

Abstract
We describe a Lab-on-a-Film disposable that detects multi-drug resistant tuberculosis (MDR-TB) from sputum extracts. The Lab-on-a-Film disposable consists of 203 gel elements that include DNA sequences (probes) for 37 mutations, deletions, or insertion elements across 5 genes (including an internal control). These gel elements are printed on a flexible film, which costs approximately 500 times less than microarray glass. The film with printed gel elements is then laminated to additional rollable materials (films) to form a microfluidic flow cell. We combined multiplex amplification and hybridization steps in a single microfluidic chamber, without buffer exchanges or other manipulations up to and throughout hybridization. This flow cell also incorporates post hybridization wash steps while retaining an entirely closed-amplicon system, thus minimizing the potential for sample or amplicon cross-contamination. We report analytical sensitivity of 32 cfu/mL across all MDR-TB markers and detection of MDR-TB positive clinical specimens using an automated TruTip workstation for extraction and the Lab-on-a-Film disposable for amplification and detection of the extracts.
Graphical Abstract
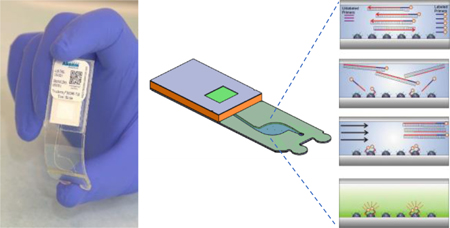
Multiplex PCR and hybridization occur within a closed-amplicon laminate, which includes gel arrays printed on unmodified and untreated plastic film.
Introduction
As of 2017, the World Health Organization (WHO) reports that tuberculosis (TB) is one of the top ten causes of death worldwide and is the leading cause from a single infectious agent, ranking ahead of HIV1. WHO estimates that one-third of the world’s population is currently infected with either active or latent TB, with a reported 10.4 million new cases and 1.3 million deaths in 20162. WHO further reports that of the 0.5 million people infected with multidrug resistant TB (MDR-TB) only 25% are positively detected. Rapid and accurate diagnosis is at the core of the international strategy to control TB, but millions remain undiagnosed or under-diagnosed and thus are either untreated or inappropriately treated.
The challenges for TB diagnostics are numerous3: (1) in order to be useful to the endemic population, the test must be low cost4, (2) there are hundreds of mutations that confer drug resistance (i.e., Sangren et al. reports 106 for rifampin [RIF] and 333 for isoniazid [INH])5 and some mutations have a geographical bias6 (3) sputum is highly heterogeneous and viscous7, (4) the use environment necessitates a simple-to-use test8, and (5) the sensitivity and specificity requirements are demanding3. Despite more focused technology and product development efforts9, there are still very few commercially-available, WHO-endorsed, Conformité Européene (CE)-marked, or Food and Drug Administration (FDA)-cleared drug-resistant TB diagnostics.
There are numerous methods for TB detection8, but there are only two manufacturers who offer WHO-endorsed drug resistance tests: Hain (Nehren, Germany) and Cepheid (Sunnyvale, CA). The Hain line probe assay, which is not an FDA-approved test, is a test for high complexity laboratories that requires skilled technicians to perform. Consequently, Hain developed an alternative Polymerase Chain Reaction (PCR) and melt-curve analysis platform to reduce the operational complexity of the line probe assay.10 Cepheid’s GeneXpert® Mycobacterium tuberculosis (MTB)/RIF cartridge, a sample-to-answer test based on real-time PCR, is widely accepted and promoted in efforts to combat TB11,12. Despite global adoption, the Xpert MTB/RIF test has a limited number of mutations that can be detected within an 81bp region in the rpoB gene and the Xpert does not report which mutations are detected, and it does not include markers for INH. The clinical algorithm has thus evolved into treating patients who test positive for RIF-resistant MTB as though they are infected with MDR-TB. However, MDR-TB is defined by the United States Center for Disease Control as resistance to RIF and INH. Thus, this clinical algorithm introduces a risk of increasing the incidence of INH mono-resistance. Therefore, there is a need for low-cost, simple-to-use tests that allow for resistance detection across more than one gene as well as detection of additional single nucleotide polymorphisms (SNPs) and insertion/deletion elements.
A number of groups are working towards a low-cost, point-of-care test that integrates nucleic acid extraction and analysis into a sample-to-answer system13–16. Liu et al., however, report a two-stage sample-to-answer system where nucleic acid extraction is separated from the amplification and detection system17. Separating extraction from analysis allows users to select their preferred extraction and analysis methods, giving them the flexibility to tailor a workflow to suits their needs.
