

Abstract
C—H functionalization is an attractive strategy to construct and diversify molecules. Heme proteins, predominantly cytochromes P450, are responsible for an array of C—H oxidations in biology. Recent work has coupled concepts from synthetic chemistry, computation, and natural product biosynthesis to engineer heme protein systems to deliver products with tailored oxidation patterns. Heme protein catalysis has been shown to go well beyond these native reactions and now accesses new-to-nature C—H transformations, including C—N and C—C bond forming processes. Emerging work with these systems moves us along the ambitious path of building complexity from the ubiquitous C—H bond.
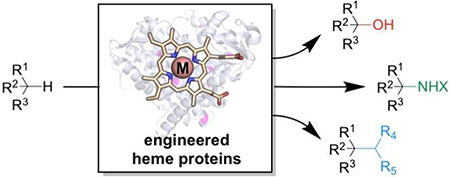
Graphical Abstract
Introduction
Selective replacement of the ubiquitous carbon—hydrogen (C—H) bond with a carbon—heteroatom or a carbon—carbon bond is an outstanding synthetic chemistry challenge to which engineered enzymes are starting to make important contributions. Collectively termed C—H functionalization, this set of reactions has the immense potential to change the logic of chemical synthesis [1, 2]. Though its development in synthetic chemistry has mainly been realized in the last few decades, nature has utilized a C—H functionalization approach to diversify molecules for eons. Most biological C—H functionalization reactions are catalyzed by cytochromes P450, a superfamily of heme-thiolate monooxygenases [3]. Inspired by the heme cofactor, early work with small-molecule transition metal catalysts for C—H hydroxylation employed the porphyrin scaffold [4]. It has since been demonstrated that porphyrin is a versatile scaffold for diverse C—H functionalization reactions (Figure 1a) [5, 6].
Figure 1.

Porphyrin is a versatile scaffold for C—H bond functionalization. (a) Porphyrin-based transition metal catalysts which functionalize C—H bonds. Examples are from ref. [4–6]. X, (1S,4R,5R,8S)-1,2,3,4,5,6,7,8-octahydro-1,4:5,8-dimethanoanthracene-9-yl. †Corresponding ketones were also formed; ketone formation is not due to further oxidation of alcohol products [4]. (b) Proteins which contain a porphyrin group have been engineered by directed evolution to perform C—H oxidation reactions with increased activity or tailored site-selectivity, C—H amination, and carbene C—H insertion. Structural models are Bacillus megaterium cytochrome P450BM3 (PDB 1JPZ, top and middle) and an engineered C—H amination enzyme derived from P450BM3 (PDB 5UCW, bottom); Y, vinyl or ethyl; Z, amino acid or organic functional group.
We believe that nature’s heme proteins have great potential for C—H functionalization, including catalyzing reactions with no biological counterparts. Enzymes could conceivably offer catalyst-controlled selectivity, high turnover numbers, or eliminate dependence on toxic noble metals, which are desirable advances for the field of C—H functionalization [1, 2]. While protein engineering has historically focused on improving the known function of an enzyme [7], a recent paradigm shift has expanded this vision. The initial focus is now on the target reaction, such as a C—H functionalization transformation, and the necessary elements to achieve that chemistry, such as the ability to bind porphyrin. Protein engineers then search through the vast collection of existing proteins, select those which have the necessary parts (it can also be beneficial to introduce artificial cofactors or computationally design a suitable protein [8]), and test for the desired activity. Once even a low level of the activity has been found, a new enzyme can be created by directed evolution, a protein engineering strategy which uses iterative cycles of mutagenesis and screening to accumulate beneficial mutations that enhance catalyst performance (Figure 1b). This approach has generated porphyrin-containing enzymes which oxidize C—H bonds with tailored site-selectivity and perform new-to-nature C—H amination and C—C bond forming reactions.
In this short opinion piece, we survey C—H functionalization transformations catalyzed by engineered heme proteins and the methods used to introduce or optimize these functions. These efforts are compelling precedents for expanding the chemistry accessible to proteins, and we anticipate that they will inform and inspire exploration of other protein systems for new catalytic functions.
