

Abstract
BACKGROUND AND PURPOSE:
Leptomeningeal inflammation is associated with the development of global cortical gray matter atrophy in multiple sclerosis. However, its association with localized loss of tissue remains unclear. The purpose of this study was to evaluate the relationship between leptomeningeal contrast enhancement, a putative marker of leptomeningeal inflammation, and focal cortical thinning in MS.
MATERIALS AND METHODS:
Forty-three patients with relapsing-remitting MS and 15 with secondary-progressive MS were imaged on a 3T scanner. Cortical reconstruction was performed with FreeSurfer. Leptomeningeal contrast-enhancement foci were visually identified on 3D-FLAIR postcontrast images and confirmed using subtraction imaging. Leptomeningeal contrast-enhancement foci were mapped onto the cortex, and ROIs were obtained by dilating along the surface multiple times (n = 5, 10, 15, 20, 25, 30, 35, 40). Resulting ROIs were then mapped onto the homologous region of the contralateral hemisphere. Paired t tests compared the thickness of the cortex surrounding individual leptomeningeal contrast-enhancement foci and the corresponding contralateral region. Results were corrected for the false discovery rate.
RESULTS:
Differences between ipsilateral and contralateral ROIs progressively decreased with larger ROIs, but no significant effects were detected when considering the entire MS sample. In patients with relapsing-remitting MS only, significantly reduced cortical thickness was found for 5 dilations (−8.53%, corrected P = .04) and 10 dilations (−5.20%, corrected P = .044).
CONCLUSIONS:
Focal leptomeningeal contrast enhancement is associated with reduced thickness of the surrounding cortex in patients with relapsing-remitting MS, but not in those with secondary-progressive MS. Our results suggest that pathology associated with the presence of leptomeningeal contrast-enhancement foci has a stronger, localized effect on cortical tissue loss earlier in the disease.
Neurodegeneration and inflammation are the 2 main pathogenetic processes in multiple sclerosis, leading to irreversible tissue loss and disability accrual with time. However, the reciprocal causal role has not been fully defined.1,2 Recently, histopathologic studies have focused on the leptomeninges after the discovery of lymphoid-like structures3 associated with cortical damage in patients with secondary-progressive MS (SPMS). A correlation among leptomeningeal inflammation, subpial cortical lesions, and cortical demyelination/degeneration already in the early stages of the disease has also recently been described.4–8 However, these findings may not be representative of the disease in general. First, postmortem studies tend to involve patients older than the general MS population. Second, biopsied samples of brain tissue likely stem from patients who have experienced nontypical symptoms or a particularly aggressive disease course. Thus, in vivo studies are attractive because they provide the opportunity to investigate cortical pathology across the spectrum of MS.
Because the cell aggregates that form the meningeal follicles are generally very small, the direct in vivo detection of leptomeningeal pathology is outside the reach of clinical MR imaging acquisitions. However, histopathologic evidence suggests that they have an impact on the permeability of the surrounding blood-CSF barrier,9 which can be exploited by high-resolution T2WI FLAIR sequences acquired after the administration of gadolinium.10–14
Foci of leptomeningeal contrast enhancement (LMCE) have been reported in patients with relapsing-remitting MS (RRMS) and SPMS,11–15 which is thought to reflect leptomeningeal inflammation as seen histopathologically.11 Increasing evidence suggests that LMCE is linked to cortical gray matter tissue loss.12,14,15 Furthermore, LMCE is apparently independent of white matter tissue volume and white matter lesion load,11,12,14,15 supporting the notion that cortical gray matter and white matter pathology are at least partially independent of each other.3,16,17
It has been postulated that the leptomeninges represent a site of chronic inflammation, characterized by the release of soluble inflammatory factors, resulting in progressive damage to the surrounding cortex, ultimately leading to tissue atrophy.4 Thus, we hypothesized that cortical areas closer to LMCE foci would be characterized by a greater degree of atrophy and that the relationship would be stronger in patients with SPMS.
