

Abstract
Glioblastoma (GBM) is the most aggressive brain tumor and resistant to current available therapeutics, such as radiation. To improve the clinical efficacy, it is important to understand the cellular mechanisms underlying tumor responses to radiation. Here, we investigated long-term cellular responses of human GBM cells to ionizing radiation. Comparing to the initial response within 12 hours, gene expression modulation at 7 days after radiation is markedly different. While genes related to cell cycle arrest and DNA damage responses are mostly modulated at the initial stage; immune-related genes are specifically affected as the long-term effect. This later response is associated with increased cellular senescence and inhibition of transcriptional coactivator with PDZ-binding motif (TAZ). Mechanistically, TAZ inhibition does not depend on the canonical Hippo pathway, but relies on enhanced degradation mediated by the β-catenin destruction complex in the Wnt pathway. We further showed that depletion of TAZ by RNAi promotes radiation-induced senescence and growth arrest. Pharmacological activation of the β-catenin destruction complex is able to promote radiation-induced TAZ inhibition and growth arrest in these tumor cells. The correlation between senescence and reduced expression of TAZ as well as β-catenin also occurs in human gliomas treated by radiation. Collectively, these findings suggested that inhibition of TAZ is involved in radiation-induced senescence and might benefit GBM radiotherapy.
Introduction
Glioblastoma (GBM) is one of the most aggressive brain tumors. Surgery followed by radiation treatment is the standard therapeutic regimen for GBM. Although the treatments could prolong survival of GBM patients, a progression of the disease always occurs after the initial treatments. The disease progression is accompanied by tumor recurrence and, in most cases, radiation-induced injury 1, 2. To improve the clinical efficacy, it is important to understand the cellular mechanisms underlying tumor responses to radiation.
Upon treatment by ionizing radiation, the primary response of GBM cells is proliferation arrest. The arrested cells then undergo premature senescence within 4–8 days after irradiation 3. Gene expression analyses of GBM cells treated by ionizing radiation have revealed that many genes are modulated after the treatment 4–9. These genes are involved in a variety of cellular processes, such as apoptosis, cell cycle, DNA replication/damage repair, cytoskeleton organization and metabolism. Most of these studies have focused on the acute (e.g. within 1 day after irradiation) responses of GBM cells to radiation and provided information to understand the initial cellular effects on GBM cells by radiation. However, the gene expression program associated with the relatively delayed responses to radiation (e.g. premature senescence) has not been investigated.
GBM is classified into several subtypes based on gene expression 10–12. Among these subtypes, the mesenchymal group associates with worst prognosis 10, 12. Transcriptional coactivator with PDZ-binding motif (TAZ) is proposed to be one of the transcriptional regulators driving the gene expression program of GBM MES differentiation 13. TAZ and its paralog, Yes-associated protein (YAP), are the two nuclear effectors of the Hippo signaling pathway. In this pathway, a core serine/threonine kinase cascade, including MST1/2 kinases and their substrates Lats1/2 kinases, is responsible for inhibiting YAP/TAZ by inducing their phosphorylation, nuclear exclusion and degradation 14.
Therapy-induced senescence has been widely described when tumor cells are treated by various therapeutic agents, including chemotherapeutic drugs and ionizing radiation. Because such cellular growth arrest can occur in tumor cells which are resistant to apoptosis stimuli, it is proposed to be an alternative avenue for cancer therapies 15, 16. Recently, inhibition of YAP was indicated to be involved in promoting senescence in fibroblast cells and hepatic stellate cells 17, 18, therefore suggesting a role of YAP/TAZ-controlled transcriptional program in preventing premature senescence.
In this study, we first used cultured human GBM cells to investigate the long-term gene expression modulation by ionizing radiation. Our studies indicated large difference of gene expression comparing to the short-term response. This later stage is associated with increased cellular senescence and reduced TAZ protein expression. Our further study found that the inhibition of TAZ is not through the canonical Hippo pathway but by activating the β-catenin destruction complex in the Wnt signaling pathway. Correspondingly, silencing TAZ expression promotes radiation-induced senescence and growth arrest in GBM cells. The correlation between senescence and reduced TAZ as well as β-catenin expression appears to also occur in gliomas treated by radiotherapy.
Results
Radiation induces cellular senescence in GBM cells
To study the responses of glioma cells to radiation, we treated LN229 human GBM cell line with gamma radiation. The colony formation assay indicated a dose-dependent growth inhibition by radiation (Figure S1a). Under the tested dosages, we found that the growth inhibition is not primarily through cell death but proliferation arrest (data not shown). These irradiated cells appear to be enlarged and flattened, which are typical appearances of senescent cells in culture (Figure S1b). We confirmed that these cells are undergoing cellular senescence by using β-gal staining (Figure S1b), a well-known senescence marker 19. To further study this radiation-induced senescence, we performed a temporal analysis of cells treated by a single 8-Gy radiation. It appears that the number of senescent cells is progressively increased within 7 days (Figure S1c). These senescent cells also show puncta of staining signals of phospho-H2AX (p-H2AX) (Figure S1d and S1e), which are frequently found in senescence-associated DNA-damage foci 19. These results were consistent with previous observations of radiation-induced senescence in other GBM cells 3 and suggested that senescence contributes radiation-induced growth inhibition of these cells.
