

Abstract
Staphylococcus aureus is a leading cause of hospital-acquired infections worldwide, and cases of community-acquired infections are becoming more prevalent. The production of numerous virulence factors in S. aureus is under the control of the accessory gene regulator (agr) quorum sensing (QS) system. S. aureus upregulates agr at high cell density by secreting a peptide pheromone, or autoinducing peptide (AIP), which is detected by its cognate transmembrane receptor, AgrC. The extracellular AIP binding site of AgrC represents an attractive target for inhibition of the agr system, and thereby, QS-controlled virulence in S. aureus. Nonnative peptides and more recently, peptidomimetics, have been reported to inhibit the AgrC receptor and represent useful chemical tools to study the role of QS in S. aureus infections. We seek to expand beyond peptide-like scaffolds to generate AgrC modulators with enhanced stability, solubility, and synthetic accessibility relative to these compounds, while maintaining their high potencies. Toward this goal, we report herein a study of the structure-activity relationships responsible for the activity of a recently reported simplified AIP mimetic and AgrC antagonist, n7OFF, and the discovery of a new AIP mimetic, Bnc3, which has low- to sub-nanomolar inhibitory activity in all four S. aureus agr specificity groups. NMR structural studies of Bnc3 revealed hydrophobic and hydrophilic faces that are likely critical for AgrC antagonism, in agreement with prior studies of peptide-derived inhibitors. Bnc3 represents an important transition compound toward the development of small-molecule AgrC antagonists.
Keywords: accessory gene regulator, autoinducing peptide, peptidomimetic, quorum sensing, Staphylococcus aureus, structure-activity relationship
Graphical Abstract
Staphylococcus aureus is a common opportunistic pathogen that colonizes approximately 30% of the world’s population.1 Most colonized individuals coexist with S. aureus as a commensal organism that inhabits the skin and nose. However, virulent S. aureus strains have been found in hospital-acquired infections for decades and are being isolated more frequently from community-acquired infections.2 Combined with the growing incidence of multidrug-resistance in these strains, S. aureus presents a significant challenge for our healthcare system. New alternatives are desperately needed to prevent and treat S. aureus infections.1–3
S. aureus produces numerous virulence factors that are responsible for many aspects of acute infections. A large proportion of these virulence factors, such as hemolysins and phenol-soluble modulins, are controlled by a cell-cell communication pathway known as quorum sensing (QS).3–5 QS is a means by which bacteria assess their local population densities and initiate group-beneficial behaviors at high cell number. Bacteria use simple chemical signals, or autoinducers, for QS that are produced at a low, but constant basal level. Once the bacterial population is sufficiently large in a given environment, the local autoinducer concentration reaches a threshold level at which it can productively bind to its cognate receptor; this signal:receptor binding event effectively signals to the bacteria that their population has reached a quorum.6 The bacterial “group” will then initiate a diversity of behaviors, ranging from bioluminescence by marine bacteria, to antibiotic production by soil dwelling bacteria, to virulence factor production by pathogens, as is the case for S. aureus.4–7
S. aureus uses the accessory gene regulator (agr) system for QS, which is considered an autocatalytic sensory transduction system.8 This system is found in many staphylococcal species, but is best understood in S. aureus and illustrated in Figure 1A.9 The agr operon encodes four proteins (AgrA−D), of which AgrC and AgrA are part of a classical two-component regulatory system. AgrD contains three domains: an amphipathic N-terminal domain that localizes the protein to the inner leaflet of the plasma membrane, a “pro-peptide” domain consisting of a linear precursor of the autoinducing peptide (AIP) QS signal, and a C-terminal recognition domain.10 AgrB is an integral membrane endopeptidase that recognizes the C-terminal domain of AgrD, cleaves this domain, and cyclizes the new C-terminal residue of AgrD to a conserved Cys residue sulfhydryl in the pro-peptide domain.11 The modified AgrD is then transported outside the plasma membrane and the N-terminal domain is cleaved, liberating the mature AIP signal outside of the cell.12 As the population grows, this newly formed QS signal accumulates in the local environment, and when a sufficient concentration of AIP is reached (and thus a quorum of S. aureus), the AIP binds to its target, AgrC, a transmembrane receptor histidine kinase. Once bound to AIP, AgrC transautophosphorylates and then phosphorylates AgrA, an intracellular transcription factor.13, 14 Binding of AgrA to the P2 promoter upregulates the agr operon and thereby amplifies the QS signal, in a typical autoinduction cycle that is a hallmark of QS systems. In turn, the binding of AgrA to the P3 and other promoters upregulates myriad virulence factors that are associated with S. aureus infections.3
Figure 1.

The agr QS system and associated AIP signals. A: Schematic of the agr system. (a) agr encodes production of AgrA−D. (b) AgrD contains the precursor for the AIP QS signal. (c) AgrB processes AgrD and liberates the mature AIP signal. (d) The AIP binds to AgrC. (e) AgrC transautophosphorylates and then phosphorylates AgrA. (f) AgrA drives transcription at the P2 and P3 promoters, which upregulates production of AgrA−D and activates virulence factor production. B: Structures of the native AIP signals used by the four groups of S. aureus, using single letter amino acid abbreviations.
The agr system of S. aureus has diverged evolutionarily into three common (I-III) and one rare (IV) specificity groups, each with a unique AIP signal (Figure 1B) and some sequence variability in the AgrA−D proteins, although the sequences of AgrA and the histidine kinase domain of AgrC are highly conserved.15–17 Among the four AIPs, the five-amino acid macrocycle and thioester bridge from the C-terminus to a Cys is conserved, and each has at least two hydrophobic amino acids at the C-terminus. Otherwise, the identity of each amino acid and the length of the AIP tail varies between the four groups.