Materials and Methods
Study Design and Patient Selection
This cross-sectional study combined data that were either acquired as part of an ongoing prospective study investigating the role of cardiovascular, environmental, and genetic factors associated with MS14,18 or from patients seen as part of their yearly routine clinical examination. All data were collected between June 2016 to October 2017. Included patients had either an RRMS or SPMS disease course according to the revised 2010 McDonald criteria.19 The study data collection was approved by the institutional review board of the University at Buffalo. All patients who were part of the prospective study provided written, informed consent. Due to the retrospective nature of the routine clinical data, we were provided an exemption for obtaining informed consent by our institutional review board.
The inclusion criteria were the following: 1) patients with an RRMS or SPMS disease course; 2) 18–75 years of age; 3) images having been acquired on the same 3T MR imaging system with a standardized protocol that included a 3D-T1WI precontrast acquisition as well as pre- and postcontrast 3D-T2WI FLAIR sequences after a single dose of gadolinium injection; 4) 3D-T2WI FLAIR positive for LMCE on pre-/postcontrast subtraction imaging; and 5) physical/neurologic examination within 30 days from MR imaging. Exclusion criteria were the following: 1) the presence of relapse and steroid treatment within the 30 days preceding study entry; 2) pre-existing medical conditions known to be associated with brain pathology (cerebrovascular disease, positive history of substance abuse); or 3) pregnancy.
Of 342 screened subjects, 58 (17%) met the inclusion criteria for LMCE positivity on 3D FLAIR pre-/postcontrast subtraction imaging. Of the included patients in the current study, 43 patients had an RRMS disease course, while the other 15 were diagnosed with SPMS.
MR Imaging Acquisition
All scans were acquired on the same 3T Signa Excite HD 12.0 MR imaging scanner (GE Healthcare, Milwaukee, Wisconsin) with an 8-channel head and neck coil. A magnetization-prepared 3D T1-weighted image (TE/TI/TR = 2.8/900/5.9 ms, flip angle = 10°, 256 × 256 matrix, voxel size = 1 mm3 isotropic, duration = 9 minutes 18 seconds) and pre- and postcontrast 3D-T2WI FLAIR sequences were acquired before and 10 minutes after an intravenous bolus of 0.1 mmol/kg of Gd-DTPA (TR/TI/TE = 9000/2420/600 ms, acquisition matrix = 256 × 192, voxel size = 1.0 × 1.3 × 1.3 mm3, duration = 12 minutes 22 seconds each). The 3D-T2WI FLAIR sequence uses variable flip angles so that the effective TE of 600 ms for the sequence is equal to a TE of 110 ms for a conventional spin-echo sequence. In addition, pre- and postcontrast spin-echo 2D-T1 sequences were acquired before and 5 minutes after Gd-DTPA injection (TE/TR = 16/600 ms, matrix = 256 × 192, voxel size = 1.0 × 10 × 3.0 mm3 without a gap, duration = 4 minutes 34 seconds each).
MR Imaging Assessment
All assessments were performed blinded to patient demographic and clinical characteristics.
Lesion Volumetry
T2 and gadolinium lesion volumes were calculated using a reliable semiautomated edge-detection contouring/thresholding technique with JIM software (www.xinapse.com) on T2WI FLAIR and 2D-T1WI postcontrast acquisitions, respectively.
LMCE Assessment
LMCE foci were identified on the 3D-T2WI FLAIR postcontrast image with the aid of a subtraction image (Fig 1).13 LMCE foci were defined as signal intensity within the subarachnoid space that was substantially greater than that of brain parenchyma and characterized as being nodular, linear, or plate-like, as previously described.11 All LMCE foci were confirmed in consensus by 2 experienced neuroimagers, each with >15 years of experience.
Fig 1.

A representative case depicting the T2WI FLAIR precontrast, T2WI FLAIR postcontrast, and T2WI FLAIR pre-/postcontrast subtraction images. The FreeSurfer-derived white and pial surfaces are shown in yellow and red, respectively. The white arrow shows a focus of leptomeningeal contrast enhancement.