We previously reported an automated extraction method for isolating MTB DNA from sputum 18,19. We also reported an integrated microfluidic amplification microarray that included a gel element array printed on a glass substrate.20 This microfluidic device does not include on-board pumps or valves, yet maintains a closed system (reducing the potential for contamination) by combining PCR and hybridization in the same chamber with the same reagent mix.21 The cost of the glass substrates, which is unlikely to meet WHO Target Product Profile (TPP) requirements, led us to a fundamental change to manufacturing gel element arrays. While others have described methods of treating and functionalizing plastics for microarray attachment22–25, we describe the implementation of an untreated, off-the-shelf flexible film, that is used as a substrate for gel element attachment as a low-cost alternative to glass. Film substrates open the possibility for manufacturing methodologies such as reel-to-reel production26,27, the assembly process used to manufacture lateral flow strips. In this work, we use the previously reported automated extraction workstation and an all-plastic Lab-on-a-Film amplification microarray and demonstrate its potential for detecting MDR-TB from sputum extracts.
Materials and Methods
Assembly
Figure 1 shows the assembly and workflow of the Lab-on-a-Film disposable, which supports amplification, microarray hybridization, washing, and imbibition (so that the reaction chamber is in a dry state for imaging). Microarray gel elements are covalently attached to the substrate of the reaction chamber, and oligonucleotide probes are covalently attached to a 2-(hydroxyethyl) methacrylamide (HEMAA) gel element network as described in Golova et al.28 Briefly, microarray gel element synthesis was performed by a UV-initiated co-polymerization process whereby oligonucleotides were incorporated into the gel network, which simultaneously polymerized and covalently attached to the substrate surface. Acrylamide gel polymer networks may also be substituted for HEMAA29–36. Whereas others have reported combined PCR and hybridization using modified primers or probes37, this scheme simply consists of asymmetric amplification in which fluorescently-labeled PCR forward primers are in higher concentration than non-labeled reverse primers. Following PCR, fluorescently-labeled amplicon hybridizes to complementary probes immobilized in a three-dimensional polymeric matrix network. A wash buffer is then introduced through a pierceable foil, which covers the inlet hole. An absorbent imbibes the PCR mastermix and wash buffer, leaving the chamber in a dry state.
Figure 1.

Amplification, hybridization, and washing take place on a Lab-on-a-Film disposable, which includes 203 hemispherical gel elements on a thin film substrate. Initially, master mix and sample are introduced into the reaction chamber. Subsequently, asymmetric PCR occurs in the chamber, resulting in a mix of fluorescently-labeled, single-stranded amplicons. Hybridization of the labeled amplicons to the gel elements follows, but this does not require a fluidic transfer step. The unbound molecules are then washed away using a pipettor, and the liquid is imbibed into an absorbent in the waste chamber. The last step is fluorescence imaging and analysis.
The assembly method for the Lab-on-a-Film disposable is described elsewhere.21 Briefly, it consists of laminating a substrate, spacer, cover, inlet spacer, and waste chamber. A distinction between the flow cell described here and the one reported previously is the length of the outlet channel. The length of the outlet channel for Cooney et al.21 is 17 mm and for these studies is 39 mm to limit evaporation to less than 5%38.
Studies to Evaluate Efficacy of Printing on Film
In order to identify a suitable film substrate for the Lab-on-a-Film disposable, a number of requirements needed to be satisfied. These include: (1) low background fluorescence, (2) allows covalent attachment of the gel elements to the film substrate, preferably without needing to pre-treat the film (3) transparency of the film to allow optical inspection with bright field imaging as a Quality Check (gel elements printed on transparent substrates reflect light at the interface allowing distinct visibility as compared to black substrates, which reflect negligible light), (4) withstands thermal cycling, (5) PCR compatible and (6) preferably can be produced in rolls to allow for reel-to-reel production of the parts.
Background fluorescence (intensity and standard deviation) was measured over a 6.5 by 4.9 mm Region of Interest (ROI) using an Akonni imager that consisted of a 530 nm nominal-wavelength excitation source, a monochrome Charged-Coupled Device (CCD) camera equipped with a 40 nm-wide band-pass emission filter with a center wavelength of 593 nm and an exposure time of 200 to 400 ms.