Because agr activity upregulates many important virulence factors in S. aureus, antagonism of this system has attracted interest as a non-bactericidal, or “anti-virulence,” strategy to block infection.18–20 Indeed, in several recent reports by Williams, Horswill, and others, antagonism of agr has reduced the infectivity of virulent S. aureus strains,21–26 underscoring the potential utility of this approach for infection control. Inhibition of agr via the AgrC receptor has been a focus of several research laboratories, including our own.23, 24, 27–32 A recognized method for antagonizing AgrC is to use AIP analogs, often with a single amino acid substitution or tail truncation, or combinations of these modifications, to competitively bind to AgrC and thereby block the native signal from binding.31–35 Most of these reported AgrC antagonists are peptides, with sizes ranging from five to nine amino acids. While some are highly potent (sub-nanomolar IC50 values), these peptides have several unfavorable attributes that impede their broad use as chemical probes. They are typically relatively large, only produced in small quantities, and hydrolytically unstable. The generation of non-peptide, or small molecule, AgrC inhibitors that circumvent these properties while maintaining high potencies would be a significant advance. Our laboratory is focused on identifying such scaffolds, and as a step toward this goal, we recently redesigned the structure of tail-truncated AIP-II (t-AIP-II), reported by the Muir lab,36 to develop a simplified, four-amino acid peptide derivative (n7OFF; Figure 2) with comparable antagonistic activity against AgrC.37 While n7OFF represents one of the first peptidomimetics of AIPs with appreciable biological activity,38, 39 it is 10−1000-fold less potent than lead peptide antagonists of AgrC. In the current study, we sought to delineate the chemical features of n7OFF that are critical to its activity in order to increase its potency. Additionally, we sought to identify structural motifs that can allow for the transition of this peptidomimetic into a non-peptide-derived AgrC antagonist, such as non-canonical amino acid motifs, which have been shown to have promise in related systems.24, 40
Figure 2.

Structures of t-AIP-II (top) and n7OFF (bottom). The nomenclature for specific motifs in n7OFF is indicated.
Herein, we report a detailed structure-activity relationship (SAR) analysis of the AgrC antagonist n7OFF and the discovery of new peptidomimetics, Bnc3 and Bn-n7OCpa(3ClF), which have low- to sub-nanomolar potencies against AgrC in all four S. aureus agr groups. An amide-linked analog of Bnc3 was also identified as a potent AgrC antagonist, with both improved solubility and stability over the parent thioester. Notably, these potency profiles are now comparable to current lead, all peptide-derived antagonists of AgrC. NMR structural studies of Bnc3 revealed hydrophobic and hydrophilic faces that appear important for AgrC antagonism. This study advances the understanding of the chemical motifs that are critical for AgrC antagonism and will inform the design of small-molecule antagonists of AgrC.
RESULTS AND DISCUSSION
Compound design and synthesis.
To define the SARs for the n7OFF scaffold, we began by examining its (i) two endocyclic Phe residues, (ii) tail region, and (iii) thioester linkage (Figure 3). We made structural changes in two phases, and combined features that engendered enhanced potencies against AgrC as the study progressed, including the use of an aliphatic linker in place of the oxa-linker (i.e., n7O2; as was examined in our previous work).37 To probe the role of sterics and electronics on the Phe aromatic rings, -Me, -OMe, -F, -Cl, and -NO2 substituents were introduced. The aromatic rings also were replaced by five- and six-member aliphatic rings. To examine the tail region, the acetyl methyl group was replaced with a set of linear aliphatic chains (ethyl, butyl, and pentyl). Two acyclic, branched aliphatic tails (isopropyl and isobutyl) and two aryl tails (benzyl and phenyl) were also evaluated. To study the thioester linkage, we replaced it with an ester and the more hydrolytically stable amide isostere. We also were interested in replacing the thioester linkage with an olefin, and first tested this replacement strategy in the known peptide-derived AgrC antagonists, t-AIP-II and t-AIP-III-D2A (Figure 4).31, 36, 38 All compounds were prepared as linear precursors on a solid support followed by solution-phase macrocyclization reactions and purification to homogeneity by HPLC, using our previously reported methods (see Methods).37, 41
Figure 3.

Structural modifications evaluated in n7OFF. Tail modifications indicated in blue at R1, side chain modifications indicated in green at R2 and R3, thioester substitutions indicated in red, and the oxa-linker modification indicated in violet.
Figure 4.

Thioester-to-olefin linkage modifications examined within known AgrC antagonists, t-AIP-II and t-AIP-III-D2A.
Biological testing.
To determine compound activities against AgrC, we utilized reporter plasmids in S. aureus strains (groups-I−IV) that permit agr activity to be measured using fluorescence (via the production of yellow-fluorescent protein (YFP)).31, 37, 42 These strains are clinical isolates, contain the native agr system, and produce AIPs at wild-type levels. Therefore, in the absence of exogenous compound, these reporter systems produce YFP once a quorum is reached. Compounds were first screened for agr antagonism (and thus a reduction in YFP production) at 10 μM, and if deemed sufficiently active, screened again at 3.3 μM (see Figures S1−S7). These screens provided initial information about compound efficacy, and compounds with near-complete antagonism of AgrC at both 10 μM and 3.3 μM were further evaluated via dose-response analyses in these same reporters to determine their IC50 values and comparisons of their relative potencies (see Tables S2−S3 for a listing of all values; key data is highlighted below).
Phase-one SAR analyses.
We first examined the effects of alterations to the Phe aromatic rings and the tail of n7OFF on potency (Table 1). Relative to the parent compound, the placement of an electronegative fluorine or chlorine at the 3-position of the C-terminal aromatic ring (Phe4) increased potency in groups-I−IV, suggesting that electronics play at least a partial role in receptor-ligand interactions. The 3-fluoro analog (n7OF(3fF)) was more potent in groups-I and -III, and the 3-chloro analog (n7OF(3ClF)) was more potent in groups-II and -IV. The same effect on potency was not universally observed with other substituents (-Me, -OMe or -NO2; Tables S2−S3), suggesting that this effect is not strictly due to an increase in steric bulk at the 3-position of Phe4.
Table 1.
