

Abstract
Cardiorespiratory disease, which includes systemic arterial hypertension, restenosis, atherosclerosis, pulmonary arterial hypertension, asthma, and chronic obstructive pulmonary disease (COPD) are highly prevalent and devastating diseases with limited therapeutic modalities. A common pathophysiological theme to these diseases is cellular remodeling, which is contributed by changes in expression and activation of ion channels critical for either excitability or growth. Calcium (Ca2+) signaling and specifically ORAI Ca2+ channels have emerged as significant regulators of smooth muscle, endothelial, epithelial, platelet, and immune cell remodeling. This review details the dysregulation of ORAI in cardiorespiratory diseases, and how this dysregulation of ORAI contributes to cellular remodeling.
Keywords: ORAI, STIM, SOCE, CRAC, ARC, Remodeling, Hypertension, Atherosclerosis, Restenosis, Pulmonary Hypertension, Asthma, COPD
Graphical abstract
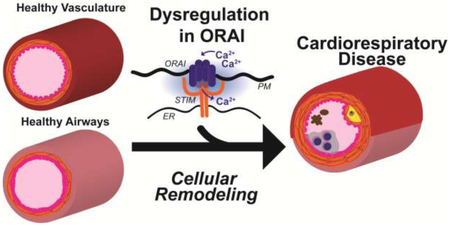
Introduction
Cardiovascular disease and chronic lower respiratory diseases are the first and fourth leading causes of death in the United States [1]. Over one in three Americans suffers from cardiovascular disease or chronic lower respiratory diseases (hereafter referred to as cardiorespiratory disease), and recent epidemiological evidence suggests that the prevalence of these diseases is on the rise [2-4]. These findings indicate that current therapeutic modalities are insufficient, and the need to identify novel molecular targets to treat these diseases.
A common theme in cardiorespiratory diseases is the molecular and cellular remodeling from a normal physiological phenotype to a dysfunctional diseased phenotype [5, 6]. The ubiquitous intracellular second messenger, calcium (Ca2+), is necessary for many cellular functions such as secretion, exocytosis, contraction, metabolism, and activation of transcriptional programs that support proliferation and migration [7]. Ca2+ signaling operates in highly specific spatiotemporal domains, and the molecular processes maintaining this specificity are exceedingly malleable and hence prone to dysfunction during cellular remodeling [8]. Dysfunction in Ca2+ signaling in cardiorespiratory disease has been previously described and is beyond the scope of this review (refer to reviews [9, 10]). One significant modulator of Ca2+ signaling that has recently emerged to be dysregulated in cardiorespiratory diseases is the ORAI family of Ca2+ channels. ORAI are highly Ca2+ selective channels located on the plasma membrane (PM) and are typically activated by the endoplasmic reticulum (ER) transmembrane proteins, Stromal Interaction Molecule (STIM), upon ER Ca2+ store depletion [11-13]. In addition, ORAI1/3 heteromeric channels have been described to be activated independently of ER Ca2+ store depletion by arachidonic acid and its metabolite, leukotriene C4 (LTC4) [14, 15]. Activation of ORAI prompts large temporal Ca2+ signals that stimulate transcription of proliferative and migratory genes[16]. Herein, we describe how dysregulation in ORAI channels contribute to cardiorespiratory disease and the potential of these channels as attractive targets for future disease therapy. We briefly summarize the mechanisms of activation of ORAI channels and discuss their role in systemic arterial hypertension, restenosis, atherosclerosis, pulmonary hypertension, asthma, and chronic obstructive pulmonary disease (COPD).
Brief Overview of ORAI Channels
Before the discovery of the molecular identity of ORAI channels, the original idea that Ca2+ influx across the plasma membrane is stimulated by the fall of Ca2+ concentration within ER lumen was first introduced by Jim Putney in 1986 and originally termed capacitative Ca2+ entry [17]. This nomenclature was subsequently abandoned in favor of stored-operated Ca2+ entry (SOCE), which implicitly indicate that activation of Ca2+ influx from the outside is controlled by the state of filling of ER Ca2+ stores. Physiologically, binding of agonists to receptors coupled to phospholipase C (PLC) isoforms causes the cleavage of the acidic lipid phosphatidylinositol 4,5-bisphosphate (PIP2) into the soluble head group inositol-1,4,5-trisphosphate (IP3) and the membrane-bound diacylglycerol (DAG). Diffusible IP3 stimulates ER Ca2+ release by activating the IP3 receptor [18]. The subsequent depletion of ER Ca2+ causes Ca2+ to dissociate from the low-affinity luminal EF-hand domains of STIM proteins (mammals have two STIM homologs, STIM1 and STIM2) [19-22] (Figure 1 and 2). This dissociation causes STIM to undergo a conformational switch, oligomerize and migrate towards the ER-PM junctions [23, 24]. At the ER-PM junctions, STIM exposes its C-terminal STIM-ORAI activating region (SOAR), which physically traps and activates ORAI channels [22,25-27] (Figure 2). Both STIM1 and STIM2 are capable of activating ORAI1; however, STIM2 is a weaker activator likely due to differences in the SOAR [28, 29] and sterile α-motif (SAM) domains [30]. Yet, the EF hand of STIM2 has a lower affinity for Ca2+, which allows STIM2 to activate ORAI1 at lower levels of agonist-induced stimulation and subsequent ER store depletion [31, 32]. In whole-cell-patch-clamp electrophysiological recordings, SOCE manifests as a highly Ca2+ selective inwardly-rectifying current termed Ca2+ release-activated current (ICRAC) [12]. Although very small in size (less than 1pA/pF), native ICRAC has been measured in a variety of cell types [33-37]. Numerous SOCE inhibitors have been used including lanthanides such as Lanthanum (La3+) and Gadolinium (Gd3+) when used at low concentrations (1-5 μM)[38], 2-Aminoethoxydiphenyl borate (2-APB), 3,5-bistrifluoromethyl pyrazole (BTP2), Synta66, and GSK-7975A [13]. However, these SOCE inhibitors have never reached the clinic mostly as a result of either their toxicity or poor specificity. They have been reported to block other Ca2+ channels including transient receptor potential (TRP) channels and IP3 receptors [39]. This highlights the need for more detailed studies focusing on ORAI channel structure and regulation, which would help in identifying specific inhibitors.
Figure 1. SOCE mediated by ORAI channels.
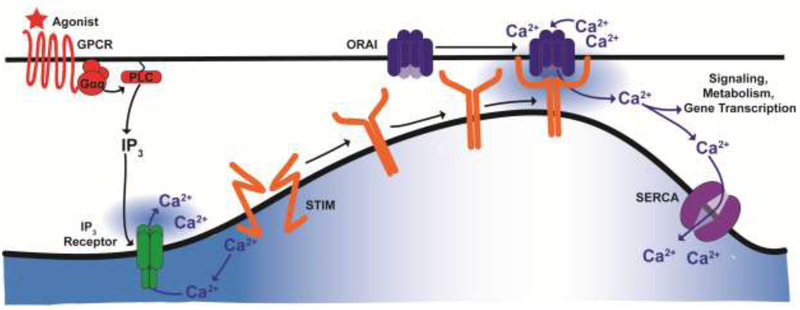
Binding of agonists to PLC-coupled receptors generates the secondary messenger IP3. IP3 induces ER Ca2+ release through the IP3 receptor. Following ER Ca2+ release, Ca2+ dissociates from the EF hand of STIM and triggers STIM to oligomerize, migrate towards the ER-PM junction, trap, and activate ORAI channels. This activation causes a large Ca2+ influx from the extracellular milieu into the cytosol, which refills ER Ca2+ stores through SERCA and create cytosolic Ca2+ microdomains that are sensed by downstream cell signaling effectors and transcription factors, activating gene programs to support metabolism, proliferation and migration.
Figure 2. Mechanism of SOCE activation.
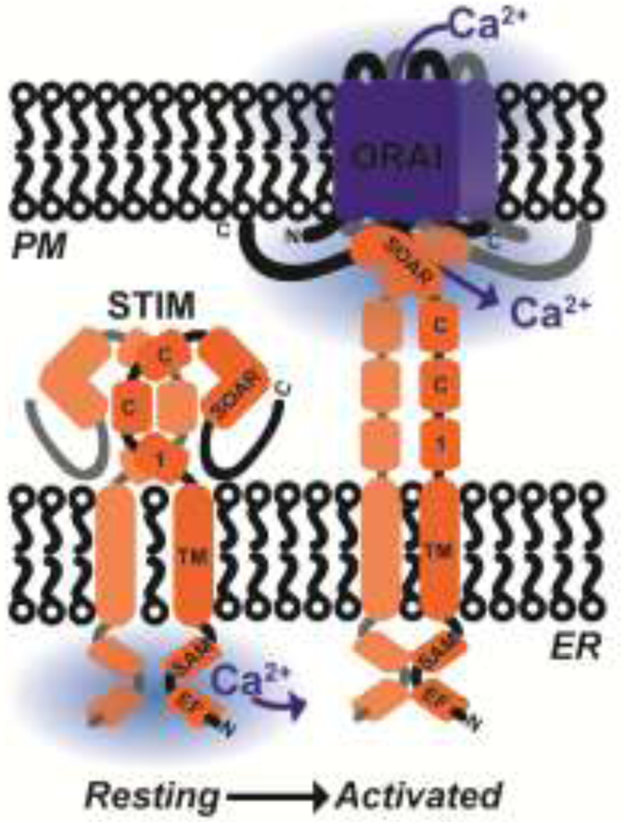
Under resting conditions, STIM proteins exist as inactive dimers in the ER membrane. STIM low-affinity luminal EF hand domains are bound to Ca2+, and the inhibitory coiled coil-1 (CC1) domain occludes the STIM-ORAI activating region (SOAR). Upon ER Ca2+ store depletion, Ca2+ dissociates from EF hand domains, which causes STIM to undergo a conformational change and gain an extended conformation, which exposes its cytosolic SOAR domain. SOAR dimers are then able to physically trap and gate PM-located ORAI Ca2+ channels leading to activation of SOCE.
