells, in vitro pre-formed CRISPR-Cas9 ribonucleoprotein (RNP) complexes were delivered into cells to knock out KAP1. We also included several controls: CXCR4 (cell surface marker allowing knockout [KO] visualization by FACS), the NF-κB p65 subunit (a positive control for reactivation assays), and a negative NT guide RNA-containing RNP complex not specific for any known human gene.
Remarkably, the KO approach was selective and effective, albeit with different efficiency levels (ranging from ~50% to 100%) (Figures 3G and 3H). After determining KO efficiency, reactivation of latent viruses was then computed using luciferase assays and p24 staining. Notably, we observed that KAP1 KO does not affect levels of proviral activation in the absence of immune stimulation (PMA) (Figures 3I and 3J), consistent with the idea that KAP1 does not control HIV transcription in steady-state conditions, in agreement with previous data (Figure S1I). However, interestingly, we found KAP1 KO dampens both lucif-erase levels and intracellular p24 expression in response to PMA, demonstrating its importance in the host cell response to immune stimulation. Remarkably, this result mirrors the KO of the master regulator NF-κB, which is key for activation of the host phase, because NF-κB KO dampened ~50% reactivation of latent HIV in response to PMA (Figures 3I and 3J). Although the levels of reactivation from latency in the primary system are low (~3% of total cells) because of the relatively low dynamic range of the assay, it must also be noticed that the reduction in reactivation efficiency after KAP1 KO is ~50%, a significant effect considering that complete gene KO in primary cells could not be achieved. More important, however, this affect approximates in magnitude the effect of NF-κB KO, arguably the most important transcription factor required for latent HIV reactivation, for which more efficient depletion was achieved. Therefore, these data support an important role for KAP1, which is on par with that of NF-κB.
In addition, importantly, we also observed that these results could be recapitulated using the SUPT1 CD4+ T cell line (Figure S2), in terms of both KO efficiency (~50%–90% depending on the target) and decreased reactivation of latent viruses in response to PMA stimulation, thus demonstrating that KAP1 has a crucial role in proviral transcription and fate in several primary and immortalized T cell models.
KAP1 Is Central for Activation of the Host Phase of the HIV Transcriptional Circuit
The previous data suggested that KAP1 facilitates HIV transcription activation in primary cells, but these experiments do not distinguish which phase of the circuit (host or viral) and which step of the transcriptional cycle (initiation or elongation) KAP1 controls. Having established this essential function, we then asked what is the contribution of KAP1 to the different phases of the HIV circuit and how does KAP1 contribute to the feedback loop?
To define in which phase of the HIV transcriptional circuit KAP1 participates, we efficiently silenced (>90%) the expression of KAP1 in two cell-based models that recapitulate the different phases of the program during infection: (1) the Jurkat HIV Tat+ clone (E4) contains a provirus that is transcribed by the sequential action of NF-κB and Tat in response to TNF, and (2) the Jurkat HIV Tat− clone (2B2D) contains a provirus that is transcribed only by NF-κB in response to TNF because of a non-functional Tat mutant (C22G) (Figures 4A and 4B).
Figure 4. KAP1 Is Required for Activation of the Host, but Not the Viral, Phase of the HIV Transcriptional Program.

(A) Scheme of proviruses containing wild-type Tat or non-functional Tat C22G mutant.
(B) Western blots of the four cell-based models.
(C–F) Fold HIV RNA induction (initiating, C and E, or elongating, D and F) in the HIV Tat− and HIV Tat+ shNT and shKAP1 cell lines in response to a time-course TNF treatment.
To test the contributions of KAP1 to the host phase (NF-κB driven) and feedback loop (sequential action of NF-κB and Tat), we measured temporal HIV expression in response to TNF in the four (shNT and shKAP1 HIV Tat− and HIV Tat+) cell lines, using amplicons that detect promoter-proximal initiating transcripts and promoter-distal elongating transcripts in real-time qPCR (Figures 4C–4F). We observed NF-κB similarly activates initiation in the host phase (~30-fold over untreated cells) in both HIV Tat− cell lines (Figure 4C). However, loss of KAP1 blunted transcription elongation (~5.5-fold less elongation in the shKAP1 cell line) (Figure 4D), indicating that KAP1 plays an important role in controlling transcription elongation in response to immune stimulation. To analyze the combined effect of KAP1’s loss on both host and viral phases, we examined the HIV Tat+ cell lines. Consistent with the previous results, although KAP1 silencing showed a minimal effect on initiation (Figure 4E), the largest effect was observed at the elongation step, as revealed by a ~6.5-fold higher level in the shNT cell line (Figure 4F).
It is worth noting that the magnitude of transcription activation in the two systems is largely different because the HIV Tat pro-virus is activated only by NF-κB (~35- to 40-fold activation), and the HIV Tat+ provirus is activated by the sequential action of NF-κB and Tat (~1,800-fold activation), leading to full activation through the feedback loop (Figures 4D and 4F). Taken together, given the fragility of the host phase (because of loss of KAP1), the Tat feedback becomes compromised.
Tat Functions in a KAP1-Independent Manner to Facilitate Transcription and Reactivation of a Latent Virus by Directly Recruiting the Host Elongation Complex to the HIV Genome
The previous data suggested a model in which KAP1 functions as a transcriptional co-activator of NF-κB to activate the host phase of the HIV transcriptional circuit in response to immune cell signaling. Given that KAP1 plays a critical role in transcription activation of the host phase, thereby influencing the magnitude of the feedback loop, none of the cell-based systems previously used allowed us to directly test the role of KAP1 on activation of the viral phase. To directly examine this, we co-transfected U2OS shNT and shKAP1 cell lines with an LTR firefly (FFL) lucif-erase (LUC) reporter, increasing amounts of Tat, and a constitutive cytomegalovirus (CMV)-Renilla (RL) as internal control, and calculated the FFL/RL ratio as previously described (D’Orso et al., 2012). We observed that Tat similarly activates both cell lines in a dose-dependent manner (Figure 5A), irrespective of the high efficiency of KAP1 KD (Figure 5B), indicating that Tat functions, at least in this assay, bypassing KAP1.
Figure 5. The Master Regulator of the Viral Phase Operates in a KAP1-Independent Manner.

(A) Quantification of luciferase activity (FFL) from an HIV reporter in the absence (0) or presence of increasing Tat (normalized to CMV-RL).
(B) Western blots of the indicated cell lines used in (A).
(C) Experimental outline in which the HIV Tat− shNT and shKAP1 cell lines were transduced with pTRIP-luciferase (LUC) or pTRIP-Tat at day 1 and collected at day 3 for their subsequent analysis.
(D) Flow cytometry-based quantitation of the percentage of GFP+ cells after transduction of the cell lines from (C) with the indicated lentiviruses.
(E) Real-time qPCR assay of RNA isolated from the indicated cell lines using the elongation amplicon (+2627). The change in HIV gene expression is shown as fold RNA induction (Tat over LUC).
(F) Top: scheme of HIV Tat. The arrow denotes the position of the TSS. Bottom: ChIP assays were performed with protein extracts from the four cells states from (C) and the indicated antibodies followed by qPCR with a series of amplicons mapping throughout the entire provirus to monitor factor interactions with the HIV genome. Values represent the percentage of input DNA immunoprecipitated (IP DNA).
For all the experiments in this figure, values plotted represent the average of three independent experiments (mean ± SEM; n = 3).
If Tat functions in a KAP1-independent manner, then the result should be independent of the model used. To test this idea, we transduced cell lines containing Tat-defective proviruses (Jurkat HIV Tat− shNT and shKAP1) with lentiviruses expressing Tat and LUC (Figure 5C) to quantitate the number of GFP+ cells and HIV expression levels (Figures 5D and 5E). Notably, we observed that Tat transduction increases the levels of GFP+ cells by ~20-fold, in agreement with the robust synthesis of viral transcripts with wild-type Tat but not a non-functional mutant (Figure S3).
If the viral phase bypasses KAP1, we would expect Tat to recruit CDK9 and promote Pol II function in the absence of KAP1. To test this model, we performed ChIP assays to measure the occupancy of KAP1, CDK9, and Pol II throughout the HIV genome in four different scenarios: with or without KAP1 and with or without Tat. By comparing the occupancy levels of CDK9 and Pol II in the absence and presence of KAP1 with Tat (shKAP1+Tat and shNT+Tat, respectively), we can interrogate whether Tat activity requires KAP1. In addition, by comparing the levels of CDK9 and Pol II in the absence and presence of KAP1 without Tat (shKAP1+LUC and shNT+LUC, respectively), we can examine changes occurring at the basal level (without immune stimulation).
First, loss of KAP1 in the absence of Tat reduced CDK9 recruitment (~2.2-fold) to the promoter, consistent with previous data (McNamara et al., 2016b). Second, Tat recruits CDK9 to the HIV promoter and inside the proviral genome with similar efficiencies, irrespective of KAP1 presence (Figure 5F). Consequently, Tat promotes the recruitment of more Pol II to the viral promoter and enhances Pol II levels inside the genome, consistent with its role in promoting transcription elongation, suggesting that Tat (viral phase) functions in a KAP1-independent manner to promote proviral activation.
We propose that this “minimalist” regulatory system that HIV evolved might explain why Tat functions as a potent transcription factor compared with cellular activators. Despite these discoveries, we are not completely ruling out the possibility that Tat could cooperate with KAP1 in the activation of proviruses integrated in other chromatin environments.
Mathematical Modeling the HIV Transcriptional Circuit Reveals a Critical Function for KAP1 in Modulating the Feedback Loop, thus Shaping Proviral Fate
HIV is efficiently activated in response to immune stimulation through the sequential action of host and viral activators. In the host phase, NF-κB rapidly translocates from its latent, cytoplasmic state into the nucleus, where it binds the proviral promoter, eliciting KAP1-dependent CDK9 activity, productive transcription elongation, and Tat synthesis. In the viral phase, Tat promotes the feedback loop to robustly activate HIV transcription through CDK9 recruitment bypassing KAP1 (Figure 6A).
Figure 6. Mathematical Model of a Complete HIV Transcriptional Program.

(A) Left: scheme of a CD4+ T cell containing a transcriptional cluster. Right: simplified model of the host and viral phases of the HIV transcriptional program.
(B and C) Computational stimulations of HIV RNA induction in cells expressing KAP1 and infected with HIV (B) Tat– or (C) Tat+ in response to a TNF time course.