IC50 values (in nM) for AgrC antagonism by selected phase-one n7OFF analogs in S. aureus groups-I−IVa. 95% confidence intervals (CI) in parentheses.
| compound | group-I | group-II | group-III | group-IV |
|---|---|---|---|---|
| n7OFF | 181 (154−213) | 583 (468−725) | 332 (280−395) | 5938 (4221−8532) |
| n7OF(3fF) | 30.6 (22.5–41.8) | 445 (217–911) | 222 (158–312) | ~3500 (ND)b |
| n7OF(3ClF) | 125 (90–173) | 167 (118–237) | 630 (364–1090) | 2240 (1150–4460) |
| n7OChaF | 439 (278–695) | 627 (317–1238) | 194 (148–255) | 1250 (990–1580) |
| n7OCpaF | 141 (107–185) | 204 (169–246) | 19.7 (14.8–26.3) | 498 (414–598) |
| Bn-n7OFF | 15.3 (12.4–19.0) | 37.8 (29.8–47.9) | 17.3 (14.1–21.2) | 564 (435–731) |
| Et-n7OFF | 129 (97.7–170) | 197 (152–255) | 531 (312–905) | --c |
| iPr-n7OFF | 45.8 (34.0–61.9) | 97.1 (65.6–144) | 316 (192–522) | --c |
| Pr-n7OFF | 57.9 (30.3–111) | 103 (68–155) | 203 (121–341) | --c |
| iBu-n7OFF | 56.0 (38.6–81.2) | 40.7 (35.3–46.9) | 55.3 (37.7–81.0) | --c |
| Bu-n7OFF | 34.7 (28.1–42.8) | 72.3 (45.6–115) | 78.1 (43.6–140) | --c |
| pentyl-n7OFF | 15.8 (13.5–18.4) | 17.0 (14.7–19.6) | 15.7 (11.8–21.0) | --c |
| Ph-n7OFF | 157 (123–201) | 23.0 (15.6–34.1) | 98.2 (75.0–129) | --c |
| n7OF(4fF) | 375 (204–689) | 354 (253–497) | 2140 (1080–4220) | --c |
Data for parent n7OFF included for comparison. See Methods section for details of strains and assays. Comprehensive assay data for all compounds tested are provided in Tables S2−S3.
The confidence interval was too wide to quantify accurately.
Minimal activity at concentrations up to 10 μM; IC50 not calculated.
Replacement of the phenyl group of Phe3 with a cyclohexyl group (n7OChaF; Cha for cyclohexylalanine) caused only a slight loss of potency relative to n7OFF in groups-I and -II and an increase in potency in groups-III and -IV (Table 1). Interestingly, contracting the ring size to a five-member ring (n7OCpaF; Cpa for cyclopentylalanine) resulted in an AgrC antagonist that was more potent than n7OFF in all four groups. This result, in combination with our earlier work with Leu3 analogs of n7OFF,37 indicates that a hydrophobic group of intermediate size with an sp3 γ-carbon (i.e., between an isobutyl and cyclohexylmethyl group) at the Phe3 position is preferred for pan-group AgrC antagonism in the n7OFF scaffold.
Addition of a benzyl group at the tail position (to give Bn-n7OFF) yielded another interesting SAR (Table 1). Relative to n7OFF, Bn-n7OFF exhibited a 10-fold improvement in potency for groups-I−IV. We note that Bn-n7OFF reduced agr activity by ~95% in groups-I−III, but only by ~70% in group-IV; other compounds discussed in this section reduced agr activity by ~95% in all four groups. Nevertheless, this result suggests that simple aryl tails, as opposed to peptide tails, can improve antagonistic activity against AgrC for AIP-type mimetics. Indeed, the addition of this third hydrophobic group, along with the two Phe groups on n7OFF, could generate a hydrophobic “triad” motif that previously has been deemed important to the activity profiles of other potent AgrC antagonists.35, 43
Additional SAR analyses in S. aureus groups-I–III.
A number of n7OFF analogs were identified in the phase-one SAR studies that were potent AgrC antagonists in S. aureus groups-I−III, but were less potent in group-IV (Table 1). As group-I−III strains are far more common than group-IV, compounds that target the former three agr groups are still of importance, and we scrutinized SARs for these additional compounds further. The most noteworthy observation was that as the aliphatic tail was increased in length on n7OFF, including the incorporation of branched chains, more potent AgrC antagonists were generated. For example, pentyl-n7OFF has an IC50 of ~16 nM in each of groups-I−III, which represents an 11- to 36-fold increase in potency relative to the parent n7OFF. Tails longer than five carbons were not evaluated as the solubility in water became problematic. The 4-fluoro derivative n7OF(4fF) was found to display potency against groups-I−III also (Table 1), albeit more modestly than the tail modified compounds. We found that introduction of a nitro group at any position of the endocyclic Phe rings yielded no potent antagonists (i.e., no IC50 < 3 μM), which is perhaps understandable if the aromatic rings bind in a hydrophobic binding site on AgrC. In addition, substitution of a methoxy group on the endocyclic Phe rings was largely not tolerated by AgrC, except for the 4-OMe Phe4 derivative that was a modest antagonist of AgrC in group-II (IC50 = 256 nM). Additional SAR trends were more subtle and are detailed in the SI (Tables S2−S3).
Phase-two SAR analyses.
The most interesting SARs identified in the phase-one analyses were combined to generate a set of second-round n7OFF analogs, and the effects on potency were largely found to be additive. Key compounds are highlighted in Table 2. First, the Cha and 3fF residues were combined to give n7OCha(3fF), which was surprising in that it was less potent than its parents n7OChaF and n7OF(3fF) in group-II, but otherwise was a more potent antagonist in groups-I, -III, and -IV than n7OChaF and more potent than n7OF(3fF) in groups-III−IV. As hydrophobicity appears to be important for AIP-AgrC interactions,31, 38 a more hydrophobic analog of n7OCha(3fF) was prepared with a saturated carbon chain in place of the n7O linkage (Figure 3), to give n7Cha(3fF). Except in group-III, this analog was less potent in each agr group, so this linker modification was not considered further. Cpa was next incorporated alongside 3fF to give n7OCpa(3fF), which yielded the most potent compound described so far. Relative to n7OFF, n7OCpa(3fF) was four-fold more potent in group-I, two-fold more potent in group-II, 60-fold more potent in group-III, and 30-fold more potent in group-IV. n7OCpa(3fF) also displays high efficacy, reducing AgrC activity by ~95% in all four groups, marking it as a promising new scaffold for further study.
Table 2.