Cortical Measures
Cortical reconstruction was performed on the 3D-T1 images using the FreeSurfer package, Version 5.3 (http://www.freesurfer.net/).20,21 Before analysis, the input image was lesion-filled to improve the reliability of cortical thickness measures.22 The 3D-T2 FLAIR precontrast image was then registered to the FreeSurfer-conformed T1 image using the bbregister tool (https://surfer.nmr.mgh.harvard.edu/fswiki/bbregister).23 Individual LMCE foci were then automatically mapped onto the nearest vertex of the pial surface using the mri_label2label tool (https://surfer.nmr.mgh.harvard.edu/fswiki/mri_label2label) in FreeSurfer. Because the putative extent of cortical thinning was not known a priori, the resulting point was morphologically dilated along the surface several times (n = 5, 10, 15, 20, 25, 30, 35, 40) to define the ROI surrounding the LMCE foci. A representative LMCE focus and corresponding set of ROIs on the cortical surface are shown in Fig 2. Next, each ROI was mapped onto the homologous region in the contralateral hemisphere via nonlinear registration of the surfaces to one another using the mris_left_right_register tool (https://surfer.nmr.mgh.harvard.edu/fswiki/) in FreeSurfer. Most important, the registration was performed separately for left to right and right to left rather than using an inverse transformation, to avoid bias in the contralateral mapping procedure. Quality control was performed at each step of the processing (eg, brain extraction, white and pial surface generation, registration), and manual corrections were made as required.
Fig 2.
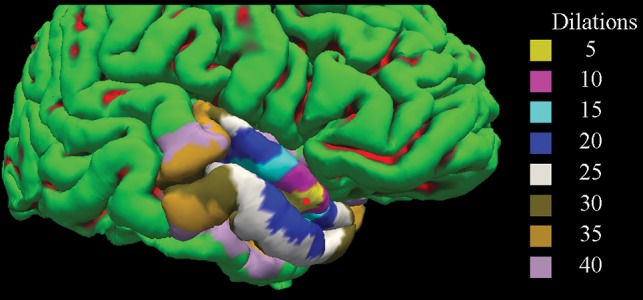
A representative cortical reconstruction from FreeSurfer is shown with dilated ROIs overlaid on the cortical surface. Gyri are shown in green, while sulci are shown in red. The red circle corresponds to the position of the mapped focus of leptomeningeal contrast enhancement. Different-sized ROIs are shown in varying colors.
To validate the primary results, we performed 2 additional analyses. First, we randomly selected 4 points on the cortical surface (2 for the left hemisphere and 2 for the right hemisphere) distant from any LMCE foci for each subject. These were then used as seeds for ROI generation. Second, we mapped the LMCE foci for a given subject to another randomly selected subject in the study. For both of these analyses, we then compared ipsilateral and contralateral thickness measures as we did in the primary analysis.
Statistical Analysis
Statistical analyses were performed using SPSS (Version 24; IBM, Armonk, New York). Differences between the RRMS and SPMS groups in terms of demographics, clinical characteristics, and lesion volumetry were analyzed using the Fisher exact, Student t, Mann-Whitney U, or χ2 test, as appropriate. Distributions of the data were tested for normality using the Shapiro-Wilk test and examination of histograms.
For each dilation, differences between ipsilateral and contralateral ROIs were examined using paired t tests. All tests assessed the entire cohort and patients with RRMS and SPMS separately. Exploratory analyses assessed whether there were any differences when comparing LMCE foci by their appearance (eg, nodular, linear, plate-like).
The Benjamini-Hochberg procedure was used to control the false discovery rate, with corrected P values (ie, Q values) < .05 considered significant.
Results
Demographic, Clinical, and MR Imaging Characteristics
Table 1 shows demographic, clinical, and MR imaging characteristics of the 2 subgroups. Patients with SPMS were older (P = .023), had a longer disease duration (P = .014), and were more disabled (P = .003). A total of 80 LMCE foci were identified.