(D and E) Computational stimulations of HIV RNA induction in cells lacking KAP1 expression and infected with HIV (D) Tat– or (E) Tat+ in response to a TNF time course.
(B–E) The y axis was set to log scale to allow better comparisons among all four simulations. Black curve denotes SDE mean. Blue dotted curve denotes ODE. Green curve denotes the SDE median. Purple dotted line and purple data points denote experimental data. Note that the duration of the two phases of the circuit is an approximation based on the length of the host phase, when the host phase starts decaying in HIV Tat− proviruses, and when the viral phase starts progressing before the decay of the host phase in cells infected with HIV Tat+.
To determine whether these previous findings can be integrated into a theoretical framework of host-viral transcriptional regulation, we developed a mathematical model that describes the minimal set of interactions in a transcriptional system. Previous computational modeling have revolved around the idea that the HIV transcriptional circuit is composed of two phases: basal and viral (Weinberger et al., 2005). However, as explained above, the complete circuit is composed of three phases: basal, host, and viral (Figure 1F). Thus, the contributions of the host phase to the feedback loop was not integrated into previous studies. We thus developed a mathematical model that enables one to investigate the individual contributions of the two phases to the HIV transcriptional circuit and thus have a complete view of the real system.
In theory, our model is based on the principle that spontaneous proviral transcription activation in response to immune simulation results from the stochastic fluctuations of host factors between the nucleoplasm and promoter interactions in chromatin territories. We propose the probability of activation is dependent on the coincidence of two events that might occur independently or simultaneously (Figure 6A). NF-κB must associate with the promoter (point 1), and a KAP1 molecule must bind near the Pol II complex-promoter (point 2). Once recruited, KAP1 delivers primed CDK9 for “on site” activation. The kinase then phosphorylates its substrates (Pol II) at the promoter (point 2). This sequence of events initiates proviral transcription and Tat synthesis (point 3), further recruiting more kinase but, in this case, bypassing the KAP1-centric host cell regulatory system (point 4), thereby promoting the feedback loop by increasing the number of elongating Pol II molecules (point 5).
In our theoretical analysis, we considered four conditions with two main variables (−/+ KAP1, in −/+Tat) to model HIV RNA synthesis response to immune stimulation. Literature values were used to estimate the rates of basal HIV RNA synthesis (α), formation and dissociation of the NF-κB-DNA complex in the host phase (kon, koff), KAP1-mediated recruitment and activation-deactivation of CDK9 in the host phase (kact(h), kdeact(h)), NF-κB-mediated HIV RNA synthesis in the host phase (ksynth(h)), Tat-mediated recruitment and activation-deactivation of CDK9 in the viral phase (kact(v), kdeact(v)), Tat-mediated HIV RNA synthesis in the viral phase (ksynth(v)), RNA translation (ktrans), RNA decay (kdecay), and Tat-positive feedback (kfb). Furthermore, experimental data were used to calibrate unknown kinetic rates.
Computational simulations resulted in an initial “boost” of TNF-induced NF-κB-mediated HIV RNA synthesis (host phase) in the presence of KAP1 but lack of feedback loop due to Tat’s absence (Figure 6B). NF-κB activated ~50-fold in silico, a value that closely resembles the measured NF-κB activation rates (Figure 4), even though the magnitude of activation is directly proportional to TNF concentration (see below), consistent with previous data (Tay et al., 2010). In addition, notably, the model gave rise to temporally decay of NF-κB activation in the absence of feedback loop, as is observed in vivo, and HIV RNA levels return to the low steady-state level of basal transcription (Figure 6B). In the presence of normal KAP1 levels and feedback loop, the initial boost is largely amplified by Tat, leading to an exponential increase (>100-fold activity) (Figure 6C), consistent with experimental data (Figure 4) and previous studies.
In cells lacking normal KAP1 levels, NF-κB activity is largely compromised (see the virtual decrease in the initial boost) (Figure 6D). With the loss of HIV RNA synthesis by NF-κB in cells lacking KAP1, some level of expression can still be observed that is accelerated by the feedback loop, albeit at a much lower rate compared with the KAP1-positive scenario (fold differences of ~2.8 at t = 480 min and ~5.5 at t = 2,000 min during the exponential growth phase) (Figure 6E).
These data indicate that the stochastic assembly of transcription elongation complexes at the proviral promoter is required to establish the initial transcriptional “boost.” Consistent with this interpretation, NF-κB was unable to activate in the absence of KAP1, despite its efficient binding to the promoter (McNamara et al., 2016b). Together, our mathematical model recapitulates the normal activation pattern of a complete HIV transcriptional circuit.
Perturbation Analysis and Model Behavior
We then investigated the model behavior during parameter perturbations. For this purpose we used the well-mixed deterministic ordinary differential equation (ODE) model, as we were interested in the overall (mean) behavior, disregarding any noise and stochastic fluctuations. As expected in the case of chemical mass action systems (Hahl and Kremling, 2016), both the ODE and the stochastic differential equation (SDE) mean show similar behavior (Figures 6B–6E). It has been shown that in linear systems, the mean after SDE and the deterministic variable of the ODE coincide (Hahl and Kremling, 2016). However, skewed fluctuations through large bursts may lead to a shift of stochastic modes away from the mean. Such bursts have not been observed in our stochastic simulations to the extent of causing deviation between the ODE model and the SDE mean. Thus, for simplicity, we used the corresponding ODE model to investigate the effect of KAP1 protein levels on the dynamic behavior. Notably, Figures 7A and 7B capture the experimental measurements and ODE model behavior in the four conditions (with or without KAP1 and with or without feedback loop; see corresponding Figures 6B–6E with additional trajectories from SDE simulations), thus demonstrating good agreement between experimental data and simulation (ODE and SDE mean) calculations.
Figure 7. KAP1 Levels Influence the Outcome of the Host Phase, Thereby Affecting the Viral Feedback Loop Potential.

(A and B) Computational simulations (ODE) of HIV RNA synthesis in cells expressing (A) or lacking (B) KAP1 and infected with HIV Tat+ (with feedback) or HIV Tat− (without feedback) in response to a temporal TNF treatment.
(C) Parameter scan computational simulations (ODE) with variable KAP1 protein levels. The black and red lines correspond to the highest and lowest KAP1 levels, respectively. The outcome of the host phase (without feedback) and host-viral phases (with feedback) is indicated.
(D) Western blots of parental (WT) and Jurkat HIV Tat+ (E4) KAP1 KD clones.
(E) Relative HIV RNA levels in the absence (−) and presence (+) of TNF (25 μg/mL) stimulation for 16 h measured by real-time qPCR (elongation amplicon, +2627). The data of the parental HIV Tat+ (E4) in the absence of TNF was set to 1 and the stimulation to 100 to allow easier comparisons. Values plotted represent the average of three independent experiments (mean ± SEM; n = 3).
(F) Correlation plot between relative KAP1 levels (normalized to levels in E4 = 1) and HIV RNA levels in the presence of TNF. A trend line was fitted to the data points, and Pearson’s correlation coefficient (R2) was calculated.
(G) Correlation plot inset (red box from F) showing all data points except WT.
(H) Simplified scheme for the KAP1-mediated recruitment of 7SK-bound P-TEFb to the HIV promoter to induce Pol II phosphorylation and host phase activation in response to TNF.
(I) Scheme of a minimal proviral promoter transcribing a LUC reporter (HIV-GAL4) in which P-TEFb is artificially recruited through GAL4.
(J) Fold activation LUC levels of the minimal reporter from (I) transfected with the indicated activators in the U2OS cell lines from Figure 5B. Values plotted represent the average of three independent experiments (mean ± SEM; n = 3).
We then performed a parameter scan by simulating the model multiple times, each time varying the value of one parameter, in this case KAP1 levels. Strikingly, the parameter scan model revealed a direct relationship between the rate of initial transcriptional “boost” and KAP1 levels and therefore potential binding to the proviral promoter (τ[KAP1]) (Figure 7C). Although the phenotype is first observed on the initial “boost,” it ultimately affects the strength of the feedback loop. Here, four states of decreasing host phase activation have proportional effects on the feedback loop. The lower the magnitude of the host phase, because of reduced KAP1 levels, the lower the magnitude of the feedback loop (Figure 7C).
Testing the Influence of Host Phase Heterogeneity and Immune Cell Signaling Strength on Transcriptional Fragility and Viral Phase Outcomes
The previous simulations indicated that oscillations of KAP1 levels during infection could generate cell-to-cell differences, thereby creating transcriptional noise in the host phase and affecting homogeneous responses to immune stimulation and latency-reversing agents (LRAs) because of alterations of the feedback loop. To test this model, HIV RNA synthesis was monitored over time in response to TNF on several Jurkat HIV Tat+ clones (created through KAP1 KD and single-cell sorting), which express variable KAP1 levels (Figure 7D). We observed that the lower KAP1 levels, the larger the reduction in HIV RNA levels in response to TNF (Figure 7E). Interestingly, correlation analysis provided direct evidence that HIV RNA levels are directly proportional to KAP1 levels in the system (Figures 7F and 7G). These results are consistent with the theory that KAP1 amplifies the initial transcriptional “boost” in the host phase and affects the outcome of the viral phase.
Given that the previous assay used different clonal cell lines, we then created an isopropyl- β-D-thio-galactoside (IPTG)-inducible KAP1 KD Jurkat cell line (HIV Tat− shKAP1) to better model the dosage effect of KAP1 levels on host phase activation in response to immune stimulation in the same system (Figure S4A). Remarkably, we observed that a dose-dependent reduction of KAP1 levels proportionally reduced proviral activation in response to TNF without affecting basal levels (Figure S4B), consistent with the idea that KAP1 is required for transcription in response to immune cell signaling. In addition, a time course of TNF-mediated activation showed a ~6-fold reduction in the magnitude of proviral activation (Figure S4C), consistent with the data in the non-inducible system (Figure 4). Together, the data reinforce the idea that KAP1 is a key regulator of the host phase and that its levels correlate with the transcriptional magnitude of the feedback loop.
The HIV transcriptional program appears to function as an “off-on” switch whereby in the absence of environmental stimulation, the system remains in the “off” state and upon activation is turned “on.” However, immune stimulation strength can affect the threshold of activation (Tay et al., 2010), and thresholds allow biological systems to respond on the basis of input strength. Given this knowledge, we hypothesized a model in which variable levels of TNF stimulation should generate “on” states with different thresholds, with a concomitant reduction in host-viral phase activation levels in the absence of KAP1.
