

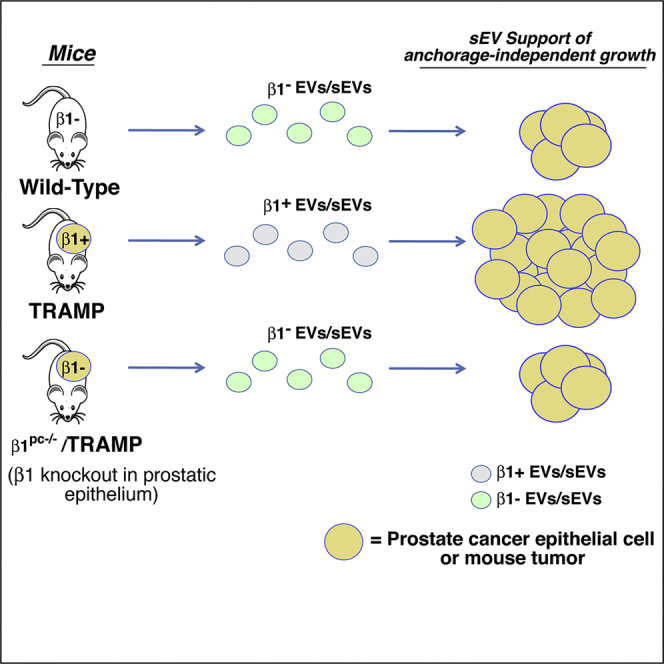
Summary
The β1 integrins, known to promote cancer progression, are abundant in extracellular vesicles (EVs). We investigated whether prostate cancer (PrCa) EVs affect anchorage-independent growth and whether β1 integrins are required for this effect. Specifically using a cell-line-based genetic rescue and an in vivo PrCa model, we show that gradient-purified small EVs (sEVs) from either cancer cells or blood from tumor-bearing TRAMP (transgenic adenocarcinoma of the mouse prostate) mice promote anchorage-independent growth of PrCa cells. In contrast, sEVs from cultured PrCa cells harboring a short hairpin RNA to β1, from wild-type mice or from TRAMP mice carrying a β1 conditional ablation in the prostatic epithelium (β1pc−/−), do not. We find that sEVs, from cancer cells or TRAMP blood, are functional and co-express β1 and sEV markers; in contrast, sEVs from β1pc−/−/TRAMP or wild-type mice lack β1 and sEV markers. Our results demonstrate that β1 integrins in tumor-cell-derived sEVs are required for stimulation of anchorage-independent growth.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
sEVs from prostate cancer stimulate anchorage-independent growth of recipient cells
-
•
sEVs from tumor bearing, but not healthy, mice contain β1 integrins
-
•
sEV stimulation of anchorage-independent growth is dependent on β1 integrins
-
•
β1 down-regulation in the prostate tumor epithelium impairs EV functions
Biological Sciences; Molecular Biology; Cell Biology; Cancer
Introduction
Prostate cancer (PrCa) is one of the most frequently diagnosed cancers among men and remains a significant clinical challenge (Siegel et al., 2018). Many factors contribute to disease progression and resistance to therapy, including deregulated interactions between integrins and extracellular matrix (ECM) proteins (Fitzgerald et al., 2008, Wang et al., 2011). Integrins are a class of heterodimeric transmembrane receptors consisting of α and β subunits that modulate cell adhesion and migration in the tumor microenvironment by binding to specific recognition peptide sequences (Lee et al., 2015, Plow et al., 2000). They signal through multiple downstream effectors such as Src and focal adhesion kinase (FAK) to modulate important functions of both normal and cancer cells. In particular, β1 integrins are known to contribute to prostate tumor growth (Goel et al., 2010, Goel et al., 2013). Our laboratory has previously shown in TRAMP (transgenic adenocarcinoma of the mouse prostate) mice that β1 integrins contribute to resistance to radiation therapy by inhibiting c-jun NH2-terminal kinase (JNK) activity (Goel et al., 2013, Sayeed et al., 2016). TRAMP mice with a conditional β1 ablation in the prostate have increased survival, decreased primary tumor burden, and decreased metastasis (Goel et al., 2013).
The β1 integrin subunit heterodimerizes with many different α subunits. In epithelial cells, β1 is often paired with α5 to function as the integrin receptor for fibronectin. The α5 subunit has been linked to cancer development and progression. In particular, there are three different studies implicating the loss of certain microRNAs in different cancers that are responsible for targeting α5 (Cimino et al., 2013, Gong et al., 2016, Yoo et al., 2016). The α5β1 integrin has been implicated in increasing cancer cell migration, invasion, and resistance to chemotherapy (Li et al., 2013, Miroshnikova et al., 2017, Paul et al., 2015, Zhu et al., 2017). However, there is less known about the role of the α5β1 integrin in anchorage-independent cell growth.