ORAI1, the canonical ORAI isoform, was identified in 2006 [40-42]. ORAI1 is a 33 kilodalton protein with four transmembrane domains where both the N- and C-termini are located in the cytosol. The functional channel is a hexamer of six ORAI subunits with the transmembrane domains arranged in concentric layers [43-45]. Mammals have three ORAI homologs (ORAI1-3), and each individual homolog is capable of mediating ICRAC when overexpressed with STIM1; however, ORAI2 and ORAI3 mediate smaller currents compared to ORAI1 [46], and with the exception of few specific instances[47-49], their role in mediating native Ca2+ entry pathways remain largely unknown[50]. ORAI1 also has two variants generated through two alternative translation-initiation sites at Methionine 1 and Methionine 64 from the same messenger RNA. While both the long (ORAI1α) and the short (ORAI1β) mediate ICRAC [51, 52], ORAI1β has a significantly smaller Ca2+ dependent inactivation (CDI) [52]. Physiologically, different ORAI homologs and translational variants appear to form heteromeric channels that are differentially expressed depending on cell types [53]. One such heteromeric channel is formed by ORAI1α and ORAI3, is activated by STIM1 independently of depletion of ER Ca2+ stores and requires instead arachidonic acid or its metabolite LTC4 [14]. These channels which are also highly Ca2+ selective are termed arachidonate-regulated Ca2+ (ARC) channels. Unlike ICRAC, IARC is not inhibited by the SOCE inhibitor 2-Aminoethoxydiphenyl borate (2-APB), and their monovalent currents measured under divalent free bath solutions do not rapidly depotentiate [54, 55].
At low physiological concentrations of agonist stimulation, the Ca2+ signal emanating from ORAI1 most notably takes the form of regenerative oscillations. These Ca2+ oscillations, which are believed to be mediated by the oscillatory activity of IP3 receptors, quickly rundown in the absence of SOCE that is needed to refill ER Ca2+ stores [56]. Changes in the frequency and amplitude of Ca2+ oscillations produce precise spatiotemporal Ca2+ mircodomains with specific signatures that activate distinct Ca2+-dependent transcription factors [57]. These include isoforms of the transcription factor, nuclear factor of activated T cells (NFAT) [58]. The Ca2+-dependent phosphatase calcineurin, which localizes to the proximity of ORAI1 channels [59], dephosphorylates NFAT isoforms, and causes their nuclear translocation and subsequent transcriptional regulation [58, 60]. Another transcriptional regulator that is specifically activated by the local Ca2+ signal generated by SOCE is c-Fos, which upon activation forms a transcriptional complex with c-Jun known as AP-1 that regulates the expression of inflammatory genes [61]. SOCE can also lead to phosphorylation of the transcription factor, cAMP response element-binding protein (CREB), which transcribes metabolic genes and indirectly induces cellular proliferation in T lymphocytes [62]. Recently, SOCE has also been shown to suppress the transcriptional coactivators of the Hippo pathway Yes-associated Protein (YAP) and PDZ-binding motif (TAZ) [63].
The physiological significance of ORAI1 is underscored with patients that have loss of function mutations in ORAI1. The most characterized mutation is R91W, which is located on the cytoplasmic side of the first transmembrane domain. These patients have drastically reduced SOCE and ICRAC and show severe combined immunodeficiency (SCID), autoimmunity, muscular myotonia, and ectodermal dysplasia [64]. Global ORAI1 knockout mice are small in size, and most die perinatally and fail to thrive [65], suggesting a crucial role for ORAI1 channels beyond their established role in specific immunity. ORAI channels are indeed ubiquitously expressed in all tissues and are increasingly implicated in the regulation of a multitude of cellular functions. Dysregulation in ORAI channels, including in their expression, heteromeric or homomeric associations, post-translational modifications and interactions with other proteins, has been associated with cardiorespiratory disease. Despite that all three ORAI isoforms are ubiquitously expressed, future targeting of ORAI in patients with cardiorespiratory diseases is possible providing we have a thorough understanding of the molecular organization and regulation of this pathway in different tissues. Potential differences in regulation, oligomerization status and isoform involvement between different tissues could potentially be exploited to achieve specificity in therapeutic targeting. While side effects from systemic treatments would likely persist, careful understanding of ORAI cannel regulation and dysregulation in a specific system combined with localized therapies may provide a therapeutic window to target ORAI in cardiorespiratory diseases. As will be discussed below, a common theme is the contribution of ORAI dysregulation to cellular remodeling in most of these diseases.
Systemic Arterial Hypertension
In the vasculature, Ca2+ signaling is essential for maintaining endothelial cell (EC) integrity, vascular smooth muscle cell (VSMC) tone, and modulating VSMC phenotype. Dysfunction in these Ca2+ signaling events have been well characterized in systemic arterial hypertension (SAH) (Figure 3A). A key pathological feature of endothelial cells (ECs) in hypertension is the impaired bioavailability of nitric oxide (NO) and increased secretion of vasoconstrictive mediators and growth factors [66]. These vasoconstrictive mediators and growth factors then directly modulate VSMC tone and remodeling. ORAI1 and STIM1 protein expression, SOCE, and native ICRAC have been characterized in endothelial cells [33, 67]. Functionally, ORAI1 has been shown to be necessary for EC proliferation, angiogenesis, but not for EC barrier function in response to activation of G protein-coupled receptor (GPCR) agonists [33, 68-70]. Decreased EC barrier function manifests with formation of interendothelial gaps, which enhance the permeability of the intimal layer to circulating cells and solutes and play a critical role in vascular disease [71]. Three different GPCR agonists (histamine, thrombin, and sphingosine-1-phosphate) that alter ECs permeability rely on STIM1 but function independently of Ca2+ release, SOCE, and ORAI1 [68, 69]. Studies describing a functional role of endothelial ORAI channels in SAH, such as those using EC-specific knockout mice, are lacking. A study showed that ORAI3 is necessary for in vitro and in vivo endothelial tube formation in response to vascular endothelial growth factor (VEGF) and that VEGF induces movement of ORAI3 to the PM of ECs through production of LTC4 and arachidonic acid [72]. Another group showed that arachidonic acid stimulates NO release by ECs through activation of Ca2+ entry through TRPV4 channels, subsequent activation of calmodulin, and Ca2+/calmodulin-dependent endothelial NO synthase, eNOS [73]. Although further studies are needed to clarify the specific role of ORAI3 in ECs, these studies suggest that TRPV4, ORAI3 (and potentially IARC) have a protective role by maintaining EC function.
Figure 3. Role of ORAI channels in cellular remodeling in cardiorespiratory disease.

Summary of functions of ORAI channels in A) SAH and PAH, B) restenosis, C) atherosclerosis, D) asthma and COPD are listed under each cell type. Marked in bold have been studied exclusively in that disease, while those marked in italic have been speculated based on other tissues or diseases. A question mark indicates that the role of ORAI in this cell type is unknown.
ORAI channels have been more extensively studied in VSMCs [74, 75]. Unlike other excitable muscle tissues such as skeletal and cardiac muscle, smooth muscle exhibits tremendous plasticity of phenotype. In healthy normotensive vasculature, VSMCs are tonically constricted and serve to maintain vascular tone. Vascular tone is primarily the result of contractility activated by increased cytosolic Ca2+ originating from PM-located L-type Ca2+ Channels (LTCCs) and ryanodine receptors (RyRs) expressed in the SR/ER. Healthy contractile VSMCs are quiescent and normally do not proliferate or migrate towards the vascular lumen [76]. However, aberrant extracellular cues including cytokines, growth factors, hormones, inflammatory mediators, or mechanical stressors causes these quiescent VSMCs to undergo a phenotypic switch to a more proliferative, migratory, or “synthetic” phenotype. Synthetic VSMCs lose their ability to contract due to transcriptional downregulation of contractile proteins including LTCCs, RyRs, smooth muscle myosin heavy chain (smMHC), and smooth muscle α-actin [76, 77]. In exchange, synthetic VSMCs increase expression of proteins that control proliferation and migration and contribute to vascular disease. The phenotypic switch from quiescent to synthetic VSMCs plays a prominent role in hypertension, atherosclerosis, diabetic vascular diseases, artery stenosis, and aneurysms [78]. Interestingly, this switch in phenotype can also be modeled in cell culture by placing freshly dissociated quiescent VSMCs in culture media containing serum [79].
In quiescent VSMCs, STIM and ORAI protein expression, SOCE, and ICRAC are barely detectable [36]. Thus, there is a lack of evidence to suggest SOCE has a physiological role in contractile smooth muscle in the systemic vasculature. In contrast, STIM1, ORAI1, SOCE and ICRAC are greatly up-regulated in synthetic VSMCs [36, 79]. The peptide hormones urotensin-II and angiotensin II, which are well-known mediators of hypertension, and other pro-proliferative and pro-migratory mediators are thought to drive the expression of ORAI1 in VSMC remodeling [80-82]. ORAI1 is necessary for VSMC proliferation and migration, and ORAI1 (but not ORAI2 or ORAI3) is necessary for platelet-derived growth factor (PDGF)-induced SOCE and VSMC migration [83]. ORAI1 associates with other Ca2+ Channels, and the expression of the Sodium (Na+)-Ca2+ exchanger (NCX) and plasma membrane Ca2+ATPase (PMCA) seems to be dependent on ORAI1 in VSMCs [84]. Takahashia et al. showed that in VSMC, phosphorylation of the pro-proliferative transcription factor CREB is dependent on STIM1 [85]. Importantly, smooth muscle-specific STIM1 knockout mice [86] are partially protected against endothelial dysfunction and development of hypertension after angiotensin II infusion[87]. These mice showed significantly blunted systolic blood pressure, ER stress, and cardiac fibrosis in response to angiotensin II infusion [87]. Similar studies using smooth muscle specific ORAI knockout mice have not been reported.
Cytosolic Ca2+ has been reported to be elevated in hypertensive rat models and patients [88, 89]. The role of ORAI in SAH was first studied in 2009 using male spontaneous hypertensive rats (SHR) [90]. Compared to normotensive Wistar Kyoto (WKY) rats, SHR rats had higher protein and mRNA expression of both ORAI1 and STIM1 in the aorta. Treatment of SHR aortic rings with 100 μM of either SOCE inhibitor 2-APB or Gd3+ or with STIM1 or ORAI1 neutralizing antibodies had reduced force generation to similar levels as WKY rats. However, these data should be interpreted with caution as neutralizing antibodies can have non-specific effects and the concentrations of 2-APB or Gd3+ used are quite high and were shown in previous studies to interfere with many channels, including IP3 receptor Ca2+ release channels and various plasma membrane TRP channels [38, 91], A similar study with WKY rats subjected to 30 days of chronic ethanol consumption demonstrated that chronic ethanol caused increased systolic blood pressure (SBP), SOCE, and STIM1 protein expression isolated from aortic tissue [92]. The SOCE inhibitors Gd3+ and SKF 96365 have also been reported to inhibit SBP in rats [93]. However, follow up studies are necessary because the concentrations of SOCE inhibitors used in these studies are relatively high and may have off-target effects. The isolated aortic rings are also a collection of a variety of cell types, and it is unclear which cell type these inhibitors are targeting.