IC50 values (in nM) for AgrC antagonism by selected phase-two n7OFF analogs in S. aureus groups-I−IVa. 95% confidence intervals (CI) in parentheses.
| compound | group-I | group-II | group-III | group-IV |
|---|---|---|---|---|
| n7OFF | 181 (154–213) | 583 (468–725) | 332 (280–395) | 5938 (4221–8532) |
| n7OCha(3fF) | 88.3 (70.9–110) | 1240 (830–1840) | 28.9 (22.5–37.2) | 432 (371–503) |
| n7Cha(3fF) | 334 (264–421) | 1850 (950–3640) | 12.1 (9.8–15.0) | 960 (780–1182) |
| n7OCpa(3fF) | 48.0 (41.1–56.2) | 207 (110–388) | 5.33 (3.84–7.39) | 190 (149–242) |
| pentyl-n7OCpa(3fF) | 2.56 (2.17–3.02) | 14.1 (11.0–18.0) | 0.148 (0.119–0.186) | 30.4 (15.7–58.9) |
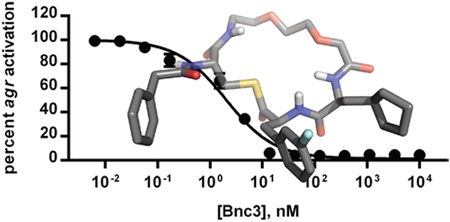
| Bn-n7OCpa(3fF) [Bnc3] | 2.01 (1.57–2.57) | 17.9 (12.9–24.9) | 0.0918 (0.0788–0.107) | 4.75 (2.98–7.59) |
| Bn-n7OCpa(3ClF) | 4.88 (3.16–7.52) | 6.47 (4.88–8.59) | 0.387 (0.325–0.461) | 3.87 (2.37–6.30) |
| Bn-n7OCpa(3fF)-amide | 81.3 (62.5–106) | 362 (137–952) | 0.634 (0.509–0.789) | 142 (79–255) |
| Bn-n7OCpa(3fF)-ester | 401 (312–516) | 870 (621–1220) | 2.08 (1.69–2.56) | 463 (298–720) |
Data for parent n7OFF included for comparison. See Methods section for details of strains and assays. Comprehensive assay data for all compounds tested are provided in Tables S2−S3.
We next examined the introduction of various tails into n7OCpa(3fF), and found the pentyl-n7OCpa(3fF) and Bn-n7OCpa(3fF) (hereafter named Bnc3) derivatives to display markedly improved potencies (18−53 fold improvements; Table 2). Notably, Bnc3 was an exceptionally potent AgrC antagonist, with IC50 values in the low- to sub-nanomolar range across the four agr groups. This activity profile was comparable to previously reported lead peptide-based AgrC inhibitors, such as AIP-I D5A and AIP-III D4A.31, 44 Critically and in support of the goals of the current study, Bnc3 was approximately 90-, 32-, 3320-, and 1250-fold more potent than n7OFF and 50-, 5-, 100-, and 30-fold more potent than t-AIP-II in groups-I−IV (Table S2),37 respectively, underscoring the value of our SAR-driven analog design. Another promising combination compound, Bn-n7OCpa(3ClF), had a chlorine in place of the fluorine of Bnc3, and was a single-digit nanomolar antagonist in each of groups-I, -II, and -IV, and a sub-nanomolar antagonist in group-III.
Examining the thioester in Bnc3 was our next target. To investigate the utility of an olefin replacement for the thioester in the Bnc3 AIP scaffold, we first examined an olefin in the known antagonists, t-AIP-II and t-AIP-III-D2A (Figure 4). We found that the olefin was an unsuitable replacement for the thioester, as these analogs were only potent antagonists in group-III (Tables S2−S3). We thus did not pursue olefin replacements in n7OFF analogs in this study. To determine if an amide linkage could be tolerated, diaminopropionic acid was incorporated in place of the thioester-forming Cys to make Bn-n7OCpa(3fF)-amide (Table 2). This compound was relatively less potent than Bnc3 in each agr group, but still a strong AgrC antagonist with potency similar to or better than t-AIP-II.33, 34, 37, 38 Further, as evidenced by its retention time from analytical HPLC (see Table S1) and previous amide versus thioester stability studies,45 Bn-n7OCpa(3fF)-amide possesses improved solubility and hydrolytic stability in aqueous media that make it a valuable new probe compound moving forward. Lastly, to further explore isosteres of the thioester, an ester linkage was incorporated to yield Bn-n7OCpa(3fF)-ester. This compound was also significantly less potent than Bnc3 in all four groups. However, in group-IV, the ester analog reduced agr activity by ~90% at the highest concentrations tested, compared to the ~70% reduction obtained by Bnc3 and other Bn-n7OFF analogs.
Summarizing the results of these two phases of n7OFF analog analysis, we found that the incorporation of a Bn tail, Cpa at position 3, 3fF or 3ClF at position 4, and an amide linkage into n7OFF generated new AgrC antagonists that should advance the design of small-molecule ligands. Bnc3, Bn-n7OCpa(3ClF), and Bn-n7OCpa(3fF)-amide represent the lead compounds from this SAR study. We note that, while highly potent, these compounds display lower efficacy in group-IV (~70% inhibition) relative to other analogs in this study. However, as Bnc3, Bn-n7OCpa(3ClF), and Bn-n7OCpa(3fF)-amide still reduce agr activity by ~95% in the other more common agr groups-I−III, this activity profile is not detrimental to their utility as probes or lead scaffolds moving forward. In addition, several analogs were identified that selectively inhibit agr in one group with minimal activity in the other groups (see Tables S2−S3); such selectivity profiles could prove useful to explore the role of these different groups in S. aureus infections.46
NMR solution structure of Bnc3.
In our past studies of non-native peptides capable of modulating AgrC, we have relied on NMR methods to characterize their solution-phase structures and develop mechanistic rationales for the modes by which these peptides could interact with AgrC receptors.37, 43, 47 As many macrocyclic peptides are found to be conformationally rigidified in aqueous media, and we could connect trends in cell-based reporter activity to specific structural features in our peptides as revealed by NMR, we reasoned that these NMR structures could be suggestive of their biologically active structures. We therefore applied these NMR techniques to Bnc3 in 1:1 CD3CN:H2O and characterized this mimetic as in our previous studies to gain insights into the structural features that may engender its high potency.