PrCa cells are capable of transferring proteins, such as integrins, to other PrCa cells via extracellular vesicles (EVs). EVs affect both cancer progression and response to cancer therapy (Minciacchi et al., 2015). There are multiple subtypes of EVs, including microvesicles (50–2,000 nm), apoptotic bodies (50–5,000 nm) (Junker et al., 2016, Kowal et al., 2016), and exosomes (exo), which range in size between 30 and 150 nm, and are secreted by the fusion of the multivesicular endosomes with the plasma membrane of the cell (Colombo et al., 2014). Exo can transfer crucial biological molecules between cells, affecting both normal and pathological processes (Colombo et al., 2014, Tkach and Thery, 2016). Several molecules implicated in PrCa progression are enriched in exo from PrCa cells, including β1 integrins, insulin-like growth factor receptor 1 (IGF-IR), and downstream signaling molecules such as c-Src and FAK (DeRita et al., 2017, Fedele et al., 2015, Krishn et al., 2018, in press; Singh et al., 2016). In addition, exo-mediated transfer of integrins between PrCa cells functionally affects recipient cancer cells (Fedele et al., 2015, Hamidi et al., 2016, Singh et al., 2016). PrCa cell exo have also been shown to increase xenograft tumor size when injected intravenously (Hosseini-Beheshti et al., 2016). Furthermore, large cancer-derived EVs called large oncosomes (1–10 μm) were reported to transfer active AKT1 and increase fibroblast Myc activity after oncosome internalization (Minciacchi et al., 2017). In addition to pro-tumorigenic molecules, tumor suppressor proteins such as maspin have also been detected in PrCa exo (Dean et al., 2017). Exo, oncosomes, and other cancer-derived EVs may be a source of biomarkers easily detectable in blood (Minciacchi et al., 2015, Minciacchi et al., 2017) and potentially linked to disease outcome and therapy response as observed for circulating tumor cells (You et al., 2016). Owing to recent updates on EV research (Thery et al., 2018), this report uses the term small EVs (sEVs) to describe the small (between 50 and 150 nm) EVs previously referred to as exo.
We demonstrate for the first time that tumor-derived β1 integrins are essential for supporting the ability to stimulate anchorage-independent growth of EVs shed by PrCa cells and circulating in the plasma of tumor-bearing mice. Although the significance of EVs in disease progression is recognized, there are no studies showing that “tumor-cell-derived” EVs are physiologically active. We demonstrate in this study, using EVs from in vitro and in vivo models, that tumor-cell-derived β1 integrins are required for EV-mediated stimulation of anchorage-independent growth. Overall, this study sheds light on the role of EVs and β1 integrins in the progression of PrCa.
Results
β1 Integrins Are Required for Extracellular-Vesicle-Stimulated Anchorage-Independent Growth of Prostate Cancer Cells
Our laboratory has previously demonstrated that integrins are expressed in PrCa-derived EVs (Fedele et al., 2015, Krishn et al., 2018, in press; Lu et al., 2018, Singh et al., 2016) and that β1 integrins promote PrCa cell growth and survival (Goel et al., 2009, Goel et al., 2010, Sayeed et al., 2012). To study β1 integrin function in PrCa EVs, we optimized our purification protocol to improve the purity and reliability of our results. In this study, we utilize small (less than 150 nm) EVs obtained from high-speed differential ultracentrifugation and EVs further purified by flotation in a density gradient. Samples that have been further purified by flotation in a density gradient have been designated small EVs (sEVs). All EVs and sEVs utilized are in the size and density range previously reported (Kowal et al., 2016). To validate the whole sEV isolation procedure, we analyzed the levels of the sEV markers CD63, CD81, TSG101, and CD9 in each iodixanol gradient fraction from a PC3-derived sEV sample and observed that their levels are the highest in the 1.112 g/mL density fraction (Figures 1A and 1B, upper panels). The input material is shown in a lighter exposure (Figures 1A and 1B, lower panels). Calnexin, an ER protein not found in EVs, is not present in these samples, confirming the absence of any contaminants (Figure 1C). Our results confirm a previous study using an iodixanol density gradient in which the sEVs are present in the 1.115 g/mL fraction (Kowal et al., 2016). However, species and biofluid of EV origin can affect the density, as we have found sEVs in the 1.14 g/mL fraction from plasma samples (Krishn et al., 2018, in press). To functionally analyze sEVs from PrCa cells, we first demonstrated that sEVs isolated from PC3 cells have the expected size range (Figure 2A) and are capable of stimulating recipient cell anchorage-independent growth, as compared with vehicle treatment (Figures 2B and 2C).
Figure 1.

Validation of Extracellular Vesicle Isolation Procedure
(A) Upper panel: A representative immunoblotting is shown for CD63 and CD81 across 10 iodixanol density gradient fractions. The two rightmost lanes represent the input material before iodixanol separation and a total cell lysate (TCL) of the EV donor cells. Lower panel: a lighter exposure of the two rightmost lanes shown in the upper panel, also representing the input material (“Input” in upper panel or EVs in lower panel) and TCL of donor cells.
(B) Upper panel: Immunoblotting is shown for TSG101 and CD9 across 10 density gradient fractions. The two rightmost lanes represent the input material before iodixanol separation and a total cell lysate (TCL) of the donor cells. Lower panel: a lighter exposure of the two rightmost lanes shown in the upper panel, also representing the input material (noted as “Input” in upper panel and “EVs” in the lower panel) and TCL of EV donor cells. Results from (A) were obtained under non-reducing conditions, and results from (B) were obtained under reducing conditions.
(C) Immunoblotting is shown for calnexin across 10 density gradient fractions. The rightmost lane represents the TCL from PC3 cells. Results were obtained under non-reducing conditions.
Figure 2.

sEVs from PrCa Cells Stimulate Recipient Cell Anchorage-Independent Growth
(A) NTA of the 1.112 g/mL fraction containing PC3-derived sEVs isolated using an iodixanol density gradient. The experiment was repeated three times.
(B) Quantification of anchorage-independent growth of PC3 cells after treatment with sEVs (1.112 g/mL fraction) from PC3 cells. Data are represented as mean ± SEM. *p < 0.000067 (Student's t test).
(C) Representative images of the data quantified in (B) are shown.