Oxidative transformations catalyzed by engineered cytochromes P450
Cytochromes P450 are nature’s most prevalent catalysts for C—H functionalization [9]. Enzymes of this vast family directly activate inert C—H bonds for a broad spectrum of oxidative transformations such as hydroxylation, desaturation, decarboxylation, and carbon-skeleton rearrangement [10, 11]. The exceptional activities of P450s have driven interest in utilizing them for diverse synthetic purposes [12]. With their remarkable capability for site-selective catalysis, P450s can be useful for late-stage molecular diversification. The use of a directing group to effect site-selectivity, a concept commonly used in small-molecule catalysis, has been combined with P450-catalysis using PikC from Streptomyces venezuelae [13]. The natural substrates of PikC are macrolides, macrocyclic lactones with a desosamine sugar; the desosamine sugar acts as an anchoring group and the N,N-dimethylamino moiety forms a salt bridge with a protein glutamate residue [14]. From substrate engineering studies, it was found that various synthetic N,N-dimethylamino and N,N-diethylamino groups are also suitable anchoring groups and affect site-selectivity in PikC catalysis [15, 16••]. In a powerful illustration of molecular diversification by enzymatic C–H functionalization, the merger of nickel and PikC catalysis accessed five different hydroxylated macrocyclic lactone products from a common linear intermediate (Figure 2a) [16••]. In this reaction, nickel-catalyzed regiodivergent cyclization converted the linear intermediate into 11- and 12-membered macrocycles; after appending anchoring groups, a single enzyme variant acted on each substrate to hydroxylate at a different position with good regioselectivity. Complementary to a substrate engineering approach, the application of homologous P450s from different natural product pathways can also access diverse outcomes available to a molecule. Starting from one intermediate, a combination of polyketide synthase (PKS) modules, in vivo glycosylation, and three different P450s delivered several tylactonebased macrolide antibiotics with varied oxygenation patterns (Figure 2b) [17•].
Figure 2.

Cytochromes P450 catalyze diverse selective oxidative transformations. (a) Synthesis of macrocyclic lactones by merging nickel-catalyzed cyclization with P450-catalyzed C—H hydroxylation [16••]. (b) PKS catalysis followed by glycosylation and P450-catalyzed oxidation affords tylactone-based macrolides [17•]. (c) Site-selective oxidation by P450 8C7 at the C7 position of an advanced intermediate in the total synthesis of nigelladine A [19••]. ADH, alcohol dehydrogenase; KPi, potassium phosphate; DMP, Dess–Martin periodinane.
P450s have also inspired new strategies for complex molecule synthesis by offering catalysts that address challenging selectivity issues [18]. A compelling example is the first enantioselective total synthesis of nigelladine A [19••]. In this synthesis, a site-selective allylic oxidation of a tricyclic intermediate at the C7 position is required. However, the presence of multiple reactive allylic C—H bonds in this intermediate significantly complicated the seemingly straightforward transformation. Indeed, a survey of a broad range of chemical oxidation methods only led to mixtures of inseparable mono-oxidation and over-oxidation products. This synthetic challenge was solved by P450BM3 variant 8C7, which was identified through screening a small set of P450BM3 variants originally engineered for oxidation of large substrates with privileged scaffolds [20]. P450 8C7 efficiently catalyzed the desired C7 oxidation with up to 1700 total turnovers and enabled a concise synthesis of nigelladine A (Figure 2c).
Computational methods such as molecular dynamics (MD) simulations have emerged as powerful tools to facilitate the laboratory evolution/engineering of P450s [21]. These in silico methods can unveil key residues involved in important dynamic interactions that are not revealed by static structural data. Employing such a computation-driven approach, Narayan et al. expanded the scope of P450 PikC to include six-membered small ring systems [22•]. In another demonstration, by combining large-scale MD simulations with site-saturation mutagenesis, Dodani et al. identified several mutations (His176Phe/Tyr/Trp) that completely redirect the regioselectivity of P450 TxtE-catalyzed nitration from the C4 to the C5 position of L-tryptophan [23•]. Additionally, there are also several studies that employ docking and MD simulation to identify important mutational hotspots for improving selectivity or substrate specificity of P450 hydroxylation [24, 25, 26].
Common to the many thousands of P450s is the conserved cysteine residue that acts as an axial ligand to the heme iron. This axial cysteine is crucial for the C—H activation activity of P450 compound I [27, 28]. In a study of the thermostable Sulfolobus acidocaldarius CYP119, mutation of the axial cysteine ligand to all other canonical amino acids created mutants which still folded and incorporated heme [29]. The crystal structure of a histidine-ligated CYP119 variant exhibited a tilted heme accompanied by a large rearrangement of the protein structure. While mutation of the axial ligand typically results in diminished or abolished hydroxylation activity, Green et al. discovered that the hydroxylation activity of CYP119 compound I could be enhanced by mutation of the axial cysteine to selenocysteine [30••]. This finding may open a new avenue for the development of robust P450 catalysts for additional challenging transformations.