To determine the efficacy of attachment of the arrays to the substrate, gel elements were printed as described in Golova et al.29 When polymerized, this composition forms a three-dimensional polymeric matrix network. Gel elements are then printed on transparent polyester film substrates according to the method of Golova et al.29, and then placed in a polymerization chamber that was purged with argon to reduce the oxygen content to less than 0.5 volume percent. To cure the gel elements, the polymerization chamber was exposed to midrange Ultraviolet (UVB) light and irradiated the substrate surface plane with at least 5 mW/cm2 generated by a lamp equipped with two 25 W low-pressure discharge tubes (6286M52, Thomas Scientific, Swedesboro, NJ) that were mounted at a height of about 60 mm above the polymerization chamber with arrays. To optimize the polymerization efficiency, the exposure time varied from 10 to 30 min in 10-min increments. Four slides per exposure condition were used, each slide having 3 arrays of 27 spots printed at a 300-micron pitch with a 19 mm distance between arrays. The substrates were then washed for 30 minutes at room-temperature and agitated in a solution of 1X SSPE and 0.01% Triton-X100. The gel-element arrays were imaged on an Akonni imager (described above) using the automated analysis method described in Linger et al.20 To determine the binding strength of the covalent bond between the gel element and the substrate, the arrays were wiped with a wet Kim wipe to assess if the gel element could be removed from the substrate surface without any traces left. We used this test to qualify the degree of gel element attachment and serve as indicator of covalent attachment to the substrate. Strong (covalent) gel element attachment results in “hydrophilic footprints” when the gel elements are removed from the substrate surface. These footprints are observable when the substrates are cooled below the dew point in a stream of air having 100% relative humidity. Under this atmospheric condition, water vapor condenses on the surface in a contiguous water film in the case when the hydrophilic residue from the gel elements coats the surface. On the other hand, well-separated condensate (water droplets) cover the unmodified substrate surface due to the hydrophobicity of the substrate.
The stability of the film when subjected to thermal cycling was initially tested in a dry state (without arrays or reagents). Blank polyester films (Melinex® 453 7 mil, DuPont Teijin Films U.S. Limited Partnership, Chester, VA; procured from the distributor Tekra, New Berlin, WI) were laser cut with a VersaLASER® 3.5 Laser Cutting System (Universal Laser Systems, Scottsdale, AZ) to dimensions of 25 × 75 mm. Note, the cost of these films were 500 times lower than the glass substrates reported previously.20 Three films were placed on a Quanta QB-96 Flat Block Thermocycler (Quanta Biotech LTD, United Kingdom) and subjected to the following protocol: initial denaturation 95°C for 15 min, 50 cycles of 95°C for 30 seconds and 55°C for 60 seconds and 65°C for 30 seconds, followed by 65°C for 3 minutes.
The following methods describe the conditions of the Lab-on-a-Film disposable for analytical and clinical tests. Analytical studies were performed at Akonni Biosystems and clinical studies were performed at Laboratorios Medicos Especializados (LME) in Juarez, Mexico.
Sample Collection and Preparation
Analytical spiking studies included quantified H37Ra MTB cells from the American Type Culture Collection (ATCC, Manassas, VA #25177) prepared as described previously19 using de-identified sputa purchased from BioIVT (Westbury, NY). Aliquots of each sputa were screened for MTB through extraction using the TruTip automated workstation and were detected by real-time PCR21. Sputa that were negative for MTB were pooled and were used for spiking studies with quantified MTB cells.
Sputum specimens for the clinical studies were collected from clinics (Clinica Medica International and Servicios Medicos de la Frontera in Juarez, Mexico), de-identified, and sent to LME for use in this study. All specimens were characterized with respect to Acid-Fast Bacilli (AFB) smear status using Ziehl-Neelson staining, Lowestein Jensen (LJ) culture, Drug Susceptibility Testing (DST) using the Agar Proportion Method39, Mycobacterium Growth Indicator Tube (MGIT™) using the BACTEC 960 (Beckton Dickinson, Sparks, MD), and bidirectional DNA sequencing (Eurofins MWG Operon, Louisville, KY) using the extracts from the TruTip automated workstation and the same primers as used in the MDR-TB assay.
Extraction using Sample Prep System
Extraction was performed using the system described in Thakore et al.18 and the protocol described in Thakore et al.19 with the exception of an additional offline heating step of 56oC for 10 minutes (in the case of the clinical studies); the system at LME did not include an onboard heater, but the system at Akonni did. The real-time PCR assay, described in Thakore et al.19, was used in these studies.
Lab-on-a-Film Reagent Preparations and Workflow
The reagents, MDR-TB primer and probe sequences, and thermal cycling protocol was the same in the analytical and clinical studies as reported in Linger et al.20 The exception was the hybridization temperature, which was 53°C. We found improved discrimination and hybridization signal intensities at 53oC hybridization as compared to 55oC, which is the optimal hybridization temperature for arrays printed on glass. For these studies, a protective mask was applied to the film sheets (605 mm x 302 mm), which were laser cut to 25 × 75 mm substrates before printing the gel elements on the film.