Table 1:
Demographic, clinical, and MR imaging characteristics of the cohort
| RRMS (n = 43) | SPMS (n = 15) | P Valuea | |
|---|---|---|---|
| Women (No.) (%) | 38 (88.4) | 11 (73.3) | .658 |
| Age (mean) (SD) (yr) | 52.8 (12.1) | 60.5 (6.7) | .023b |
| Disease duration (mean) (SD) (yr) | 15.2 (8.9) | 21.9 (8.3) | .014b |
| EDSS (median) (IQR) | 3.0 (2.0–3.5) | 6.0 (3.6–6.5) | .003b |
| Age at onset (mean) (SD) (yr) | 37.6 (12.0) | 38.7 (8.5) | .428 |
| Disease-modifying therapy (No.) (%) | |||
| Interferon β-1a I.M. | 14 (32.6) | 2 (13.3) | .111 |
| Interferon β-1a S.C. | 3 (9.3) | 0 (0) | |
| Glatiramer acetate | 8 (18.6) | 2 (13.3) | |
| Natalizumab | 4 (9.3) | 1 (6.7) | |
| Dimethyl fumarate | 2 (4.7) | 0 (0) | |
| Fingolimod | 3 (7.0) | 0 (0) | |
| Teriflunomide | 1 (2.3) | 3 (20.0) | |
| Rituximab | 1 (2.3) | 0 (0) | |
| Ocrelizumab | 0 (0) | 1 (6.7) | |
| IVIG | 1 (2.3) | 1 (6.7) | |
| No therapy | 6 (14.0) | 5 (33.3) | |
| No. of LMCE foci | 1 (n = 33) | 1 (n = 8) | .292 |
| 2 (n = 8) | 2 (n = 4) | ||
| 3 (n = 2) | 3 (n = 3) | ||
| LMCE shape (No.) | .637 | ||
| Nodular | 36 | 16 | |
| Linear | 7 | 5 | |
| Plate-like | 12 | 4 | |
| T2 lesion volume (mean) (SD) (mL) | 16.0 (16.2) | 15.8 (11.4) | .965 |
| Gd lesion volume (median) (IQR) (mL) | 0 (0–0) | 0 (0–0) | .097 |
Note:—EDSS indicates Expanded Disability Status Scale; I.M., intramuscular; S.C., subcutaneous; IVIG, intravenous immunoglobulin; Gd, gadolinium.
P value represents differences between the patients with RRMS and SPMS. The differences between the groups were analyzed using the Fisher exact, Student t, Mann-Whitney U, or χ2 test.
P values < .05.
Cortical Thickness Comparison between the Area Surrounding LMCE Foci and the Contralateral Area
Table 2 shows the results comparing cortical thickness measurements in the region surrounding LMCE foci and in the contralateral region. For LMCE in the entire MS sample and in the RRMS cohort, the percentage differences between the 2 regions were greater for more focal ROIs (ie, fewer dilations), but this was not the case for patients with SPMS. When we considered the entire cohort, significant reductions in cortical thickness were detected for 10 and 15 dilations, though these results did not survive multiple-comparison correction. However, when we limited the analysis to only the LMCE foci in the patients with RRMS, significant differences were found for 5 dilations (−8.53%, corrected P = .04) and 10 dilations (−5.20%, corrected P = .044). No differences were found in the SPMS cohort. When we compared LMCE type, no significant differences were detected for the entire cohort or when assessing patients with RRMS and SPMS separately (results not shown).