Advancing biocatalytic C—H amination using directed evolution
The frequent presence of nitrogen in natural products and drug molecules drives the search for methods to form C—N bonds. Biological systems typically rely on enzymatic functional group manipulation of pre-oxidized substrates to forge this bond. This approach has been applied in the design of multi-enzyme biocatalytic cascades for formal C—H amination (Figure 3a) [31, 32]. As a complement to nature’s biosynthetic logic, recent work with engineered heme proteins has identified enzymes which directly install a C—N bond in place of an sp3-hydridized C—H bond.
Figure 3.

Representative examples of engineered heme proteins used for C—H amination. (a) Biocatalytic cascade for formal C—H amination [31]. (b) Cytochrome P450-catalyzed reactions of lidocaine [33]. Distribution between N-dealkylation and cyclization products is controlled by mutations to the protein scaffold. (c) Intermolecular C—H amination catalyzed by an engineered cytochrome P450 [43••]. This reaction proceeds through a putative iron-nitrene intermediate. Four beneficial mutations, whose positions are shown as spheres in the structural model (PDB 5UCW), were accumulated in the directed evolution of a C—H amination enzyme. †The initial protein was an engineered variant of P450BM3 which differs from wild-type by seventeen mutations.
In a study to produce metabolites of drug molecules, mutants of cytochrome P450BM3 were discovered to perform an unusual cyclization reaction on lidocaine (Figure 3b) [33]. The cyclization, an intramolecular C—H amination reaction, competes with N-dealkylation, a known reaction of P450s. The distribution of the two products is entirely controlled by the protein scaffold: two variants with divergent selectivity for N-dealkylation and cyclization differed by a single amino acid. Further work created a set of P450BM3 variants which performed α-functionalization of diverse 2-aminoacetamides and thioamides [34•]. In the proposed mechanism, P450 compound I is involved in the formation of an iminium species which subsequently undergoes cyclization. Examples of this transformation are limited to functionalization of α-amino C—H bonds, in agreement with the proposed mechanism. Wild-type P450BM3 did not catalyze the cyclization reaction or gave only low conversion (<5%) on model substrates, demonstrating that the discovered mutations promote the chemistry [34•].
Heme proteins are not limited to the reactions of iron-oxo intermediates. When given the opportunity to interact with certain nitrogen-containing substrates, heme proteins can putatively form iron-nitrene species, which can then perform C—H amination. Though first demonstrated nearly 35 years ago [35], it was only with the advent of modern directed evolution techniques that variants of P450BM3 adopted the C—H amination function with synthetically useful levels of activity [36, 37]. Remarkably, these enzymes can be engineered to alter the regioselectivity of amination in an intramolecular system and override substrate reactivity patterns [38]. The creation of an intramolecular C—H amination enzyme does not necessitate use of an enzyme scaffold: Physeter catodon myoglobin, which has no known natural catalytic function, was also engineered to perform this chemistry [39]. Additional testing of engineered heme proteins [40] and directed evolution of heme protein-derived catalysts, including a CYP119 derivative in which the heme group was replaced with Ir(Me)-porphyrin [41], further expanded the scope of enzymatic C—H amination.
Intermolecular C—H amination affords increased synthetic flexibility. However, changing from an intramolecular to an intermolecular reaction is a fundamental challenge in catalysis [42], and to date there is only one report of heme protein-catalyzed intermolecular C—H amination [43••]. In the intermolecular reaction, C—H insertion must compete with iron-nitrene decomposition pathways without the implicit proximity advantage of an intramolecular arrangement. Nonetheless, directed evolution found a protein scaffold which overcame these challenges: a serine-ligated P450BM3 variant delivered seventeen different chiral amine compounds with good turnovers and high enantioselectivity (Figure 3c).
Previously thought to be absent from natural enzyme mechanisms, an iron-nitrene has been put forth as a possible intermediate for recently discovered cytochrome P450 BezE involved in benzastatin biosynthesis [44•]. This is an excellent demonstration of how findings from biocatalysis can inform mechanistic possibilities for enzymes in complex biosynthetic pathways. At the same time, the discovery of new enzymes provides biocatalysis with an increasing repertoire of starting points for the implementation of new chemistry.