To test this model, we compared the levels of HIV RNA synthesis produced by HIV Tat− proviruses in response to three TNF in puts (high, medium, and low) and observed the graded decrease of HIV RNA signal intensity in control cells (shNT) as a function of reduced immune stimulation strength (Figure S4D). Furthermore, we detected proportionally reduced RNA synthesis in the shKAP1 cell line compared with shNT, indicating that both KAP1 levels and stimulation input strength control HIV provirus transcriptional output in the host phase. Similarly, we observed that the reduced HIV RNA synthesis in response to a decreased graded TNF levels in the host phase directly affects the magnitude of feedback loop activation in HIV Tat+ proviruses and again much reduced levels (~4- to 6-fold) in the shKAP1 compared with shNT cell lines (Figure S4E).
Given that TNF is a strong immune stimulus, we then asked whether known LRAs that function through different mechanisms such as bryostatin (a PKC agonist) and suberoylanilide hydroxamic acid (SAHA) (a pan-histone deacetylase inhibitor) would show similar KAP1-dependent activation mechanisms. We thus treated the Jurkat HIV Tat− and HIV Tat+ shNT and shKAP1 cell lines with bryostatin or SAHA and observed that loss of KAP1 also affected latency reversal of the host phase and the threshold of activation of the viral phase, respectively (Figures S4F and S4G), implying that KAP1 plays a key role in activation of the host phase in response to strong immune modulators as well as commonly used LRAs.
Collectively, the host phase is subject to tight control by host factors whose activity is indispensable to maintain active proviral transcription and avoid the establishment of latency.
Forced Elongation Complex Recruitment to the HIV Promoter Bypasses KAP1 Requirement for Transcription Activation, as in the Viral Program
Previous studies suggested that KAP1 enables recruitment of 7SK RNA-bound CDK9 to the proviral genome to promote NF-κB-dependent transcription elongation in response to immune stimulation (“normal kinase recruitment”) (Figure 7H). Loss of KAP1 could then abolish transcription elongation because CDK9 is not properly recruited to the promoter despite normal NF-κB binding kinetics (McNamara et al., 2016b). If KAP1 functions as a co-activator of NF-κB by assisting CDK9 recruitment, then we would expect that forced CDK9 recruitment to the promoter (“forced kinase recruitment”) (Figure 7I) should facilitate transcription activation in a KAP1-independent manner and functioning in a manner similar to Tat (like in the viral program).
To test this model, we delivered P-TEFb directly to the promoter through the heterologous yeast GAL4 DNA binder (Figure 7I). In this context, GAL4 mimics KAP1 to deliver the kinase to the promoter for transcription activation. To test this model, we used a U2OS cell line in which KAP1 has been efficiently knocked down (Figure 5B), or a control cell line, to transfect a minimal promoter containing five copies of the GAL4 binding site fused to LUC (GAL4-LUC reporter), along with increasing amounts of GAL4 and GAL4-fused or unfused P-TEFb subunits. Notably, we found that GAL4-CycT1 similarly activates the HIV reporter in both the NT and KAP1 KD cell lines, with no or minimal effect of GAL4 or unfused CycT1 (Figure 7J). Supporting these data, GAL4-CDK9, but not a non-functional kinase (T186A) or the unfused CDK9, largely activates the promoter despite KAP1 expression (Figure 7J), indicating that, at least in this artificial system, forced recruitment of CDK9 to a promoter bypasses the requirement for KAP1 toward transcription activation.
Collectively, one key function of KAP1 is to recruit CDK9 to the promoter for viral activation. By rewiring the circuit to operate through promoter-bound CDK9, KAP1 becomes dispensable for activation, as in the viral phase of the program. Nonetheless, because the reporter system used is artificial, this result does not provide quantitative evidence that KAP1 scales with CDK9 recruitment and does not rule out KAP1 could play other essential roles in the transcriptional cycle in proviruses integrated into different chromatin territories.
DISCUSSION
Viruses have evolved unique strategies to regulate gene expression, rewire host cell programs, and evade immune system responses to survive within their hosts. In this work, we found that HIV hijacks the host transcription elongation complex to regulate transcription of its genome in a unique way. For more than two decades, it was assumed the host and virus use similar regulatory strategies to stimulate transcription from the proviral genome. Here, we demonstrate that although the two phases require CDK9 for activation, the kinase is recruited differently. In the host phase, KAP1 assists the process of transcription by cellular activators by recruiting CDK9 to the promoter. In contrast, HIV evolved Tat, which recruits CDK9 bypassing KAP1. However, despite the minimalistic nature of the viral phase, malfunction of the host phase (which primes HIV for activation) directly affects the extent of the feedback loop. This phenomenon, which we call “transcriptional circuit fragility,” proposes that activation of the circuit in the host phase in response to immune stimulation does not follow a deterministic trajectory and experiences stochastic outcomes. This concept differs from the stochastic variability described for the circuit in the basal state (Weinberger et al., 2005) and may help explain how proviral phenotypic diversity arises.
Transcriptional circuit fragility implies that the regulatory program of the host phase is “fragile,” meaning that protein level fluctuations or malfunction of host factors implicated in key regulatory pathways (e.g., cell signaling, co-activator function) would generate an abnormal activation threshold in the program, leading to variable cell-to-cell transcriptional outcomes. Multiple lines of evidence indicate that the cellular state is a critical determinant for proviral transcription and escape from latency establishment. Despite recent theories that the virus may function as a cell-autonomous unit (Razooky et al., 2015), the consensus in the field is that the status of the infected cell (Shan et al., 2017) ultimately determines the level of proviral transcription activation and fate. In fact, it is believed that proviral transcription remains in the “off” state without immune stimulation, which generates signaling events inducing host master regulators required for proviral transcription. This is because efficient formation of fully elongated and mature HIV transcripts requires sustained induction by the cellular activators, which will then promote de novo synthesis of Tat (Karn, 2011; Williams et al., 2007). Thus, importantly, without proper activation of the host phase (“fragility”), the magnitude of the viral phase, and consequently the Tat feedback loop, gets largely compromised.
Given KAP1 operates in primary T cells, it is possible that, as a consequence of the system’s transcriptional fragility, fluctuations in KAP1 levels in patient samples could blunt activation of the host phase, and ultimately affect the extent of the Tat feedback loop and the magnitude of HIV latency reversal (thereby leading to proviral fate divergence). As such, single-cell heterogeneity in host phase responses could thus account for the large variations in latency reversal observed both in different primary models and patient samples ex vivo (Spina et al., 2013).
Despite mechanistic evidence in CD4+ T cell lines and primary cells, the proposed “fragility” model will have to be tested in patient-derived cells to provide in vivo relevance. However, to do so, future approaches will need to overcome current technical hurdles to simultaneously measure KAP1 protein expression levels and HIV reactivation at the single-cell level. Nonetheless, our discoveries have direct implications for HIV cure efforts in individuals who have full suppression of viral replication on ART. Besides the possible fluctuations in activation of the viral phase (Weinberger et al., 2005), we argue that the main component that must be considered in any approach to eradicating the latent reservoir is the level of activation of the host phase (Shan et al., 2017), which sets the threshold for activation of the cell-autonomous viral phase. Furthermore, given the large number of latent reservoirs harboring replication-competent proviruses within one patient and their clonal expansion capacity (Maldarelli et al., 2014; Hughes and Coffin, 2016), several factors beyond host transcriptional circuit fragility could contribute to the pleiotropy in heterogeneous transcriptional responses including the integration landscape. Thus, future work will be needed to precisely uncouple the contribution of these various factors to latency establishment and/or maintenance.
Even though our data clearly favor a model in which KAP1 is critical for activation of the host phase, we are not formally excluding the involvement of additional co-factors, including master regulators, co-activators, and pioneer factors required for chromatin accessibility. So by no means we are proposing KAP1 is the “major factor” but a key co-activator of the host phase, because it helps relieve the elongation blockage through recruitment of primed CDK9. Despite our study focused on KAP1 role on activation of the host phase by NF-κB in T cells in response to TNF, the concept of transcriptional fragility is broadly applicable to different cell types, master regulators, inducers and LRAs.
Intriguingly, beyond its proviral activating role, KAP1 appears to regulate other phases of the viral life cycle, such as integration (Allouch et al., 2011). Thus, it seems reasonable to think it would be beneficial for the virus to integrate in sites of higher chromatin accessibility bound by KAP1, which can then facilitate proviral activation (either in steady-state conditions or in response to immune signaling), thereby guaranteeing active replication. These possibilities open up a new area of investigation to test a potential KAP1 role in coupling both viral life cycle regulatory steps.
In addition to its activating role, KAP1 has been previously linked to epigenetic silencing of retroelements, genes in progenitor cells and viruses. Loss of KAP1 releases human CMV from latency in CD34+ hematopoietic stem cells (HSC), but not in permissive cells such as fibroblasts (Rauwel et al., 2015). Thus, KAP1 plays a dual role as a repressor and an activator, depending on the cell type and interacting protein complexes (Iyengar and Farnham, 2011; Bunch et al., 2014; McNamara et al., 2016b), and could only confer the repressive phenotype in a context-dependent manner. It is worth noting KAP1-mediated silencing of retroelements and genes during development is a mechanism that has been acquired during millions of years of evolution (Imbeault et al., 2017). As such, it makes sense that HIV (a relatively young virus in the evolutionary timescale) would not have undergone this suppressive regulatory mechanism by host cell factors such as the KRAB-ZnF DNA-binding proteins.
It remains unclear why KAP1 does not fulfill the expected repressive function in immortalized and primary T cell models of latency. It is possible the lack of KRAB-ZnF factors required for KAP1’s repressive role, and/or the nature of the cellular systems used (permissive or committed cell lineages) both contribute to the lack of epigenetic silencing. Recent studies, however, proposed that KAP1 contributes to HIV latency by sumoylating CDK9 (Ma et al., 2019) and imposing repressive chromatin modifications at the viral promoter (Taura et al., 2019). Although we are not completely ruling out the possibility that KAP1 represses some HIV proviruses integrated in unique chromatin territories, it is evident from our studies that efficient KAP1 KD (both sustained and acute) does not appear to promote robust latent HIV reactivation. Given the critical role of KAP1 in DNA repair, future studies will be needed to rule out the possibility that sustained KAP1 loss indirectly induces host programs, leading to the observed changes in promoter-associated chromatin signatures and latent provirus reactivation.