Differential gene expression associates with the short-term and long-term responses after irradiation
Our observations above suggested that senescence is a longer-term inhibitory effect of irradiation. To understand how such response was achieved, we conducted gene expression analysis by RNA-sequencing (RNA-seq). At 12 hours after irradiation, expression of 209 genes is upregulated, whereas 149 genes are downregulated comparing to non-irradiated cells (>2 folds, p<0.05, one-way ANOVA test). At 7 days after irradiation, expression of 556 genes is upregulated, and 206 genes are downregulated comparing to passage-matched non-irradiated cells. Therefore, much more genes have been affected at 7 days after irradiation than 12 hours after irradiation (especially those genes whose expression is upregulated) (Figure 1a and 1b). Ingenuity pathway analysis of the differentially expressed genes at each time point suggested that distinct cellular pathways are involved. Pathways related to cell cycle, such as mitotic roles of Polo-like kinase (Z-score=−2; p=0.014) and Cyclins & cell cycle regulation (Z-score=−2; p=0.026), appear to be specifically inhibited at 12 hours after irradiation. G2/M DNA damage checkpoint regulation pathway is specifically activated (Z-score=2; p=0.0048). However, a S-phase entry pathway appears to be specifically inhibited at 7 days after irradiation (Z-score=−2; p=0.014). These results suggested that irradiated cells at different stages are arrested at distinct cell cycle phases. Remarkably, most of the pathways specifically associated with cells at 7 days after irradiation are related to immune responses (Figure 1c). Considering that a large portion of cells at this stage are undergoing senescence, these immune response pathways may be due to the senescence-associated secretory phenotype (SASP) 20. Interestingly, p53 signaling appears to be similarly activated at both 12 hours and 7 days after irradiation (Z-score=2.24 and 2.11, respectively; p=0.00096 and p=0.000014, respectively), suggesting it is involved in both short-term and long-term responses to irradiation.
Figure 1.

Differential gene expression associates with the short-term and the long-term response after irradiation. (a) LN229 cells were treated with or without ionizing radiation (8 Gy), cultured for 12 hours or 7 days, and subjected to RNA-seq gene expression analysis. Cells from two independent petri dishes were collected. (a) Hierarchical clustering of all samples was shown. (b) Scatter plot comparisons of irradiated (IR) and non-irradiated (No-IR) cells. Ingenuity pathway analysis (c) and upstream analysis (d) of differentially expressed genes at each time point after irradiation. (e) Top predicted upstream regulators from the upstream analysis were shown.
Multiple transcriptional regulators could be involved in the long-term response to irradiation
To understand how the differential gene expression was achieved, we analyzed the potential transcriptional regulators, which may control the gene expression program. Comparing to 12 hours after irradiation, more transcriptional regulators were predicted to be involved in cells 7 day after irradiation (Figure 1d). At 12 hours after irradiation, 8 transcription regulators (FOXM1, MED1, HLX, SMAD7, NR3C1, TRIM24, STAT5A, KLF2) were predicted to be inhibited, whereas 21 regulators (TP53, HIF1A, HMGB1, CDKN2A, PARP1, CTNNB1, BRCA1, NFKB1, etc) were predicted to be activated (Z-score cut-off = ±1.5; p<0.01). At 7 days after irradiation, 67 transcription regulators were predicted to be activated, whereas 41 regulators were inhibited (Z-score cut-off = ±1.5; p<0.01). Those regulators showed the most significant correlation (p<10e-10) were listed (Figure 1e). Consistent with activation of p53 signaling, TP53 was predicted to be activated in both situations. CDKN2A, a master senescence activator, is predicted to be more active at 7 days after irradiation. Among these transcriptional regulators, TAZ (Z-score=−1.66; p=4.14e-12) was predicated to be inhibited. Although YAP did not satisfy our cut off criteria, it was also predicted to be inhibited to a modest extent (Z-score=−1.183; p=2.12e-10). Based on the above analysis, we concluded that transcription programs associated with the long-term response to irradiation might involve multiple transcription regulators.
Reduced TAZ expression enhances radiation-induced senescence in GBM cells
The in silico studies above promoted us to further examine whether YAP and TAZ are inhibited in irradiated tumor cells. We found that the expression of CTGF and Cyr61, two well-characterized YAP/TAZ target genes, is reduced in irradiated LN229 cells (Figure 2a). Correspondently, the protein level of TAZ is progressively reduced, whereas YAP protein level is not clearly changed (Figure 2a). Therefore, these results confirmed our in silico studies and suggested that TAZ is specifically inhibited as a long-term response to irradiation. The correlation between TAZ inhibition and cellular senescence associating with the long-term response of irradiation suggested that TAZ inhibition might be involved in senescence. To test this, we used two different shRNAs to silence TAZ expression in LN229 cells (Figure 2b). TAZ knockdown reduces the expression of CTGF and Cyr61 in these cells (Figure 2b). Under this circumstance, more LN229 cells undergo senescence after irradiation (Figure 2c and 2d). Notably, reduction of TAZ expression per se is not sufficient to mimic radiation in inducing senescence, although a modestly increased senescence frequency was observed (Figure 2c and 2d). Therefore, inhibition of TAZ could facilitate tumor cells to undergo senescence after irradiation.
Figure 2.