Engineering heme proteins for C—C bond formation
A prevalent belief in biocatalysis is that proteins cannot access the diversity of chemical transformations available to synthetic chemistry. Some of this comes from the observation that biological systems appear to use just a small set of elements from the periodic table; for instance, known natural enzymes access their powerful and varied chemistry using predominantly earth-abundant first-row transition metals. In contrast, small-molecule catalysts for certain classes of reactions, such as sp3 C—H functionalization, commonly employ noble metals [1, 2].
The creation of artificial metalloenzymes which contain noble metal complexes [45, 46], including the replacement of the heme group in heme proteins with porphyrins containing alternative metals, is one strategy to expand the scope of reactions accessible to enzymes. Replacing the iron-porphyrin cofactor with an iridium-porphyrin creates artificial metalloenzymes which install a new C—C bond in place of an sp3 C—H bond (Figure 4a). First demonstrated using Physeter catodon myoglobin, protein variants containing the Ir(Me)-porphyrin complex and engineered by directed evolution catalyze enantioselective intramolecular carbene C—H insertion to deliver cyclic ether products [47••]. In agreement with previously observed reactivity patterns [6], the free Ir(Me)-porphyrin complex catalyzes the model reaction with a higher reaction rate than an Ir(Me)-porphyrin myoglobin enzyme. Changing the protein scaffold to apo-CYP119 and subsequent directed evolution, however, delivered an artificial metalloenzyme with 23-fold higher turnover frequency compared with the free cofactor [48•]. Intermolecular C—H functionalization of phthalan was accomplished using evolved variants of Ir(Me)-porphyrin CYP119 [48•] and variants of P. catodon apo-myoglobin equipped with porphyrins containing alternative metals, including iridium [49]. In contrast, the alkylation of sp2-hybridized C—H bonds of unprotected indole substrates has been achieved using variants of myoglobin which retain their native heme group. Chemoselective for C3 functionalization, the alkylation occurs through electrophilic aromatic substitution rather than a C—H insertion mechanism which is expected for sp3 C—H functionalization [50•].
Figure 4.

C—H to C—C bond transformations catalyzed by artificial metalloenzymes and cytochromes P450. (a) Replacement of heme in myoglobin with Ir(Me)-mesoporphyrin IX results in an artificial metalloenzyme which catalyzes carbene C—H insertion, a reaction that the iron-based enzyme does not catalyze [47••]. Cartoons were created using PDB 1MBN; porphyrin cofactors have been enlarged (not to scale). (b) The IPC intermediate has been captured by X-ray crystallography in two poses, end-on (left, PDB 6CUN) and Fe–C– N(pyrrole) bridging (right, PDB 6G5B), in engineered cytochrome c and myoglobin carbene transferases, respectively [51•, 52•]. (c) P450 catalyzed C—C bond forming reactions in natural product biosynthesis. Representative examples are from ref. [10], [56], and [57] (left to right).
The described non-natural C—C bond-forming reactions proceed via the intermediacy of an electrophilic metal-carbene species. Remarkably, iron porphyrin carbene (IPC) intermediates in two engineered proteins have now been captured by X-ray crystallography and studied spectroscopically. Two recent reports show that the IPC can exist as either an end-on adduct [51•], which is responsible for carbene transfer activity, or have a Fe–C–N(pyrrole) bridging configuration, which (if formed) is in equilibrium with the end-on adduct (Figure 4b) [52•]. These reports illustrate that the protein scaffold affects bonding and the electronic state of the IPC, and can even dictate the orientation of this intermediate [51•]. Together with knowledge gained from quantum chemical calculations on iron-porphyrin carbene systems [53, 54] and experimental work with small-molecule systems [55], these studies can inform new approaches to expand the limits of heme protein-catalyzed C—H functionalization.
Natural heme proteins have their own strategies to mediate C—H to C—C bond conversions [10]. Many of these transformations are catalyzed by cytochromes P450 and result from radical coupling (e.g. synthesis of salutaridine from reticuline in morphine biosynthesis [10], phenol coupling in fungal natural products [56]) or rearrangements of substrate radicals followed by quenching (e.g. transformation littorine to hyoscyamine aldehyde in tropane alkaloid biosynthesis [57]) (Figure 4c). The potentials of these enzymes for novel reactions have yet to be explored.