Using an all-atom alignment, the average RMSD for the NMR solution structure ensemble of Bnc3 was 0.87 Å, and using a heavy-atom alignment, the average RMSD for the ensemble was 0.68 Å (see Figure S8 for complete ensemble). The RMSD value is smaller than that previously observed for n7OFF,37 suggesting that Bnc3 is more rigid than n7OFF. We suspect that the Bn tail constrains the amide position and corresponding sidechain of Cys1, reducing the conformational freedom of Bnc3 relative to n7OFF.37 A representative 3D NMR structure that most closely matches the ensemble “average” structure is shown in Figure 5B, alongside its 2D chemical structure in Figure 5A.
Figure 5.

2D structure and 3D NMR structure of Bnc3. A: Chemical structure of Bnc3, with tail and amino acid residues identified. B: Representative NMR structure of Bnc3, with the Connolly surface shown. Sulfur is gold, oxygen is red, nitrogen is blue, fluorine is cyan, amide hydrogens are white, and carbon is grey.
In the NMR structure of Bnc3, we clearly see its amphipathic nature, with one face comprised of lipophilic side chains and the opposing face comprised of numerous heteroatoms. As the ligand binding site of AgrC has been proposed to be a hydrophobic cleft in the extracellular transmembrane segments,33, 34, 48 it is logical to envisage such an amphipathic compound to engage with AgrC via its lipophilic face. This ampipathicity for Bnc3 is very similar to that observed in NMR structures of its parent compound, n7OFF, and other peptide-derived AIP mimetics.37, 43, 47 Indeed, the relative orientation of the side chains was found to coincide with patterns previously observed in our NMR structures of other AIP mimetics;37, 43, 47 for example, the two endocyclic hydrophobic residues of Bnc3 overlay with the two endocyclic hydrophobic residues of t-AIP-II and n7OFF (Figure 6A and 6B), and the orientation of the Cys residues of Bnc3 and t-AIP-II are also similar (Figure 6A). Additionally, a comparison of the hydrophilic Ser residues of t-AIP-II and the hydrophilic n7O linker of Bnc3 shows that they occupy similar spaces (Figure 6A). These structural trends support the hypothesis that one face of Bnc3 interacts with the proposed hydrophobic cleft of the receptor site, while the other face participates in favorable solvent interactions, thus stabilizing the ligand-AgrC complex. It is plausible that if an AgrC ligand does not have such favorable solvent interactions, it would likely pay an entropic penalty for perturbing the hydrogen bonding network of the surrounding water and interact less strongly with the receptor. This may partially explain the observation that n7Cha(3fF), which lacks the same hydrophilic face as n7OCha(3fF) due to the absence of the n7O linker, is less potent than n7OCha(3fF) (Table 2). We note that the structure adopted in solution of course may not be the same as the structure adopted when the ligand binds to AgrC, but for these rigidified systems, it is very likely that the conformations would be similar.
Figure 6.

NMR structure overlays. A: Overlay of representative NMR structures for t-AIP-II and Bnc3, using the nine most similar atoms for alignment (0.86 Å RMS difference). B: Overlay of representative NMR structures for n7OFF and Bnc3, using the 34 most similar atoms for alignment (1.49 Å RMS difference). Sulfur is gold, oxygen is red, nitrogen is blue, fluorine is cyan, amide hydrogens are white, and carbon is magenta (t-AIP-II), grey (Bnc3, or cyan (n7OFF).
CONCLUSION
This study reports our progress toward the goal of developing simplified, small molecule AgrC antagonists through the analysis of a reported peptidomimetic scaffold (n7OFF) with moderate potency against AgrC. The design, synthesis, and biological testing of a series of close n7OFF analogs revealed SARs important to its activity: namely, amino acids with highly lipophilic side chains at the 3 and 4 positions (with sp3 and sp2 carbons at the respective γ-carbons), a bulky hydrophobic tail group, and an oxa-linker that creates an amphipathic nature are necessary elements for strong inhibitory potency against AgrC in all four groups of S. aureus (Figure 7). Critically, this work revealed new antagonists, such as Bnc3, which have low- to sub-nanomolar potencies against all four agr groups of S. aureus. We also discovered that an amide analog, Bn-n7OCpa(3fF)-amide, was only slightly less potent and should have utility as a probe compound with increased stability and solubility.
Figure 7.

The evolution of n7OFF to Bnc3 and key SAR features discovered in this study. The new AIP mimetic Bnc3 has a phenyl group added to the tail (in blue), 3-fluorophenylalanine adjacent to the thioester (in green), and cyclopentylalanine (in red) in place of Phe3 of n7OFF.
A solution NMR structure of Bnc3 revealed its amphipathic nature, with the relative orientation of the side chains overlapping with patterns previously seen in peptide AIP mimetics. This structural data illuminates the directions that functionality should protrude from the Bnc3 scaffold to effectively antagonize AgrC, and provides a 3D framework from which to begin to design non-peptide, small molecule modulators of AgrC. At a minimum, we reason that a thioester or suitable isostere and a hydrophilic ring structure need to be maintained in the structure, with lipophilic functionality present on the opposite side. The design and synthesis of compounds with such features is currently in progress and will be reported in due course.
METHODS
Reagents and general methods.
Chemical reagents and solvents were purchased from commercial sources (Sigma-Aldrich and Chem-Impex International) and used without further purification, with the exception of dichloromethane, which was distilled and stored over activated 3 Å molecular sieves. Dawson Dbz AM and Wang solid-phase resins were purchased from Novabiochem/EMD Millipore. 18 MΩ water was prepared using a Sartorius Arium Pro water purifier. Brain-heart infusion (BHI) medium powder was purchased from Teknova.
Peptide synthesis.