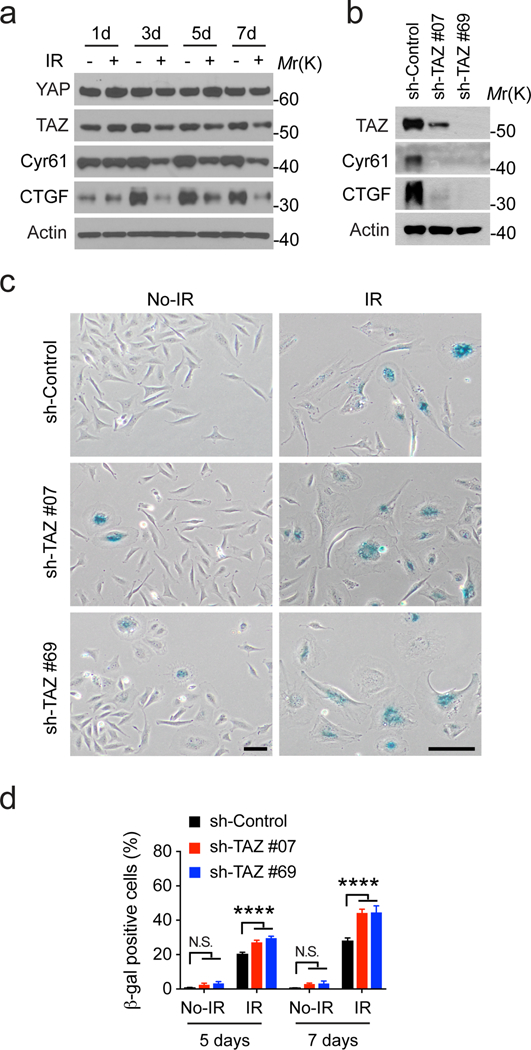
Reduced TAZ expression enhances radiation-induced senescence in LN229 cells. (a) Cells were treated with or without ionizing radiation (8 Gy), cultured for indicated days, and subjected to western blotting as indicated. Representative blots from two independent experiments were shown. (b) Cells stably transduced by shRNA control (sh-Co.) or against TAZ (sh-#07 and 69) were subjected to western blotting. Representative blots from three independent experiments were shown. (c) Cells stably transduced by shRNA control (sh-Co.) or against TAZ (sh-#07 and 69) were treated by irradiation (IR, 8 Gy) or not (No-IR). 7 days after irradiation, β-gal staining was conducted as shown. Scale bars=200 μM. (d) β-gal staining from (b) as well as 5 days after irradiation was quantified and shown. 2-way ANOVA multiple comparisons was performed. N=3. ****P<0.01. N.S. P>0.05.
Radiation-induced TAZ inhibition is not through the canonical Hippo pathway but relies on inhibition of the Wnt signaling
To understand the mechanism by which expression of TAZ is reduced in irradiated tumor cells, we first examined its mRNA by q-RT-PCR. It appears that the mRNA level of TAZ does not change in response to irradiation (Figure 3a). In the Hippo pathway, TAZ degradation is induced by Lats1/2-mediated phosphorylation 21. Therefore, we examined if Lats1/2 mediate TAZ inhibition in response to irradiation. Surprisingly, depletion of both Lats1 and Lats2 cannot rescue the reduction of TAZ in irradiated cells (Figure 3b). Correspondingly, expression of CTGF and Cyr61 is still suppressed by radiation. These results suggested that phosphorylation by Lats1/2 is not required for TAZ inhibition in this condition. Because ubiquitylation-mediated degradation is a mechanism for inhibiting TAZ 22, we used the proteasome inhibitor MG132 to block this protein degradation pathway. Under this circumstance, the protein level of TAZ is recovered in irradiated cells (Figure 3c). These results suggested that TAZ is controlled by ubiquitylation-mediated degradation.
Figure 3.
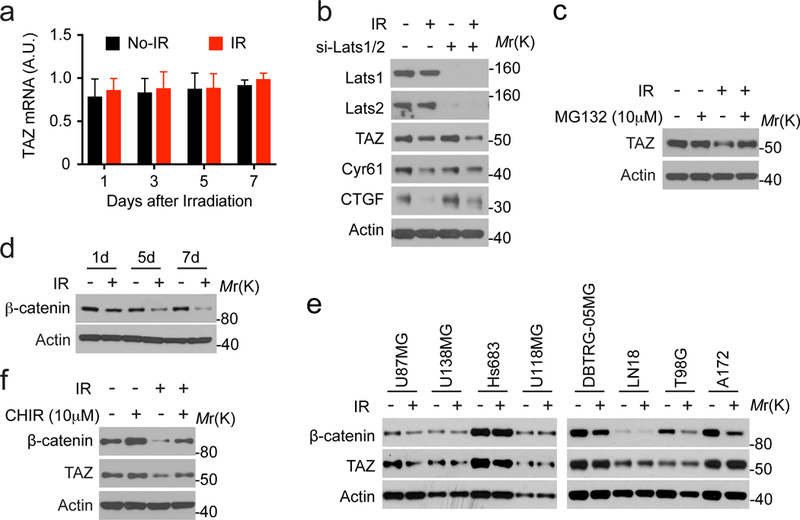
Radiation-induced TAZ inhibition relies on inhibition of the Wnt signaling. (a) LN229 cells were treated with or without ionizing radiation (8 Gy), cultured for indicated days, and subjected to q-RT-PCR. N=2. (b) LN229 cells transfected by siRNAs targeting Lats1 and Lats2 were treated with or without ionizing radiation (8 Gy). 7 days after irradiation, cell lysates were analyzed by western blotting. Representative blots from two independent experiments were shown. (c) LN229 cells treated with or without ionizing radiation (8 Gy) were cultured for 7 days. Cells were treated by MG132 for 12 hours before being analyzed by western blotting. Representative blots from two independent experiments were shown. (d) LN229 cells were treated with or without ionizing radiation (8 Gy), cultured for indicated days, and were subjected to western blotting as indicated. Representative blots from three independent experiments were shown. (e) Indicated cells were treated with or without ionizing radiation (8 Gy), cultured for 7 days, and were subjected to western blotting as indicated. Representative blots from two independent experiments were shown. (f) LN229 cells treated with or without ionizing radiation (8 Gy) were cultured for 7 days. Cells were treated by CHIR for 12 hours before being analyzed by western blotting. Representative blots from two independent experiments were shown.