Next, to elucidate the role of EV β1 integrins in stimulating anchorage-independent growth of PrCa cells, we conducted in vitro and in vivo experiments. In the in vitro approach, we used PC3 cells with a knockdown of the β1 integrin subunit (designated shβ1 PC3 cells; Goel et al., 2010). As previously reported (Goel et al., 2010), endogenous levels of β1 integrins in PC3 cells are required for the formation of large colonies in a soft agar matrix (Figure 3A). We then isolated EVs from these cells using differential ultracentrifugation. Nanoparticle tracking analysis (NTA) reveals that there are no significant differences in the average particle size upon β1 downregulation (Figure 3B). Analysis of colonies ≥25 μm in diameter, expressed as a percentage of the total number of colonies, shows that the EVs from shβ1 PC3 cells lose the ability to stimulate large colony growth compared with their counterparts from mock PC3 cells (Figures 3C and 3D). As PC3 cells are a naturally aggressive cell line and have an innate ability to form small colonies in standard media alone, we decided to evaluate larger colonies with size ≥25 μm in diameter rather than the total colony number. The β1 integrin subunit has many different binding partners in the cell, particularly α5 in epithelial cells, which has been previously associated with the progression of cancer (Cimino et al., 2013, Gong et al., 2016, Yoo et al., 2016). We therefore analyzed the levels of α5 in PC3 cells and EVs released by these cells upon β1 knockdown. Although the levels of an EV marker, TSG101, are not modified upon β1 integrin downregulation (Figure 3F), α5 levels are reduced in those EVs (Figure 3E), indicating that the α5 and β1 integrin subunits, as expected, are linked. We additionally examined EV levels of FAK, a downstream effector of the α5β1 integrin, and find that it is also downregulated in EVs upon short hairpin RNA (shRNA) targeting of β1 (Figure 3F). Furthermore, we observe that FAK is increased in cells treated with EVs from β1-positive PC3 cells (Figure 3G). EVs from shβ1 cells do not increase recipient cell FAK levels. Owing to the decreased levels of FAK in shβ1 EVs, it is likely that the EVs are transferring FAK to the recipient cells (Figure 3G).
Figure 3.

β1 Integrins from PrCa Cells Are Required for EV-Mediated Stimulation of Anchorage-Independent Growth
(A) Quantification of anchorage-independent growth of shβ1 or mock PC3 cells with no other treatments. The total number of colonies were counted per field (325 × 250 μm) after 4 weeks in soft agar. p < 0.0000013.
(B) NTA profiles of the EV population isolated from PC3 cells transfected either with shRNA to β1 (shβ1) or an empty vector (mock). Three technical replicates were performed on each sample. The area under the curve represents all detected particles in the sample and an average of three independent readings.
(C) Quantification of anchorage-independent growth of PC3 cells after treatment with EVs from either shβ1 or mock PC3 cells. Data are represented as mean ± SEM. *p < 0.045, **p < 0.0045 (Student's t test).
(D) Representative images of the data quantified in (C) are shown. Two technical and biological replicates were performed.
(E) Immunoblotting of both EVs and total cell lysates (TCL) from shβ1 and mock PC3 cells for β1, TSG101, and calnexin (left panel); and for α5 (right panel); 15 μg protein was loaded per lane, and results were obtained in reducing conditions.
(F) Immunoblotting of EVs from shβ1 or mock PC3 cells for FAK and TSG101; 15 μg protein was loaded per lane.
(G) Immunoblotting for FAK and ERK of TCL from PC3 cells incubated with EVs from shβ1 PC3 cells, mock PC3 cells, or PBS; 40 μg protein was loaded per lane.
See also Figure S1.
We then investigated whether the significant functional differences between EVs from shβ1 and mock PC3 cells are due to differences in EV internalization. We treated DU145 PrCa cells with EVs labeled with PKH26 red dye and analyzed internalization by confocal microscopy. We observe that there is no difference in the number of cells that internalized EVs from shβ1 and mock PC3 cells (Figure S1). This indicates that EV-derived β1 integrins play a functional role as signaling molecules rather than influencing EV internalization. When cells were treated with only PKH26 dye without EVs, there was no evidence of red dye internalization in recipient cells (unpublished data). Together, these results demonstrate the requirement of β1 integrins for the ability of PrCa EVs to stimulate anchorage-independent growth.
Rescue of Functional β1 Integrins Restores the Stimulatory Function of Secreted EVs on Anchorage-Independent Growth
To further demonstrate the importance of β1 integrin expression in PrCa cells for the anchorage-independent function of EVs, we re-transfected shβ1 PC3 cells with chicken β1 (Hayashi et al., 1990), which would not be targeted by the shRNA against human β1. We first analyzed the total cell lysates (TCL) for chicken β1 expression after transfection and confirm specific expression of chicken β1 in transfected cells (Figure 4A). To reach detectable levels of relevant proteins in TCL samples, we loaded nearly three times the amount of protein (Figure 4A) as we did when analyzing proteins in EV samples (Figure 3E). Flow cytometric analysis of those transfected cells also confirms the expression of chicken β1 on the cell surface (Figure 4B). Next, we analyzed the EVs released by those cells. NTA shows that there are no significant differences between the size and abundance of EVs from shβ1 PC3 cells transfected with chicken β1 and those transfected with empty vector (Figure 4C). We observe that the EV markers CD63, CD81, TSG101, and CD9 are also enriched in the EVs compared with TCL (Figures 4D and 4E). The levels of total β1 are increased in EVs from shβ1 PC3 cells transfected with chicken β1, along with the downstream signaling protein c-Src (Figure 4E).
Figure 4.

Rescue of β1 Expression Restores the Stimulatory Function of Secreted Extracellular Vesicles on Anchorage-Independent Growth
(A) Immunoblotting for chicken β1, total β1, and α5 of TCL from shβ1 PC3 cells transfected with either full-length chicken β1 (chick β1) or empty vector (mock) alone. Results for chicken-specific β1 (chick β1) were obtained under non-reducing conditions, whereas the results for total β1 (β1 total) were obtained under reducing conditions. AKT is used as a loading control; 50 μg protein was loaded per lane.
(B) Flow cytometric analysis of shβ1 cells transfected with either chicken β1 or empty vector using an Ab specific to chicken β1 integrin.
(C) NTA of EVs derived from shβ1 cells transfected with either chicken β1 or vector. Three technical replicates were performed on each sample.