Outlook
New enzymes (some even with new cofactors [58]) are being discovered every day. Considering only cytochromes P450, there are more than 206,000 genes known in 2018 [10] vs. only 18,000 just five years ago [59]. The considerable diversity of heme proteins and their proven evolvability supply a fertile landscape for the discovery and optimization of new reactions. Although new-to-nature C—N and C—C bond-forming processes have not yet been applied in the context of complex molecule synthesis or diversification, the achievements of P450-catalyzed site-selective hydroxylation provide a roadmap. Looking forward, the successes of engineering heme proteins for diverse chemistry should stimulate experimentation with other groups of proteins for non-native catalysis [60]. Approaching nature’s protein diversity with a new chemical perspective and a powerful set of protein engineering tools, we see a bright future for creating new enzymes for selective C—H bond functionalization.
Highlights.
Interplay between synthetic chemistry and enzyme discovery/creation.
Cytochromes P450 for selective hydroxylation of complex molecules.
Engineered heme proteins catalyze non-natural C—H functionalization reactions.
Acknowledgments
This work was supported by the NSF, Division of Molecular and Cellular Biosciences (grant MCB-1513007). R. K. Z. acknowledges support from the NSF Graduate Research Fellowship (grant DGE-1144469) and the Donna and Benjamin M. Rosen Bioengineering Center. X. H. is supported by an NIH pathway to independence award (grant K99GM129419).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
• • of outstanding interest
- [1].Hartwig J: Evolution of C—H bond functionalization from methane to methodology. J Am Chem Soc 2016, 138: 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yamaguchi J, Yamaguchi AD, Itami K: C—H bond functionalization: Emerging synthetic tools for natural products and pharmaceuticals. Angew Chem Int Ed 2012, 51: 8960–9009. [DOI] [PubMed] [Google Scholar]
- [3].Poulos TL: Heme enzyme structure and function. Chem Rev 2014, 114: 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Groves JT, Viski P: Asymmetric hydroxylation, epoxidation, and sulfoxidation catalyzed by vaulted binaphthyl metalloporphyrins. J Org Chem 1990, 55: 3628–3634. [Google Scholar]
- [5].Liang J-L, Yuan S-X, Huang J-S, Yu W-Y, Che C-M: Highly diastereo‐ and enantioselective intramolecular amidation of saturated C—H bonds catalyzed by ruthenium porphyrins. Angew Chem Int Ed 2002, 41: 3465–3468. [DOI] [PubMed] [Google Scholar]
- [6].Wang J-C, Zhang Y, Xu Z-J, Lo VK, Che C-M: Enantioselective intramolecular carbene C—H insertion catalyzed by a chiral iridium(III) complex of D4-symmetric porphyrin ligand. ACS Catal 2013, 3: 1144–1148. [Google Scholar]
- [7].Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K: Engineering the third wave of biocatalysis. Nature 2012, 485: 185–194. [DOI] [PubMed] [Google Scholar]
- [8].Kries H, Blomberg R, Hilvert D: De novo enzymes by computational design. Curr Opin Chem Biol 2013, 17: 221–228. [DOI] [PubMed] [Google Scholar]
- [9].Lewis JC, Coelho PS, Arnold FH: Enzymatic functionalization of carbon—hydrogen bonds. Chem Soc Rev 2011, 40: 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guengerich FP, Yoshimoto FK: Formation and cleavage of C—C bonds by enzymatic oxidation–reduction reactions. Chem Rev 2018, 118: 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang X, Groves JT: Oxygen activation and radical tansformations in heme proteins and metalloporphyrins. Chem Rev 2018, 118: 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Girvan HM, Munro AW: Applications of microbial cytochrome P450 enzymes in biotechnology and synthetic biology. Curr Opin Chem Biol 2016, 31: 136–145. [DOI] [PubMed] [Google Scholar]