The β-catenin destruction complex (APC/Axin/GSK3) is able to trigger TAZ degradation together with β-catenin when the Wnt signaling is turned off 23. This process requires GSK3 kinase to phosphorylate β-catenin, which then transfers TAZ to the ubiquitin ligase β-TrCP. Both β-catenin and TAZ are degraded through the ubiquitylation-mediated degradation. This process appears to be independent to Lats1/2-mediated TAZ phosphorylation 23. To test if radiation-induced TAZ degradation is through inhibiting the Wnt signaling, we first examined the expression of β-catenin. Similar to TAZ, the protein level of β-catenin was also reduced progressively in response to irradiation in LN229 cells (Figure 3d).
To examine whether the correlated inhibition of TAZ and β-catenin also occurs in other irradiated GBM cells, we used a panel of 9 human GBM cell lines. First, we evaluated the relative baseline signaling activity of YAP/TAZ in these cells by examining the mRNA levels of Cyr61 and CTGF (Figure S2b and S2c). Both of these markers suggested that the activity of YAP/TAZ is the lowest in U87MG and U138MG cells, whereas higher in LN229, Hs683, LN18 and A172 cells. We also evaluated the proneural and mesenchymal status of these cells by using two mesenchymal markers Fibronectin-1 and CD44 13 (Figure S2a). The expression patterns of these two markers do not correlate with each other. This suggested that the proneural and mesenchymal status of the established GBM cell lines might not be simply determined through these markers. We then treated these cells by radiation. Interestingly, TAZ protein is reduced in three of the irradiated GBM cells (U87MG, U138MG and Hs683), whereas β-catenin protein is reduced in five of these irradiated cells (U87MG, DBTRG-05MG, LN18, T98G and A172) (Figure 3e). Notably, three of the tested cell lines (LN229, U87MG and U138MG) were indicated to possess certain p53 activity, whereas four of the cell lines (LN18, T98G, Hs683 and A172) have no detectable p53 activity 24. These results indicated that the co-inhibition of TAZ and β-catenin in response to irradiation occurs in certain cellular context and p53 activity may not be sufficient to predict such co-inhibition.
To explore the mechanism of the co-inhibition, we used LN229 cells, in which the co-inhibition appears to be most evident. We used a small-molecule inhibitor CHIR99021 (CHIR) to inhibit GSK3 23. Treatment by CHIR was able to inhibit radiation-induced reduction of β-catenin and TAZ in LN229 cells (Figure 3f). These results suggested that the β-catenin destruction complex (APC/Axin/GSK3) is involved in radiation-induced degradation of TAZ.
Inhibition of the Wnt pathway promotes TAZ degradation and radiation-induced growth suppression
Since inhibition of TAZ contributes radiation-induced senescence (Figure 2c and 2d), we thought to examine if TAZ inhibition could enhance the growth suppression by irradiation. Indeed, we found that silencing TAZ expression by two different shRNAs (Figure 2b) enhances the inhibitory effects by irradiation on LN229 cells (Figure 4a).
Figure 4.

Inhibition of the Wnt pathway promotes TAZ degradation and radiation-induced growth suppression. (a) LN229 cells stably transduced by shRNA control (sh-Control) or against TAZ (sh-#07 and 69) were treated with indicated doses of ionizing radiation and subjected to colony formation assay. 2-way ANOVA multiple comparisons was performed. N=3. (b) LN229 cells treated with or without ionizing radiation (8 Gy) were cultured for 7 days. Cells were treated by XAV939 for 24 hours before being analyzed by western blotting. Representative blots from three independent experiments were shown. (c)-(e) densitometry of each protein blot was performed. N=3. (f) LN229 cells treated with or without ionizing radiation (8 Gy) were cultured for 7 days. Cells were treated by IWP-2 for 24 hours before being analyzed by western blotting. Representative blots from two independent experiments were shown. (g) LN229 cells were treated with indicated doses of ionizing radiation and cultured for 7 days. In the last three days, cells were treated by XAV939 at different concentrations and subjected to the MTT assay. N=3. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, N.S. P>0.05.
To further test this notion, we thought to leverage our above finding that radiation-induced TAZ degradation is through the β-catenin destruction complex. The small molecule XAV939 is able to stimulate β-catenin degradation by stabilizing Axin, the concentration-limiting component of the destruction complex 25. We found that the protein level of Axin is higher in irradiated LN229 cells than non-irradiated cells (Figure 4b and 4c, compare No-IR to IR cells without the treatment of XAV939). This is accompanied by reduction of β-catenin and TAZ in irradiated cells (Figure 4b, 4d and 4e, compare NO-IR to IR cells without the treatment of XAV939). Treatment by XAV939 led to a dose-dependent increase of Axin in both irradiated and non-irradiated cells (Figure 4b and 4c). Concurrently, the protein levels of β-catenin and TAZ were reduced by XAV939 treatment in a dose-dependent manner in both cells (Figure 4b, 4d and 4e). These results further supported that the β-catenin destruction complex is involved in radiation-induced TAZ degradation. The Wnt signaling can inhibit the β-catenin destruction complex 23. To activate the β-catenin destruction complex through an alternative way, we use a small molecule IWP-2, which can inhibit the production of Wnt, thereby releasing the β-catenin destruction complex from suppression by Wnt signals 26. Similar to XAV939, treatment by IWP-2 (10 μM) enhances the reduction of β-catenin and TAZ by irradiation in LN229 cells (Figure 4f).