Epidemiological evidence supports the idea that males are more susceptible to hypertension than females [94]. Giachini et al. observed that male SHR aortic rings which generate more contractile force, had enhanced ORAI1 and STIM1 protein and mRNA expression in aortic tissue compared to female SHR rats [95]. Treatment of male SHR aortic rings with ORAI1 or STIM1 neutralizing antibodies restored force contraction to similar levels as SHR females. An ovariectomy in female SHR also increased aortic ORAI1 protein expression and force contraction in aortic rings to similar levels as male SHR, suggesting that estrogen or progesterone can regulate ORAI protein expression and protect against SAH [95]. Interestingly, estrogen has been shown to increase ORAI3 but not ORAI1 protein expression in estrogen-positive breast cancer cells [48]. By virtue of ORAI3 being uniquely involved as an essential component of heteromeric ARC channels, ORAI3 might be required for activating downstream signaling pathways distinct from those activated downstream SOCE [14]. Determination of ORAI3 protein expression and ARC channel activity in female versus male SHR rats has not been undertaken.
Sympathetic nerve fibers maintain vascular homeostasis; they innervate arteriolar VSMCs and secrete norepinephrine (NE) to stimulate α1 adrenergic receptors on VSMCs to induce vasoconstriction. Hyperactivity of the sympathetic nervous system is a well-known characteristic of hypertension [96]. However, the potential role of ORAI or STIM in sympathetic nerve function is currently unknown. Inflammation has also emerged as a crucial player in the pathogenesis of hypertension [97]. Numerous markers of inflammation are augmented in hypertension including an elevation of the acute phase protein C-reactive peptide (CRP), Interleukin-6 (IL-6), and tumor necrosis factor α (TNF-α) [98]. These inflammatory cytokines recruit leukocytes to the vasculature to further propagate inflammation, endothelial dysfunction, and VSMC remodeling [99, 100]. ORAI has been thoroughly described in the context of immunity [40, 101-103]. ORAI1 channels are the main source of cytosolic Ca2+ required for activation of the calcineurin-NFAT pathway, which is critical for clonal expansion and secretion of cytokines by immune cells [104]. SOCE is required for T cell, neutrophil, B cell, and natural killer cell function [105]. Neutrophil recruitment to sites of inflamed endothelium is an important step in vascular disease. SOCE mediated by ORAI1 is necessary for neutrophil polarization and arrest at sites of inflamed endothelium [106]. Clinical studies have demonstrated an association between treatment with immunosuppressants (azathioprine, myclophenolic acid, and anti-TNF-α therapy) and a lower SBP [107, 108], suggesting that targeting ORAI1 may reduce inflammation to the vasculature and counteract hypertension. T regulatory cells (Tregs), which are circulating immune cells that suppress inflammation in both immune and nonimmune cells through anti-inflammatory cytokines such as Interleukin-10 (IL-10) and transforming growth factor β (TGFβ), are reduced in hypertension [109-111]. Adoptive transfer of Tregs into angiotensin II-infused mice enhanced vasodilator response and reduced SBP, vascular stiffness, and aortic macrophage recruitment [112, 113], supporting the protective role of Tregs in hypertension. Any potential differences in the mechanisms of activation and regulation of ICRAC between Tregs and effector T cells that could be exploited for selective therapy of hypertension remain uncertain.
Restenosis and Thrombosis
Restenosis is the pathological remodeling and re-narrowing of arteries (most often coronary arteries) after percutaneous angioplasty and stenting, which occurs in 1-3% of patients and often leads to acute mycocardial infarction (MI) or acute cardiac arrest [114, 115]. Restenosis is the result of neointima formation, which results from VSMC proliferation and migration into the lumen of vessels (Figure 3B). ORAI1 and STIM1 are upregulated in animal models of neointimal hyperplasia, including the rat carotid balloon injury model [83, 116] and the carotid ligation model in mice [116]. Knockdown of either STIM1 or ORAI1 using shRNA-encoding lentiviral constructs inhibited VSMC neointimal hyperplasia [116] and this coincided with decreased expression of CamKIIδ2 isoform and NFAT nuclear translocation [116]. Both CamKIIδ2 and NFAT are important for transcribing pro-proliferative and pro-migratory genes [117, 118]. Similar results were obtained with STIM1 in vivo knockdown [116, 119, 120], as well as with STIM1 smooth muscle-specific knockout mice subjected to ligation injury [121]. Gonzalez-Cobos et al. showed that ORAI3 was also upregulated in VSMC after balloon injury and is necessary for driving neointimal hyperplasia [122]. In neointimal VSMC, ORAI3 association with ORAI1 produces a heteromeric channel that was not activated upon ER Ca2+ store depletion but by cytosolic LTC4 produced from arachidonic acid metabolism downstream receptor activation [15, 55, 123]. Subsequent studies revealed that this LTC4-activated heteromeric ORAI1/ORAI3 Ca2+ channel is in fact IARC, encoded by the same populations of ORAI1 and ORAI3 in both VSMCs and HEK293 cells [124].
The initial pathological injury in restenosis is denudation of the ECs in the intimal layer. ECs are important for secreting factors like growth factors and NO to maintain the integrity of the basement membrane and to preserve VSMCs in the quiescent state [125]. Loss of this endothelial layer causes a sudden increase in VSMC remodeling [126]. As described above, ORAI and SOCE are essential for endothelial cell proliferation and angiogenesis [33]. The role of EC ORAI channels in restenosis is currently unknown. Whether promoting the expression of ORAI in ECs can salvage the formation of the intimal layer and prevent intimal hyperplasia is an intriguing idea that remains untested. Likewise, endothelial progenitor cells (EPCs) are circulating hemopoietic cells, which can migrate to injured intimal layers. At these sites, EPCs differentiate into mature ECs. Accelerating EPC migration and differentiation can accelerate intimal healing and may prevent restenosis [127]. SOCE has been measured in EPCs, and the SOCE inhibitor BTP-2 caused reduced EPC proliferation [128]. ORAI1 and SOCE were decreased in EPCs during atherosclerosis, which is another disease contributed by endothelial dysfunction [129]. In the context of restenosis, it might be worthwhile to determine ORAI expression and SOCE activation in EPCs as restoring or promoting ORAI in EPCs may have beneficial effects in restenosis.
In most cases of percutaneous angioplasty and stenting, thrombosis is an early and major contributor to myocardial infarction. A necessary step in thrombosis is the binding of agonists like thrombin, thromboxane A2 (TXA2), and ADP to PLC-coupled receptors, which causes an elevation in cytosolic Ca2+ in platelets [130]. Through the ORAI1 chimera mouse model (an irradiated wildtype mouse infused with the bone marrow of an ORAI1 knockout mouse), ORAI1 channels were shown to be necessary for SOCE in response to thapsigargin, ADP, thrombin, and TXA2 in platelets [131]. However, platelet aggregation was similar to controls when stimulated with thrombin, ADP, CRP, and collagen, although stimulation with low concentrations of CRP, convulxin, and collagen showed reduced platelet aggregation in ORAI1 chimera mice. Similarly, the activation and exposure of the platelet activation markers glycoprotein IIb/IIIa and P-selectin were identical between ORAI1 chimeras and controls when stimulated with ADP and thrombin; yet, the activation and exposure of these markers were reduced in ORAI1 chimeras when stimulated with CRP and convulxin. Under shear perfusion on a collagen-coated surface, ORAI1 chimera failed to form stable thrombi. Unlike thrombin and ADP, which activate PLCβ, CRP, convulxin (CVX), and collagen activate PLCγ through the receptor Glycoprotein VI. Perhaps ORAI1 differentially modulates platelet activation and aggregation based on the specific receptor activated. In addition, ORAI1 chimera mice were protected from cerebral ischemia [131]. The same group reported similar findings with STIM1 chimera mice [132]. Treating mice with the SOCE inhibitors 2-APB, Synta66, and GSK-7975A also reduced thrombosis from cerebral ischemia [133]. Another group observed that chimeric mice bearing the mutation found in SCID patients, ORAI1R93W, had reduced SOCE in platelets. Only low concentrations of the agonists PAR4p and CVX induced platelet activation and exposure of glycoprotein IIb/IIIa and P-selectin. The platelet activation marker phosphatidylserine (PS) was also reduced in ORAI1R93W chimera mice[134]. However, Bergmeier et al. found platelet aggregation to be identical between ORAI1R93W chimera mice and controls in response to all agonists. One gain of function (GOF) STIM1 mutant mice (STIM1D84G) showed premature platelet activation and macrothrombocytopenia [135] while another GOF mutant STIM1R304W had impaired platelet activation possibly due to reduced STIM1 expression in platelets in these mutant mice [132, 136]. While these results are quite confusing, what might be the consensus from these studies is that ORAI1 is important for SOCE in platelets but perhaps not for physiological coagulation. However, from the cerebral ischemia studies [131, 133], SOCE may play a role in pathological coagulation such as during restenosis, which warrants a comprehensive study. This also suggests there may be a therapeutic window for SOCE inhibitors in thrombosis. The other ORAI homologs are also expressed in platelets but have not been studied in the context of pathological thrombosis.
There is a known inflammatory component to thrombosis as neutrophils, monocytes, and lymphocytes are quickly recruited to the injured site once a thrombus has formed [137]. As mentioned above for the case of SAH, these active immune cells further stimulate VSMC remodeling and endothelial dysfunction [99, 100, 138]. Inhibition of ORAI in immune cells is expected to prevent immune cell differentiation and activation [105], suggesting that ORAI in immune cells might be a contributor to restenosis.