Finally, our findings provide a mechanistic explanation for the importance of the host phase of the HIV transcriptional program to ensure that the virus is readily and robustly activated during infection to complete the pathogenic cycle. Although HIV drives molecular innovation to fuel robust gene activation, it suffers from host phase fragility, thereby influencing latent proviral transcription and homogeneous reactivation potential. Taken together, our discoveries have important implications for disease control and targeting in patients, and our experimental-mathematical modeling framework should provide a resource to guide the discovery of alternative HIV cure approaches.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information and reagents may be directed to the Lead Contact, Dr. Iván D’Orso, at the University of Texas Southwestern Medical Center (ivan.dorso@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Jurkat CD4+ T and J-Lat cells (10.6, 6.3, 8.4, 9.2, E4 and 2B2D) (Jordan et al., 2003; Pearson et al., 2008) and derivative cell lines (see Table S1 for a complete list) were cultured in RPMI 1640 media supplemented with 10% Fetal Bovine Serum (FBS) and 1X Penicillin/Streptomycin at 37°C with 5% CO2 at an optimal seeding density of 500,000 cells/mL. The E4 and 2B2D clones derive from HIV-1 NL4–3 infectious molecular clone (Pearson et al., 2008) and 10.6, 6.3, 8.4 and 9.2 clones derive from the R7/3/GFP molecular clone and contain an env frameshift and GFP in place of nef (R7/E-/GFP) (Jordan et al., 2003). Jurkat cells stably expressing shRNAs were grown as above, but selected with the addition of 1 μg/mL of puromycin in case of the pLKO.1 cell lines or cell sorted (mCherry+), as indicated below, in the case of the pLVTHM cell lines. SUPT1 cells were cultured in RPMI 1640 media containing 10% FBS and 2 mM L-Glu. U2OS and derivatives (shNT and shKAP1), HEK293T and HEK293FT cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1X Penicillin/Streptomycin at 37°C with 5% CO2. The Jurkat clones were treated with TNF-α, SAHA, Bryostatin, Triptolide or Flavopiridol for the indicated time points and concentrations. Primary CD4+ T cells were isolated and cultured as indicated below.
Bacterial Strains
DH5α and STBL3 cells were obtained from Thermo Fisher, stored at −80°C, grown in Luria Broth (LB) media at 37°C, and used to propagate plasmid DNAs.
METHOD DETAILS
Lentiviral Transduction and shRNA-mediated Knockdown
pLKO.1 NT (SHC002) and KAP1 (SHCLND-NM_005762) directed shRNA’s were obtained from Sigma. NT and KAP1 shRNAs were cloned into the ClaI and MluI restriction sites of pLVTHM (see Table S2 for a complete list of shRNA plasmids) using standard molecular biology procedures. pLVTHM-expressing mCherry instead of GFP was previously described (Jadlowsky et al., 2014). The empty and NELF-E shRNA expressing pLVTHM vectors were kindly provided by J. Karn (Case Western Reserve University, Cleveland, OH). The pLKO.1 and pLVTHM shRNA-containing vectors were transfected along with gag/pol (psPAX2) and VSV-G (pMD2.G) into HEK293T cells for expression of competent lentiviruses. Cell supernatants were collected two days post-transfection. Viral transduction was done by spinoculation using 2×105 cells, 8 μg/mL polybrene, and unsupplemented RPMI 1640 to a final volume of 0.2 mL per well of a 96-well plate at room temperature for 2 hr at 400 g. Transduced cells were selected with puromycin (1 μg/mL) 2 days post-infection (for pLKO.1) or cell sorted on mCherry(+) cells (for pLVTHM). Cells were monitored for KD efficiency through standard western blot and RT-qPCR assays. For Figure 5, each cell line (Jurkat HIV Tat- shNT and Jurkat HIV Tat- shKAP1) was transduced with pTRIP-LUC or pTRIP-Tat (see Table S5 for a complete list of plasmids) in 96-well plates (2 plates per cell line at 0.2 mL lentiviral mix/well). Cells were then used in flow cytometry and RT-qPCR assays as indicated below. For Figures S4A–S4C, Jurkat HIV Tat- cell lines containing IPTG-inducible NT and KAP1 shRNAs (pLKO.1-IPTG-3xLacO) (Table S2) were created by transducing Jurkat HIV Tat- with the corresponding lentiviruses and selecting with puromycin (1 μg/mL) for 1 week. Selected cell lines were then treated for 2 days with three different IPTG concentrations (1, 10, and 100 mM) to model the dosage effect of KAP1 protein levels on proviral transcription activation.
Flow Cytometry Analysis
5×105 cells per sample were transferred to an uncoated V-bottom 96-well plate (Nunc). The samples were spun down at 750 g for 5 min at room temperature and washed with 1X PBS. Washed cells were spun down again and the 1X PBS was aspirated. Cells were fixed using 20 μL of 1% paraformaldehyde (PFA) at room temperature for 10 min. The PFA was washed with 100 μL of PBS, spun down, buffer aspirated, and cell pellets resuspended in 100 μL of 1X PBS. A 96-well plate reader (A600 HTAS, Stratedigm) was used to run the samples; lasers with a wavelength of 615 nm and 530 nm were used to measure mCherry and GFP, respectively. CellCapTure (Stratedigm) was used to visualize the running samples. 20,000 set count cells were analyzed per sample. For CXCR4 detection, cells were stained with anti-human CXCR4, PE conjugated for 30 min. For viability, cells were stained with Fixable Viability Dye eFluor 450. HIV-1NL4–3-infected cells were analyzed by flow cytometry first by staining with Fixable Viability Dye eFluor 450 and then with anti-human CD4, APC conjugated for 30 min. After washing with 1X PBS containing 3% FBS, cells were fixed and permeabilized using Cytofix/Cytoperm for 30 min and then stained with anti-HIV p24, FITC conjugated. Flow cytometry was performed with a BD LRS Fortessa X-20 flow cytometer using FACSDiva acquisition software. Data analysis was performed with FlowJo version 10.1.
Cell Sorting
One day post-transduction, cells were collected for sorting, washed with sterile 1X PBS, and resuspended in 10% RPMI 1640 media containing 10% FBS in 1X PBS. Cells were then transferred to 5 mL sterile polypropylene collection tubes (Falcon) containing 1 mL of 10% complete RPMI 1640 media in PBS, and analyzed directly or kept at 4°C until sorting (within 1 hr). A BD FACS Aria II (Becton Dickinson) was used (UTSW Flow Cytometry Core Facility) to sort live mCherry+/GFP- cells. A purity check was run after 1×106 cells had been sorted. The cells were spun down and resuspended in 5 mL of complete RPMI 1640 media and grown in T-25 flasks (Corning) for use in western blots and RT-qPCR assays.
RNA Extraction and RT-qPCR Assay
Isolation of total RNA was done using the Quick-RNA miniprep kit (Zymo). RNA quality was assessed by computing the RIN index (RNA Integrity Number) by running the samples on a 2200 Tapestation (Agilent) and was always RIN > 9.5. First strand cDNA synthesis was done using M-MuLV Reverse Transcriptase with oligo(dT)18 and random decamers. Quantitative PCR was performed with a SybrGreen master mix on an ABI7500 instrument (Applied Biosystems). Ct values were obtained as previously described in detail (McNamara et al., 2013). The fold change of the target mRNA levels relative to control was calculated as 2−ΔΔCt. A list of DNA oligonucleotides used in RT-qPCR assays can be found in Table S3.
ChIP-qPCR Assays
ChIP assays in Jurkat cells were performed as previously described (McNamara et al., 2016b). Purified cell nuclei were sonicated 60 cycles (30 s on/30 s off) on a Bioruptor UCD-300 water bath (Diagenode) to obtain DNA fragments of an average size of ~300 bp. 5 mg of antibody were conjugated to 50 μL of 50% slurry protein G Dynabeads at 4°C for 2 hr and added to purified sonicated cell nuclei as follows: 1×107 cell nuclei for Pol II, and 2.5×107 cell nuclei for CDK9, KAP1, and IgG (see Table S4 for complete list of antibodies and dilutions used). ChIP assays were performed with protein extracts from the indicated cells and using the antibodies indicated followed by qPCR with a series of amplicons mapping throughout the entire provirus mentioned at the top of the schematic to monitor factor interactions with the HIV genome. The ChIP-qPCR data was normalized using the “Percent Input Method,” which includes normalization for background and Input chromatin used for each ChIP. ChIP signals were divided by signals obtained from the Input sample (1% of starting chromatin), which signifies the amount of chromatin used per ChIP. Values represent the percentage (%) of input DNA immunoprecipitated (IP DNA) presented after background (normal IgG) substraction, and are the average of three independent experiments.
GAL4 Plasmid DNAs and Luciferase Assay
U2OS shNT and U2OS shKAP1 cell lines were seeded onto 48-well plates and transfected with a mix of DNAs (250 ng total DNA/well) and 0.5 μL Polyjet per well. For the experiment in Figure 5A, both cell lines were transfected with a pcDNA3.1-HIV-LTR-FFL LUC reporter (25 ng/well) and increasing amounts of a pcDNA4/TO-Tat:Strep plasmid and carrier DNA (pBluescript II KS+) to complete 250 ng total DNA per well. For the experiment in Figure 7, the luciferase reporter plasmid is a pcDNA3.1+ vector containing a minimal LTR promoter plus 5xGAL4 binding sites and the activator plasmid containing yeast GAL4 DNA-binding domain alone or fused to the indicated P-TEFb subunit (CycT1, Cdk9, and Cdk9T186A non-functional mutant) as previously described (Table S5). Firefly luciferase reporter activities were normalized to a constitutive CMV Renilla (RL) luciferase expressor using the Dual-Luciferase Reporter Assay System (D’Orso et al., 2012). Luciferase of cell supernatants in SUPT1 and primary CD4+ T cell infections was measured using Nano-Glo Luciferase Assay System.
Western blot Assays
Total protein extracts from 0.2×106 cells (~20 μg, as quantitated using the Pierce BCA protein assay kit) were electrophoresed on home-made 10% polyacrylamide SDS-PAGE gels using 1X Tris-Glycine-SDS running buffer prepared from a 10X stock, and then transferred onto 0.45 μM nitrocellulose membranes using a standard Towbin transfer buffer (20% methanol, 25 mM Tris-Base, 192 mM glycine, pH 8.3). Once transfer was complete, membranes were blocked in Tris-buffered saline (TBS) containing 0.2% Tween-20 and 5% non-fat dry milk for 2 hr, and incubated with primary antibodies at 4°C from 1 hr to overnight. See list of all primary and secondary antibodies and their concentrations used in Table S4. Once the blotting was complete, membranes were incubated for 5 min with Clarity Western ECL substrate and exposed to film. Films were then scanned, cropped in Adobe Photoshop and directly used to make the figures in Illustrator (Adobe) without any further manipulation. When indicated (Figure 7), signal intensities in western blots were quantified using ImageJ 1.43r (Schneider et al., 2012).