Analysis
Result reporting followed the same algorithm as Linger et al.20 Briefly, gene targets that resulted in a Signal-to-Noise Ratio (SNR) greater than 3 were considered detected. If gene targets are detected, ratios were calculated to determine mutant (Mut) from wild-type (WT) probes. Mutants are determined by the following: (WT-Mut)/(WT+Mut) <0.
Results and Discussion
As part of our investigations, we surveyed a number of plastic films and identified a transparent film that did not require pre-treatment or functionalization, had acceptable background fluorescence and uniformity, allowed for covalent gel element attachment, withstood thermal cycling temperatures, and was compatible with standard PCR reactions.
The background fluorescence of the film substrates when measured within the Cy3 emission bandwidth for the typical excitation conditions was eight times higher than that of the microarray-grade glass slides, and the background non-uniformity (evaluated in terms of standard deviation) was twice that of the glass slides. In general, background fluorescence increases with the thickness of the plastic, thus the use of thin films is expected to have a lower background fluorescence intensity than thicker injection-molded plastics. However, the film extrusion process introduces effects whereby long polymer chains migrate from the bulk to the surface in the form of particulates, which create fluorescent non-uniformities. With the obvious caveat, non-uniformity of the fluorescence background may be more of an issue than high, but uniform, background. The latter is characterized by spatial frequencies close to zero, which are easy to attenuate with a simple high-pass digital filter, whereas the spatial Fourier spectrum of a highly non-uniform background (for instance the one due to the presence of small-scale particulates), may essentially overlap with the Fourier spectrum of gel elements, which makes such an interference impossible to filter out. We, therefore, sought to remove particulates from the surface of the film, and indeed we found that soaking the films in ethanol or sonication in ethanol at room temperature for 15 minutes followed by rinsing with water and drying with compressed air effectively removes them. With this procedure, the coefficient of variation (CV) of the Cy3 fluorescence signal intensity of 1620 gel elements, as determined by Cy3-labeled oligonucleotides immobilized within the gel element, was 10.7% across 6 replicate substrates. Alternatively, and preferably, we serendipitously discovered that the use of a protective mask, following extrusion, drastically reduces the presence of these particulates – even over the course of months. A hypothesis for the absence of these particulates is their electrostatic attraction to the static (non-stick) protective mask and subsequent removal when releasing the mask. The analytical and clinical studies that follow included the use of the protective mask because of the simplicity of manufacturing, the reduced cost compared to sonication in ethanol, and the protection from dust and scratches.
Following the selection of the film, we evaluated the robustness of the attachment of the gel elements to the film by attempting to physically remove the gel elements. Figure 2 shows that UVB polymerization resulted in satisfactory morphology of the gel elements and good performance in hybridization. Following 30 minutes of UVB exposure, gel element residues remained on the film after physically attempting to remove them with an abrasive Kim wipe, indicating robust (covalent) attachment to the film surface. The fluorescent image obtained after hybridization further confirmed that the immobilized oligonucleotide is indeed present in the gel network, and the network is sufficiently porous to allow the target to permeate and hybridize to the probe. We also evaluated gel element polymerization with UVA. While the Cy3 signals were three to five times higher for UVA compared to UVB polymerization, the gel element attachment was poor and not robust. The higher Cy3 signals were observed for arrays polymerized using UVA light and may be attributed to the lower Cy3 photobleaching rate, which is a predictable result of using the longer-wavelength, less reactive UV light.40 Note, this does not affect unlabeled probes that rely on hybridization with fluorescently-labeled amplicons.
Figure 2.
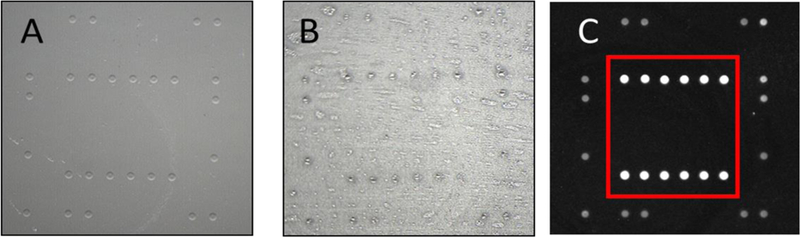
(A) Bright field image showing the morphology of the gel elements on a transparent film after UVB polymerization for 30 minutes, (B) “hydrophilic footprints” (residue) of gel elements after attempting to physically remove the gel elements; the remaining residues indicate that 30 minutes of UVB polymerization resulted in covalent attachment to the film substrate and (C) fluorescence images of gel elements after hybridization; gel elements outside of the red rectangle contain immobilized Cy3-labeled markers, which serve as fidicuals for the automated image analysis algorithm, and gel elements inside the red rectangle contain immobilized probes, which hybridized to complementary Cy3-labeled target.