We then examined if enhancing the inhibition of β-catenin and TAZ could promote radiation-induced growth suppression of LN229 cells. XAV939 treatment per se inhibits the growth of LN229 cells in a dose-dependent manner (Figure 4g). In irradiated cells, XAV939 also displays a dose-dependent inhibition. Although there is no synergistic effect observed, the results did suggest that treatments by both radiation and XAV939 could have additive effects (Figure 4g).
Increased senescence and reduced TAZ as well as β-catenin expression associate with patient gliomas treated by radiotherapy.
Our above studies found a correlation between senescence and reduced expression of TAZ as well as β-catenin in irradiated GBM cells. To examine whether these phenomena also occur in gliomas treated by radiation, we collected 61 human glioma samples (Table S1). 6 tumors, which have identifiable para-tumor tissues (PT), were from the first surgery (Figure 5a). 27 tumors were from the first surgery before radiotherapy (Primary). 20 tumors were from the second surgery of recurrent tumors after radiotherapy (IR-R). 8 tumors were from the second surgery of recurrent tumors without radiotherapy (No-IR-R). In patients with recurrent tumors, those were treated by radiotherapy showed longer progression-free survival (PFS) (data not shown). In samples stained by H&E, tumors in the IR-R group contain high and low cellularity regions (Figure 5b, marked by T and N, respectively). The former are likely due to the recurrent tumor growth, whereas the later are likely associated with radiation necrosis 1, 2, 27. Ki67 staining showed that tumors in each group have significantly more Ki67 positive cells than the PT group (Figure 5b and 5c). Notably, the Ki67 scoring in the IR-R group was focused on the high cellularity regions (T), since these regions likely represent the recurrent tumors. To examine senescent cells, we used p-H2AX as a marker 19. p-H2AX positive cells are readily detected in the IR-R group of tumors (Figure 5b and 5d). Notably, these p-H2AX positive cells appear to mostly localize at the interface between regions of high and low cellularity (Figure 5b, inset). In contrast, p-H2AX positive cells were rarely found in other groups (Figure 5b and 5d). These results suggested that senescent cells likely exist in tumors that were treated by radiation. We then examined expression of TAZ and β-catenin in these tumor samples. While whole tumor specimens were analyzed in all of other groups, areas in between high and low cellularity regions were focused in the IR-R group. This is because that our studies suggested that senescent cells are likely enriched in these areas. Interestingly, TAZ and β-catenin protein levels at these regions are significantly lower than tumors in the primary and No-IR-R groups (Figure 5b, 5e and 5f). We also assessed the nuclear and cytoplasmic distributions of TAZ and β-catenin. It appears that TAZ is more readily detected in the nucleus in the Primary and No-IR-R groups than the PT and IR-R groups (P<0.0001, Fisher’s exact test) (Table S2). However, nuclear β-catenin is most readily detected in the No-IR-R group (P<0.0001, Fisher’s exact test) (Table S3). Overall, these results suggested that senescence as well as reduced expression of YAP and β-catenin associate with gliomas treated by radiotherapy.
Figure 5.

Increased senescence and reduced TAZ expression associate with patient gliomas treated by radiotherapy. (a) A diagram showing patient treatment procedures. (b) H&E and IHC of indicated proteins in each group of tumors. Scale bar=100 μM. (c)-(f) semiquantification of each protein immunoreactivity from the IHC specimen as being conducted in (b). NPT=6, NPrimary=27, NIR-R=20, NNo-IR-R=8. One-Way ANOVA (Nonparametric) Kruskal-Wallis test. FDR *q<0.05, **q<0.01, ***q<0.001, ****q<0.0001. N.S. q>0.1.
Discussion
In this study, we found that the gene expression modulation in GBM cells as a long-term response to ionizing radiation is markedly different from that as an initial response. The long-term gene modulation associates with increased cellular senescence and could involve multiple transcription regulators, including inhibition of TAZ. Depletion of TAZ by RNAi is able to promote radiation-induced senescence and growth arrest in GBM cells. TAZ inhibition under this circumstance is through enhanced degradation mediated by the β-catenin destruction complex in the Wnt pathway. Pharmacological activation of the β-catenin destruction complex is able to promote radiation-induced TAZ inhibition and growth arrest in these tumor cells. Importantly, the correlation between senescence and reduced expression of YAP as well as β-catenin also occurs in human gliomas treated by radiation. Collectively, these findings suggested that inhibition of TAZ might benefit GBM radiotherapy.
Consistent with previous studies, our gene expression analysis confirmed that genes related to DNA damage responses, cell cycle arrest and apoptosis are major modulated genes at the initial stage after irradiation. It was shown that the G2 phase delay is the predominant cell cycle response to irradiation, although delays in G1 and S phases also exist 28. Activation of the G2/M damage checkpoint and inhibition of mitotic roles of polo-like kinase suggested in our studies are consistent with a G2 phase delay. Our results also suggested activation of p53 signaling, which may contribute to the G1 delay 28. In contrast, as cells go through the 7-day progression after irradiation, the gene expression program markedly shifts. Other than cell cycle regulations related to the G2 delay, the G1 delay (indicated by inhibition of estrogen-mediated S-phase entry and activation of p53 signaling) appears to be the major cell cycle feature at this stage. This is likely due to increased frequency of senescent cells, which are predominantly arrested at the G1 phase 19. Another gene expression feature associated with the long-term effect is expression of immune-related genes. This gene expression program may contribute to the SASP by senescent cells. Consistent with the activated gene expression program, multiple transcriptional regulators were suggested to be involved in tumor cells at 7 days after irradiation. Some of these factors are well known to be involved in cellular senescence, such as TP53, CDKN2A and SIRT1. However, involvement in radiation-induced senescence by most other regulators, such as EZH2 and TRIM24, which have been implicated to be important for glioma development and therapeutic resistance 29–32, still needs to be studied.