Table 2:
Cortical thickness measurements in the ipsilateral region surrounding focal areas of leptomeningeal contrast enhancement and in the contralateral regiona
| Entire Cohort (80 LMCE Foci) |
Patients with RRMS (55 LMCE Foci) |
Patients with SPMS (25 LMCE Foci) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dilations | Ipsilateral | Contralateral | Pct. Diff. | P/FDR-P | Ipsilateral | Contralateral | Pct. Diff. | P/FDR-P | Ipsilateral | Contralateral | Pct. Diff. | P/FDR-P |
| 5 | 2.043 (.515) | 2.149 (.556) | −5.06 | .07/.187 | 2.031 (.534) | 2.212 (.567) | −8.53 | .005/.04b | 2.070 (.480) | 2.000 (.510) | 3.44 | .562/.823 |
| 10 | 2.063 (.437) | 2.160 (.476) | −4.59 | .016/.128 | 2.099 (.455) | 2.211 (.501) | −5.20 | .011/.044b | 1.979 (.387) | 2.039 (.396) | −2.99 | .493/.823 |
| 15 | 2.088 (.411) | 2.153 (.413) | −3.07 | .039/.156 | 2.141 (.414) | 2.208 (.432) | −3.08 | .055/.147 | 1.964 (.383) | 2.023 (.339) | −2.96 | .384/.823 |
| 20 | 2.114 (.286) | 2.136 (.385) | −1.04 | .409/.554 | 2.167 (.394) | 2.196 (.395) | −1.33 | .365/.417 | 1.989 (.342) | 2.000 (.325) | −.55 | .895/.895 |
| 25 | 2.116 (.362) | 2.131 (.368) | −.71 | .485/.554 | 2.167 (.376) | 2.194 (.377) | −1.24 | .306/.408 | 1.996 (.301) | 1.983 (.306) | 0.65 | .749/.856 |
| 30 | 2.113 (.348) | 2.131 (.354) | −.85 | .342/.554 | 2.162 (.363) | 2.195 (.361) | −1.51 | .137/.274 | 1.997 (.284) | 1.980 (.291) | 0.85 | .617/.823 |
| 35 | 2.115 (.338) | 2.127 (.342) | −.57 | .483/.554 | 2.165 (.353) | 2.190 (.347) | −1.15 | .191/.306 | 1.998 (.273) | 1.977 (.284) | 1.06 | .509/.823 |
| 40 | 2.117 (.327) | 2.120 (.332) | −.14 | .882/.882 | 2.168 (.341) | 2.180 (.336) | −.55 | .529/.529 | 1.998 (.261) | 1.979 (.282) | 0.95 | .485/.823 |
Note:—Pct. Diff. indicates percentage difference; FDR, false discovery rate.
Ipsilateral and contralateral columns represent mean (standard deviation) cortical thickness measures in millimeters.
Paired t tests comparing the ipsilateral and contralateral regions were used to calculate P values. The Benjamini-Hochberg procedure was used to control the false discovery rate.
Corrected P values < .05.
Validation Analyses
No significant results were found for either of the validation analyses, as expected. For the first analysis in which non-LMCE points were used for ROI generation, the minimum corrected P values were .878, .912, and .955 for the entire cohort, patients with RRMS, and those with SPMS, respectively. For the second analysis in which LMCE foci were mapped to another randomly selected subject, the minimum corrected P values were .782, .825, and .881 for the entire cohort, patients with RRMS, and those with SPMS, respectively.
Discussion
In this cross-sectional study, we demonstrated that LMCE foci are associated with localized cortical atrophy in RRMS, suggesting a direct link between leptomeningeal inflammation and focal thinning of the cortex in these patients. Moreover, we found that the relative decrease of cortical thickness in the area surrounding LMCE foci followed a clear gradient with larger regions progressively reflecting smaller differences. We also performed validation analyses to bolster the overall confidence in our findings. These results suggest that our findings are not driven by age-related atrophy or cortical asymmetries nor are they a consequence of imaging or postprocessing artifacts. However, contrary to our initial hypothesis, we did not find an apparent relationship between focal thinning and LMCE presence in the SPMS cohort.
Histopathologic studies have revealed strong associations between meningeal inflammation and cortical damage in the form of numerous features, including demyelination4,24 and loss of neurons, astrocytes, and oligodendrocytes.24 A more focal topographic association between leptomeningeal inflammation and cortical demyelination was reported compared with a relatively wider pattern of neuronal loss in the surrounding cortex.24 These findings support the notion that the meninges play a role in the development and maintenance of an inflammatory milieu.25 In this context, it is likely that cytotoxic cytokines, originating from ectopic lymphoid-like structures in the meninges,9 diffuse through the subarachnoid space and have a direct, causative role in the development of cortical pathology. Although the exact reason for the apparently greater effect on neuronal loss is not yet clear, it might be related to increased sensitivity of neurons to cytotoxic damage.24 Another possibility is that areas farther away from focal meningeal inflammation may have a better chance at successful remyelination due to the reduced frequency of direct contact with diffusing cytokines. Nevertheless, it is perhaps surprising that we were unable to find evidence of increased cortical thinning in the areas surrounding LMCE foci in the SPMS cohort.