Atherosclerosis
The pathogenesis of atherosclerosis begins as a subendothelial accumulation of lipids in the vasculature known as “fatty streaks.” Fatty streaks stimulate the migration of monocytes and macrophages into the plaques between the ECs and VSMCs (Figure 3C). At these fatty streaks, these immune cells engulf excessive amounts of lipids and become “foam cells,” which propagate a chronic inflammatory environment in the vasculature. This chronic inflammation activates VSMC remodeling and further induces endothelial dysfunction, which ultimately cause narrowing of the vascular lumen [139]. The apolipoprotein E (apoE) knockout mouse model spontaneously develops hyperlipidemia and atherosclerosis. Assche et al. reported apoE knockout mice have higher ATP induced SOCE in endothelial denuded aortic rings. A substantial SOCE was measured in four month old apoE KO mice, which are too young to develop significant atherosclerotic plaques [140]. This suggests SOCE and VSMC remodeling might be an early step in atherosclerosis. Another group reported ORAI1 protein and mRNA expression to be upregulated in aortic tissue isolated from apoE knockout mice when fed a high fat diet. Knockdown of ORAI1 with siRNA or use of the SOCE inhibitor, SKF96365, decreased atherosclerotic plaque size [141]. Again these studies have not specifically examined one particular cell type and warrants further clarification. Similarly, STIM1 and ORAI1 protein expression and SOCE were reported to be elevated in VSMCs isolated from pigs fed a high-calorie diet. Interestingly, exercise had reduced STIM1 expression and SOCE [142], which suggests that diet and excercise modulate SOCE and STIM1 protein expression. Further studies are necessary to identify the mechanisms of how risk factors of atherosclerosis regulate STIM and ORAI1 expression (and those of other STIM/ORAI isoforms) and their role in smooth muscle remodeling during atherosclerosis.
Endothelial dysfunction is an early step in the development of atherosclerotic plaques. As is the case for SAH and restenosis, STIM and ORAI are important for maintaining EC integrity, proliferation, and angiogenesis. It is unclear what role EC proliferation and angiogenesis may play in atherosclerosis [143]. Similar to systemic hypertension and restenosis, ECs are clearly essential for maintaining VSMCs in the quiescent state through mediators like NO [125]. EPCs are also important for regenerating healthy intima, and apoE knockout mice have decreased EPC proliferation and migration [129]. The EPCs from apoE knockout mice also have reduced SOCE, STIM1, and ORAI1 protein expression [129]. EPC proliferation and migration was also attenuated with the SOCE inhibitors 2-APB, ML-9, and shRNA targeting STIM1. The same group also reported that SOCE serves to protect EPCs from decreased cellular proliferation induced by oxidized LDL [144]. Oxidized LDL increased STIM1 protein expression, increased intracellular Ca2+ in EPCs through SOCE and induced autophagy in EPCs through activation of calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and inactivation of mammalian target of rapamycin (mTOR). This mechanism was proposed to ultimately spare EPCs from deleterious effects of oxidized LDL [144].
Because ORAI1 is necessary for neutrophil migration into inflamed endothelium [106], neutrophils and monocytes migration could be a contributor to atherosclerosis, although this idea remain untested. ORAI has been proposed as necessary for the formation of foam cells from lipid-laden macrophages [141]. Liang et al. observed that oxidized LDL activated Ca2+ entry through ORAI1 in macrophages. However, the molecular mechanisms of how oxidized LDL activates ORAI1 remain unknown. These authors proposed that oxidized LDL activation of ORAI1-mediated Ca2+ entry activates calcineurin, which leads to the expression of scavenger receptor A through c-Jun N-terminal kinase (JNK) and p38 kinase. The upregulated expression of scavenger receptor A would cause further uptake of LDL and promote the transition to a foam cell [141].
Pulmonary Arterial Hypertension
Although a relatively rare disease, pulmonary arterial hypertension (PAH) causes major complications including dyspnea and heart failure. Like SAH, much of the pathogenesis of PAH is idiopathic; however, the two diseases often have distinct etiologies. The pathological hallmark of PAH are smooth muscle and endothelial proliferation and migration, as well as thrombosis (Figure 3A) [145]. While the endothelium only plays a supporting role in SAH, endothelial dysfunction is believed to drive PAH. The role of endothelial ORAI in PAH remain unknown. Pulmonary artery smooth muscle (PASMC) remodeling in PAH is quite similar to VSMC remodeling in the systemic arteries. ORAI1 mediated SOCE has been measured in isolated PASMCs [146], and Fernandez et al. observed ORAI2 protein expression and SOCE to be increased in cultured synthetic PASMCs. In contrast, acutely isolated PASMCs expressed contractile proteins like smooth muscle 22-α actin and myosin heavy chain [147]. Others observed ORAI1 and ORAI2 (but not ORAI3) expression and increased SOCE in PASMCs under chronic hypoxia [148, 149]. This upregulation under hypoxia was also unique to PASMCs since it was not observed with coronary VSMCs. The upregulation of ORAI2 (but not ORAI1) was dependent on the transcription factor hypoxia-inducible factor 1α (HIF1α) [149]. PASMCs isolated from patients with PAH also had increased expression of ORAI2 and STIM2 [150]. Further studies are necessary to dissect the relative role of ORAI2 versus ORAI1 in PASMC remodeling in PAH mouse models and how HIF1α regulates the expression of these proteins. ORAI3 protein expression has been measured in PASMCs [149]; however, ORAI3 role in PAH is unknown. The vasoconstrictor serotonin, which induces PASMC remodeling [151], has been shown to activate a store-independent Ca2+ channel that resembled the pharmacological profile of IARC [152], suggesting that serotonin might regulate PASMC remodeling through activation of IARC and ORAI3.[122].
Asthma
Asthma is a chronic inflammatory disease in the airways that affects 25.7 million American per year [153]. Inflammatory T-helper type 2 (TH2) cells inappropriately infiltrate the airways and secrete cytokines like IL-4, IL-5, and IL-13, which activate B cells, basophils, mast cells, and eosinophils to secrete inflammatory mediators like histamine, trypsin, thrombin, and bradykinin (Figure 3D). These inflammatory mediators act on PLC-coupled receptors and stimulate airway hyper-responsiveness (AHR) to physiological stimuli [154]. Similar to the vascular diseases discussed above, this chronic inflammation also causes significant structural remodeling in the airways. This airway remodeling includes goblet cell hyperplasia, airway smooth muscle cell (ASMC) proliferation and migration, fibrosis, and angiogenesis [75, 155]. Airway remodeling causes a significant loss of pulmonary function and is most prevalent in severe and difficult to treat asthmatics [156, 157].
ORAI1 and STIM1 protein expression were upregulated in airway smooth muscle tissue isolated from the asthmatic mouse model challenged with the allergen ovalbumin [158]. β1 integrin and TGFβ are inflammatory mediators that are elevated in asthmatic airways, and these mediators have been shown to modulate the expression of ORAI1 in the mouse airways and rat ASMCs respectively, although the mechanism is unclear [159, 160]. ORAI protein expression, SOCE, and ICRAC have also been measured in human airway smooth muscle cells (ASMCs) [158, 161]. Secreted PDGF from inflammation also activated ORAI1 dependent ICRAC in human ASMCs. Knockdown of ORAI1 in human ASMCs inhibited PDGF-induced proliferation and migration [162]. Similarly, ORAI1 and STIM1 were necessary for mouse ASMC SOCE, ICRAC, proliferation, and migration [158]. Animal studies of asthma using ASMC tissue-specific ORAI knockout mice have not been reported. Sutovska et al. showed that asthmatic mice treated with the SOCE inhibitor 3-fluoropyridine-4-carboxylic acid (FPCA) had significant bronchodilation and cough suppression [163]. Treatment of mouse ASMCs with the SOCE inhibitors GSK7975A and GSK5498A inhibited methacholine induced-oscillations and lung slice contractions [138], while voltage-gated Ca2+channel inhibitors were not effective. Although ORAI3-mediated IARC has not been directly measured in ASMCs, arachidonic acid-induced Ca2+ oscillations were detected in cultured ASMCs. These oscillations were unaffected by SOCE inhibitors but were inhibited by knockdown of ORAI3 with siRNA [164].
The lumen of the airways is lined with epithelial cells, which serve as a barrier to pathogens, chemicals, and inflammatory cells from the environment. A special type of epithelial cell known as goblet cells secrete mucus in the airways, which acts as a lubricant and an innate immune defense mechanism for the airways. In airway remodeling, the epithelium cycles from epithelium damage, repair, and metaplasia. Indeed, goblet cell metaplasia and hyperplasia are pathological hallmarks of airway remodeling [165]. Epithelial cells are also important for maintaining ASMCs in the quiescent state, and epithelial cell injury induces the secretion of cytokines such as IL-6, IL-8, matrix metalloproteinase 9 (MMP-9), which stimulate ASMC proliferation[166, 167]. Jairaman et al. has investigated the role of ORAI in airway epithelial cells and reported that allergens such as house dust mite extracts were able to activate ORAI1-dependent SOCE through the protease-activated receptor type 2 (PAR2) in a bronchial epithelial cell line. SOCE inhibitors caused decreased allergen-induced cytokine production from epithelial cells[168].
Ashmole et al. showed that human lung mast cells, an important cell type in development of asthma, express ORAI1, ORAI2, and ORAI3 mRNA and protein and develop ICRAC upon FCεR1 activation. The SOCE inhibitors GSK-7975A and Synta-66 inhibited mast cell secretion of histamine, LTC4, IL-5, IL-13, TNF-α, and IL-8 [169]. Knockdown or dominant negative strategies against ORAI1 or ORAI2 inhibited human lung mast cell SOCE and LTC4 secretion [170]. In vivo studies on the contribution of ORAI channels from a specific cell type (e.g. mast cells, TH2 cells, Tregs or endothelial cells) to development of asthma using tissue-specific knockout mice are currently lacking.
Chronic Obstructive Pulmonary Disorder (COPD)
COPD is highly prevalent and estimated to affect over 384 million people worldwide. The dyspnea and cough in COPD are progressive and very difficult to treat since many patients become resistant to corticosteroids [171]. ASMC remodeling due to chronic inflammation is central to the pathogenesis of COPD. Much of the mechanisms previously discussed involving ORAI channels have not been studied in COPD. Unlike asthma, Cigarette smoke is the primary etiology of COPD. Wylam et al. observed that cigarette smoke enhanced the expression of ORAI1 as well as increase ORAI1-dependent SOCE in cultured ASMCs. Cigarette smoke also enhanced ASMC growth [172]. Since COPD has many similar pathological features to asthma and other vascular diseases, ORAI channels involvement in the pathogenesis of COPD warrants investigation.