Our studies suggested that the β-catenin destruction complex is activated by radiation, probably through inhibition of the Wnt singaling. Interestingly, the Wnt pathway did not come up in the Ingenuity Pathway Analysis. In our Upstream Analysis, we found that β-catenin is predicted to be moderately active (Z-score=0.272; p=2.28e-14), whereas TCF7L1, one of the transcription factors that function together with Ctnnb1, is predicted to be inactive (Z-score=−2; p=0.0251). However, TCF7L2, another TCF family member, is predicated to be moderately active (Z-score=0.133; p=0.0135). These results suggested that the transcriptional program controlled by Ctnnb1/TCF might be complex and determined by specific effectors. In this case, it is likely that a prediction of the Wnt pathway status might not be simply achievable. This may explain why the Wnt pathway did not come up in our Canonical Pathways Analysis.
TAZ is one of the transcriptional regulators suggested to be inactivated in tumor cells 7 days after irradiation. We confirmed this notion and found that the β-catenin destruction complex in the Wnt pathway mediates its inhibition. Although TAZ is inhibited by this way, the stability of YAP did not change. These results suggested that radiation-induced inhibition is specific to TAZ. It was previously shown that the protein level of YAP is not regulated by the Wnt signaling 23. Therefore, it is likely that the β-catenin destruction complex can only affect TAZ, but not YAP in irradiated cells. The role of YAP and TAZ signaling in anti-apoptosis has been well recognized. However, their role and regulation in senescence is still not clear. Recently, inhibition of YAP has been shown to be involved in promoting senescence in non-tumor cells 17, 18. Here, we found a link between inhibition of TAZ and radiation-induced senescence in tumor cells. Therefore, YAP and TAZ signaling may also be important to prevent cells from entering premature senescence.
Therapy-induced senescence has been proposed to be an avenue for cancer therapies 15, 16. Our studies suggested that suppression of TAZ might enhance therapy-induced senescence and benefit radiotherapies.
Materials & Methods
Patient tumor samples
Brain tumor samples were collected from patients who underwent surgical resection at Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (HUST) at Wuhan, China. The procedure of human sample collection was approved by the Ethic Committee of Tongji Hospital, Tongji Medical College, HUST. Informed consent was obtained from all subjects.
All available pathological materials were evaluated by two neuropathologists blinded to the original diagnoses. Malignant tumors were defined by high-grade features including mitotic figures, vascular proliferation, and pseudopalisading necrosis. For IHC, samples showing signals in more than 1% of all cells in one slide were defined as positive-staining. Expression of Ki67, p-H2AX, TAZ and β-catenin were detected by immunohistochemistry. Staining was scored in a semiquantitative manner. The positive percentage (PP) was scored as follows: 0, 1 (< 25%), 2 (25–50%), 3 (50–75%) and 4 (>75%). The staining intensity (SI) was scored as follows: none-0, weak-1, moderate-2, and strong-3. For Ki67 staining, only PP was scored. For p-H2AX staining, only SI was scored. For TAZ and β-catenin, a final immunoreactivity score (IRS) was calculated for each case as: IRS = PP x SI.
Cells
LN229 (CRL-2611), U87 MG (HTB-14), U138 MG (HTB-16), Hs683 (HTB-138), DBTRG-05MG (CRL-2020), LN18 (CRL-2610), T98G (CRL-1690), A172 (CRL-1620) and U118MG (HTB15) human glioblastoma cell lines were from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Corning, 10–013-CV) supplemented with 10% Fetal Bovine Serum (Gibco, 10437028) and 1% Antibiotic-Antimycotic Solution (Corning, 30–004-CI) at 37°C with 5% CO2. The cell lines were not independently authenticated in this study. The cell lines were examined to be mycoplasma negative before experiments. Unless otherwise indicated, experiments were performed with cells grown to 50% confluency.
Antibodies and Compounds
Antibodies for Phospho-Histone H2A.X (Ser139) (2577), YAP (12395), TAZ (4883), Lats1 (3477), Lats2 (5888), β-catenin (9562), Axin1 (3323), rabbit-IgG-HRP (7074), mouse-IgG-HRP (7076) antibodies are from Cell Signaling Technology. Anti-β-Actin (A5316) antibody is from Sigma-Aldrich. Anti-CTGF (sc-14939) and anti-Cyr61 (sc-13100) antibodies are from Santa Cruz Biotechnology. Anti-Ki67 (GB13030) is from Servicebio. CHIR-99021 (S2924) is from Selleck Chemicals. XAV939 (X3004) and IWP-2 (I0536) is from Sigma-Aldrich.
Radiation Treatment
For radiation treatment, cells were seeded in complete medium on petri dish at a density of 5×104/cm2 24 hours before the treatment. The cells were then exposed to γ-ray once using a GammaCell 220 Cobalt 60 irradiator (Nordion International) with indicated doses. After irradiation, the medium was replaced with fresh complete medium.
Colony formation assay
Cells were seeded in 96-well plates by serials dilution. After incubation at 37 °C for 7–10 days, the number of cell colonies containing at least 50 cells was determined in each well. The colony formation efficiencies (CFE) were calculated using the formula CFE = (number of colonies scored)/(number of cells seeded). Viable cell colonies were caculated by nomalizing CFE of irradiated cells to non-irradiated cells (100%). Four replicated wells were scored for each time. We performed three independent experiments for each dose of irradiation.
Senescence-associated beta-galactosidase (SA-β-Gal) assay
SA-β-Gal staining was done with a Senescence β-Galactosidase Staining Kit (9860, Cell Signaling Technology) according to manufacturer’s protocol. Briefly, cells were washed twice with cold PBS and fixed in the fixative solution for 10–15 min. After washing with PBS twice again, cells were then incubated in SA-β-gal staining solution overnight at 37 °C.