(D) Immunoblotting for calnexin and the EV markers CD63 and CD81.
(E) Immunoblotting for α5, total β1, c-Src, TSG101, and CD9 of EVs and TCL from shβ1 cells transfected with either chicken β1 or empty vector. For data in (D and E), 15 μg protein per lane was loaded.
(F) Left panel: quantification of anchorage-independent growth of PC3 cells after treatment with EVs derived from shβ1 cells transfected with either chicken β1 or empty vector. The number of colonies ≥25 μm per field (325 × 250 μm) was quantified and reported as an absolute number after 2 weeks in soft agar. *p < 0.002. Right panel: quantification of anchorage-independent growth of PC3 cells after treatment with EVs derived from shβ1 cells transfected with either chicken β1 or mock vector. The number of colonies ≥25 μm per field (325 × 250 μm) was quantified and reported as percentage of the total after 2 weeks in soft agar. Data are represented as mean ± SEM. *p < 0.002 (Student's t test).
(G) Representative images of data quantified in (F) are shown.
We then treated PC3 cells with EVs from shβ1 PC3 cells transfected with chicken β1 or empty vector and analyzed their anchorage-independent growth. We clearly demonstrate that transfection of shβ1 cells with chicken β1 rescues the ability of EVs to stimulate large colony growth of recipient PC3 cells (Figure 4F). Once again, colonies ≥25 μm were counted 2 weeks after seeding in soft agar and expressed as either an absolute number or a percentage of total number of colonies (Figure 4F, left and right panels). EVs from shβ1 PC3 cells transfected with an empty vector caused significantly less colony growth than EVs from the shβ1 PC3 cells that were transfected with chicken β1 (Figures 4F and 4G).
Prostate-Specific Ablation of β1 Integrins in Mice Alters the Protein Content, Physical Properties, and Function of Circulating EVs
The in vitro results prompted us to analyze circulating plasma sEVs from the TRAMP mouse model. After sEV isolation from the plasma of TRAMP mice (n = 6), we demonstrate that the sEV markers CD63 and CD9 are present in the expected sEV density fraction (1.14 g/mL, based on previous study from our laboratory using human plasma; Krishn et al., 2018, in press) (Figure 5A, right panel)]. We had previously used sucrose density gradient separation to demonstrate enriched levels of β1 integrins and c-Src in sEVs from PrCa cells. Here we confirm that both β1 and c-Src are present predominantly in the same iodixanol density fraction (1.14 g/mL) of TRAMP sEVs as markers CD63 and CD9 (DeRita et al., 2017) (Figure 5A, right panel). Calnexin is absent from these samples (unpublished data). Conditional ablation of β1 from the prostatic epithelium in TRAMP mice (β1pc−/−/TRAMP) alters the protein composition and density distribution of sEVs from the blood of these mice (n = 8). The sEV marker CD9 is undetectable in the 1.14 g/mL density fraction. β1 and the downstream signaling protein c-Src, which we have previously shown to be enriched in PrCa EVs (DeRita et al., 2017), are also absent (Figure 5A, left panel). We performed analysis on non-tumor-bearing wild-type mice (n = 6) as well and observe that there is no detectable β1, CD63, or CD9 in either the 1.14 g/mL fraction or any of the other nine density gradient fractions (Figure 5A, middle panel). In addition, NTA of the 1.14 g/mL fraction shows that the amount of sEVs is approximately two times higher in TRAMP mice versus β1pc−/−/TRAMP mice, whereas the difference between wild-type and TRAMP was less pronounced (Figure 5B and Table 1). However, the tumor masses at the age of 20 weeks are statistically similar between TRAMP and β1pc−/−/TRAMP mice (Table 1); this is consistent with our previous findings that β1 plays a role in PrCa progression beyond 20 weeks in TRAMP mice (Goel et al., 2013). The reduction in the number of sEVs in the 1.14 g/mL fraction between TRAMP and β1pc−/−/TRAMP mice is not due to a reduction in tumor size, but rather due to the fact that β1 integrins in the tumor epithelium may have a role specifically in sEV formation and secretion. Altogether, these data suggest that the β1-positive PrCa tumor epithelial cells are the predominant source of circulating β1 in sEVs and that β1 integrins in the prostatic epithelium contribute to sEV processing into circulation.
Figure 5.

Prostate-Specific Ablation of β1 Integrins Alters the Protein Content and Physical Properties of Circulating Extracellular Vesicles
(A) Immunoblotting is shown for β1, c-Src, CD63, and CD9 across 10 iodixanol density gradient fractions obtained from β1pc−/−/TRAMP (left panel), wild-type (middle panel), and TRAMP (right panel) mouse EVs. For adequate input material, EV samples from three mice were pooled before gradient separation. The rightmost lane on the middle and right panels represents a positive control EV sample from a wild-type (middle panel) or TRAMP (right panel) mouse, i.e., the pellet obtained after initial EV precipitation.
(B) Graphical representation of NTA of the 1.14 g/mL sEV fraction of the iodixanol density gradient from β1pc−/−/TRAMP (left panel), wild-type (middle panel), or TRAMP (right panel) mice. The experiment was repeated twice, and results were obtained from pooled samples from three mice for each condition. A total of six TRAMP, eight β1pc−/−/TRAMP, and six wild-type mice were analyzed.
Table 1.
Analysis of EV Abundance from TRAMP, β1pc−/−/TRAMP, and Wild-type Mice
| Mouse Genotype | Average Tumor Mass (Age 20 Weeks) | Average Plasma sEV Concentration (1010 sEV per mL Plasma) |
|---|---|---|
| TRAMP | 132.2 mg | 3.1 |
| β1pc−/−/TRAMP | 207.5 mg | 1.7 |
| Wild-type | No tumor present | 2.3 |
The average values of the sEV concentration (particles from the 1.14 g/mL iodixanol density gradient fraction) are reported. For average tumor mass between TRAMP and β1pc−/−/TRAMP mice, p > 0.05 (non-significant). TRAMP: n = 6; β1pc−/−/TRAMP: n = 8; wild-type: n = 6.