Transduction of Jurkat HIV Tat- Cell Lines for Flow Cytometry, RNA, Protein and ChIP Assays
pTRIP-LUC (Schoggins et al., 2011), pTRIP-Tat, and pTRIP-Tat C22G plasmid DNAs (Table S5) were transfected into HEK293T cells for lentiviral production as previously mentioned. The NT and KAP1 shRNA-expressing Jurkat HIV Tat- cell lines were transduced with the pTRIP lentiviruses indicated above. Cells were collected on day one, three, and five days post transduction for FACs analysis (GFP, RFP), or for the indicated assays as previously mentioned.
Virus Production
For Figures 3 and S2, pseudotyped viruses (pNL4.3-deltaEnv-nLuc-2ANef-VSVG) were produced by co-transfecting pNL4.3-deltaEnv-nLuc-2ANef (containing NanoLuc (Promega)) (Martins et al., 2016) and pCMV-VSV-G (Stewart et al., 2003) (in a 2.5:1 plasmid DNA ratio) into HEK293FT cells using calcium phosphate. After 2 days, cell supernatants were collected and filtered with a Millex-GP syringe filter unit, 0.22 μm, polyethersulfone, 33 mm, gamma sterilized (Millipore Sigma). Viruses were tittered on SUPT1 cells and stored at −80°C when needed. SUPT1 cells were infected by viruses in a series of amounts. P24 expression was checked by flow cytometry 2 days post-infection.
Generation of CRISPR-Cas9 Knockout on HIV Latency Models
For Figures 3 and S2, cells were infected with pseudotyped viruses (pNL4.3-deltaEnv-nLuc-2ANef-VSVG) for 2 days. After amplification as indicated in the Figures, CD4+ cells were isolated using Dynabeads CD4 Positive Isolation Kit. TracrRNA and guide RNAs (IDT, scrambled gRNA; CXCR4 gRNA: 5′-GAAGCGTGATGACAAAGAGG-3′; NF-κB p65 subunit gRNA: 5′-GAGGGGGAACAGTTCTGAAA-3′; KAP1 gRNA: 5′-ACGTTCACCATCCCGAGACT-3′) were mixed and heated at 95°C for 5 min. Cas9 was added to the RNA mixture [3.23 μg Cas9 protein and 21.6 pmol gRNA] and incubated for 20 min. CD4+ cells were washed with PBS and resuspended in 10 μL of Buffer R (for SUPT1) or Buffer T (for primary CD4+ T cells) of Neon Transfection System kit. Pre-assembled Cas9-gRNA ribonucleoprotein (RNP) complexes were electroporated into cells using Neon Transfection System. After 2 days, cell viability and CXCR4 staining were performed. Cells were then seeded into 96-well plates, treated with 10 ng/mL PMA or vehicle (DMSO 99.7%) for 2 days. Luciferase and intracellular p24 levels were recorded by luciferase assays and flow cytometry, respectively.
Generation of the Latency Model in Primary Central Memory T cells (TCM) and Analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donors. Naive CD4+ T cells were isolated and TCM cells were generated and infected as previously described (Bosque and Planelles, 2009). Briefly, naive CD4+ T cells were obtained by magnetic isolation using the EasySep Human Naive CD4+ T cell Isolation kit from healthy donor blood samples (Gulf Coast Regional Blood Center). Naive CD4+ T cells were activated using human anti-CD3/CD28-coated magnetic beads in the presence of anti-IL4 (2 μg/106 cells), anti-IL12 (4 μg/106 cells) and tumor growth factor (TGF-b1) (0.8 μg/106 cells). After 3 days, magnetic beads were removed, cells were washed and maintained at a concentration of 106 cells/mL in media containing 30 IU of human IL2. HIV-1NL4–3 was generated in HEK293FT cells using calcium phosphate. TCM cells were then infected with HIV-1NL4–3 by spinoculation at a multiplicity of infection (MOI) of 0.6 using a concentration of 2×106 cells/mL and centrifuged for 2 hr at 37°C and 162 g. Following infection, cells were cultured in 96-well round bottom plates (105 cells/100 μL/well) for 3 days (from day 7 to 10). At day 10, cells were cultured in standard tissue culture flasks at a cell density of 106 cells/mL. At day 13, 1 μM of nelfinavir was added to the cells for viral suppression.
Crosslinking of Primary TCM Cells for ChIP Assays
TCM cells (~2×107) were pelleted by centrifugation (600 g for 5 min at room temperature) and resuspended at a density of 1×107 cells/mL in 0.5% methanol-free formaldehyde diluted in 1X PBS. Cells were nutated for 5 min at room temperature. Glycine (0.15 M) was added to quench crosslinking and cells nutated for 10 min at room temperature. Cells were then pelleted at 750 g for 5 min at 4°C and 2X with cold PBS. Snap-frozen cell pellets were kept at −80°C until sonication as indicated above. Briefly, TCM cells were processed for ChIP assays like Jurkat cells. 2×107 cell nuclei were used per ChIP assay with IgG, Pol II, CDK9, and KAP1 antibodies.
Mathematical Modeling
Introduction
We seek to understand the functional interplay between host cell factors such as KAP1 and the cellular (NF-κB) and viral (Tat) transcriptional activators during HIV RNA synthesis and latency-reversal in response to immune stimulation. Notably, KAP1 allows for the initial NF-κB–mediated transcriptional “boost,” which facilitates robust Tat positive feedback loop. Conversely, loss of KAP1 blunts the initial “boost” thereby dampening Tat function and latency-reversal. Although the viral-driven phase of the transcriptional program is “minimalist” (because of the bypass of host cell requirements), the strict dependence of cellular factors for the host phase makes the complete circuit “fragile,” thus revealing key information that must be contemplated for HIV cure strategies.
Model Overview
We are considering four components to model HIV RNA synthesis by the combined action of the host and viral phases. In cells expressing normal KAP1 levels (i) with and (ii) without feedback loop, and in cells where KAP1 expression is lost (iii) with and (iv) without feedback loop. In normal conditions (cells expressing normal KAP1 levels), one can distinguish between an early KAP1-dependent “boost” and a later KAP1-independent phase of HIV RNA synthesis.
Step (0): Involving multiple feedback loops, NF-κB translocates from the cytosol to the nucleus after binding of TNF to its receptor and following activation of the IkB kinase (IKK).
Step (1): RNA synthesis is initiated by NF-κB binding to the viral promoter (kon), which uses KAP1/CDK9 as co-activator for HIV proviral transcription activation.
Step (2): Once at the viral promoter, KAP1 promotes CDK9 delivery and activation (kact(h)). However, KAP1/CDK9 activity decays quickly (kdeact(h)) as a consequence of NF-κB dissociation from the promoter (koff).
Step (3): This initial “boost” promotes the synthesis of HIV transcripts (ksynth(h)), which can be degraded (kdecay).
Step (4): Alternatively, HIV RNAs serve as templates for translation (ktrans) leading to Tat synthesis.
Step (5): Although NF-κB concentration at the promoter decreases quickly as a consequence of its dissociation from the template DNA (koff), Tat itself takes the place of KAP1 for recruitment and activation-deactivation of CDK9 in the viral phase with kinetic activation and deactivation parameters kact(v) and kdeact(v), respectively.
Step (6): As a consequence of Tat activity (Step (5)), the positive feedback loop (kfb) becomes dominant leading to robust and sustained HIV RNA synthesis (ksynth(v)).
Without feedback loop, HIV RNA synthesis receives an initial KAP1-dependent “boost” by NF-κB. However, with NF-κB diminishing as a consequence of its dissociation from the promoter (koff) and re-translocation to the cytoplasm, HIV transcription soon returns to the low steady-state level of the basal transcription rate; and as a consequence, the feedback loop (kfb) does not operate normally. Loss of KAP1 virtually abolished the initial transcriptional “boost” mediated by NF-κB; consequently, the feedback loop is largely reduced in magnitude compared with the KAP1 positive scenario. Furthermore, without feedback loop, the levels of HIV RNA synthesis remain extremely low and indistinguishable from basal activity.
Assumptions
Basal HIV RNA synthesis remains constant over time and is very low compared to RNA synthesis induced in response to NF-κB and Tat activation (thereby becoming depreciable).
Basal HIV RNA does not contribute to the pool of molecules that generate fully mature HIV RNAs leading to viral products to perpetuate the infection.
The overall effect of NF-κB activation involves the canonical positive/negative feedback loop due to NF-κB binding to the promoter (leading to activation) followed by its release (leading to deactivation).
NF-κB–promoter association and dissociation is induced after activation by TNF.
KAP1 is already bound to CDK9 and dissociates from the promoter with the given rate.
Tat translation explicitly requires NF-κB–mediated HIV RNA synthesis in response to immune stimulation.
Molecular processes involved in the Tat positive feedback loop are non-limiting and can be reduced to overall rates.
Variables
Time is simulated in discrete steps according to the simulation implementations used (deterministic or stochastic). [RNA](t), [Tat](t), [NF-κB](t), [KAP1](t) and [TNF](t) describe the amount of HIV RNA, Tat, NF-κB, KAP1 (in the nucleus) and TNF, respectively.
Parameters
See Table S6.
α is the rate of basal HIV RNA synthesis
kon and koff represent the spontaneous association and dissociation of transcription factor and co-activators to the proviral promoter, respectively.
kact(h), kdeact(h), describe the rates of KAP1-mediated recruitment and activation-deactivation of CDK9 in the host phase, respectively.
ksynth(h) describes the rate of HIV RNA synthesis by NF-κB (initial transcriptional boost; host phase).
ktrans and kdecay describe the rate of Tat RNA translation and decay in the absence of feedback loop, respectively.
kact(v), kdeact(v), describe the corresponding rates of Tat–CDK9 association and dissociation from the promoter, respectively.
ksynth(v) describes the rate of HIV RNA synthesis by Tat (feedback loop; viral phase).
kfb describes the rate of Tat positive feedback loop induced by Tat recruiting new CDK9 to the promoter and activating transcription elongation.