As for the weaker gel attachment to substrates observed in the experiment under discussion, it is worth noting that both the UVA and UVB tubes used in our photopolymerization facility are, in fact, polychromatic light sources emitting within relatively wide bands. Thus, the UVA tube spectrum may comprise a certain fraction of photons with energies high enough for breaking chemical bonds in both the gel element and film substrate. This may explain why the arrays polymerized using the UVA illumination did show a certain degree of covalent attachment to the substrates sufficient for the arrays to survive the post-polymerization washing. The reaction rate of such binding, however, was apparently too low for the binding to be reliable. Thus, to ensure robust gel element attachment, we polymerized the gel elements with 30 minutes of UVB exposure for the remainder of these studies.
Whereas gel element attachment to the native polyester film requires UVB polymerization, silane-coated glass does not have this limitation, and thus we standardly polymerize gel elements on glass slides with lower-energy UVA. However, the significantly lower cost and potential for scaling the manufacture of the film substrates certainly outweighs the benefits of UVA polymerization.
We next performed a temperature stability study using the thermal cycling protocol described in the Methods section and did not observe damage (melting or warping) to the films. While the thermal conductivity of glass (~1 W/mK) is higher than polyester (0.15 to 0.4 W/mK), microscope-slide glass is also much thicker (~1 mm) than the film substrate (0.175 mm). Thus, the heat transfer through the film substrate is approximately equivalent to the glass substrate, and therefore did not warrant a change to the thermal cycling protocol reported in previous studies20.
The flow cell design included a long and narrow outlet channel to minimize evaporation through an outlet vent. For a given spacer thickness, the length and width of the channel was calculated using the water-vapor diffusion model described by Wang et al.31 We first replicated their model calculations and verified that the output to our model agreed with their reports. We then further validated the model using the same thermal cycling temperatures and durations as well as their reported constants for vapor pressure and diffusivity. The analytical detection studies were performed at a relative humidity ranging from 35 to 50% and a temperature ranging from 21 to 22°C. According to the diffusion model, these conditions were expected to result in less than 5% liquid loss.
The workflow includes a step that renders the gel elements in a somewhat “dry” state by using an absorbent in the waste chamber that imbibes the entire wash buffer volume. This step is important because we found that Cy-3 labeled DNA in “wet” gel elements emit weaker fluorescence than in dry gel elements. These observations are in agreement with the model proposed by Sanborn et al. whereby the relaxation of a Cy3 molecule from the first excited singlet state may occur with or without photon emission41. In the latter case, the molecule undergoes a trans – cis isomerization reaction, and the activation energy of such photoisomerization “depends strongly on the rigidity of the microenvironment in which the dye is located.” In other words, the photoisomerization competes with fluorescence, and the rigidity of environment determines the fluorescence quantum yield. In practice, gel elements with immobilized Cy3 can yield 10 times more fluorescence than wet gel elements.
Using the aforementioned thermal cycling protocol and flow cell design, we tested six Lab-on-a-Film disposables at concentrations of 1 pg of H37Rv DNA, and all six correctly reported “wild-type” for all MDR-TB markers, which includes 37 mutations, deletions or insertion elements across 4 genes.
Analytical Studies
The columns in the bar chart shown in Figure 3 represent the average of the integrated fluorescence from three replicate gel elements for probes that are considered “universal.” These universal probes were designed for highly-conserved regions of the gene that are not specific to wild-type or mutant strains of MTB. Thus, these universal probes served as indicators of a combination of amplification efficiency and target concentration for all strains of MTB. The lowest titer for which all replicates, gene targets (inhA, katG, rpoB, and IS6110), and mutations were correct was 32 cfu/mL. Extracts from these titers were also processed with a qPCR real-time assay, and the yield of DNA was 5.9 × 105 , 4.1 × 104, 1.4 × 104, 4.3 × 103, 5.16 × 102, and 116 genomic copies for titers of 105, 2 ×104, 4000, 800, 160, and 32 cfu/mL, respectively. Note that 1 cfu ranges from 10 to 100 bacilli42. The M13 internal control showed consistent amplification, even in the absence of target.
Figure 3.