Stimulation of Anchorage-Independent Growth by Tumor-Derived Circulating sEVs Is Dependent on β1 Integrins
To see if the effects of cancer-cell-derived EVs are tumor specific and transferrable to an in vivo setting, we next analyzed if there was a difference in function between β1-positive sEVs from TRAMP mice and β1-negative sEVs from wild-type mice. PC3 cells were treated with equal numbers of sEVs from the plasma of either TRAMP or wild-type mice and then analyzed for anchorage-independent growth. It is clear that the β1-positive sEVs from TRAMP mice are able to stimulate anchorage-independent growth, whereas β1-negative sEVs from wild-type (non-tumor-bearing) mice do not (Figures 6A and 6B).
Figure 6.

Stimulation of Anchorage-Independent Growth by Tumor-Derived Circulating EVs Is Dependent on β1 Integrins
(A) Quantification of anchorage-independent growth of PC3 cells after treatment with sEVs from TRAMP and wild-type mice. *p < 0.0004.
(B) Representative images of the data quantified in (A) are shown. The experiment was repeated three times. All mice were aged 20 weeks at the time of sacrifice, blood collection, and EV analysis.
(C) Immunoblotting of gradient-purified sEVs of the indicated densities for β1, α5, c-Src, and CD63. For β1, α5, and c-Src 7.5 × 108 sEVs were loaded per lane and analyzed under reducing conditions. For CD63, 7.5 × 107 sEVs were loaded per lane and analyzed under non-reducing conditions.
(D) Transmission electron microscopy of mouse-derived, gradient-purified sEVs obtained from a pool of either three wild-type (upper panel) or three TRAMP (lower panel) mice. Upper panel, 26K magnification; lower panel, 32K magnification; scale bar, 50 nm.
(E) Quantification of anchorage-independent growth of PC3 cells incubated with TRAMP mouse plasma-derived EVs, pre-treated with ATN-161, an α5β1 inhibitory peptide; control peptide GRGESP (RGE); or PBS vehicle. Data are represented as mean ± SEM. **p < 0.0000072, *p < 0.0015 (Student's t test). Two technical and biological replicates were performed.
(F) Representative images of the data quantified in (E).
(G) NTA of EVs from β1pc−/−/TRAMP mice.
(H) Quantification of anchorage-independent growth of PC3 cells after treatment with β1pc−/−/TRAMP or TRAMP EVs. *p < 1.4 × 10−6. Two technical and biological replicates were performed.
Given that the functional studies were performed by treating recipient cells with equal numbers of particles from TRAMP and wild-type sEV samples, we wanted to biochemically examine gradient-purified sEV content in the same manner. Immunoblotting on equal numbers of vesicles was performed (Figure 6C). We observe, similar to when the samples were loaded based on volume in Figure 5A, that sEVs from wild-type mice do not exhibit the sEV marker CD63, β1, or c-Src. They also lack α5. This shows that even when examining equal numbers of vesicles, there is a dramatic difference in sEV cargo between wild-type and TRAMP mice. Transmission electron microscopy displays standard size and morphology of sEVs isolated from the wild-type mice (Figure 6D) despite not exhibiting the standard protein signature seen in TRAMP mice sEVs (Figure 6C) and PrCa cell-line-derived sEVs (Figure 1).
To validate the function of EV-derived α5β1 observed ex vivo, we first pre-treated EVs from TRAMP mice with ATN-161, an inhibitory peptide to the α5β1 integrin, before incubating them with PC3 cells and assessing their anchorage-independent growth. The concentration of ATN-161 (100 μg/mL) has been previously validated by another group while studying mouse α5β1 function in vitro (Sundaram et al., 2017). Colonies whose sizes were ≥25 μm were quantified and expressed as a percent of the total number of colonies. ATN-161 pre-treatment of TRAMP mouse-derived EVs significantly reduces the colony size of EV recipient cells compared with RGE-containing control peptide and vehicle pre-treatment (Figures 6E and 6F). These results show that the α5β1 integrin plays a crucial role in EV-mediated stimulation of anchorage-independent growth of PrCa cells. The effects observed with ATN-161 on the EVs is not due to any residual free-floating inhibitor binding directly to the cells because a separate experiment shows that ATN-161 treatment of the cells in the absence of sEVs does not significantly alter their anchorage-independent growth (unpublished data).
As the circulating β1 integrin-containing EVs can originate from a plethora of organs and tissues, we next sought to analyze the role of prostate-derived EV β1 integrins in anchorage-independent growth of cancer cells. We utilized tumor-bearing β1pc−/−/TRAMP mice. After isolating plasma-derived EVs, we evaluated EV abundance and size distribution by NTA. The average EV size from β1pc−/−/TRAMP mice was 121.9 nm and the average EV concentration was 4.7 × 1010 EVs/mL of plasma (Figure 6G). The average size of TRAMP EVs was similar (118.0 nm), whereas the concentration (10.5 × 1010 EVs/mL of plasma) was twice the amount of β1pc−/−/TRAMP EVs. This is to be expected given our previous results examining gradient-purified sEVs (Table 1). To determine if prostate-specific β1 is affecting EV function, we analyzed PC3 anchorage-independent growth in response to prostate-derived TRAMP EVs. EVs from β1pc−/−/TRAMP mice do not stimulate anchorage-independent growth of PC3 cells, indicating the importance of β1 expression in the prostate tumor epithelium to the oncogenic ability of secreted EVs (Figure 6H).