Concluding Remarks
The cardiorespiratory diseases discussed above are highly prevalent diseases that cause significant morbidity and mortality. A major theme of these diseases is cellular remodeling, which coincides with a remodeling of the Ca2+ signaling machinery. Among these, ORAI Ca2+ channels, which are activated either as a result of ER/SR Ca2+ store depletion (ICRAC) or independently of store depletion by inflammatory metabolites (IARC), have emerged as major regulators of cellular remodeling through their ability to modulate transcriptional programs that drive proliferation, hypertrophy, migration and secretion of cytokines [16, 53, 75]. We have just begun to understand the role ORAI channels play in various cell types involved in cardiorespiratory diseases. Studies on isolated cells, including smooth muscle, endothelial, and immune cells have been useful in understanding the contribution of ORAI channels to Ca2+ signaling in response to various disease-relevant receptor agonists. These cellular studies have also shed light on the contribution of various STIM and ORAI isoforms to cell function. However, there is much more to understand and future studies need to address the downstream targets and mechanisms modulated by ORAI activation as well as the upstream regulators leading to changes in ORAI expression and function during disease. Much of the previous work has focused on the canonical ORAI1 isoform, and future studies should dissect the contribution of other ORAI isoforms, including ORAI2, ORAI3, and ORAI1β. This includes their ability to heteromultimerize and to be subject to post-translational and regulatory modifications. The contribution of various ORAI isoforms to the makeup of channels with unique biophysical properties could offer hope for specific targeting of these channels in future therapy of cardiorespiratory disease. ORAI1 glycosylation among other post-translational modifications have been shown to modulate channel function [173], and perhaps different modifications occur for other ORAI isoforms that can drive cellular remodeling in cardiorespiratory diseases. However, one hindrance to such studies is the current lack of specific and reliable antibodies against ORAI2 and ORAI3. Most importantly, more studies with animal models of disease, especially those using cell type-specific knockout mice of different ORAI isoforms, are needed to demonstarte the translational potential of this important class of Ca2+ channels in cardiorespiratory disease.
Highlights.
Cellular remodeling is a common theme in cardiorespiratory diseases, including systemic hypertension, restenosis, thrombosis, atherosclerosis, pulmonary arterial hypertension, asthma, and chronic obstructive pulmonary disease (COPD).
ORAI channels are calcium (Ca2+) selective channels, which are typically activated by endoplasmic reticulum (ER) Ca2+ store depletion, have emerged as significant regulators of cellular remodeling during disease.
This review details how dysregulation in ORAI channel expression and activation contributes to cellular remodeling in cardiorespiratory diseases.
ORAI channels may be potential pharmaceutical targets in cardiorespiratory diseases.
ACKNOWLEDGMENTS
Research in the authors’ laboratories is supported by grants R01HL123364, R01HL097111 and R21AG050072 from the National Institutes of Health and by grant NPRP8-110-3-021 from the Qatar National Research Fund (QNRF) to M.T. and by a training grant TL1TR002016 from the National Center for Advancing Translational Sciences of the National Institute of Health to M.J..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- [1].Murphy SL, Kochanek KD, Xu J, Arias E, Mortality in the United States, 2014, NCHS data brief, (2015) 1–8. [PubMed] [Google Scholar]
- [2].Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Mackey RH, Matsushita K, Mozaffarian D, Mussolino ME, Nasir K, Neumar RW, Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA, Rosamond WD, Sasson C, Towfighi A, Tsao CW, Turner MB, Virani SS, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, C. American Heart Association Statistics, S. Stroke Statistics, Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association, Circulation, 135 (2017) e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beasley R, Semprini A, Mitchell EA, Risk factors for asthma: is prevention possible?, Lancet (London, England), 386 (2015) 1075–1085. [DOI] [PubMed] [Google Scholar]
- [4].Taichman DB, Mandel J, Epidemiology of pulmonary arterial hypertension, Clinics in chest medicine, 34 (2013) 619–637. [DOI] [PubMed] [Google Scholar]
- [5].Heusch G, Libby P, Gersh B, Yellon D, Böhm M, Lopaschuk G, Opie L, Lancet Seminar: Cardiovascular Remodelling in Coronary Artery Disease and Heart Failure(), Lancet (London, England), 383 (2014) 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bergeron C, Tulic MK, Hamid Q, Airway remodelling in asthma: from benchside to clinical practice, Canadian respiratory journal, 17 (2010) e85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Berridge MJ, Bootman MD, Roderick HL, Calcium signalling: dynamics, homeostasis and remodelling, Nat Rev Mol Cell Biol, 4 (2003) 517–529. [DOI] [PubMed] [Google Scholar]
- [8].Berridge MJ, Calcium signalling remodelling and disease, Biochemical Society transactions, 40 (2012) 297–309. [DOI] [PubMed] [Google Scholar]
- [9].Berridge MJ, The inositol trisphosphate/calcium signaling pathway in health and disease, Physiological reviews, 96 (2016) 1261–1296. [DOI] [PubMed] [Google Scholar]
- [10].Landstrom AP, Dobrev D, Wehrens XH, Calcium signaling and cardiac arrhythmias, Circulation research, 120 (2017) 1969–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Putney JW Jr., Capacitative calcium entry revisited, Cell Calcium, 11 (1990) 611–624. [DOI] [PubMed] [Google Scholar]
- [12].Hoth M, Penner R, Depletion of intracellular calcium stores activates a calcium current in mast cells, Nature, 355 (1992) 353–356. [DOI] [PubMed] [Google Scholar]
- [13].Prakriya M, Lewis RS, Store-Operated Calcium Channels, Physiol Rev, 95 (2015) 1383–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mignen O, Thompson JL, Shuttleworth TJ, Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels, The Journal of physiology, 586 (2008) 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang W, Zhang X, Gonzalez-Cobos JC, Stolwijk JA, Matrougui K, Trebak M, Leukotriene-C4 synthase, a critical enzyme in the activation of store-independent Orai1/Orai3 channels, is required for neointimal hyperplasia, The Journal of biological chemistry, 290 (2015) 5015–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Trebak M, STIM/Orai signalling complexes in vascular smooth muscle, J Physiol, 590 (2012) 4201–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Putney JW Jr., A model for receptor-regulated calcium entry, Cell Calcium, 7 (1986) 1–12. [DOI] [PubMed] [Google Scholar]
- [18].Streb H, Irvine RF, Berridge MJ, Schulz I, Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate, Nature, 306 (1983) 67–69. [DOI] [PubMed] [Google Scholar]
- [19].Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr., Meyer T, STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx, Curr Biol, 15 (2005) 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA, STIM1, an essential and conserved component of store-operated Ca2+ channel function, J Cell Biol, 169 (2005) 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M, Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry, The Journal of biological chemistry, 281 (2006) 35855–35862. [DOI] [PubMed] [Google Scholar]
- [22].Zhou Y, Cai X, Nwokonko RM, Loktionova NA, Wang Y, Gill DL, The STIM-Orai coupling interface and gating of the Orai1 channel, Cell Calcium, 63 (2017) 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS, Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation, Nature, 454 (2008) 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD, STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane, Nature, 437 (2005) 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yuan JP, Zeng W, Dorwart MR, Choi YJ, Worley PF, Muallem S, SOAR and the polybasic STIM1 domains gate and regulate Orai channels, Nat Cell Biol, 11 (2009) 337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS, STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1, Cell, 136 (2009) 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feske S, Prakriya M, Conformational dynamics of STIM1 activation, Nat Struct Mol Biol, 20 (2013) 918–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, Gill DL, Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site, Nat Commun, 5 (2014) 3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zheng S, Ma G, He L, Zhang T, Li J, Yuan X, Nguyen NT, Huang Y, Zhang X, Gao P, Nwokonko R, Gill DL, Dong H, Zhou Y, Wang Y, Identification of molecular determinants that govern distinct STIM2 activation dynamics, PLoS Biol, 16 (2018) e2006898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng L, Stathopulos PB, Schindl R, Li GY, Romanin C, Ikura M, Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry, Proceedings of the National Academy of Sciences of the United States of America, 108 (2011) 1337–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brandman O, Liou J, Park WS, Meyer T, STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels, Cell, 131 (2007) 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Subedi KP, Ong HL, Son GY, Liu X, Ambudkar IS, STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions, Cell Rep, 23 (2018) 522–534. [DOI] [PubMed] [Google Scholar]
- [33].Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M, Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation, Circ Res, 103 (2008) 1289–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lewis RS, Cahalan MD, Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells, Cell regulation, 1 (1989) 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu X, Groschner K, Ambudkar IS, Distinct Ca(2+)-permeable cation currents are activated by internal Ca(2+)-store depletion in RBL-2H3 cells and human salivary gland cells, HSG and HSY, The Journal of membrane biology, 200 (2004) 93–104. [DOI] [PubMed] [Google Scholar]