Analytical sensitivity study of Lab-on-a-Film disposables, showing correct detection down to 32 cfu/ml. Error bars are standard deviations for n=6 replicates per titer. NTC is the no template control. M13 is an internal control. UN is a universal probe on the array that represents amplification and hybridization efficiency for each of the gene targets. All probes for mutations, deletions, and insertion elements on the array correctly reported wild-type for all titers and all replicates.
The lower titers (32 and 160 cfu/mL) resulted in more variability compared to the higher titers (800 to 105 cfu/mL). This is to be expected. For end-point reactions, when amplification is strong (i.e., high titers), the end-point reaction nears completion. However, when the target concentration is low, the reaction does not have sufficient time to complete and/or spurious primer-dimer formation ensues. Additionally, lower titers are more susceptible to pipetting inaccuracies. Thus, in some cases, amplitudes are lower for 32 and 160 cfu/mL compared to 800 to 105 cfu/mL.
Clinical Studies
DNA extraction from TB-positive sputa specimens can be more challenging than extraction from negative sputa spiked with H37Ra cells. In positive TB cases, patient sputa specimens form mucoid around the MTB bacilli and encapsulate it43. Releasing the MTB DNA from this encapsulation requires an effective extraction technique. Additionally, the cell wall integrity differs across different strains of MTB, thus H37Ra bacilli are expected to behave differently than other (especially resistant) strains of MTB. We thus sought to evaluate the behaviour of the Lab-on-a-Film disposable with clinical specimen (sputum) extracts.
Supplement Table 1 is a summary of a clinical data set from 27 sputa. Of these 27, all were MTB positive according to MGIT, and 11 were drug resistant according to DST (3 were MDR-TB, 7 were INH mono-resistant, and 1 was RIF mono-resistant). All but one of the Lab-on-a-Film MTB detection results were concordant with MGIT. The one discordant sample (PSR 12) resulted in poor amplification for both sequencing and the Lab-on-a-Film test, and it had a “no growth” condition for the LJ culture. This sample may be an example of highly mucoidal sputa preventing efficient and/or high-purity extraction since there does not appear to be amplification either from our Lab-on-a-Film test or the sequencing preparation. Since this sample showed no LJ growth, it also likely had a low MTB bacillary load.
Five samples were initially discordant with DST and three were indeterminate but after repeating the test, all but one (PSR 12) were concordant with DST, and all were concordant with sequencing. Sequencing reported two samples as mixtures of wild-type and INH resistance, and the Lab-on-a-Film test reported them as INH resistant with the same mutations as found with sequencing. Of note is that 5 of the 27 samples were LJ negative for MTB, but MTB positive for both the Lab-on-a-Film test and MGIT. PSR 11 was reported as contaminated for LJ and positive for MGIT. LJs are inherently more susceptible to contamination as compared to MGITs, which includes a BBL™ MGIT PANTA™ antibiotic additive.
This preliminary study included only three unique drug-conferring mutations: rpoB D516V, rpoB S531L, or katG S315T. A larger sample set with more diversity would be of interest. While the “n” in this clinical study is too limited to adequately report positive or negative predictive values (or sensitivity/specificity), it does suggest that this potentially low-cost test is capable of detecting drug-resistance in clinical sputa specimens.
In summary, after repeats, all Lab-on-a-Film genotypes were concordant with sequencing, and all but one were concordant with MGIT. Of note is that these MTB-positive samples included smear negatives and even culture (LJ) negatives.
Future Work
We now plan to integrate our previously-reported automated sample preparation workstation13 and this Lab-on-a-Film disposable into a sample-to-answer automated system. This sample preparation workstation, which was used to extract MTB DNA from disinfected and liquefied sputa specimens14, utilizes an automated pipetting method. Automated pipetting methods such as this one and that reported by Lu et al.44 are expected to integrate well with the Lab-on-a-Film disposable reported here. The interface required for this sample-to-answer integration is a pierceable and re-sealable septum. We intend that the platform will maintain an open architecture (while keeping it closed amplicon) to allow users the freedom to select the automated workstation for extraction, the Lab-on-a-Film disposable for analysis, or the combination of the two for an uninterrupted workflow.