- [13].Li SY, Chaulagain MR, Knauff AR, Podust LM, Montgomery J, Sherman DH: Selective oxidation of carbolide C—H bonds by an engineered macrolide P450 mono-oxygenase. Proc Natl Acad Sci U S A 2009, 106: 18463–18468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li SY, Ouellet H, Sherman DH, Podust LM: Analysis of transient and catalytic desosamine-binding pockets in cytochrome P450 PikC from Streptomyces venezuelae. J Biol Chem 2009, 284: 5723–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Negretti S, Narayan ARH, Chiou KC, Kells PM, Stachowski JL, Hansen DA, Podust LM, Montgomery J, Sherman DH: Directing group-controlled regioselectivity in an enzymatic C—H bond oxygenation. J Am Chem Soc 2014, 136: 4901–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••[16].Gilbert MM, DeMars MD, Yang S, Grandner JM, Wang S, Wang H, Narayan ARH, Sherman DH, Houk KN, Montgomery J: Synthesis of diverse 11- and 12-membered macrolactones from a common linear substrate using a single biocatalyst. ACS Cent Sci 2017, 3: 1304–1310. Describes a modular strategy for enabling site-selective C—H oxidation using a single engineered P450 and a set of linkers which modify the substrate. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[17].Lowell AN, DeMars MD, Slocum ST, Yu F, Anand K, Chemler JA, Korakavi N, Priessnitz JK, Park SR, Koch AA, et al. : Chemoenzymatic total synthesis and structural diversification of tylactone-based macrolide antibiotics through latestage polyketide assembly, tailoring, and C—H functionalization. J Am Chem Soc 2017, 139: 7913–7920. A synthetic intermediate processed through polyketide synthase modules and in vivo glycosylation is diversified to eight products with different oxidation patterns by a combination of P450 catalysis and a small-molecule oxidation system. Suitable P450s to access divergent oxidations were identified from several biosynthetic pathways. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].King-Smith E, Zwick CR, Renata H: Applications of oxygenases in the chemoenzymatic total synthesis of complex natural products. Biochemistry 2018, 57: 403–412. [DOI] [PubMed] [Google Scholar]
- ••[19].Loskot SA, Romney DK, Arnold FH, Stoltz BM: Enantioselective total synthesis of nigelladine A via late-stage C—H oxidation enabled by an engineered P450 enzyme. J Am Chem Soc 2017, 139: 10196–10199. The first enantioselective total synthesis of nigelladine A was accomplished using an engineered P450 for site-selective oxidation of an advanced intermediate; chemical methods tested for this step either did not form the desired product or delivered a mixture of inseparable oxidized products. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong C-H, Arnold FH: Combinatorial alanine substitution enables rapid optimization of cytochrome P450BM3 for selective hydroxylation of large substrates. ChemBioChem 2010, 11: 2502–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Romero-Rivera A, Garcia-Borras M, Osuna S: Computational tools for the evaluation of laboratory-engineered biocatalysts. Chem Commun 2017, 53: 284–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[22].Narayan ARH, Jiménez-Osés G, Liu P, Negretti S, Zhao W, Gilbert MM, Ramabhadran RO, Yang Y-F, Furan LR, Li Z, et al. : Enzymatic hydroxylation of an unactivated methylene C—H bond guided by molecular dynamics simulations. Nat Chem 2015, 7: 653–660. Molecular dynamics simulations identified amino acid residues in P450 PikC for mutagenesis to promote catalysis. Mutations at these positions created an enzyme with improved turnover and high regioselectivity for oxidation of a methylene C—H bond distal to the anchoring group, overriding inherent substrate reactivity patterns. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[23].Dodani SC, Kiss G, Cahn JKB, Su Y, Pande VS, Arnold FH: Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models. Nat Chem 2016, 8: 419–425. Molecular dynamics aids in the discovery of a single amino acid position which controls both protein conformation and the regioselectivity of tryptophan nitration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Petrović D, Bokel A, Allan M, Urlacher VB, Strodel B: Simulation-guided design of cytochrome P450 for chemo- and regioselective macrocyclic oxidation. J Chem Inf Model 2018, 58: 848–858. [DOI] [PubMed] [Google Scholar]
- [25].Acevedo-Rocha CG, Gamble CG, Lonsdale R, Li A, Nett N, Hoebenreich S, Lingnau JB, Wirtz C, Fares C, Hinrichs H, et al. : P450-catalyzed regio- and diastereoselective steroid hydroxylation: efficient directed evolution enabled by mutability landscaping. ACS Catal 2018, 8: 3395–3410. [Google Scholar]
- [26].Ebert MCCJC, Guzman Espinola J, Lamoureux G, Pelletier JN: Substrate-specific screening for mutational hotspots using biased molecular dynamics simulations. ACS Catal 2017, 7: 6786–6797. [Google Scholar]