Discussion
β1 integrins are expressed in many different cell types and contribute to prostate tumor growth. Our study is the first to demonstrate that β1 integrins in EVs, including sEVs, specifically from prostate tumor epithelial cells, or PrCa cultured cells, mediate the stimulatory function of secreted EVs on anchorage-independent growth. Through the use of a genetic mouse model and cell-line-based genetic rescue, we demonstrate that β1 integrins are required for the stimulatory effects of EVs derived from cancer cells on anchorage-independent growth.
β1 integrins may affect the anchorage-independent functions of EVs on recipient cells in multiple ways. β1 integrins are heavily trafficked between the cell surface and endosomal pathway (De Franceschi et al., 2015, Huet-Calderwood et al., 2017) and are known to signal through endosomes (Alanko et al., 2015); thus they are likely to influence the biology of secreted EVs. Another possible role for β1 in EV functions is that they may influence the composition of EVs secreted from cancer cells as shown in a previous study from our laboratory on other integrins (Lu et al., 2018). Finally, as it is known that a single cell type can produce many different subsets of EVs, it is possible that β1 integrins are contributing to the biogenesis of, and are carried in, a specific subset of EVs that exhibit tumorigenic effects on recipient cells. Our results indicate that β1 may be playing its role in stimulating anchorage-independent growth at the cell surface, but does not affect the ability of EVs to be internalized by recipient cells.
We have established a requirement for β1 in circulating EVs to stimulate in vitro anchorage-independent growth, which suggests that these EVs are able to potentially modulate both primary and metastatic tumor growth. Furthermore, there is evidence of integrins from cancer-derived EVs altering the function of other non-epithelial, non-cancerous cell types. For example, exo from myeloid leukemia cells have been shown to stimulate angiogenic behavior of recipient endothelial cells (Mineo et al., 2012). Another study has shown that pancreatic cancer-derived exo contribute to pre-metastatic niche formation in the liver (Costa-Silva et al., 2015). Furthermore, we have previously demonstrated the transfer of β6-positive EVs to monocytes, causing their M2 polarization (Lu et al., 2018).
β1 integrin-mediated signaling occurs through a complex network of proteins, including JNK, Src, IGF-IR (Goel et al., 2013, Sayeed et al., 2013, Sayeed et al., 2016, Varkaris et al., 2014), and FAK, a non-receptor tyrosine kinase that, with Src, coordinates cell adhesion and cytoskeletal dynamics and regulates cancer cell migration and invasion (Varkaris et al., 2014). Our previous studies have shown that FAK, Src, and IGF-IR are detected in PrCa EVs and thus may be involved in EV-mediated tumorigenic cell growth. We have previously demonstrated a functional interdependence between the α5β1 integrin and IGF-IR in PrCa cells and that this interaction influences androgen receptor activity and cancer cell growth (Sayeed et al., 2012). IGF-IR specifically stabilizes the α5β1 integrin by preventing proteasomal degradation but has no effect on the α2, α4, α6, and α7 subunits (Sayeed et al., 2013). Given the importance of integrin function and integrin-IGF-IR cross talk in cancer cell growth (Sayeed et al., 2012, Sayeed et al., 2013, Sayeed et al., 2016), and IGF-IR enrichment in PrCa EVs (DeRita et al., 2017), IGF-IR may support α5β1 stability and promote EV functions. FAK is also associated with β1 signaling and cancer cell survival in the absence of ECM attachment (Alanko et al., 2015). As we see that EV FAK levels correlate with β1 and that recipient cells have increased FAK upon transfer of β1 and FAK-positive EVs, it is possible that FAK is also a driver of EV-mediated stimulation of cancer cell anchorage-independent growth.
Our findings implicate that β1 integrins in the prostate tumor epithelium may be involved in the formation and secretion of EVs. This was evidenced by the fact that the total number of circulating EVs was twice as high in the TRAMP mice compared with the β1pc−/−/TRAMP mice, which at 20 weeks show comparable tumor sizes (Table 1). We have also demonstrated that the prostate tumor epithelial cells are responsible for nearly all the circulating β1-positive EVs that express canonical markers such as CD9 and CD63. Overall, this study demonstrates that circulating EV β1 integrins shed by prostate tumors are activators of cancer cell growth, thus opening new perspectives in translational medicine and cancer treatment.
Limitations of the Study
This study performed using a mouse model needs to be translated in human subjects. Furthermore, at this time, although the results may be informative for early-stage cancer diagnosis, they may not provide insightful information on advanced disease.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was supported by NIH R01 CA-109874, CA-224769, P01 CA-140043; Thomas Jefferson University Dean's Transformational Science Award. This project is also funded, in part, under a Commonwealth University Research Enhancement Program grant with the Pennsylvania Department of Health (H.R.); the Department specifically disclaims responsibility for any analyses, interpretations or conclusions. Dr. Dicker's research is supported by a Challenge Grant Award from the Prostate Cancer Foundation. Dr. Rodeck would like to acknowledge support from the DoD PCRP (W81XWH-16-1-0679 and W81XWH12-1-0477). Research reported in this publication utilized the shared flow cytometry and bioimaging core facilities at Sidney Kimmel Cancer Center at Jefferson Health and was supported by the National Cancer Institute of the National Institutes of Health under Award Number P30CA056036. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We also acknowledge the use of instruments at the Electron Microscopy Resource Laboratory at the University of Pennsylvania Perelman School of Medicine. We would like to thank Dr. Richard Hynes, MIT, for chicken β1 cDNA, and Drs. Andrew Aplin, Jeffery Benovic, Maria Yolanda Covarrubia, James Keen, Huimin Lu, My Mahoney, Vera Moiseenkova-Bell, and Andrea Morrione, for thoughtful discussion on this work. We would like to thank Veronica Robles for administrative assistance with the preparation of the manuscript.