The combination of other pathologic processes along with a less localized effect of long-lasting leptomeningeal inflammation might, in part, explain the apparent lack of an association in the advanced stage of MS. Another possibility is that the largest effect of leptomeningeal inflammation on localized neurodegeneration occurs during the early-to-midterm period once an LMCE focus develops. Subsequently, other pathologic features, such as retrograde and anterograde degeneration following axonal transection due to white matter lesions,26 may end up playing a greater role in the development of cortical atrophy. Diffusion tensor imaging studies have revealed a clear link between thinning of the cortex and damage in the so-called normal-appearing white matter of connected fiber tracts.27,28 Tract-specific measures of white matter bundles connected to cortical areas surrounding the LMCE foci may be useful to explore this possibility. Finally, it has been reported that even healthy controls may present with nodular LMCE foci; thus, this subtype might not be MS-specific.12 We did not find, however, any cortical thickness differences when comparing the type of LMCE focus.
An alternative explanation for the lack of significant differences in the SPMS cohort is that the overall degree of leptomeningeal inflammation is greater in these patients compared with those with an RRMS disease course. In such a scenario, one could hypothesize that the contralateral side is affected to a similar degree; thus, no differences in cortical thickness are observed. While the detection of leptomeningeal inflammation, as evidenced by LMCE, may be poorer with 3T MR imaging compared with 7T,12,29 such an explanation seems unlikely. A recent study found that global cortical atrophy was numerically greater, but not statistically different, during 5 years of follow-up in patients with SPMS with LMCE foci compared with those without them.14 In line with the current findings, that same study found a significantly greater rate of cortical atrophy in patients with RRMS with LMCE foci compared with those without them. One other possibility is that because the overall extent of gray matter loss in patients with SPMS is so extensive throughout the cortex, any focal effects surrounding individual LMCE foci are lost. Finally, LMCE foci might not reflect the exact same underlying pathology throughout the course of the disease, leading to differential effects on cortical atrophy between patients with RRMS and those with SPMS. Currently, this possibility remains purely speculative.
Our study has limitations. First, the data were retrospectively analyzed, and the study was cross-sectional. Although most LMCE foci appear to remain stable with time, at least during the midterm,11 it is unclear how quickly they effect the development of cortical thinning. The study sample was relatively small, especially for the SPMS group, which had only 15 patients. This drawback is somewhat offset, however, because the analysis was performed at the level of individual LMCE foci rather than subject-wise. In this regard, about 31% (25 of 80) of all foci were from patients with SPMS. Moreover, differences between ipsilateral/contralateral ROIs in patients with SPMS did not follow the clear progression that was found in patients with RRMS with respect to ROI size. Nevertheless, future studies are warranted with larger sample sizes. In addition, our study protocol did not include a double inversion recovery sequence to aid in the visualization of cortical lesions. Subpial lesions, which histopathologic data suggest are linked to leptomeningeal inflammation,4 are generally not visible even with double inversion recovery at 3T.30 However, a recent 7T study found a limited association between individual LMCE foci and the presence of cortical lesions, suggesting that the role of leptomeningeal inflammation on the development of cortical lesions might actually be weak.31
Conclusions
Our in vivo MR imaging study supports previous histopathologic findings, suggesting that leptomeningeal inflammation, as evidenced by LMCE, is anatomically related to focal cortical thinning in patients with RRMS. These findings, especially the apparent lack of an association in the SPMS phenotype, should be confirmed in prospective studies with larger sample sizes.