- [27].Green MT: C—H bond activation in heme proteins: the role of thiolate ligation in cytochrome P450. Curr Opin Chem Biol 2009, 13: 84–88. [DOI] [PubMed] [Google Scholar]
- [28].Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT: Iron(IV)hydroxide pK(a) and the role of thiolate ligation in C—H bond activation by cytochrome P450. Science 2013, 342: 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McIntosh JA, Heel T, Buller AR, Chio L, Arnold FH: Structural adaptability facilitates histidine heme ligation in a cytochrome P450. J Am Chem Soc 2015, 137: 13861–13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••[30].Onderko EL, Silakov A, Yosca TH, Green MT: Characterization of a selenocysteineligated P450 compound I reveals direct link between electron donation and reactivity. Nat Chem 2017, 9: 623–628. Changing the native cysteine axial ligand of CYP119 to selenocysteine delivered an enzyme which forms compound I with increased activity for C—H oxidation. [DOI] [PubMed] [Google Scholar]
- [31].Both P, Busch H, Kelly PP, Mutti FG, Turner NJ, Flitsch SL: Whole-cell biocatalysts for stereoselective C—H amination reactions. Angew Chem Int Ed 2016, 55: 1511–1513. [DOI] [PubMed] [Google Scholar]
- [32].Schrewe M, Ladkau N, Bühler B, Schmid A: Direct terminal alkylamino-functionalization via multistep biocatalysis in one recombinant whole-cell catalyst. Adv Synth Catal 2013, 355: 1693–1697. [Google Scholar]
- [33].Ren X, Yorke JA, Taylor E, Zhang T, Zhou W, Wong LL: Drug oxidation by cytochrome P450BM3: Metabolite synthesis and discovering new P450 reaction types. Chem Eur J 2015, 21: 15039–15047. [DOI] [PubMed] [Google Scholar]
- •[34].Ren X, O’Hanlon JA, Morris M, Robertson J, Wong LL: Synthesis of imidazolidin-4-ones via a cytochrome P450-catalyzed intramolecular C—H amination. ACS Catal 2016, 6: 6833–6837. Example of an alternative reaction accessible to the iron-oxo intermediate of P450; includes mechanistic hypothesis. [Google Scholar]
- [35].Svastits EW, Dawson JH, Breslow R, Gellman SH: Functionalized nitrogen atom transfer catalyzed by cytochrome P-450. J Am Chem Soc 1985, 107: 6427–6428. [Google Scholar]
- [36].McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH: Enantioselective intramolecular C—H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew Chem Int Ed 2013, 52: 9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Singh R, Bordeaux M, Fasan R: P450-catalyzed intramolecular sp3 C—H amination with arylsulfonyl azide substrates. ACS Catal 2014, 4: 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hyster TK, Farwell CC, Buller AR, McIntosh JA, Arnold FH: Enzyme-controlled nitrogen-atom transfer enables regiodivergent C—H amination. J Am Chem Soc 2014, 136: 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bordeaux M, Singh R, Fasan R: Intramolecular C(sp3)—H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg Med Chem 2014, 22: 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Singh R, Kolev JN, Sutera PA, Fasan R: Enzymatic C(sp3)—H amination: P450-catalyzed conversion of carbonazidates into oxazolidinones. ACS Catal 2015, 5: 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dydio P, Key HM, Hayashi H, Clark DS, Hartwig JF: Chemoselective, enzymatic C—H bond amination catalyzed by a cytochrome P450 containing an Ir(Me)-PIX cofactor. J Am Chem Soc 2017, 139: 1750–1753. [DOI] [PubMed] [Google Scholar]
- [42].Bruice TC, Lightstone FC: Ground state and transition state contributions to the rates of intramolecular and enzymatic reactions. Acc Chem Res 1999, 32: 127–136. [Google Scholar]
- ••[43].Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S, Arnold FH: Enantioselective, intermolecular benzylic C—H amination catalysed by an engineered iron-haem enzyme. Nat Chem 2017, 9: 629–634. Synthetically useful and chemically challenging nitrene C—H insertion reaction achieved by directed evolution of a cytochrome P450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[44].Tsutsumi H, Katsuyama Y, Izumikawa M, Takagi M, Fujie M, Satoh N, Shin-ya K, Ohnishi Y: Unprecedented cyclization catalyzed by a cytochrome P450 in benzastatin biosynthesis. J Am Chem Soc 2018, 140: 6631–6639. Identified a cytochrome P450, BezE, which may perform nitrene transfer as its native function in the biosynthesis of benzastatins. [DOI] [PubMed] [Google Scholar]