Immunoblotting
For western blotting, cells were seeded in complete medium on petri dish at a density of 4×104/cm2 24 hours before collection. Immunoblotting procedure was described previously 33. Briefly, cells were lysed in SDS-lysis buffer (10 mM Tris pH 7.5, 1% SDS, 50 mM NaF, 1 mM NaVO4) and subjected to SDS-PAGE on 4–12% Bis-Tris SDS-PAGE gels (Invitrogen) and transferred to Immobilon-P membranes (Millipore). Membranes were incubated in blocking buffer (5% skim milk, 0.1% Tween, 10 mM Tris at pH 7.6, 100 mM NaCl) for 1 hour at room temperature and then with primary antibodies diluted in blocking buffer overnight at 4°C. After three washes, the membranes were incubated with goat anti-rabbit HRP-conjugated antibody or goat anti-mouse HRP-conjugated antibody at room temperature for 2 hour and subjected to chemiluminescence using ECL (Pierce #1856136).
Immunofluorescent Staining
Immunofluorescent Staining was described previously 33. Briefly, cells were fixed with 4% paraformaldehyde in PBS for 20 min and incubated in permeabilization buffer (PDT: 0.3% sodium deoxycholate, 0.3% Triton X-100 in PBS) for 30 min on ice. They were then blocked with 5% BSA/PBS at 4°C for 1hr and followed by incubating overnight at 4°C with primary antibodies diluted in 2.5% BSA/0.05% Triton X-100/PBS. After washing with 0.1% Triton X-100/PBS, cells were incubated with secondary antibodies diluted in 2.5% BSA/0.05% Triton X-100/PBS for 2 hours at 4°C. Cells were washed with 0.1% Triton X-100/PBS, rinsed with PBS, and mounted in ProLong Gold Mountant (Invitrogen #P10144). When indicated, nuclei were stained with DAPI.
Gene Expression and Silencing
Lentiviral vectors encoding shRNAs targeting TAZ (#07: TRCN0000370007; #69: TRCN0000019469) were from Sigma-Aldrich and described previously 34. ON-TARGETplus SMARTpool siRNAs against Lats1, Lats2 or ON-TARGETplus siCONTROL were from Dharmacon.
RNA-sequencing and Data Processing
RNA sequencing was performed on an Illumina HiSeq 2500 for 50 cycles using a single-read recipe according to the manufacturer’s instructions. Sequencing data were analyzed using Strand NGS. Briefly, reads were aligned to reference human genome and annotation file (GRCh38, build 38, RefSeq genes and transcripts, 2017_01_13). One-way ANOVA was performed using p-value threshold=0.05. For Hierarchical clustering analysis, genes showing changes above 2 folds were used. For ingenuity pathway analysis, 1.8-fold was used as the cut-off of downregulated genes, whereas 2-fold was used for upregulated genes. Direct relationships were chosen. For upstream analysis, the p-value cut-off is 0.01.
Quantitative RT-PCR
Cells were seeded in complete medium on petri dish at a density of 4×104/cm2 24 hours before collection. q-RT-PCR was carried out according to standard protocols. Briefly, total RNA was extracted using TRIzol reagent (Invitrogen). cDNAs were synthesized with iScript cDNA Synthesis kit (Bio-Rad, 1708891), and qPCR was carried out on a CFX96 Touch Real-Time PCR Detection System with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, 1725271). GAPDH was used as an internal reference to normalize the input cDNA.
MTT Cell viability assay
Irradiated or non-irradiated cells were seeded in complete medium at a density of 3000 cells per well in 96-well plates. After culturing for three days, cells were treated with XAV939 at indicated concentrations for additional three days. Viable cells were determined using the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) (Invitrogen #M6494) reagent according to the manufacturer’s protocols (Invitrogen). Each assay consisted of two or three replicate wells. Data were expressed as ratios to control cells (treated by DMSO).
Statistical Methods
Statistical significance was determined by unpaired two-tailed student’s t-test unless indicated otherwise. For statistical analyses, samples sizes were chosen based on if the differences between groups are biologically meaningful and are statistically significant. No data were excluded from the analyses. For cell experiments, all cells in one experiment were from the same pooled parental cells. For data collection relying on objective instruments, such as plate reader, q-PCR, Microscopy software and western blotting, the investigators were not blinded to group allocation during data collection. The variance is similar between the groups that are being statistically compared. All error bars shown were standard error of the mean. All statistical calculations and plotting were performed using Graphpad Prism 7.