ABBREVIATIONS:
- LMCE
leptomeningeal contrast enhancement
- RRMS
relapsing-remitting MS
- SPMS
secondary-progressive MS
Footnotes
Disclosures: Bianca Weinstock-Guttman—UNRELATED: Board Membership: Biogen, TEVA Neuroscience, Novartis, Celgene, EMD Serono, Genentech; Consultancy: Biogen, TEVA Neuroscience, Novartis, Celgene, EMD Serono, Genentech; Grants/Grants Pending: Biogen, TEVA Neuroscience, EMD Serono; Payment for Lectures Including Service on Speakers Bureaus: Biogen, TEVA, Novartis, Genentech. Robert Zivadinov—UNRELATED: Consultancy: Novartis, Sanofi, Celgene, EMD Serono*; Grants/Grants Pending: Novartis, Sanofi, Protembo*; Payment for Lectures Including Service on Speakers Bureaus: Novartis, Sanofi, Celgene, EMD Serono.* David Hojnacki—UNRELATED: Consultancy: Biogen, Genentech, EMD Serono, Novartis; Payment for Lectures Including Service on Speakers Bureaus: Biogen, Genentech, EMD Serono, Novartis. *Money paid to the institution.
Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under award No. UL1TR001412.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015;14:183–93 10.1016/S1474-4422(14)70256-X [DOI] [PubMed] [Google Scholar]
- 2. Herranz E, Gianni C, Louapre C, et al. . Neuroinflammatory component of gray matter pathology in multiple sclerosis. Ann Neurol 2016;80:776–90 10.1002/ana.24791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Magliozzi R, Howell O, Vora A, et al. . Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007;130:1089–104 [DOI] [PubMed] [Google Scholar]
- 4. Lucchinetti CF, Popescu BF, Bunyan RF, et al. . Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 2011;365:2188–97 10.1056/NEJMoa1100648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haider L, Zrzavy T, Hametner S, et al. . The topography of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016;139:807–15 10.1093/brain/awv398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Choi SR, Howell OW, Carassiti D, et al. . Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012;135:2925–37 10.1093/brain/aws189 [DOI] [PubMed] [Google Scholar]
- 7. Howell OW, Reeves CA, Nicholas R, et al. . Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011;134:2755–71 10.1093/brain/awr182 [DOI] [PubMed] [Google Scholar]
- 8. Popescu BF, Lucchinetti CF. Meningeal and cortical grey matter pathology in multiple sclerosis. BMC Neurol 2012;12:11 10.1186/1471-2377-12-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Serafini B, Rosicarelli B, Magliozzi R, et al. . Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 2004;14:164–74 10.1111/j.1750-3639.2004.tb00049.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zurawski J, Lassmann H, Bakshi R. Use of magnetic resonance imaging to visualize leptomeningeal inflammation in patients with multiple sclerosis: a review. JAMA Neurol 2017;74:100–09 10.1001/jamaneurol.2016.4237 [DOI] [PubMed] [Google Scholar]
- 11. Absinta M, Vuolo L, Rao A, et al. . Gadolinium-based MRI characterization of leptomeningeal inflammation in multiple sclerosis. Neurology 2015;85:18–28 10.1212/WNL.0000000000001587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harrison DM, Wang KY, Fiol J, et al. . Leptomeningeal enhancement at 7T in multiple sclerosis: frequency, morphology, and relationship to cortical volume. J Neuroimaging 2017;27:461–68 10.1111/jon.12444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zivadinov R, Ramasamy DP, Hagemeier J, et al. . Evaluation of leptomeningeal contrast enhancement using pre-and postcontrast subtraction 3D-FLAIR imaging in multiple sclerosis. AJNR Am J Neuroradiol 2018;39:642–47 10.3174/ajnr.A5541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zivadinov R, Ramasamy DP, Vaneckova M, et al. . Leptomeningeal contrast enhancement is associated with progression of cortical atrophy in MS: a retrospective, pilot, observational longitudinal study. Mult Scler 2017;23:1336–45 10.1177/1352458516678083 [DOI] [PubMed] [Google Scholar]