- [45].Yang H, Swartz AM, Park HJ, Srivastava P, Ellis-Guardiola K, Upp DM, Lee G, Belsare K, Gu Y, Zhang C, Moellering RE, Lewis JC: Evolving artificial metalloenzymes via random mutagenesis. Nat Chem 2018, 10: 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jeschek M, Reuter R, Heinisch T, Trindler C, Klehr J, Panke S, Ward TR: Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537: 661–665. [DOI] [PubMed] [Google Scholar]
- ••[47].Key HM, Dydio P, Clark DS, Hartwig JF: Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534: 534–537. Myoglobin containing Ir(Me)-porphyrin is optimized by active site engineering for increased activity and selectivity for intramolecular carbene C—H insertion reactions. Heme proteins (without iridium) were found to be inactive for this challenging transformation. [DOI] [PubMed] [Google Scholar]
- •[48].Dydio P, Key HM, Nazarenko A, Rha JY-E, Seyedkazemi V, Clark DS, Hartwig JF: An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354: 102–106. Higher catalytic efficiency of intramolecular carbene C—H insertion was obtained when Ir(Me)-porphyrin was introduced to apo-CYP119, compared with previous report on Ir(Me)-porphyrin myoglobin. Subsequent directed evolution created variants of Ir(Me)-porphyrin CYP119 which catalyze an extended set of carbene C—H insertion reactions. [DOI] [PubMed] [Google Scholar]
- [49].Sreenilayam G, Moore EJ, Steck V, Fasan R: Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts. Adv Synth Catal 2017, 359: 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[50].Vargas D, Tinoco A, Tyagi V, Fasan R: Myoglobin‐catalyzed C—H functionalization of unprotected indoles. Angew Chem Int Ed 2018, 57: 9911–9915. Describes the discovery, substrate scope, and mechanistic characterization of engineered myoglobin variants which catalyze C3 alkylation of unprotected indoles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[51].Lewis RD, Garcia-Borràs M, Chalkley MJ, Buller AR, Houk KN, Kan SBJ, Arnold FH: Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase. Proc Natl Acad Sci U S A 2018, 115: 7308–7313. Iron porphyrin carbene intermediate was captured in the reactive end-on conformation in an engineered cytochrome c using X-ray crystallography and studied with spectroscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •[52].Hayashi T, Tinzl M, Mori T, Krengel U, Proppe J, Soetbeer J, Klose D, Jeschke G, Reiher M, Hilvert D: Capture and characterization of a reactive haem–carbenoid complex in an artificial metalloenzyme. Nat Catal 2018, 1: 578–584. The iron porphyrin carbene intermediate can adopt a Fe–C–N(pyrrole) bridging conformation, as shown in this engineered myoglobin containing an axial N-methyl histidine residue. [Google Scholar]
- [53].Khade RL, Zhang Y: C‒H insertions by iron porphyrin carbene: Basic mechanism and origin of substrate selectivity. Chem Eur J 2017, 23: 17654–17658. [DOI] [PubMed] [Google Scholar]
- [54].Sharon DA, Mallick D, Wang B, Shaik S: Computation sheds insight into iron porphyrin carbenes’ electronic structure, formation, and N—H insertion reactivity. J Am Chem Soc 2016, 138: 9597–9610. [DOI] [PubMed] [Google Scholar]
- [55].Liu Y, Xu W, Zhang J, Fuller W, Schulz CE, Li J: Electronic configuration and ligand nature of five-coordinate iron porphyrin carbene complexes: An experimental study. J Am Chem Soc 2017, 139: 5023–5026. [DOI] [PubMed] [Google Scholar]
- [56].Mazzaferro LS, Hüttel W, Fries A, Müller M: Cytochrome P450-catalyzed regio- and stereoselective phenol coupling of fungal natural products. J Am Chem Soc 2015, 137: 12289–12295. [DOI] [PubMed] [Google Scholar]
- [57].Nasomjai P, Reed DW, Tozer DJ, Peach MJG, Slawin AMZ, Covello PS, O’Hagan D: Mechanistic insights into the cytochrome P450-mediated oxidation and rearrangement of littorine in tropane alkaloid biosynthesis. ChemBioChem 2009, 10: 2382–2393. [DOI] [PubMed] [Google Scholar]
- [58].Desguin B, Zhang T, Soumillion P, Hols P, Hu J, Hausinger RP: A tethered niacinderived pincer complex with a nickel-carbon bond in lactate racemase. Science 2015, 349: 66–69. [DOI] [PubMed] [Google Scholar]
- [59].Guengerich FP, Munro AW: Unusual cytochrome P450 enzymes and reactions. J Biol Chem 2013, 288:17065–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Emmanuel MA, Greenberg NR, Oblinsky DG, Hyster TK: Accessing non-natural reactivity by irradiating nicotinamide-dependent enzymes with light. Nature 2016, 540: 414–417 [DOI] [PubMed] [Google Scholar]