Physical Basis for Parameters
We assume a well-mixed scenario for our simulations. Kinetic parameters have been either taken from published data together with the underlying experimental conditions, or have been fitted using measured RNA concentrations and JSim’s Simplex non-linear steepest-descent algorithm (Butterworth et al., 2013).
Ordinary Differential Equations (ODE)
Table S7 shows a deterministic approximation of the model as system of six ODEs and a seventh equation of the total concentration of RNA in the system. We used a set of different initial conditions for KAP1 to assess its effect on the dynamics of the model. We consider basal and stimulated RNA expression separately. The dynamics of the non-basal RNA is dependent on activation by NF-κB, KAP1 and Tat. The dynamics of stimulated RNA synthesis in our dynamic model (Table S7) is described by ODE (1):
| (1) |
The first term describes an overall, Michaelis-Menten type, dynamics regulated by NF-κB, with μRNA = kcat[NFκB]0 describing the maximal reaction velocity with [NFκB]0denoting the initial concentration of NF-κB, and kMm = (koff + kcat)/kon being the corresponding Michaelis-Menten constant.
The second term denotes the additional effect of KAP1 on transcription together with NF-κB. Overall activation rate is tRNA with the implicit inclusion of activation rate kact(h) (see below). The third and fourth terms refer to KAP1-initiated and KAP1-independent; respectively, Tat translation, and further contribution by Tat through the feedback loop. Given the stimulated RNA, which is further translated into Tat, and its concentration [RNA], the dynamic of Tat translation with [Tat] denoting the concentration of expressed Tat protein is then given by the following equation:
Tat is known to establish a positive feedback loop via binding to the TAR RNA stem-loop formed at the 5′ end of nascent viral premRNAs. Following (Razooky and Weinberger, 2011), we ‘lump’ many of the detailed molecular interactions known to take place in this process into two parameters to generate a minimal model of HIV provirus trans-activation. The resulting minimal model can be described by ODE (2):
| (2) |
We use the same terms as in Equations (1) and (2); however, the middle term represents a saturable positive-feedback loop, where mTat represents the positive-feedback strength, and kMTat is the saturation constant of the system. Similar to the Michaelis-Menten approach we employed for the NF-κB–regulated dynamics in Equation (1), we use a Michaelis-Menten dynamics to model the Tat feedback loop in Equation (2). The corresponding maximal reaction velocity (μTat) is described by μTat = kfb [Tat]0 and kMTat = (kdeact(v) + kfb)/kact(v) being the Michaelis-Menten constant. dTat refers to the degradation rate of Tat.
Other reactions included in the model are the activation of NF-κB by TNF, transport of KAP1 to/from the nucleus and TNF signaling. We simplified the complex feedback loop between TNF, the inhibitor of NF-κB kinase (IKK), other kinases (such as TAK1), IκBα, A20 and NF-κB into a linear module capturing the overall dynamics of NF-κB activation by TNF.
| (3) |
Kinetic parameter b describes the overall NF-κB activation (including translocation to the nucleus), whereas dNFκB denotes the deactivation rate.
Similar to the simplification of the TNF ↔ 4 NF-κB feedback loop, we streamline the translocation of KAP1 from the nucleoplasm to the promoter/chromatin, activation-deactivation of the promoter by KAP1, involving CDK9, and KAP1 relocation to the nucleoplasm by overall reactions with the implicit activation rate in the host phase kact(h) (see explanation to Equation (1)) and deactivation rate in the host phase kdeact(h):
| (4) |
with import rate r and export-deactivation rate dKAP1 (implicitly including kdeact(h)), yielding a stationary state concentration in the nucleus of [KAP1] = ρ/dKAP1.
The activation by TNF is modeled by a simple exponential decay from a finite value at t = 0 with decay rate dTNF.
| (5) |
The differential equation that describes the basal expression is:
| (6) |
yielding a steady state of [RNAbasal] = α/dRNAbasal: dRNAbasal denotes the degradation rate of basal RNA.
Together with the non-basal RNA, the total, measured RNA is then:
| (7) |
Stochastic Description
RNA, Tat, KAP1, TNF, Pol II and NF-κB are actually simulated as discrete molecules in a well stirred mixture in which stochastic Poisson processes act. Such Poisson processes are well described by the Chemical Master Equation (Hahl and Kremling, 2016) capturing the corresponding reaction system.
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
| (20) |
KAP1 initiation denotes the contribution of KAP1 to the host phase and NF-κB initiation denotes activation of the host phase.
Simulations
Both the deterministic as well as the stochastic simulation use standard procedures according to the implementation of the corresponding simulation software (see section “Implementation” below). In the case of the stochastic simulations, 100 trajectories for each run have been calculated.
Implementation
We implemented the deterministic simulation of the corresponding ODEs [Equations (1) – (7)] within JSim v2.15 in JSim’s own Mathematical Modeling Language (MML). The Dormand-Prince explicit Runge-Kutta method of order 5(4) for non-stiff equations (Dopri5) was used for simulation, with a fallback option to the implicit Runge-Kutta method of variable order (Radau; solver setting to “auto”). Parameter optimization for unknown parameters was performed using the simplex method. The stochastic version of the dynamic model was implemented in StochSS (Drawert et al., 2016) [see Equations (8) – (20)]. StochSS provides implementations of several exact stochastic simulation algorithms (SSA), including the direct method, optimized direct method and composition-rejection method. These approaches all generate exact samples (trajectories) from the chemical master equation. After model analysis, StochSS automatically chooses the appropriate algorithm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were processed and visualized using GraphPad Prism. All statistical details can be found in the figure legends. All quantified data (n) represent the number of biological replicates. Plotted values are the average of three independent experiments (mean ± SEM; n = 3). Student’s t test was used to determine statistical significance. We considered p < 0.05 to be statistically significant. Western blot band intensities were quantified using ImageJ.
DATA AND SOFTWARE AVAILABILITY
All software used in this study is listed in the Key Resources Table. The original data has been deposited to Mendeley Data: http://doi:10.17632/kvtryf67vg.1
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal beta-actin (clone C4) | Santa Cruz | Cat# sc-47778; RRID: AB_626632 |
| Rabbit polyclonal NELF-E (clone H-140) | Santa Cruz | Cat# sc-32912; RRID: AB_2177858 |
| Mouse monoclonal KAP1 (clone 20C1) | Abcam | Cat# ab22553; RRID: AB_447151 |
| Rabbit polyclonal RNA polymerase II (clone N-20) | Santa Cruz | Cat# sc-899; RRID: AB_632359 |
| Rabbit polyclonal CDK9 (clone C-20) | Santa Cruz | Cat# sc-484; RRID: AB_2275986 |
| Rabbit polyclonal NF-κB p65 (clone C-20) | Santa Cruz | Cat# sc-372; RRID: AB_632037 |
| Mouse monoclonal CXCR4, PE conjugated | BD Biosciences | Cat# 555974; RRID: AB_396267 |
| Mouse monoclonal CD4, APC conjugated | Thermo Fisher | S3.5, MHCD0405; RRID: AB_10373698 |
| Mouse monoclonal p24, FITC conjugated | Beckman Coulter | Cat# 6604665; RRID: N/A |
| Rabbit polyclonal anti-human IL-4 | PeproTech | Cat# 500-P24; RRID: AB_1479023 |
| Goat polyclonal anti-human IL-12 | PeproTech | Cat# 500-P154; RRID: AB_148144 |
| Mouse IgG | Santa Cruz | Cat# sc-2025; RRID: AB_737182 |
| Donkey anti-rabbit IgG, HRP conjugated | Santa Cruz | Cat# sc-2313; RRID: AB_641181 |
| Goat anti-mouse IgG, HRP conjugated | Santa Cruz | Cat# sc-2005; RRID: AB_631736 |
| Deposited Data | ||
| Western blot original scans | This paper | http://doi:10.17632/kvtryf67vg.1 |
| Bacterial and Virus Strains | ||
| Escherichia coli DH5α | Thermo Fisher | Cat# 18265017 |
| Escherichia coli STBL3 | Thermo Fisher | Cat# C737303 |
| HIV-1 NL4–3 | NIH AIDS Reagent Program | Cat# 114 |
| HIV-1 NL4–3-deltaEnv-nLuc-2ANef | Martins et al., 2016 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin dihydrochloride | Millipore Sigma | Cat# P8833 |
| Triptolide | Santa Cruz | Cat# sc-200122 |
| Flavopiridol | Millipore Sigma | Cat# F3055 |
| Bryostatin | Millipore Sigma | Cat# B7431 |
| Random decamers | Thermo Fisher | Cat# AM5722G |
| Fast SYBR green master mix | Thermo Fisher | Cat# 4385616 |
| Polyjet | SignaGen | Cat# SL100688 |
| Dynabeads human T activator CD3/CD28 | Thermo Fisher | Cat# 11161D |
| Vorinostat (SAHA) | ApexBio | Cat# MK0683 |
| Human TNF-α | Millipore Sigma | Cat# T6674 |
| PMA | Millipore Sigma | Cat# P8139 |
| DMSO | ACROS Organics | Cat# 610420010 |
| Human TGF-β1 | PeproTech | Cat# 100–21 |
| Human IL-2 | NIH AIDS Reagent Program | Cat# 126 |
| Nelfinavir | NIH AIDS Reagent Program | Cat# 4621 |
| RPMI 1640 | HyClone | Cat# SH30027.FS |
| DMEM | HyClone | Cat# SH30022.FS |
| Fetal Bovine Serum | Millipore Sigma | Cat# F4135 |
| Penicillin-Streptomycin | MP Biomedicals | 091670049 |
| Polybrene | Millipore Sigma | Cat# H9268 |
| PBS | HyClone | Cat# SH30028.02 |
| Paraformaldehyde | Millipore Sigma | Cat# P6148 |
| Cytofix/Cytoperm | Becton Dickinson | Cat# 554714 |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1960 |
| Nano-Glo Luciferase Assay System | Promega | Cat# N1110 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 233225 |