- [36].Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M, Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 23 (2009) 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Motiani RK, Abdullaev IF, Trebak M, A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells, J Biol Chem, 285 (2010) 19173–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Trebak M, Bird GS, McKay RR, Putney JW Jr., Comparison of human TRPC3 channels in receptor-activated and store-operated modes. Differential sensitivity to channel blockers suggests fundamental differences in channel composition, J Biol Chem, 277 (2002) 21617–21623. [DOI] [PubMed] [Google Scholar]
- [39].Tian C, Du L, Zhou Y, Li M, Store-operated CRAC channel inhibitors: opportunities and challenges, Future Med Chem, 8 (2016) 817–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A, A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function, Nature, 441 (2006) 179–185. [DOI] [PubMed] [Google Scholar]
- [41].Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP, CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry, Science (New York, N.Y.), 312 (2006) 1220–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD, Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity, Proceedings of the National Academy of Sciences of the United States of America, 103 (2006) 9357–9362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cai X, Zhou Y, Nwokonko RM, Loktionova NA, Wang X, Xin P, Trebak M, Wang Y, Gill DL, The Orai1 Store-operated Calcium Channel Functions as a Hexamer, The Journal of biological chemistry, 291 (2016) 25764–25775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hou X, Pedi L, Diver MM, Long SB, Crystal structure of the calcium release-activated calcium channel Orai, Science (New York, N.Y.), 338 (2012) 1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Yen M, Lokteva LA, Lewis RS, Functional Analysis of Orai1 Concatemers Supports a Hexameric Stoichiometry for the CRAC Channel, Biophys J, 111 (2016) 1897–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW Jr., Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1, The Journal of biological chemistry, 281 (2006) 24979–24990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Motiani RK, Stolwijk JA, Newton RL, Zhang X, Trebak M, Emerging roles of Orai3 in pathophysiology, Channels (Austin), 7 (2013) 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M, Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 27 (2013) 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vaeth M, Yang J, Yamashita M, Zee I, Eckstein M, Knosp C, Kaufmann U, Karoly Jani P, Lacruz RS, Flockerzi V, Kacskovics I, Prakriya M, Feske S, ORAI2 modulates store-operated calcium entry and T cell-mediated immunity, Nat Commun, 8 (2017) 14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Trebak M, Kinet JP, Calcium signalling in T cells, Nat Rev Immunol, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fukushima M, Tomita T, Janoshazi A, Putney JW, Alternative translation initiation gives rise to two isoforms of Orai1 with distinct plasma membrane mobilities, Journal of cell science, 125 (2012) 4354–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Desai PN, Zhang X, Wu S, Janoshazi A, Bolimuntha S, Putney JW, Trebak M, Multiple types of calcium channels arising from alternative translation initiation of the <em>Orai1</em> message, Science Signaling, 8 (2015) ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Trebak M, Putney JW, ORAI Calcium Channels, Physiology, 32 (2017) 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Thompson JL, Shuttleworth TJ, Exploring the unique features of the ARC channel, a store-independent Orai channel, Channels (Austin, Tex.), 7 (2013) 364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang X, Gonzalez-Cobos JC, Schindl R, Muik M, Ruhle B, Motiani RK, Bisaillon JM, Zhang W, Fahrner M, Barroso M, Matrougui K, Romanin C, Trebak M, Mechanisms of STIM1 activation of store-independent leukotriene C4-regulated Ca2+ channels, Mol Cell Biol, 33 (2013) 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Putney JW, Bird GS, Cytoplasmic calcium oscillations and store-operated calcium influx, The Journal of physiology, 586 (2008) 3055–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Smedler E, Uhlen P, Frequency decoding of calcium oscillations, Biochim Biophys Acta, 1840 (2014) 964–969. [DOI] [PubMed] [Google Scholar]
- [58].Gwack Y, Feske S, Srikanth S, Hogan PG, Rao A, Signalling to transcription: store-operated Ca2+ entry and NFAT activation in lymphocytes, Cell Calcium, 42 (2007) 145–156. [DOI] [PubMed] [Google Scholar]
- [59].Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML, Hogan PG, Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling, Nat Struct Mol Biol, 19 (2012) 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Loh C, Shaw KT, Carew J, Viola JP, Luo C, Perrino BA, Rao A, Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity, The Journal of biological chemistry, 271 (1996) 10884–10891. [DOI] [PubMed] [Google Scholar]
- [61].Di Capite J, Ng SW, Parekh AB, Decoding of cytoplasmic Ca(2+) oscillations through the spatial signature drives gene expression, Curr Biol, 19 (2009) 853–858. [DOI] [PubMed] [Google Scholar]
- [62].Maus M, Cuk M, Patel B, Lian J, Ouimet M, Kaufmann U, Yang J, Horvath R, Hornig-Do HT, Chrzanowska-Lightowlers ZM, Moore KJ, Cuervo AM, Feske S, Store-Operated Ca(2+) Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism, Cell Metab, 25 (2017) 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liu Z, Wei Y, Zhang L, Yee PP, Johnson M, Zhang X, Gulley M, Atkinson JM, Trebak M, Wang HG, Li W, Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ, Oncogene, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lacruz RS, Feske S, Diseases caused by mutations in ORAI1 and STIM1, Ann N Y Acad Sci, 1356 (2015) 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A, Hair loss and defective T- and B-cell function in mice lacking ORAI1, Mol Cell Biol, 28 (2008) 5209–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bleakley C, Hamilton PK, Pumb R, Harbinson M, McVeigh GE, Endothelial Function in Hypertension: Victim or Culprit?, J Clin Hypertens (Greenwich), 17 (2015) 651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Trebak M, STIM1/Orai1, ICRAC, and endothelial SOC, Circ Res, 104 (2009) e56–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Shinde AV, Motiani RK, Zhang X, Abdullaev IF, Adam AP, Gonzalez-Cobos JC, Zhang W, Matrougui K, Vincent PA, Trebak M, STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry, Sci Signal, 6 (2013) ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Stolwijk JA, Zhang X, Gueguinou M, Zhang W, Matrougui K, Renken C, Trebak M, Calcium Signaling Is Dispensable for Receptor Regulation of Endothelial Barrier Function, The Journal of biological chemistry, 291 (2016) 22894–22912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, Majeed Y, Tumova S, Seymour VA, Taylor H, Stacey M, O'Regan D, Foster R, Porter KE, Kearney MT, Beech DJ, Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation, Circ Res, 108 (2011) 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ochoa CD, Stevens T, Studies on the cell biology of interendothelial cell gaps, Am J Physiol Lung Cell Mol Physiol, 302 (2012) L275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Li J, Bruns AF, Hou B, Rode B, Webster PJ, Bailey MA, Appleby HL, Moss NK, Ritchie JE, Yuldasheva NY, Tumova S, Quinney M, McKeown L, Taylor H, Prasad KR, Burke D, O'Regan D, Porter KE, Foster R, Kearney MT, Beech DJ, Orai3 Surface Accumulation and Calcium Entry Evoked by Vascular Endothelial Growth Factor, Arterioscler Thromb Vasc Biol, 35 (2015) 1987–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zuccolo E, Dragoni S, Poletto V, Catarsi P, Guido D, Rappa A, Reforgiato M, Lodola F, Lim D, Rosti V, Guerra G, Moccia F, Arachidonic acid-evoked Ca(2+) signals promote nitric oxide release and proliferation in human endothelial colony forming cells, Vascul Pharmacol, 87 (2016) 159–171. [DOI] [PubMed] [Google Scholar]
- [74].Ruhle B, Trebak M, Emerging roles for native Orai Ca2+ channels in cardiovascular disease, Curr Top Membr, 71 (2013) 209–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Spinelli AM, Trebak M, Orai channel-mediated Ca2+ signals in vascular and airway smooth muscle, Am J Physiol Cell Physiol, 310 (2016) C402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].House SJ, Potier M, Bisaillon J, Singer HA, Trebak M, The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease, Pflugers Arch, 456 (2008) 769–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Halayko AJ, Solway J, Invited Review: Molecular mechanisms of phenotypic plasticity in smooth muscle cells, Journal of Applied Physiology, 90 (2001) 358–368. [DOI] [PubMed] [Google Scholar]
- [78].Owens GK, Kumar MS, Wamhoff BR, Molecular regulation of vascular smooth muscle cell differentiation in development and disease, Physiol Rev, 84 (2004) 767–801. [DOI] [PubMed] [Google Scholar]
- [79].Berra-Romani R, Mazzocco-Spezzia A, Pulina MV, Golovina VA, Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture, Am J Physiol Cell Physiol, 295 (2008) C779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Guo RW, Yang LX, Li MQ, Pan XH, Liu B, Deng YL, Stim1- and Orai1-mediated store-operated calcium entry is critical for angiotensin II-induced vascular smooth muscle cell proliferation, Cardiovasc Res, 93 (2012) 360–370. [DOI] [PubMed] [Google Scholar]
- [81].Papadopoulos P, Bousette N, Giaid A, Urotensin-II and cardiovascular remodeling, Peptides, 29 (2008) 764–769. [DOI] [PubMed] [Google Scholar]
- [82].Rodriguez-Moyano M, Diaz I, Dionisio N, Zhang X, Avila-Medina J, Calderon-Sanchez E, Trebak M, Rosado JA, Ordonez A, Smani T, Urotensin-II promotes vascular smooth muscle cell proliferation through store-operated calcium entry and EGFR transactivation, Cardiovasc Res, 100 (2013) 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, Alzawahra WF, Barroso M, Singer HA, Jourd'heuil D, Trebak M, Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration, Am J Physiol Cell Physiol, 298 (2010) C993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA, Cross talk between plasma membrane Na(+)/Ca (2+) exchanger-1 and TRPC/Orai-containing channels: key players in arterial hypertension, Adv Exp Med Biol, 961 (2013) 365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Takahashi Y, Watanabe H, Murakami M, Ono K, Munehisa Y, Koyama T, Nobori K, Iijima T, Ito H, Functional role of stromal interaction molecule 1 (STIM1) in vascular smooth muscle cells, Biochem Biophys Res Commun, 361 (2007) 934–940. [DOI] [PubMed] [Google Scholar]