Supplementary Material
Acknowledgements
We thank Brian J. Lorah, Kim Powell, Bryan E. Achey and Steven P. Leibig in the Division of Health Physics, Yuka Imamura Kawasawa in the Institute for Personalized Medicine of Penn State College of Medicine for technical support. We thank Dong Kuang and Yan-Bing Chen in Department of Pathology of Tongji Hospital, Tongji Medical College, HUST for pathologic analysis. This work was supported by the National Natural Science Foundation of China 81472364 (to B.W.), the National Institutes of Health Grant K22 5K22CA190440 (to W.L.), the American Cancer Society-Institutional Research Grant 124171-IRG-13-043-01 (to W.L.), and the Four Diamonds Fund for Pediatric Cancer Research (to W.L.).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Burger PC, Dubois PJ, Schold SC Jr., Smith KR Jr., Odom GL, Crafts DC et al. Computerized tomographic and pathologic studies of the untreated, quiescent, and recurrent glioblastoma multiforme. J Neurosurg 1983; 58: 159–169. [DOI] [PubMed] [Google Scholar]
- 2.Zamorano L, Katanick D, Dujovny M, Yakar D, Malik G, Ausman JI. Tumour recurrence vs radionecrosis: an indication for multitrajectory serial stereotactic biopsies. Acta Neurochir Suppl (Wien) 1989; 46: 90–93. [DOI] [PubMed] [Google Scholar]
- 3.Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J Neurosurg 2006; 105: 111–118. [DOI] [PubMed] [Google Scholar]
- 4.Godoy PR, Mello SS, Magalhaes DA, Donaires FS, Nicolucci P, Donadi EA et al. Ionizing radiation-induced gene expression changes in TP53 proficient and deficient glioblastoma cell lines. Mutat Res 2013; 756: 46–55. [DOI] [PubMed] [Google Scholar]
- 5.Camphausen K, Purow B, Sproull M, Scott T, Ozawa T, Deen DF et al. Orthotopic growth of human glioma cells quantitatively and qualitatively influences radiation-induced changes in gene expression. Cancer Res 2005; 65: 10389–10393. [DOI] [PubMed] [Google Scholar]
- 6.Tsai MH, Cook JA, Chandramouli GV, DeGraff W, Yan H, Zhao S et al. Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation. Cancer Res 2007; 67: 3845–3852. [DOI] [PubMed] [Google Scholar]
- 7.Ma H, Rao L, Wang HL, Mao ZW, Lei RH, Yang ZY et al. Transcriptome analysis of glioma cells for the dynamic response to gamma-irradiation and dual regulation of apoptosis genes: a new insight into radiotherapy for glioblastomas. Cell Death Dis 2013; 4: e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bassi C, Mello SS, Cardoso RS, Godoy PD, Fachin AL, Junta CM et al. Transcriptional changes in U343 MG-a glioblastoma cell line exposed to ionizing radiation. Hum Exp Toxicol 2008; 27: 919–929. [DOI] [PubMed] [Google Scholar]
- 9.Otomo T, Hishii M, Arai H, Sato K, Sasai K. Microarray analysis of temporal gene responses to ionizing radiation in two glioblastoma cell lines: up-regulation of DNA repair genes. J Radiat Res 2004; 45: 53–60. [DOI] [PubMed] [Google Scholar]
- 10.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157–173. [DOI] [PubMed] [Google Scholar]
- 11.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010; 17: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017; 32: 42–56 e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhat KP, Salazar KL, Balasubramaniyan V, Wani K, Heathcock L, Hollingsworth F et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev 2011; 25: 2594–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015; 163: 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst 2010; 102: 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011; 11: 503–511. [DOI] [PubMed] [Google Scholar]
- 17.Xie Q, Chen J, Feng H, Peng S, Adams U, Bai Y et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res 2013; 73: 3615–3624. [DOI] [PubMed] [Google Scholar]
- 18.Jin H, Lian N, Zhang F, Bian M, Chen X, Zhang C et al. Inhibition of YAP signaling contributes to senescence of hepatic stellate cells induced by tetramethylpyrazine. Eur J Pharm Sci 2017; 96: 323–333. [DOI] [PubMed] [Google Scholar]
- 19.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–740. [DOI] [PubMed] [Google Scholar]
- 20.Schosserer M, Grillari J, Breitenbach M. The Dual Role of Cellular Senescence in Developing Tumors and Their Response to Cancer Therapy. Front Oncol 2017; 7: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol 2008; 28: 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu CY, Zha ZY, Zhou X, Zhang H, Huang W, Zhao D et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem 2010; 285: 37159–37169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S et al. Role of TAZ as Mediator of Wnt Signaling. Cell 2012;151:1443–56. [DOI] [PubMed] [Google Scholar]
- 24.Van Meir EG, Kikuchi T, Tada M, Li H, Diserens AC, Wojcik BE et al. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res 1994; 54: 649–652. [PubMed] [Google Scholar]
- 25.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009; 461: 614–20. [DOI] [PubMed] [Google Scholar]
- 26.Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 2009; 5: 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tihan T, Barletta J, Parney I, Lamborn K, Sneed PK, Chang S. Prognostic value of detecting recurrent glioblastoma multiforme in surgical specimens from patients after radiotherapy: should pathology evaluation alter treatment decisions? Hum Pathol 2006; 37: 272–282. [DOI] [PubMed] [Google Scholar]
- 28.Bernhard EJ, Maity A, Muschel RJ, McKenna WG. Effects of ionizing radiation on cell cycle progression. A review. Radiat Environ Biophys 1995; 34: 79–83. [DOI] [PubMed] [Google Scholar]
- 29.Lv D, Li Y, Zhang W, Alvarez AA, Song L, Tang J et al. TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat Commun 2017; 8: 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang LH, Yin AA, Cheng JX, Huang HY, Li XM, Zhang YQ et al. TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene 2015; 34: 600–610. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Cheng P, Pavlyukov MS, Yu H, Zhang Z, Kim SH et al. Targeting NEK2 attenuates glioblastoma growth and radioresistance by destabilizing histone methyltransferase EZH2. J Clin Invest 2017; 127: 3075–3089. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Jin X, Kim LJY, Wu Q, Wallace LC, Prager BC, Sanvoranart T et al. Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nat Med 2017; 23: 1352–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014; 26: 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Wei Y, Zhang L, Yee PP, Johnson M, Zhang X et al. Induction of store-operated calcium entry (SOCE) suppresses glioblastoma growth by inhibiting the Hippo pathway transcriptional coactivators YAP/TAZ. Oncogene 2018. 10.1038/s41388-018-0425-7. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.