Thioester-, ester-, and amide-containing peptidomimetics were prepared on solid-phase resin, purified using reverse-phase semi-preparative HPLC, and characterized using high-resolution mass spectrometry (HRMS) and analytical HPLC as reported previously.37 The olefin-containing peptidomimetics were prepared by solution-phase coupling of a linear peptide precursor prepared on solid support with a C-terminal amino olefin building block, followed by cyclization via ring-closing metathesis using Grubbs’ Generation-II catalyst and established procedures.41, 49 See SI for additional experimental details and characterization data.
Fluorescent reporter assay.
Compounds were evaluated for activity in AgrC using S. aureus YFP agr reporter strains generously provided by A. Horswill (strains AH1677, AH430, AH1747, and AH1872 for groups-I−IV, respectively, containing the pDB59 plasmid with YFP10B gene under control of the P3 promoter of agr).42 Overnight cultures were diluted 1 to 50 into fresh BHI medium and added to a 96-well microtiter plate containing the compound (1% DMSO), as a single-concentration or serial dilution series, with DMSO and growth medium controls. The plates were incubated for 24 h with shaking at 200 rpm and 37 °C. OD600 and YFP fluorescence (excitation 500 nm, emission 540 nm) were read with a BioTek Synergy 2 plate reader. Compounds were screened at a single concentration and, if deemed interesting, over a 1 to 3 serial dilution with at least 3 biological replicates. Each biological replicate was the average of 3 or 4 technical replicates and prepared from a fresh overnight culture. Raw fluorescence measurements were processed by subtracting the background (BHI medium), correcting for OD600, and normalizing to the DMSO control to obtain percent agr activity for each compound concentration. A four-parameter sigmoidal dose response curve was fitted using GraphPad Prism 6, with the top of the curve set to 100% and the bottom of the curve constrained to remain above 0%, and IC50 values with the 95% confidence intervals and Hill slopes (h) were calculated (see Tables S2 and S3, respectively).
Solution NMR structural studies.
Bnc3 was dissolved to a concentration of 2 mM in 1:1 CD3CN:H2O at pH 5 and characterized by NMR to assign chemical shifts and obtain interproton distances using ROESY and NOESY spectra, with scripts written for NIH Xplor (v 2.35) to determine a solution NMR structure as previously described (see SI for additional details).37, 43, 47 Structural images and statistics were generated using PyMOL v4.3.0, with the representative structure shown in Figure 5B having the most similarity to the average structure and lowest relative energy selected from the ensemble.50
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by the Office of Naval Research (N00014-16-1-2185) and the NSF (CHE-1708714). J.K.V. was supported in part by the UW−Madison NIH Chemistry−Biology Interface Training Program (T32GM008505), the UW−Madison Office of the Vice Chancellor for Research and Graduate Education (with funding from the Wisconsin Alumni Research Foundation), and an NSF Graduate Research Fellowship (DGE-1747503). MS instrumentation in the UW−Madison Department of Chemistry was supported by the NIH (1S10 OD020022-1) and a gift from Paul Bender. NMR instrumentation in the UW−Madison Department of Chemistry was supported by the NIH (S10 OD012245). This study made use of the National Magnetic Resonance Facility at Madison (NMRFAM), which is supported by the NIH (P41 GM103399 and P41 GM66326). Additional equipment for NMRFAM was purchased with funds from the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), the USDA, and UW–Madison. We thank A. Horswill (CU Denver) for providing the S. aureus reporter strains.
Footnotes
Supporting Information: Additional experimental details, MS spectral data and HPLC traces, biological assay data, Xplor information, NMR spectra, and structural data.
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- [1].Tong SY, Davis JS, Eichenberger E, Holland TL, and Fowler VG Jr. (2015) Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management, Clin. Microbiol. Rev 28, 603–661 DOI 10.1128/CMR.00134-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Centers for Disease Control and Prevention. (2013) Antibiotic Resistance Threats in the United States
- [3].Thoendel M, Kavanaugh JS, Flack CE, and Horswill AR (2011) Peptide Signaling in the Staphylococci, Chem. Rev 111, 117–151 DOI 10.1021/cr100370n [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Novick RP, and Geisinger E (2008) Quorum Sensing in Staphylococci, Annu. Rev. Genet 42, 541–564 DOI 10.1146/annurev.genet.42.110807.091640 [DOI] [PubMed] [Google Scholar]
- [5].Tan L, Li SR, Jiang B, Hu XM, and Li S (2018) Therapeutic Targeting of the Staphylococcus aureus Accessory Gene Regulator (agr) System, Front. Microbiol 9, 55 DOI 10.3389/fmicb.2018.00055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rutherford ST, and Bassler BL (2012) Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control, Cold Spring Harbor Perspect. Med 2:a012427 DOI 10.1101/cshperspect.a012427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kong K-F, Vuong C, and Otto M (2006) Staphylococcus quorum sensing in biofilm formation and infection, Int. J. Med. Microbiol 296, 133–139 DOI 10.1016/j.ijmm.2006.01.042 [DOI] [PubMed] [Google Scholar]
- [8].Novick RP, Projan SJ, Kornblum J, Ross HF, Ji G, Kreiswirth B, Vandenesch F, and Moghazeh S (1995) The agr P2 operon—an autocatalytic sensory transduction system in Staphylococcus aureus, Mol. Gen. Genet 248, 446–458 DOI 10.1007/BF02191645 [DOI] [PubMed] [Google Scholar]