- 15. Makshakov G, Magonov E, Totolyan N, et al. . Leptomeningeal contrast enhancement is associated with disability progression and grey matter atrophy in multiple sclerosis. Neurol Res Int 2017;2017:8652463 10.1155/2017/8652463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bö L, Geurts JG, van der Valk P, et al. . Lack of correlation between cortical demyelination and white matter pathologic changes in multiple sclerosis. Arch Neurol 2007;64:76–80 10.1001/archneur.64.1.76 [DOI] [PubMed] [Google Scholar]
- 17. Calabrese M, Seppi D, Romualdi C, et al. . Gray matter pathology in MS: a 3-year longitudinal study in a pediatric population. AJNR Am J Neuroradiol 2012;33:1507–11 10.3174/ajnr.A3011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kappus N, Weinstock-Guttman B, Hagemeier J, et al. . Cardiovascular risk factors are associated with increased lesion burden and brain atrophy in multiple sclerosis. J Neurol Neurosurg Psychiatry 2016;87:181–87 10.1136/jnnp-2014-310051 [DOI] [PubMed] [Google Scholar]
- 19. Polman CH, Reingold SC, Banwell B, et al. . Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302 10.1002/ana.22366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis, I: segmentation and surface reconstruction. Neuroimage 1999;9:179–94 10.1006/nimg.1998.0395 [DOI] [PubMed] [Google Scholar]
- 21. Fischl B, Salat DH, Busa E, et al. . Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002;33:341–55 10.1016/S0896-6273(02)00569-X [DOI] [PubMed] [Google Scholar]
- 22. Magon S, Gaetano L, Chakravarty MM, et al. . White matter lesion filling improves the accuracy of cortical thickness measurements in multiple sclerosis patients: a longitudinal study. BMC Neurosci 2014;15:106 10.1186/1471-2202-15-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Greve DN, Fischl B. Accurate and robust brain image alignment using boundary-based registration. Neuroimage 2009;48:63–72 10.1016/j.neuroimage.2009.06.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Magliozzi R, Howell OW, Reeves C, et al. . A gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010;68:477–93 10.1002/ana.22230 [DOI] [PubMed] [Google Scholar]
- 25. Schläger C, Körner H, Krueger M, et al. . Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 2016;530:349–53 10.1038/nature16939 [DOI] [PubMed] [Google Scholar]
- 26. Sepulcre J, Goñi J, Masdeu JC, et al. . Contribution of white matter lesions to gray matter atrophy in multiple sclerosis: evidence from voxel-based analysis of T1 lesions in the visual pathway. Arch Neurol 2009;66:173–79 10.1001/archneurol.2008.562 [DOI] [PubMed] [Google Scholar]
- 27. Bergsland N, Laganà MM, Tavazzi E, et al. . Corticospinal tract integrity is related to primary motor cortex thinning in relapsing-remitting multiple sclerosis. Mult Scler 2015;21:1771–80 10.1177/1352458515576985 [DOI] [PubMed] [Google Scholar]
- 28. Steenwijk MD, Daams M, Pouwels PJ, et al. . Unraveling the relationship between regional gray matter atrophy and pathology in connected white matter tracts in long-standing multiple sclerosis. Hum Brain Mapp 2015;36:1796–807 10.1002/hbm.22738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jonas SN, Izbudak I, Frazier AA, et al. . Longitudinal persistence of meningeal enhancement on postcontrast 7T 3D-FLAIR MRI in multiple sclerosis. AJNR Am J Neuroradiol 2018;39:1799–805 10.3174/ajnr.A5796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nielsen AS, Kinkel RP, Tinelli E, et al. . Focal cortical lesion detection in multiple sclerosis: 3 Tesla DIR versus 7 Tesla FLASH-T2. J Magn Reson Imaging 2012;35:537–42 10.1002/jmri.22847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harrison D, Jonas S, Izbudak I. Investigating the relationship between meningeal enhancement on 7T MRI and cortical gray matter lesions in multiple sclerosis. In: Proceedings of the Annual Congress of the European Committee for Treatment and Research in Multiple Sclerosis, Berlin, Germany October 10–12, 2018; 489 [Google Scholar]