- [86].Kassan M, Zhang W, Aissa KA, Stolwijk J, Trebak M, Matrougui K, Differential role for stromal interacting molecule 1 in the regulation of vascular function, Pflugers Arch, 467 (2015) 1195–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kassan M, Ait-Aissa K, Radwan E, Mali V, Haddox S, Gabani M, Zhang W, Belmadani S, Irani K, Trebak M, Matrougui K, Essential Role of Smooth Muscle STIM1 in Hypertension and Cardiovascular Dysfunction, Arterioscler Thromb Vasc Biol, 36 (2016) 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Bendhack LM, Sharma RV, Bhalla RC, Altered signal transduction in vascular smooth muscle cells of spontaneously hypertensive rats, Hypertension, 19 (1992) II142–148. [DOI] [PubMed] [Google Scholar]
- [89].Bohr DF, Webb RC, Vascular smooth muscle membrane in hypertension, Annu Rev Pharmacol Toxicol, 28 (1988) 389–409. [DOI] [PubMed] [Google Scholar]
- [90].Giachini FR, Chiao CW, Carneiro FS, Lima VV, Carneiro ZN, Dorrance AM, Tostes RC, Webb RC, Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction, Hypertension, 53 (2009) 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM, 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release, FASEB J, 16 (2002) 1145–1150. [DOI] [PubMed] [Google Scholar]
- [92].Souza Bomfim GH, Mendez-Lopez I, Arranz-Tagarro JA, Ferraz Carbonel AA, Roman-Campos D, Padin JF, Garcia AG, Jurkiewicz A, Jurkiewicz NH, Functional Upregulation of STIM-1/Orai-1-Mediated Store-Operated Ca2+ Contributing to the Hypertension Development Elicited by Chronic EtOH Consumption, Curr Vasc Pharmacol, 15 (2017) 265–281. [DOI] [PubMed] [Google Scholar]
- [93].Xu YJ, Elimban V, Dhalla NS, Reduction of blood pressure by store-operated calcium channel blockers, J Cell Mol Med, 19 (2015) 2763–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gillis EE, Sullivan JC, Sex Differences in Hypertension: Recent Advances, Hypertension, 68 (2016) 1322–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Giachini FR, Lima VV, Filgueira FP, Dorrance AM, Carvalho MH, Fortes ZB, Webb RC, Tostes RC, STIM1/Orai1 contributes to sex differences in vascular responses to calcium in spontaneously hypertensive rats, Clin Sci (Lond), 122 (2012) 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Mancia G, Grassi G, The autonomic nervous system and hypertension, Circ Res, 114 (2014) 1804–1814. [DOI] [PubMed] [Google Scholar]
- [97].De Miguel C, Rudemiller NP, Abais JM, Mattson DL, Inflammation and hypertension: new understandings and potential therapeutic targets, Curr Hypertens Rep, 17 (2015) 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bautista LE, Vera LM, Arenas IA, Gamarra G, Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension, J Hum Hypertens, 19 (2005) 149–154. [DOI] [PubMed] [Google Scholar]
- [99].Zhang C, The role of inflammatory cytokines in endothelial dysfunction, Basic Res Cardiol, 103 (2008) 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Chen Q, Jin M, Yang F, Zhu J, Xiao Q, Zhang L, Matrix metalloproteinases: inflammatory regulators of cell behaviors in vascular formation and remodeling, Mediators Inflamm, 2013 (2013) 928315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich-Siebelt F, Clancy R, Unutmaz D, Feske S, Store-Operated Ca(2+) Entry in Follicular T Cells Controls Humoral Immune Responses and Autoimmunity, Immunity, 44 (2016) 1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Vaeth M, Feske S, Ion channelopathies of the immune system, Curr Opin Immunol, 52 (2018) 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Vaeth M, Maus M, Klein-Hessling S, Freinkman E, Yang J, Eckstein M, Cameron S, Turvey SE, Serfling E, Berberich-Siebelt F, Possemato R, Feske S, Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming, Immunity, 47 (2017) 664–679 e666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Feske S, Skolnik EY, Prakriya M, Ion channels and transporters in lymphocyte function and immunity, Nat Rev Immunol, 12 (2012) 532–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Shaw PJ, Feske S, Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity, The Journal of physiology, 590 (2012) 4157–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Schaff UY, Dixit N, Procyk E, Yamayoshi I, Tse T, Simon SI, Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow, Blood, 115 (2010) 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Maki-Petaja KM, Hall FC, Booth AD, Wallace SM, Yasmin, Bearcroft PW, Harish S, Furlong A, McEniery CM, Brown J, Wilkinson IB, Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-alpha therapy, Circulation, 114 (2006) 1185–1192. [DOI] [PubMed] [Google Scholar]
- [108].Ferro CJ, Edwards NC, Hutchison C, Cockwell P, Steeds RP, Savage CO, Townend JN, Harper L, Does immunosuppressant medication lower blood pressure and arterial stiffness in patients with chronic kidney disease? An observational study, Hypertens Res, 34 (2011) 113–119. [DOI] [PubMed] [Google Scholar]
- [109].Kassan M, Galan M, Partyka M, Trebak M, Matrougui K, Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice, Arterioscler Thromb Vasc Biol, 31 (2011) 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kassan M, Wecker A, Kadowitz P, Trebak M, Matrougui K, CD4+CD25+Foxp3 regulatory T cells and vascular dysfunction in hypertension, J Hypertens, 31 (2013) 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Regulatory T cells: how do they suppress immune responses?, Int Immunol, 21 (2009) 1105–1111. [DOI] [PubMed] [Google Scholar]
- [112].Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL, T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury, Hypertension, 57 (2011) 469–476. [DOI] [PubMed] [Google Scholar]
- [113].Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez-Villalobos RA, Chentoufi AA, Kadowitz P, Belmadani S, Partyka M, Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice, Am J Pathol, 178 (2011) 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Daemen J, Wenaweser P, Tsuchida K, Abrecht L, Vaina S, Morger C, Kukreja N, Juni P, Sianos G, Hellige G, van Domburg RT, Hess OM, Boersma E, Meier B, Windecker S, Serruys PW, Early and late coronary stent thrombosis of sirolimus-eluting and paclitaxel-eluting stents in routine clinical practice: data from a large two-institutional cohort study, Lancet (London, England), 369 (2007) 667–678. [DOI] [PubMed] [Google Scholar]
- [115].Iakovou I, Schmidt T, Bonizzoni E, Ge L, Sangiorgi GM, Stankovic G, Airoldi F, Chieffo A, Montorfano M, Carlino M, Michev I, Corvaja N, Briguori C, Gerckens U, Grube E, Colombo A, Incidence, predictors, and outcome of thrombosis after successful implantation of drug-eluting stents, JAMA, 293 (2005) 2126–2130. [DOI] [PubMed] [Google Scholar]
- [116].Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, Singer HA, Matrougui K, Trebak M, Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury, Circ Res, 109 (2011) 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].House SJ, Singer HA, CaMKII-delta isoform regulation of neointima formation after vascular injury, Arterioscler Thromb Vasc Biol, 28 (2008) 441–447. [DOI] [PubMed] [Google Scholar]
- [118].Hamada N, Miyata M, Eto H, Shirasawa T, Akasaki Y, Nagaki A, Tei C, Tacrolimus-eluting stent inhibits neointimal hyperplasia via calcineurin/NFAT signaling in porcine coronary artery model, Atherosclerosis, 208 (2010) 97–103. [DOI] [PubMed] [Google Scholar]
- [119].Guo RW, Wang H, Gao P, Li MQ, Zeng CY, Yu Y, Chen JF, Song MB, Shi YK, Huang L, An essential role for stromal interaction molecule 1 in neointima formation following arterial injury, Cardiovasc Res, 81 (2009) 660–668. [DOI] [PubMed] [Google Scholar]
- [120].Aubart FC, Sassi Y, Coulombe A, Mougenot N, Vrignaud C, Leprince P, Lechat P, Lompre AM, Hulot JS, RNA interference targeting STIM1 suppresses vascular smooth muscle cell proliferation and neointima formation in the rat, Mol Ther, 17 (2009) 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Mancarella S, Potireddy S, Wang Y, Gao H, Gandhirajan RK, Autieri M, Scalia R, Cheng Z, Wang H, Madesh M, Houser SR, Gill DL, Targeted STIM deletion impairs calcium homeostasis, NFAT activation, and growth of smooth muscle, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 27 (2013) 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Gonzalez-Cobos JC, Zhang X, Zhang W, Ruhle B, Motiani RK, Schindl R, Muik M, Spinelli AM, Bisaillon JM, Shinde AV, Fahrner M, Singer HA, Matrougui K, Barroso M, Romanin C, Trebak M, Store-independent Orai1/3 channels activated by intracrine leukotriene C4: role in neointimal hyperplasia, Circ Res, 112 (2013) 1013–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Trebak M, Zhang W, Ruhle B, Henkel MM, Gonzalez-Cobos JC, Motiani RK, Stolwijk JA, Newton RL, Zhang X, What role for store-operated Ca(2)(+) entry in muscle?, Microcirculation, 20 (2013) 330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zhang X, Zhang W, Gonzalez-Cobos JC, Jardin I, Romanin C, Matrougui K, Trebak M, Complex role of STIM1 in the activation of store-independent Orai1/3 channels, J Gen Physiol, 143 (2014) 345–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kipshidze N, Dangas G, Tsapenko M, Moses J, Leon MB, Kutryk M, Serruys P, Role of the endothelium in modulating neointimal formation: vasculoprotective approaches to attenuate restenosis after percutaneous coronary interventions, J Am Coll Cardiol, 44 (2004) 733–739. [DOI] [PubMed] [Google Scholar]
- [126].Gimbrone MA Jr., Vascular endothelium, hemodynamic forces, and atherogenesis, Am J Pathol, 155 (1999) 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Lan H, Wang Y, Yin T, Wang Y, Liu W, Zhang X, Yu Q, Wang Z, Wang G, Progress and prospects of endothelial progenitor cell therapy in coronary stent implantation, J Biomed Mater Res B Appl Biomater, 104 (2016) 1237–1247. [DOI] [PubMed] [Google Scholar]
- [128].Sanchez-Hernandez Y, Laforenza U, Bonetti E, Fontana J, Dragoni S, Russo M, Avelino-Cruz JE, Schinelli S, Testa D, Guerra G, Rosti V, Tanzi F, Moccia F, Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells, Stem Cells Dev, 19 (2010) 1967–1981. [DOI] [PubMed] [Google Scholar]
- [129].Wang LY, Zhang JH, Yu J, Yang J, Deng MY, Kang HL, Huang L, Reduction of Store-Operated Ca(2+) Entry Correlates with Endothelial Progenitor Cell Dysfunction in Atherosclerotic Mice, Stem Cells Dev, 24 (2015) 1582–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Mazzucato M, Pradella P, Cozzi MR, De Marco L, Ruggeri ZM, Sequential cytoplasmic calcium signals in a 2-stage platelet activation process induced by the glycoprotein Ibalpha mechanoreceptor, Blood, 100 (2002) 2793–2800. [DOI] [PubMed] [Google Scholar]