- [9].Otto M (2009) Staphylococcus epidermidis — the ‘accidental’ pathogen, Nat. Rev. Microbiol 7, 555 DOI 10.1038/nrmicro2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Thoendel M, and Horswill AR (2009) Identification of Staphylococcus aureus AgrD Residues Required for Autoinducing Peptide Biosynthesis, J. Biol. Chem 284, 21828–21838 DOI 10.1074/jbc.M109.031757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang B, Zhao A, Novick RP, and Muir TW (2015) Key driving forces in the biosynthesis of autoinducing peptides required for staphylococcal virulence, Proc. Natl. Acad. Sci. U. S. A 112, 10679–10684 DOI 10.1073/pnas.1506030112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kavanaugh JS, Thoendel M, and Horswill AR (2007) A role for type I signal peptidase in Staphylococcus aureus quorum sensing, Mol. Microbiol 65, 780–798 DOI 10.1111/j.1365-2958.2007.05830.x [DOI] [PubMed] [Google Scholar]
- [13].Wang B, Zhao A, Novick RP, and Muir TW (2014) Activation and inhibition of the receptor histidine kinase AgrC occurs through opposite helical transduction motions, Mol. Cell 53, 929–940 DOI 10.1016/j.molcel.2014.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang B, Zhao A, Xie Q, Olinares PD, Chait BT, Novick RP, and Muir TW (2017) Functional Plasticity of the AgrC Receptor Histidine Kinase Required for Staphylococcal Virulence, Cell Chem. Biol 24, 76–86 DOI 10.1016/j.chembiol.2016.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ji G, Beavis R, and Novick RP (1997) Bacterial Interference Caused by Autoinducing Peptide Variants, Science 276, 2027–2030 DOI 10.1126/science.276.5321.2027 [DOI] [PubMed] [Google Scholar]
- [16].Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick RP, and Muir TW (1999) Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence, Proc. Natl. Acad. Sci. U. S. A 96, 1218–1223 DOI 10.1073/pnas.96.4.1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jarraud S, Lyon GJ, Figueiredo AMS, Gérard L, Vandenesch F, Etienne J, Muir TW, and Novick RP (2000) Exfoliatin-Producing Strains Define a Fourth agr Specificity Group in Staphylococcus aureus, J. Bacteriol 182, 6517–6522 DOI 10.1128/JB.06355-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Le KY, and Otto M (2015) Quorum-sensing regulation in staphylococci-an overview, Front. Microbiol 6, 1174 DOI 10.3389/fmicb.2015.01174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dickey SW, Cheung GYC, and Otto M (2017) Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance, Nat. Rev. Drug Discov 16, 457–471 DOI 10.1038/nrd.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Salam AM, and Quave CL (2018) Targeting Virulence in Staphylococcus aureus by Chemical Inhibition of the Accessory Gene Regulator System In Vivo, mSphere 3 DOI 10.1128/mSphere.00500-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Todd DA, Parlet CP, Crosby HA, Malone CL, Heilmann KP, Horswill AR, and Cech NB (2017) Signal Biosynthesis Inhibition with Ambuic Acid as a Strategy To Target Antibiotic-Resistant Infections, Antimicrob. Agents Chemother 61, e00263–00217 DOI 10.1128/aac.00263-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, Sklar LA, Horswill AR, Hall PR, and Gresham HD (2014) Selective Chemical Inhibition of agr Quorum Sensing in Staphylococcus aureus Promotes Host Defense with Minimal Impact on Resistance, PLoS Path 10, e1004174 DOI 10.1371/journal.ppat.1004174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Murray EJ, Crowley RC, Truman A, Clarke SR, Cottam JA, Jadhav GP, Steele VR, O’Shea P, Lindholm C, Cockayne A, Chhabra SR, Chan WC, and Williams P (2014) Targeting Staphylococcus aureus quorum sensing with nonpeptidic small molecule inhibitors, J. Med. Chem 57, 2813–2819 DOI 10.1021/jm500215s [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nielsen A, Mansson M, Bojer MS, Gram L, Larsen TO, Novick RP, Frees D, Frokiaer H, and Ingmer H (2014) Solonamide B inhibits quorum sensing and reduces Staphylococcus aureus mediated killing of human neutrophils, PLoS One 9, e84992 DOI 10.1371/journal.pone.0084992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hall PR, Elmore BO, Spang CH, Alexander SM, Manifold-Wheeler BC, Castleman MJ, Daly SM, Peterson MM, Sully EK, Femling JK, Otto M, Horswill AR, Timmins GS, and Gresham HD (2013) Nox2 Modification of LDL Is Essential for Optimal Apolipoprotein B-mediated Control of agr type III Staphylococcus aureus Quorum-sensing, PLoS Path 9, e1003166 DOI 10.1371/journal.ppat.1003166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Paharik AE, Parlet CP, Chung N, Todd DA, Rodriguez EI, Van Dyke MJ, Cech NB, and Horswill AR (2017) Coagulase-Negative Staphylococcal Strain Prevents Staphylococcus aureus Colonization and Skin Infection by Blocking Quorum Sensing, Cell Host Microbe 22, 746–756e745 DOI 10.1016/j.chom.2017.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim MK, Zhao A, Wang A, Brown ZZ, Muir TW, Stone HA, and Bassler BL (2017) Surface-attached molecules control Staphylococcus aureus quorum sensing and biofilm development, Nat. Microbiol 2, 17080 DOI 10.1038/nmicrobiol.2017.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tsuchikama K, Shimamoto Y, and Anami Y (2017) Truncated Autoinducing Peptide Conjugates Selectively Recognize and Kill Staphylococcus aureus, ACS Infect. Dis 3, 406–410 DOI 10.1021/acsinfecdis.7b00013 [DOI] [PubMed] [Google Scholar]
- [29].Kratochvil MJ, Tal-Gan Y, Yang T, Blackwell HE, and Lynn DM (2015) Nanoporous Superhydrophobic Coatings that Promote the Extended Release of Water-Labile Quorum Sensing Inhibitors and Enable Long-Term Modulation of Quorum Sensing in Staphylococcus aureus, ACS Biomater. Sci. Eng 1, 1039–1049 DOI 10.1021/acsbiomaterials.5b00313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Melamed Yerushalmi S., Buck ME, Lynn DM, Lemcoff NG, and Meijler MM (2013) Multivalent alteration of quorum sensing in Staphylococcus aureus, Chem. Commun 49, 5177–5179 DOI 10.1039/C3CC41645C [DOI] [PubMed] [Google Scholar]