Author Contributions
R.M.D, A.S., and L.R.L. designed experiments. R.M.D., and A.S. performed experiments. R.M.D., A.S., and L.R.L. analyzed corresponding results. V.G., S.R.K., A.F., and C.D.S. assisted in performing experiments and generating mice and cell lines. V.G., S.S., and A.S. managed mouse colonies and handling. R.M.D., A.S., and L.R.L. wrote the manuscript. A.P.D., U.R., and P.M. provided guidance on the study. U.R. assisted in writing revisions. S.K.M. performed EM analysis.
Declaration of Interests
The authors declare no competing interests.
Published: April 26, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.03.022.
Supplemental Information
References
- Alanko J., Mai A., Jacquemet G., Schauer K., Kaukonen R., Saari M., Goud B., Ivaska J. Integrin endosomal signalling suppresses anoikis. Nat. Cell Biol. 2015;17:1412–1421. doi: 10.1038/ncb3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino D., De Pitta C., Orso F., Zampini M., Casara S., Penna E., Quaglino E., Forni M., Damasco C., Pinatel E. miR148b is a major coordinator of breast cancer progression in a relapse-associated microRNA signature by targeting ITGA5, ROCK1, PIK3CA, NRAS, and CSF1. FASEB J. 2013;27:1223–1235. doi: 10.1096/fj.12-214692. [DOI] [PubMed] [Google Scholar]
- Colombo M., Raposo G., Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- Costa-Silva B., Aiello N.M., Ocean A.J., Singh S., Zhang H., Thakur B.K., Becker A., Hoshino A., Mark M.T., Molina H. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi N., Hamidi H., Alanko J., Sahgal P., Ivaska J. Integrin traffic - the update. J. Cell Sci. 2015;128:839–852. doi: 10.1242/jcs.161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean I., Dzinic S.H., Bernardo M.M., Zou Y., Kimler V., Li X., Kaplun A., Granneman J., Mao G., Sheng S. The secretion and biological function of tumor suppressor maspin as an exosome cargo protein. Oncotarget. 2017;8:8043–8056. doi: 10.18632/oncotarget.13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRita R.M., Zerlanko B., Singh A., Lu H., Iozzo R.V., Benovic J.L., Languino L.R. c-Src, insulin-like growth factor I receptor, G-protein-coupled receptor kinases and focal adhesion kinase are enriched into prostate cancer cell exosomes. J. Cell. Biochem. 2017;118:66–73. doi: 10.1002/jcb.25611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele C., Singh A., Zerlanko B.J., Iozzo R.V., Languino L.R. The αvβ6 integrin is transferred intercellularly via exosomes. J. Biol. Chem. 2015;290:4545–4551. doi: 10.1074/jbc.C114.617662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald T.J., Wang T., Goel H.L., Huang J., Stein G., Lian J., Davis R.J., Doxsey S., Balaji K.C., Aronowitz J. Prostate carcinoma and radiation therapy: therapeutic treatment resistance and strategies for targeted therapeutic intervention. Expert Rev. Anticancer Ther. 2008;8:967–974. doi: 10.1586/14737140.8.6.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel H.L., Moro L., Murphy-Ullrich J.E., Hsieh C.C., Wu C.L., Jiang Z., Languino L.R. β1 integrin cytoplasmic variants differentially regulate expression of the antiangiogenic extracellular matrix protein thrombospondin 1. Cancer Res. 2009;69:5374–5382. doi: 10.1158/0008-5472.CAN-09-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel H.L., Sayeed A., Breen M., Zarif M.J., Garlick D.S., Leav I., Davis R.J., Fitzgerald T.J., Morrione A., Hsieh C.C. β1 integrins mediate resistance to ionizing radiation in vivo by inhibiting c-Jun amino terminal kinase 1. J. Cell. Physiol. 2013;228:1601–1609. doi: 10.1002/jcp.24323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel H.L., Underwood J.M., Nickerson J.A., Hsieh C.C., Languino L.R. β1 integrins mediate cell proliferation in three-dimensional cultures by regulating expression of the sonic hedgehog effector protein, GLI1. J. Cell. Physiol. 2010;224:210–217. doi: 10.1002/jcp.22116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C., Yang Z., Wu F., Han L., Liu Y., Gong W. miR-17 inhibits ovarian cancer cell peritoneal metastasis by targeting ITGA5 and ITGB1. Oncol. Rep. 2016;36:2177–2183. doi: 10.3892/or.2016.4985. [DOI] [PubMed] [Google Scholar]
- Hamidi H., Pietila M., Ivaska J. The complexity of integrins in cancer and new scopes for therapeutic targeting. Br. J. Cancer. 2016;115:1017–1023. doi: 10.1038/bjc.2016.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y., Haimovich B., Reszka D., Boettiger D., Horwitz A. Expression and function of chicken integrin β1 subunit and its cytoplasmic domain mutants in mouse NIH 3T3 cells. J. Cell Biol. 1990;110:175–194. doi: 10.1083/jcb.110.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini-Beheshti E., Choi W., Weiswald L.B., Kharmate G., Ghaffari M., Roshan-Moniri M., Hassona M.D., Chan L., Chin M.Y., Tai I.T. Exosomes confer pro-survival signals to alter the phenotype of prostate cells in their surrounding environment. Oncotarget. 2016;7:14639–14658. doi: 10.18632/oncotarget.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet-Calderwood C., Rivera-Molina F., Iwamoto D.V., Kromann E.B., Toomre D., Calderwood D.A. Novel ecto-tagged integrins reveal their trafficking in live cells. Nat. Commun. 2017;8:570. doi: 10.1038/s41467-017-00646-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker K., Heinzelmann J., Beckham C., Ochiya T., Jenster G. Extracellular vesicles and their role in urologic malignancies. Eur. Urol. 2016;70:323–331. doi: 10.1016/j.eururo.2016.02.046. [DOI] [PubMed] [Google Scholar]
- Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Thery C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. U S A. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishn S.R., Singh A., Bowler N., Duffy A.N., Friedman A., Fedele C., Kurtoglu S., Tripathi S.K., Wang K., Hawkins A. Prostate cancer sheds the alphavbeta3 integrin in vivo through exosomes. Matrix Biol. 2018 doi: 10.1016/j.matbio.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.C., Lin S.C., Yu G., Cheng C.J., Liu B., Liu H.C., Hawke D.H., Parikh N.U., Varkaris A., Corn P. Identification of bone-derived factors conferring de novo therapeutic resistance in metastatic prostate cancer. Cancer Res. 2015;75:4949–4959. doi: 10.1158/0008-5472.CAN-15-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.F., Gemenetzidis E., Marshall F.J., Davies D., Yu Y., Frese K., Froeling F.E., Woolf A.K., Feakins R.M., Naito Y. RhoC interacts with integrin alpha5beta1 and enhances its trafficking in migrating pancreatic carcinoma cells. PLoS One. 2013;8:e81575. doi: 10.1371/journal.pone.0081575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Bowler N., Harshyne L.A., Hooper D.C., Krishn S.R., Kurtoglu S., Fedele C., Liu Q., Tang H.Y., Kossenkov A.V. Exosomal ανβ6 integrin is required for monocyte m2 polarization in prostate cancer. Matrix Biol. 2018;70:20–35. doi: 10.1016/j.matbio.2018.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi V.R., Freeman M.R., Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015;40:41–51. doi: 10.1016/j.semcdb.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi V.R., Spinelli C., Reis-Sobreiro M., Cavallini L., You S., Zandian M., Li X., Mishra R., Chiarugi P., Adam R.M. MYC mediates large oncosome-induced fibroblast reprogramming in prostate cancer. Cancer Res. 2017;77:2306–2317. doi: 10.1158/0008-5472.CAN-16-2942. [DOI] [PubMed] [Google Scholar]
- Mineo M., Garfield S.H., Taverna S., Flugy A., De Leo G., Alessandro R., Kohn E.C. Exosomes released by K562 chronic myeloid leukemia cells promote angiogenesis in a Src-dependent fashion. Angiogenesis. 2012;15:33–45. doi: 10.1007/s10456-011-9241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroshnikova Y.A., Rozenberg G.I., Cassereau L., Pickup M., Mouw J.K., Ou G., Templeman K.L., Hannachi E.I., Gooch K.J., Sarang-Sieminski A.L. alpha5beta1-Integrin promotes tension-dependent mammary epithelial cell invasion by engaging the fibronectin synergy site. Mol. Biol. Cell. 2017;28:2958–2977. doi: 10.1091/mbc.E17-02-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul N.R., Allen J.L., Chapman A., Morlan-Mairal M., Zindy E., Jacquemet G., Fernandez del Ama L., Ferizovic N., Green D.M., Howe J.D. alpha5beta1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. J. Cell Biol. 2015;210:1013–1031. doi: 10.1083/jcb.201502040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plow E.F., Haas T.A., Zhang L., Loftus J., Smith J.W. Ligand binding to integrins. J. Biol. Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- Sayeed A., Alam N., Trerotola M., Languino L.R. Insulin-like growth factor 1 stimulation of androgen receptor activity requires β1A integrins. J. Cell. Physiol. 2012;227:751–758. doi: 10.1002/jcp.22784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayeed A., Fedele C., Trerotola M., Ganguly K.K., Languino L.R. IGF-IR promotes prostate cancer growth by stabilizing α5β1 integrin protein levels. PLoS One. 2013;8:e76513. doi: 10.1371/journal.pone.0076513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayeed A., Lu H., Liu Q., Deming Ii D., Duffy A., McCue P., Dicker A.P., Davis R.J., Gabrilovich D., Rodeck U. β1 integrin- and JNK-dependent tumor growth upon hypofractionated radiation. Oncotarget. 2016;7:52618–52630. doi: 10.18632/oncotarget.10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Singh A., Fedele C., Lu H., Nevalainen M.T., Keen J.H., Languino L.R. Exosome-mediated transfer of ανβ3 integrin from tumorigenic to non-tumorigenic cells promotes a migratory phenotype. Mol. Cancer Res. 2016;14:1136–1146. doi: 10.1158/1541-7786.MCR-16-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram A., Chen C., Khalifeh-Soltani A., Atakilit A., Ren X., Qiu W., Jo H., DeGrado W., Huang X., Sheppard D. Targeting integrin alpha5beta1 ameliorates severe airway hyperresponsiveness in experimental asthma. J. Clin. Invest. 2017;127:365–374. doi: 10.1172/JCI88555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thery C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M., Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- Varkaris A., Katsiampoura A.D., Araujo J.C., Gallick G.E., Corn P.G. Src signaling pathways in prostate cancer. Cancer Metastasis Rev. 2014;33:595–606. doi: 10.1007/s10555-013-9481-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Languino L.R., Lian J., Stein G., Blute M., Fitzgerald T.J. Molecular targets for radiation oncology in prostate cancer. Front. Oncol. 2011;1:1–11. doi: 10.3389/fonc.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo H.I., Kim B.K., Yoon S.K. MicroRNA-330-5p negatively regulates ITGA5 expression in human colorectal cancer. Oncol. Rep. 2016;36:3023–3029. doi: 10.3892/or.2016.5092. [DOI] [PubMed] [Google Scholar]
- You S., Knudsen B.S., Erho N., Alshalalfa M., Takhar M., Al-Deen Ashab H., Davicioni E., Karnes R.J., Klein E.A., Den R.B. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016;76:4948–4958. doi: 10.1158/0008-5472.CAN-16-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Chen A., Li S., Tao X., Sheng B., Chetry M., Zhu X. Predictive role of galectin-1 and integrin alpha5beta1 in cisplatin-based neoadjuvant chemotherapy of bulky squamous cervical cancer. Biosci. Rep. 2017;37 doi: 10.1042/BSR20170958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.