Conclusions
This work shows that the Lab-on-a-Film disposable is capable of very sensitive detection from sputum extracts, can genotype by computing ratios of mutant to wild-type hybridization signals from gel elements printed on unmodified and untreated film substrates, supports PCR without active valves, and is made of laminate materials that can be produced with reel-to-reel manufacturing. While we initially suspected that the background fluorescence of film would be unsuitable for sensitive fluorescence discrimination because of the increased background fluorescence compared to glass, the analytical data shows that the combination of Akonni’s TruTip automated workstation and Lab-on-a-Film disposables is indeed capable of very sensitive detection. Fluorescence discrimination of one gel element with respect to another (i.e., to determine mutant to wild-type ratios) increased the stringency of the requirement for a uniform background. However, the serendipitous result of using a protective mask for handling and storage of the films drastically improved the homogeneity and allowed for correct discrimination at low titers without needing to wash, treat, or prepare the film substrates for use. We were also initially pessimistic about the PCR compatibility of the laminate and the film substrate, but again found that these materials allowed for acceptable assay behaviour. In general, the advantage of the array, as compared with real-time PCR, is the ability to discriminate and report many more mutations (tens to hundreds vs <10) from a single sample, which is particularly important for detecting MTB drug resistance. In summary, this work demonstrates that adhesive laminates and film substrates for gel element array printing can be used for sensitive PCR and molecular detection of mutations, deletions, and insertion elements, and its form factor is compatible with reel-to-reel manufacturing.
Supplementary Material
Acknowledgements
The authors wish to thank the National Science Foundation (Grant Number 1230152) and the National Institutes of Health (U19 AI109755, R44 EB011274, R01 AI111435, R44 AI138903, R44 AI085650 and RC3 AI089106) for supporting this work.
Footnotes
Conflicts of interest
All Akonni-affiliated authors are employees of Akonni Biosystems Inc. AK, AP, CK, YL, DPC and CGC are also Akonni shareholders.
References
- 1.World Health Organization, Global tuberculosis report, 2018. [Google Scholar]
- 2.World Health Organization, Global tuberculosis report, 2016. [Google Scholar]
- 3.World Health Organization, High priority target product profiles for new tuberculosis diagnostics: report of a consensus meeting, 2014. [Google Scholar]
- 4.Pantoja A, Kik S, and Denkinger C, J. Infect. Dis, 2015, 211, S67–S77. [DOI] [PubMed] [Google Scholar]
- 5.Sandgren A, Strong M, Muthukrishnan P, Weiner B, Church G and Murray M, PLoS Med, 2009, 6, e1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoshide M, Qian L, Rodrigues C, Warren R, Victor T, Evasco H II, Tupasi T, Crudu V, and Douglas J, J. Clin. Microbiol, 2014, 52, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholas B and Djukanović R, Biochem. Soc. Tr, 2009, 37, 868–872. [DOI] [PubMed] [Google Scholar]
- 8.Denkinger C, Nicolau L, Ramsay A, Chedore A,P and Pai M, Eur Respir J, 2013, 42, 544–547. [DOI] [PubMed] [Google Scholar]
- 9.Pai M, M. and Schito, J. Infect. Dis, 2015, 211, S21–S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vos M, Derendinger B, Dolby T, Simpson J, van Helden P, Rice J, Wangh L, Theron G and Warren R, J. Clin. Microbiol, 2018, 56, e00531–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helb D, Jones M, Story M,E, Boehme C, Wallace E, Ho K, Kop J, Owens M, Rodgers R, Banada P, and Safi H, J. Clin. Microbiol, 2010, 48, 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boehme C, Nabeta P, Hillemann D, Nicol M, Shenai S, Krapp F, Allen J, Tahirli R, Blakemore R, Rustomjee R, and Milovic A, New Engl. J. Med, 2010, 363, 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi J, Yong K, Tang R, Gong Y, Wen T, Li F, Pingguan-Murphy B, Bai D, and Xu F, TrAC, Trends Anal. Chem, 2017, 93, 37–50. [Google Scholar]
- 14.Tang R, Yang H, Gong Y, You M, Liu Z, Z., Choi J, Wen T, Qu Z, Mei Q, and Xu F, Lab Chip, 2017, 17, 1270–1279. [DOI] [PubMed] [Google Scholar]