| 10XTris/Glycine/SDS Running Buffer | Bio-Rad | Cat# 1610732 |
| 0.45 μM Nitrocellulose Membrane | Bio-Rad | Cat# 1620115 |
| Tween-20 | Fisher Chemical | Cat# BP337–500 |
| Non-fat Dry Milk | LabScientific | Cat# M0841 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 1705061 |
| 5” × 7” Blue X-Ray Film | Phenix Research Products | Cat# F-BX57 |
| 8” × 10” Blue X-Ray Film | Phenix Research Products | Cat# F-BX810 |
| Dynabeads CD4 Positive Isolation Kit | Thermo Fisher | Cat# 11331D |
| Neon Transfection System 10 ml Kit | Thermo Fisher | Cat# MPK1096 |
| Neon Transfection System | Thermo Fisher | Cat# MPK5000 |
| Methanol-free formaldehyde | Thermo Fisher | Cat# 28908 |
| PMSF | Millipore Sigma | Cat# P7626 |
| cOmplete, Mini EDTA-free Protease Inhibitor Cocktail | Millipore Sigma | Cat# 04693159001 |
| NP-40 | Thermo Fisher | Cat# 28324 |
| Proteinase K | Thermo Fisher | Cat# 03115828001 |
| RNase A, DNase and Protease-free | Thermo Fisher | Cat# EN0531 |
| Alt-R Cas9 Nuclease V3 | IDT | Cat# 1081058 |
| Critical Commercial Assays | ||
| Dynabeads Protein G | Thermo Fisher | Cat# 10004D |
| M-MuLV Reverse Transcriptase | New England Biolabs | Cat# M0253 |
| Mycoplasma Detection Kit | SouthernBiotech | Cat# 13100–01 |
| Fixable Viability Dye eFluor 450 | Thermo Fisher | Cat# 65-0863-14 |
| Quick-RNA Miniprep Kit | Zymo | Cat# R1055 |
| EasySep Human Naive CD4+ T Cell Isolation kit | Stemcell | Cat# 19555 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| Deposited Data | ||
| Western blot original scans | This paper | Uploaded |
| Experimental Models: Cell Lines | ||
| Jurkat, Clone E6–1 | ATCC | Cat# TIB-152 |
| SUP-T1 | ATCC | Cat# CRL-1942 |
| HEK293FT | Thermo Fisher | Cat# 70007 |
| HEK293T/17 | ATCC | Cat# CRL-11268 |
| U2 OS | ATCC | Cat# HTB-96 |
| J-Lat 6.3 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9846 |
| J-Lat 8.4 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9847 |
| J-Lat 9.2 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9848 |
| J-Lat 10.6 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9849 |
| Jurkat, E4 Clone | Pearson et al., 2008 | N/A |
| Jurkat, 2B2D Clone | Pearson et al., 2008 | N/A |
| Engineered Cell Lines, see Table S1 | This paper | N/A |
| Oligonucleotides | ||
| Primers for RNAi, see Table S2 | Millipore Sigma | N/A |
| Primers for RT-qPCR and ChIP, see Table S3 | Millipore Sigma | N/A |
| TracrRNA | IDT | Cat# 1072532 |
| Scrambled guide RNA | IDT | Cat# 1072544 |
| CXCR4 guide RNA | IDT | N/A |
| NF-κB p65 guide RNA | IDT | N/A |
| KAP1 guide RNA | IDT | N/A |
| Recombinant DNA | ||
| pcDNA4/TO (empty vector) | Thermo Fisher | Cat# V102020 |
| pcDNA3.1+ (empty vector) | Thermo Fisher | Cat# V79020 |
| pBluescript II KS+ | Agilent | Cat# X52327 |
| psPAX2 | Unpublished | Addgene Cat# 12260 |
| pMD2.G | Unpublished | Addgene Cat# 12259 |
| pCMV-VSV-G | Stewart et al., 2003 | Addgene Cat# 8454 |
| pTRIP-LUC | Schoggins et al., 2011 | N/A |
| pLVTHM mCherry | Jadlowsky et al., 2014 | N/A |
| pLKO.1 shRNA control plasmid DNA | Millipore Sigma | Cat# SHC002 |
| pLKO.1-IPTG-3xLacO shRNA control plasmid DNA | Millipore Sigma | Cat#SHC332V |
| DNA Clones Generated in This Study | This paper | See Table S5 |
| Software and Algorithms | ||
| JSim v2.15 (developed by the NSR Physiome Project) | Butterworth et al., 2013) https://www.physiome.org/jsim/ | RRID:SCR_007379 |
| Chemical Master Equation | Hahl and Kremling, 2016 | N/A |
| StochSS 1.9 (using Docker) | (Drawert et al., 2016) http://stochss.org/ | N/A |
| Docker | https://www.docker.com/ | RRID: SCR_016445 |
| FlowJo | TreeStar Inc. | RRID: SCR_008520 |
| FACSDiva | Becton Dickinson | RRID: SCR_001456 |
| Prism (version 7) | GraphPad | RRID: SCR_002798 |
| ImageJ | Schneider et al., 2012 | RRID: SCR:003070 |
Supplementary Material
Highlights.
The two phases of the HIV transcriptional circuit have distinct functional requirements
HIV evolved a minimalist program to robustly bypass host cell regulatory checkpoints
Mathematical modeling reveals the host phase is subject to transcriptional fragility
Transcriptional fragility influences the viral feedback and latency reversal potential
ACKNOWLEDGMENTS
We are grateful to J. Karn and E. Verdin for generously sharing the J-Lat clones. We apologize to many colleagues whose work could not be cited because of space constrains. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH under award numbers R01AI114362 and R33AI116222 and Welch Foundation grant I-1782 (to I.D.) and by NIAID under award numbers R21AI123035–01 and R33AI122377 (to V.P.) and grant U01AI111598 (to C.V.F.). N.-G.P.R was supported by NIH Pharmacological Sciences Training grant GM007062 and a 2016 pre-doctoral fellowship from the Ford Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.007.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Allouch A, Di Primio C, Alpi E, Lusic M, Arosio D, Giacca M, and Cereseto A (2011). The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 9, 484–495. [DOI] [PubMed] [Google Scholar]
- Bacon CW, and D’Orso I (2018). CDK9: a signaling hub for transcriptional control. Transcription September 19, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosque A, and Planelles V (2009). Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch H, Zheng X, Burkholder A, Dillon ST, Motola S, Birrane G, Ebmeier CC, Levine S, Fargo D, Hu G, et al. (2014). TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat. Struct. Mol. Biol 21, 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterworth E, Jardine BE, Raymond GM, Neal ML, and Bassingthwaighte JB (2013). JSim, an open-source modeling system for data analysis. F1000Res. 2, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, and Siliciano RF (1995). In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat. Med 1, 1284–1290. [DOI] [PubMed] [Google Scholar]
- D’Orso I (2016). 7SKiing on chromatin: move globally, act locally. RNA Biol. 13, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orso I, Jang GM, Pastuszak AW, Faust TB, Quezada E, Booth DS, and Frankel AD (2012). Transition step during assembly of HIV Tat:P-TEFb transcription complexes and transfer to TAR RNA. Mol. Cell. Biol 32, 4780–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawert B, Hellander A, Bales B, Banerjee D, Bellesia G, Daigle BJ Jr., Douglas G, Gu M, Gupta A, Hellander S, et al. (2016). Stochastic simulation service: bridging the gap between the computational expert and the biologist. PLoS Comput. Biol 12, e1005220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, et al. (1999). Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med 5, 512–517. [DOI] [PubMed] [Google Scholar]
- Hahl SK, and Kremling A (2016). A comparison of deterministic and stochastic modeling approaches for biochemical reaction systems: on fixed points, means, and modes. Front. Genet 7, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SH, and Coffin JM (2016). What integration sites tell us about HIV persistence. Cell Host Microbe 19, 588–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbeault M, Helleboid PY, and Trono D (2017). KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 543, 550–554. [DOI] [PubMed] [Google Scholar]
- Iyengar S, and Farnham PJ (2011). KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem 286, 26267–26276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadlowsky JK, Wong JY, Graham AC, Dobrowolski C, Devor RL, Adams MD, Fujinaga K, and Karn J (2014). Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol 34, 1911–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, and Verdin E (2003). HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22, 1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J (2011). The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 6, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita S, Su L, Amano M, Timmerman LA, Kaneshima H, and Nolan GP (1997). The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 6, 235–244. [DOI] [PubMed] [Google Scholar]
- Ma X, Yang T, Luo Y, Wu L, Jiang Y, Song Z, Pan T, Liu B, Liu G, Liu J, et al. (2019). TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Agarwal S, Driscoll S, Lettieri K, Wang J, Andrews SE, Franco L, Rosenfeld MG, Ren B, and Pfaff SL (2011). Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 25, 594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, et al. (2014). HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins LJ, Bonczkowski P, Spivak AM, De Spiegelaere W, Novis CL, DePaula-Silva AB, Malatinkova E, Trypsteen W, Bosque A, Vanderkerckhove L, and Planelles V (2016). Modeling HIV-1 latency in primary T cells using a replication-competent virus. AIDS Res. Hum. Retroviruses 32, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara RP, McCann JL, Gudipaty SA, and D’Orso I (2013). Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 5, 1256–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara RP, Bacon CW, and D’Orso I (2016a). Transcription elongation control by the 7SK snRNP complex: Releasing the pause. Cell Cycle 15, 2115–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara RP, Reeder JE, McMillan EA, Bacon CW, McCann JL, and D’Orso I (2016b). KAP1 recruitment of the 7SK snRNP complex to promoters enables transcription elongation by RNA polymerase II. Mol. Cell 61, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel G, and Baltimore D (1987). An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326, 711–713. [DOI] [PubMed] [Google Scholar]