- [31].Tal-Gan Y, Stacy DM, Foegen MK, Koenig DW, and Blackwell HE (2013) Highly Potent Inhibitors of Quorum Sensing in Staphylococcus aureus Revealed Through a Systematic Synthetic Study of the Group-III Autoinducing Peptide, J. Am. Chem. Soc 135, 7869–7882 DOI 10.1021/ja3112115 [DOI] [PubMed] [Google Scholar]
- [32].Gless BH, Peng P, Pedersen KD, Gotfredsen CH, Ingmer H, and Olsen CA (2017) Structure–Activity Relationship Study Based on Autoinducing Peptide (AIP) from Dog Pathogen S. schleiferi, Org. Lett 19, 5276–5279 DOI 10.1021/acs.orglett.7b02550 [DOI] [PubMed] [Google Scholar]
- [33].Lyon GJ, Wright JS, Christopoulos A, Novick RP, and Muir TW (2002) Reversible and Specific Extracellular Antagonism of Receptor-Histidine Kinase Signaling, J. Biol. Chem 277, 6247–6253 DOI 10.1074/jbc.M109989200 [DOI] [PubMed] [Google Scholar]
- [34].Lyon GJ, Wright JS, Muir TW, and Novick RP (2002) Key Determinants of Receptor Activation in the agr Autoinducing Peptides of Staphylococcus aureus, Biochemistry 41, 10095–10104 DOI 10.1021/bi026049u [DOI] [PubMed] [Google Scholar]
- [35].McDowell P, Affas Z, Reynolds C, Holden MT, Wood SJ, Saint S, Cockayne A, Hill PJ, Dodd CE, Bycroft BW, Chan WC, and Williams P (2001) Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus, Mol. Microbiol 41, 503–512 DOI 10.1046/j.1365-2958.2001.02539.x [DOI] [PubMed] [Google Scholar]
- [36].Lyon GJ, Mayville P, Muir TW, and Novick RP (2000) Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase, AgrC, Proc. Natl. Acad. Sci. U. S. A 97, 13330–13335 DOI 10.1073/pnas.97.24.13330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Vasquez JK, Tal-Gan Y, Cornilescu G, Tyler KA, and Blackwell HE (2017) Simplified AIP-II Peptidomimetics Are Potent Inhibitors of Staphylococcus aureus AgrC Quorum Sensing Receptors, ChemBioChem 18, 413–423 DOI 10.1002/cbic.201600516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].George EA, Novick RP, and Muir TW (2008) Cyclic Peptide Inhibitors of Staphylococcal Virulence Prepared by Fmoc-Based Thiolactone Peptide Synthesis, J. Am. Chem. Soc 130, 4914–4924 DOI 10.1021/ja711126e [DOI] [PubMed] [Google Scholar]
- [39].Johnson JG, Wang B, Debelouchina GT, Novick RP, and Muir TW (2015) Increasing AIP Macrocycle Size Reveals Key Features of agr Activation in Staphylococcus aureus, ChemBioChem 16, 1093–1100 DOI 10.1002/cbic.201500006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].McBrayer DN, Cameron CD, Gantman BK, and Tal-Gan Y (2018) Rational Design of Potent Activators and Inhibitors of the Enterococcus faecalis Fsr Quorum Sensing Circuit, ACS Chem. Biol 13, 2673–2681 DOI 10.1021/acschembio.8b00610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Blackwell HE, Sadowsky JD, Howard RJ, Sampson JN, Chao JA, Steinmetz WE, O’Leary DJ, and Grubbs RH (2001) Ring-Closing Metathesis of Olefinic Peptides: Design, Synthesis, and Structural Characterization of Macrocyclic Helical Peptides, J. Org. Chem 66, 5291–5302 DOI 10.1021/jo015533k [DOI] [PubMed] [Google Scholar]
- [42].Malone CL, Boles BR, Lauderdale KJ, Thoendel M, Kavanaugh JS, and Horswill AR (2009) Fluorescent reporters for Staphylococcus aureus, J. Microbiol. Methods 77, 251–260 DOI 10.1016/j.mimet.2009.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tal-Gan Y, Ivancic M, Cornilescu G, Cornilescu CC, and Blackwell HE (2013) Structural Characterization of Native Autoinducing Peptides and Abiotic Analogues Reveals Key Features Essential for Activation and Inhibition of an AgrC Quorum Sensing Receptor in Staphylococcus aureus, J. Am. Chem. Soc 135, 18436–18444 DOI 10.1021/ja407533e [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chan WC, Coyle BJ, and Williams P (2004) Virulence Regulation and Quorum Sensing in Staphylococcal Infections: Competitive AgrC Antagonists as Quorum Sensing Inhibitors, J. Med. Chem 47, 4633–4641 DOI 10.1021/jm0400754 [DOI] [PubMed] [Google Scholar]
- [45].Tal-Gan Y, Ivancic M, Cornilescu G, Yang T, and Blackwell HE (2016) Highly Stable, Amide-Bridged Autoinducing Peptide Analogues that Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus, Angew. Chem. Int. Ed 55, 8913–8917 DOI 10.1002/anie.20160297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tal-Gan Y, Stacy DM, and Blackwell HE (2014) N-Methyl and peptoid scans of an autoinducing peptide reveal new structural features required for inhibition and activation of AgrC quorum sensing receptors in Staphylococcus aureus, Chem. Commun 50, 3000–3003 DOI 10.1039/C4CC00117F [DOI] [PubMed] [Google Scholar]
- [47].Tal-Gan Y, Ivancic M, Cornilescu G, and Blackwell HE (2016) Characterization of structural elements in native autoinducing peptides and non-native analogues that permit the differential modulation of AgrC-type quorum sensing receptors in Staphylococcus aureus, Org. Biomol. Chem 14, 113–121 DOI 10.1039/C5OB01735A [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wright JS, Lyon GJ, George EA, Muir TW, and Novick RP (2004) Hydrophobic interactions drive ligand-receptor recognition for activation and inhibition of staphylococcal quorum sensing, Proc. Natl. Acad. Sci. U. S. A 101, 16168–16173 DOI 10.1073/pnas.0404039101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Das P, and McNulty J (2010) Dichotomous Reactivity in the Reaction of Triethyl- and Triphenylphosphane HBr Salts with Dimethyl Acetals: A Novel Entry to α-Alkoxy-Functionalized Ylides and General Synthesis of Vinyl Ethers and Alkoxy Dienes, Eur. J. Org. Chem 2010, 3587–3591 DOI 10.1002/ejoc.201000601 [DOI] [Google Scholar]
- [50].(2015), The PyMOL Molecular Graphics System, Version 1.7, Schrödinger, LLC
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.