- 15.Magro L, Escadafal C, Garneret P, Jacquelin B, Kwasiborski A, Manuguerra J, Monti F, Sakuntabhai A, Vanhomwegen J, Lafaye P, and Tabeling P, Lab Chip, 2017, 17, 2347–2371. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez N, Wong W, Liu L, Dewar R, and Klapperich C, Lab Chip, 2016, 16, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Q, Nam J, Kim S, Lim C, Park M, and Shin Y, Biosens. Bioelectron, 2016, 82, 1–8. [DOI] [PubMed] [Google Scholar]
- 18.Thakore N, Garber S, Bueno A, Qu P, Norville R, Villanueva M, Chandler D, Holmberg R, and Cooney C, J. Microbiol. Meth, 2018, 148, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thakore N, Norville R, Franke M, Calderon R, Lecca L, Villanueva M, Murray M, Cooney C, Chandler D, and Holmberg R, PloS One, 2018, 13, e0199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linger Y, Knickerbocker C, Sipes D, Golova J, Franke M, Calderon R, Lecca L, Thakore N, Holmberg R, Qu P, Kukhtin A, Murray M, Cooney C, and Chandler D, J. Clin. Microbiol, 2018, JCM-01652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooney C, Sipes D, Thakore N, Holmberg R, and Belgrader P, Biomed. Microdevices, 2012, 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Z, Peytavi R, Diaz-Quijada G, Picard F, Huletsky A, Leblanc É, Frenette J, Boivin G, Veres T, Dumoulin M, and Bergeron M, J. Clin. Microbiol, 2008, 46, 3752–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holden M, Carter M, Wu C, Wolfer J, Codner E, Sussman M, Lynn D, and Smith L, Anal. Chem, 2015, 87, 11420–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo D, Wu H, Wu L, Zheng B, Small Molecule Microarrays, 2017, 1518, 19–28. [DOI] [PubMed] [Google Scholar]
- 25.Bañuls M, Morais S, Tortajada-Genaro L and Maquieira A, Á., Microarray Technology, 2016, 37–51. [DOI] [PubMed] [Google Scholar]
- 26.Cooney C, US Patent 8,828,912, 2014. [Google Scholar]
- 27.Bose I, Ohlander A, Kutter C, and Russom A, A., Sensor Acuat B-Chem, 2018, 259, 917–925. [Google Scholar]
- 28.Golova J, Chernov B, Perov A, Reynolds J, Linger Y, Kukhtin A, and Chandler D, Anal. Biochem, 2012, 421, 526 –533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bavykin S, Akowski J, Zakhariev V, Barsky V, Appl. Environ. Microb, 2001, 67, 922–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu W, Mirzabekov A, Stahl D, Environ. Microbiol, 2001, 3, 619–629. [DOI] [PubMed] [Google Scholar]
- 31.Arenkov P, Kukhtin A, Gemmell A, Voloshchuk S, V, Anal. Biochem, 2000, 278, 123–131. [DOI] [PubMed] [Google Scholar]
- 32.Dubiley S, Kirillov E, Mirzabekov A, Nucleic Acids Res, 1999, 7, e19–i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikhailovich V, Lapa S, Gryadunov D, Strizhkov B, Sobolev A, Skotnikova O, Irtuganova O, Moroz A, Litvinov V, Shipina L, and Vladimirskii M, B. Exp. Biol. Med, 2001, 131, 94–98. [DOI] [PubMed] [Google Scholar]
- 34.Strizhkov B, Drobyshev A, Mikhailovich V, and Mirzabekov A, Biotechniques, 2000, 29, 844–857. [DOI] [PubMed] [Google Scholar]
- 35.Yershov G, Barsky V, Belgovskiy A, Kirillov E, Kreindlin E, Ivanov I, Parinov D S. Guschin, Drobishev A, Dubiley S and Mirzabekov A, P. Natl. Acad. Sci. USA, 93, 4913–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasiliskov A, Timofeev E, Surzhikov S, Drobyshev A, Shick V, and Mirzabekov A, Biotechniques, 1999, 27, 592–606. [DOI] [PubMed] [Google Scholar]
- 37.Girard L, Boissinot K, Peytavi R, Boissinot M, M. and Bergero, Analyst, 2015, 140, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang F, Yang M, and Burns M, Lab Chip, 2008, 8, 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clinical and Laboratory Standards Institute, 31, M24–A2, Standard Second Edition [Google Scholar]
- 40.Jouonang L, Didier P, and Mely Y, Phys. Chem. Chem. Phys, 2012, 14, 1585–1588 [DOI] [PubMed] [Google Scholar]
- 41.Sanborn M, Connolly B, Gurunathan K, and Levitus M, J. Phys. Chem. B, 2007, 111, 11064–11074. [DOI] [PubMed] [Google Scholar]
- 42.Brodie D and Schluger N, Clinics in chest medicine, 2005, 26, 247–271. [DOI] [PubMed] [Google Scholar]
- 43.Clinical and Laboratory Standards Institute 38, M48–A. [Google Scholar]
- 44.Lu W, Wang J, Wu Q, Sun J, Chen Y, Zhang L, Zheng C, Gao W, Liu Y, and Jiang X, Biosens. Bioelectron, 2016, 75, 28–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.