- Ott M, Geyer M, and Zhou Q (2011). The control of HIV transcription: keeping RNA polymerase II on track. Cell Host Microbe 10, 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pache L, Dutra MS, Spivak AM, Marlett JM, Murry JP, Hwang Y, Maestre AM, Manganaro L, Vamos M, Teriete P, et al. (2015). BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by Smac mimetics to promote reversal of viral latency. Cell Host Microbe 18, 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, and Karn J (2008). Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol 82, 12291–12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauwel B, Jang SM, Cassano M, Kapopoulou A, Barde I, and Trono D (2015). Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. eLife 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky BS, and Weinberger LS (2011). Mapping the architecture of the HIV-1 Tat circuit: A decision-making circuit that lacks bistability and exploits stochastic noise. Methods 53, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razooky BS, Pai A, Aull K, Rouzine IM, and Weinberger LS (2015). A hardwired HIV latency program. Cell 160, 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B, and Yin J (1999). Quantitative intracellular kinetics of HIV type 1. AIDS Res. Hum. Retroviruses 15, 273–283. [DOI] [PubMed] [Google Scholar]
- Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, et al. (2010). KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463, 237–240. [DOI] [PubMed] [Google Scholar]
- Ruelas DS, and Greene WC (2013). An integrated overview of HIV-1 latency. Cell 155, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, and Bushman F (2002). HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529. [DOI] [PubMed] [Google Scholar]
- Shan L, Deng K, Gao H, Xing S, Capoferri AA, Durand CM, Rabi SA, Laird GM, Kim M, Hosmane NN, et al. (2017). Transcriptional reprogramming during effector-to-memory transition renders CD4(+) T cells permissive for latent HIV-1 infection. Immunity 47, 766–775.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, et al. (2013). An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 9, e1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taura M, Song E, Ho YC, and Iwasaki A (2019). Apobec3A maintains HIV-1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc. Natl. Acad. Sci. USA 116, 2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, and Covert MW (2010). Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 466, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, and Schaffer DV (2005). Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 122, 169–182. [DOI] [PubMed] [Google Scholar]
- Williams SA, Kwon H, Chen LF, and Greene WC (2007). Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol 81, 6043–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, and Goff SP (2007). TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131, 46–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software used in this study is listed in the Key Resources Table. The original data has been deposited to Mendeley Data: http://doi:10.17632/kvtryf67vg.1
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal beta-actin (clone C4) | Santa Cruz | Cat# sc-47778; RRID: AB_626632 |
| Rabbit polyclonal NELF-E (clone H-140) | Santa Cruz | Cat# sc-32912; RRID: AB_2177858 |
| Mouse monoclonal KAP1 (clone 20C1) | Abcam | Cat# ab22553; RRID: AB_447151 |
| Rabbit polyclonal RNA polymerase II (clone N-20) | Santa Cruz | Cat# sc-899; RRID: AB_632359 |
| Rabbit polyclonal CDK9 (clone C-20) | Santa Cruz | Cat# sc-484; RRID: AB_2275986 |
| Rabbit polyclonal NF-κB p65 (clone C-20) | Santa Cruz | Cat# sc-372; RRID: AB_632037 |
| Mouse monoclonal CXCR4, PE conjugated | BD Biosciences | Cat# 555974; RRID: AB_396267 |
| Mouse monoclonal CD4, APC conjugated | Thermo Fisher | S3.5, MHCD0405; RRID: AB_10373698 |
| Mouse monoclonal p24, FITC conjugated | Beckman Coulter | Cat# 6604665; RRID: N/A |
| Rabbit polyclonal anti-human IL-4 | PeproTech | Cat# 500-P24; RRID: AB_1479023 |
| Goat polyclonal anti-human IL-12 | PeproTech | Cat# 500-P154; RRID: AB_148144 |
| Mouse IgG | Santa Cruz | Cat# sc-2025; RRID: AB_737182 |
| Donkey anti-rabbit IgG, HRP conjugated | Santa Cruz | Cat# sc-2313; RRID: AB_641181 |
| Goat anti-mouse IgG, HRP conjugated | Santa Cruz | Cat# sc-2005; RRID: AB_631736 |
| Deposited Data | ||
| Western blot original scans | This paper | http://doi:10.17632/kvtryf67vg.1 |
| Bacterial and Virus Strains | ||
| Escherichia coli DH5α | Thermo Fisher | Cat# 18265017 |
| Escherichia coli STBL3 | Thermo Fisher | Cat# C737303 |
| HIV-1 NL4–3 | NIH AIDS Reagent Program | Cat# 114 |
| HIV-1 NL4–3-deltaEnv-nLuc-2ANef | Martins et al., 2016 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Puromycin dihydrochloride | Millipore Sigma | Cat# P8833 |
| Triptolide | Santa Cruz | Cat# sc-200122 |
| Flavopiridol | Millipore Sigma | Cat# F3055 |
| Bryostatin | Millipore Sigma | Cat# B7431 |
| Random decamers | Thermo Fisher | Cat# AM5722G |
| Fast SYBR green master mix | Thermo Fisher | Cat# 4385616 |
| Polyjet | SignaGen | Cat# SL100688 |
| Dynabeads human T activator CD3/CD28 | Thermo Fisher | Cat# 11161D |
| Vorinostat (SAHA) | ApexBio | Cat# MK0683 |
| Human TNF-α | Millipore Sigma | Cat# T6674 |
| PMA | Millipore Sigma | Cat# P8139 |
| DMSO | ACROS Organics | Cat# 610420010 |
| Human TGF-β1 | PeproTech | Cat# 100–21 |
| Human IL-2 | NIH AIDS Reagent Program | Cat# 126 |
| Nelfinavir | NIH AIDS Reagent Program | Cat# 4621 |
| RPMI 1640 | HyClone | Cat# SH30027.FS |
| DMEM | HyClone | Cat# SH30022.FS |
| Fetal Bovine Serum | Millipore Sigma | Cat# F4135 |
| Penicillin-Streptomycin | MP Biomedicals | 091670049 |
| Polybrene | Millipore Sigma | Cat# H9268 |
| PBS | HyClone | Cat# SH30028.02 |
| Paraformaldehyde | Millipore Sigma | Cat# P6148 |
| Cytofix/Cytoperm | Becton Dickinson | Cat# 554714 |
| Dual-Luciferase Reporter Assay System | Promega | Cat# E1960 |
| Nano-Glo Luciferase Assay System | Promega | Cat# N1110 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 233225 |
| 10XTris/Glycine/SDS Running Buffer | Bio-Rad | Cat# 1610732 |
| 0.45 μM Nitrocellulose Membrane | Bio-Rad | Cat# 1620115 |
| Tween-20 | Fisher Chemical | Cat# BP337–500 |
| Non-fat Dry Milk | LabScientific | Cat# M0841 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 1705061 |
| 5” × 7” Blue X-Ray Film | Phenix Research Products | Cat# F-BX57 |
| 8” × 10” Blue X-Ray Film | Phenix Research Products | Cat# F-BX810 |
| Dynabeads CD4 Positive Isolation Kit | Thermo Fisher | Cat# 11331D |
| Neon Transfection System 10 ml Kit | Thermo Fisher | Cat# MPK1096 |
| Neon Transfection System | Thermo Fisher | Cat# MPK5000 |
| Methanol-free formaldehyde | Thermo Fisher | Cat# 28908 |
| PMSF | Millipore Sigma | Cat# P7626 |
| cOmplete, Mini EDTA-free Protease Inhibitor Cocktail | Millipore Sigma | Cat# 04693159001 |
| NP-40 | Thermo Fisher | Cat# 28324 |
| Proteinase K | Thermo Fisher | Cat# 03115828001 |
| RNase A, DNase and Protease-free | Thermo Fisher | Cat# EN0531 |
| Alt-R Cas9 Nuclease V3 | IDT | Cat# 1081058 |
| Critical Commercial Assays | ||
| Dynabeads Protein G | Thermo Fisher | Cat# 10004D |
| M-MuLV Reverse Transcriptase | New England Biolabs | Cat# M0253 |
| Mycoplasma Detection Kit | SouthernBiotech | Cat# 13100–01 |
| Fixable Viability Dye eFluor 450 | Thermo Fisher | Cat# 65-0863-14 |
| Quick-RNA Miniprep Kit | Zymo | Cat# R1055 |
| EasySep Human Naive CD4+ T Cell Isolation kit | Stemcell | Cat# 19555 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| Deposited Data | ||
| Western blot original scans | This paper | Uploaded |
| Experimental Models: Cell Lines | ||
| Jurkat, Clone E6–1 | ATCC | Cat# TIB-152 |
| SUP-T1 | ATCC | Cat# CRL-1942 |
| HEK293FT | Thermo Fisher | Cat# 70007 |
| HEK293T/17 | ATCC | Cat# CRL-11268 |
| U2 OS | ATCC | Cat# HTB-96 |
| J-Lat 6.3 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9846 |
| J-Lat 8.4 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9847 |
| J-Lat 9.2 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9848 |
| J-Lat 10.6 | Jordan et al., 2003 | NIH AIDS Reagent Program Cat# 9849 |
| Jurkat, E4 Clone | Pearson et al., 2008 | N/A |
| Jurkat, 2B2D Clone | Pearson et al., 2008 | N/A |
| Engineered Cell Lines, see Table S1 | This paper | N/A |
| Oligonucleotides | ||
| Primers for RNAi, see Table S2 | Millipore Sigma | N/A |
| Primers for RT-qPCR and ChIP, see Table S3 | Millipore Sigma | N/A |
| TracrRNA | IDT | Cat# 1072532 |
| Scrambled guide RNA | IDT | Cat# 1072544 |
| CXCR4 guide RNA | IDT | N/A |
| NF-κB p65 guide RNA | IDT | N/A |
| KAP1 guide RNA | IDT | N/A |
| Recombinant DNA | ||
| pcDNA4/TO (empty vector) | Thermo Fisher | Cat# V102020 |
| pcDNA3.1+ (empty vector) | Thermo Fisher | Cat# V79020 |
| pBluescript II KS+ | Agilent | Cat# X52327 |
| psPAX2 | Unpublished | Addgene Cat# 12260 |
| pMD2.G | Unpublished | Addgene Cat# 12259 |
| pCMV-VSV-G | Stewart et al., 2003 | Addgene Cat# 8454 |
| pTRIP-LUC | Schoggins et al., 2011 | N/A |
| pLVTHM mCherry | Jadlowsky et al., 2014 | N/A |
| pLKO.1 shRNA control plasmid DNA | Millipore Sigma | Cat# SHC002 |
| pLKO.1-IPTG-3xLacO shRNA control plasmid DNA | Millipore Sigma | Cat#SHC332V |
| DNA Clones Generated in This Study | This paper | See Table S5 |
| Software and Algorithms | ||
| JSim v2.15 (developed by the NSR Physiome Project) | Butterworth et al., 2013) https://www.physiome.org/jsim/ | RRID:SCR_007379 |
| Chemical Master Equation | Hahl and Kremling, 2016 | N/A |
| StochSS 1.9 (using Docker) | (Drawert et al., 2016) http://stochss.org/ | N/A |
| Docker | https://www.docker.com/ | RRID: SCR_016445 |
| FlowJo | TreeStar Inc. | RRID: SCR_008520 |
| FACSDiva | Becton Dickinson | RRID: SCR_001456 |
| Prism (version 7) | GraphPad | RRID: SCR_002798 |
| ImageJ | Schneider et al., 2012 | RRID: SCR:003070 |