- [131].Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, Bosl M, Stoll G, Nieswandt B, Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation, Blood, 113 (2009) 2056–2063. [DOI] [PubMed] [Google Scholar]
- [132].Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, Renne T, Stoll G, Nieswandt B, The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction, J Exp Med, 205 (2008) 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].van Kruchten R, Braun A, Feijge MA, Kuijpers MJ, Rivera-Galdos R, Kraft P, Stoll G, Kleinschnitz C, Bevers EM, Nieswandt B, Heemskerk JW, Antithrombotic potential of blockers of store-operated calcium channels in platelets, Arterioscler Thromb Vasc Biol, 32 (2012) 1717–1723. [DOI] [PubMed] [Google Scholar]
- [134].Bergmeier W, Oh-Hora M, McCarl CA, Roden RC, Bray PF, Feske S, R93W mutation in Orai1 causes impaired calcium influx in platelets, Blood, 113 (2009) 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Grosse J, Braun A, Varga-Szabo D, Beyersdorf N, Schneider B, Zeitlmann L, Hanke P, Schropp P, Muhlstedt S, Zorn C, Huber M, Schmittwolf C, Jagla W, Yu P, Kerkau T, Schulze H, Nehls M, Nieswandt B, An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice, J Clin Invest, 117 (2007) 3540–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Gamage TH, Gunnes G, Lee RH, Louch WE, Holmgren A, Bruton JD, Lengle E, Kolstad TRS, Revold T, Amundsen SS, Dalen KT, Holme PA, Tjonnfjord GE, Christensen G, Westerblad H, Klungland A, Bergmeier W, Misceo D, Frengen E, STIM1 R304W causes muscle degeneration and impaired platelet activation in mice, Cell Calcium, 76 (2018) 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Inoue T, Croce K, Morooka T, Sakuma M, Node K, Simon DI, Vascular inflammation and repair: implications for re-endothelialization, restenosis, and stent thrombosis, JACC Cardiovasc Interv, 4 (2011) 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Chen J, Sanderson MJ, Store-operated calcium entry is required for sustained contraction and Ca(2+) oscillations of airway smooth muscle, The Journal of physiology, 595 (2017) 3203–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Lusis AJ, Atherosclerosis, Nature, 407 (2000) 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Van Assche T, Fransen P, Guns PJ, Herman AG, Bult H, Altered Ca2+ handling of smooth muscle cells in aorta of apolipoprotein E-deficient mice before development of atherosclerotic lesions, Cell Calcium, 41 (2007) 295–302. [DOI] [PubMed] [Google Scholar]
- [141].Liang SJ, Zeng DY, Mai XY, Shang JY, Wu QQ, Yuan JN, Yu BX, Zhou P, Zhang FR, Liu YY, Lv XF, Liu J, Ou JS, Qian JS, Zhou JG, Inhibition of Orai1 Store-Operated Calcium Channel Prevents Foam Cell Formation and Atherosclerosis, Arterioscler Thromb Vasc Biol, 36 (2016) 618–628. [DOI] [PubMed] [Google Scholar]
- [142].Edwards JM, Neeb ZP, Alloosh MA, Long X, Bratz IN, Peller CR, Byrd JP, Kumar S, Obukhov AG, Sturek M, Exercise training decreases store-operated Ca2+entry associated with metabolic syndrome and coronary atherosclerosis, Cardiovasc Res, 85 (2010) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Gimbrone MA Jr., Garcia-Cardena G, Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis, Circ Res, 118 (2016) 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Yang J, Yu J, Li D, Yu S, Ke J, Wang L, Wang Y, Qiu Y, Gao X, Zhang J, Huang L, Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure, Autophagy, 13 (2017) 82–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Farber HW, Loscalzo J, Pulmonary arterial hypertension, N Engl J Med, 351 (2004) 1655–1665. [DOI] [PubMed] [Google Scholar]
- [146].Ng LC, Ramduny D, Airey JA, Singer CA, Keller PS, Shen XM, Tian H, Valencik M, Hume JR, Orai1 interacts with STIM1 and mediates capacitative Ca2+ entry in mouse pulmonary arterial smooth muscle cells, Am J Physiol Cell Physiol, 299 (2010) C1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Fernandez RA, Wan J, Song S, Smith KA, Gu Y, Tauseef M, Tang H, Makino A, Mehta D, Yuan JX, Upregulated expression of STIM2, TRPC6, and Orai2 contributes to the transition of pulmonary arterial smooth muscle cells from a contractile to proliferative phenotype, Am J Physiol Cell Physiol, 308 (2015) C581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].He X, Song S, Ayon RJ, Balisterieri A, Black SM, Makino A, Wier WG, Zang WJ, Yuan JX, Hypoxia selectively upregulates cation channels and increases cytosolic [Ca(2+)] in pulmonary, but not coronary, arterial smooth muscle cells, Am J Physiol Cell Physiol, 314 (2018) C504–C517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Wang J, Xu C, Zheng Q, Yang K, Lai N, Wang T, Tang H, Lu W, Orai1, 2, 3 and STIM1 promote store-operated calcium entry in pulmonary arterial smooth muscle cells, Cell Death Discov, 3 (2017) 17074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Song MY, Makino A, Yuan JX, STIM2 Contributes to Enhanced Store-operated Ca Entry in Pulmonary Artery Smooth Muscle Cells from Patients with Idiopathic Pulmonary Arterial Hypertension, Pulm Circ, 1 (2011) 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].MacLean MMR, The serotonin hypothesis in pulmonary hypertension revisited: targets for novel therapies (2017 Grover Conference Series), Pulm Circ, 8 (2018) 2045894018759125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Guibert C, Marthan R, Savineau JP, 5-HT induces an arachidonic acid-sensitive calcium influx in rat small intrapulmonary artery, Am J Physiol Lung Cell Mol Physiol, 286 (2004) L1228–1236. [DOI] [PubMed] [Google Scholar]
- [153].Akinbami LJ, Moorman JE, Bailey C, Zahran HS, King M, Johnson CA, Liu X, Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010, NCHS data brief, (2012) 1–8. [PubMed] [Google Scholar]
- [154].Lambrecht BN, Hammad H, The immunology of asthma, Nat Immunol, 16 (2015) 45–56. [DOI] [PubMed] [Google Scholar]
- [155].Shifren A, Witt C, Christie C, Castro M, Mechanisms of remodeling in asthmatic airways, J Allergy (Cairo), 2012 (2012) 316049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Lange P, Parner J, Vestbo J, Schnohr P, Jensen G, A 15-year follow-up study of ventilatory function in adults with asthma, N Engl J Med, 339 (1998) 1194–1200. [DOI] [PubMed] [Google Scholar]
- [157].Pepe C, Foley S, Shannon J, Lemiere C, Olivenstein R, Ernst P, Ludwig MS, Martin JG, Hamid Q, Differences in airway remodeling between subjects with severe and moderate asthma, J Allergy Clin Immunol, 116 (2005) 544–549. [DOI] [PubMed] [Google Scholar]
- [158].Spinelli AM, Gonzalez-Cobos JC, Zhang X, Motiani RK, Rowan S, Zhang W, Garrett J, Vincent PA, Matrougui K, Singer HA, Trebak M, Airway smooth muscle STIM1 and Orai1 are upregulated in asthmatic mice and mediate PDGF-activated SOCE, CRAC currents, proliferation, and migration, Pflugers Arch, 464 (2012) 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Qiu C, Liu W, Shi F, Fen M, Ren L, Qi H, Silencing of beta1 integrin regulates airway remodeling by regulating the transcription of SOCEassociated genes in asthmatic mice, Mol Med Rep, 16 (2017) 2645–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Gao Y.-d., Zheng J.-w., Li P, Cheng M, Yang J, Store-Operated Ca2+ Entry is involved in Transforming Growth Factor-β1 Facilitated Proliferation of Rat Airway Smooth Muscle Cells, Journal of Asthma, 50 (2013) 439–448. [DOI] [PubMed] [Google Scholar]
- [161].Peel SE, Liu B, Hall IP, ORAI and store-operated calcium influx in human airway smooth muscle cells, Am J Respir Cell Mol Biol, 38 (2008) 744–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Suganuma N, Ito S, Aso H, Kondo M, Sato M, Sokabe M, Hasegawa Y, STIM1 regulates platelet-derived growth factor-induced migration and Ca2+ influx in human airway smooth muscle cells, PLoS One, 7 (2012) e45056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Sutovska M, Kocmalova M, Adamkov M, Vybohova D, Mikolka P, Mokra D, Hatok J, Antosova M, Franova S, The long-term administration of Orai 1 antagonist possesses antitussive, bronchodilatory and anti-inflammatory effects in experimental asthma model, Gen Physiol Biophys, 32 (2013) 251–259. [DOI] [PubMed] [Google Scholar]
- [164].Thompson MA, Prakash YS, Pabelick CM, Arachidonate-regulated Ca(2+) influx in human airway smooth muscle, Am J Respir Cell Mol Biol, 51 (2014) 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Davies DE, The role of the epithelium in airway remodeling in asthma, Proc Am Thorac Soc, 6 (2009) 678–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Freishtat RJ, Watson AM, Benton AS, Iqbal SF, Pillai DK, Rose MC, Hoffman EP, Asthmatic airway epithelium is intrinsically inflammatory and mitotically dyssynchronous, Am J Respir Cell Mol Biol, 44 (2011) 863–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Malavia NK, Raub CB, Mahon SB, Brenner M, Panettieri RA Jr., George SC, Airway epithelium stimulates smooth muscle proliferation, Am J Respir Cell Mol Biol, 41 (2009) 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Jairaman A, Maguire CH, Schleimer RP, Prakriya M, Allergens stimulate store-operated calcium entry and cytokine production in airway epithelial cells, Sci Rep, 6 (2016) 32311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Ashmole I, Duffy SM, Leyland ML, Morrison VS, Begg M, Bradding P, CRACM/Orai ion channel expression and function in human lung mast cells, J Allergy Clin Immunol, 129 (2012) 1628–1635 e1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Ashmole I, Duffy SM, Leyland ML, Bradding P, The contribution of Orai(CRACM)1 and Orai(CRACM)2 channels in store-operated Ca2+ entry and mediator release in human lung mast cells, PLoS One, 8 (2013) e74895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Rabe KF, Watz H, Chronic obstructive pulmonary disease, Lancet (London, England), 389 (2017) 1931–1940. [DOI] [PubMed] [Google Scholar]
- [172].Wylam ME, Sathish V, VanOosten SK, Freeman M, Burkholder D, Thompson MA, Pabelick CM, Prakash YS, Mechanisms of Cigarette Smoke Effects on Human Airway Smooth Muscle, PLoS One, 10 (2015) e0128778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Dorr K, Kilch T, Kappel S, Alansary D, Schwar G, Niemeyer BA, Peinelt C, Cell type-specific glycosylation of Orai1 modulates store-operated Ca2+ entry, Sci Signal, 9 (2016) ra25. [DOI] [PubMed] [Google Scholar]