

ABSTRACT
Diet and nutrition contribute to both beneficial and harmful aspects of oxidative processes. The harmful processes, termed oxidative stress, occur with many human diseases. Major advances in understanding oxidative stress and nutrition have occurred with broad characterization of dietary oxidants and antioxidants, and with mechanistic studies showing antioxidant efficacy. However, randomized controlled trials in humans with free-radical-scavenging antioxidants and the glutathione precursor N-acetylcysteine have provided limited or inconsistent evidence for health benefits. This, combined with emerging redox theory, indicates that holistic models are needed to understand the interplay of nutrition and oxidative stress. The purpose of this article is to highlight how recent advances in redox theory and the development of new omics tools and data-driven approaches provide a framework for future nutrition and oxidative stress research. Here we describe why a holistic approach is needed to understand the impact of nutrition on oxidative stress and how recent advances in omics and data analysis methods are viable tools for systems nutrition approaches. Based on the extensive research on glutathione and related thiol antioxidant systems, we summarize the advancing framework for diet and oxidative stress in which antioxidant systems are a component of a larger redox network that serves as a responsive interface between the environment and an individual. The feasibility for redox network analysis has been established by experimental models in which dietary factors are systematically varied and oxidative stress markers are linked through integrated omics (metabolome, transcriptome, proteome). With this framework, integrated redox network models will support optimization of diet to protect against oxidative stress and disease.
Keywords: exposome, high-resolution metabolomics, redox signaling, redox interface, thiol-disulfide redox state
Introduction
Nutrition science includes approaches for both population and individual health and is an inherently holistic discipline. Despite this, nutrition has largely relied upon the logic of Descartes (Cartesian reductionism), namely, that the whole can be understood by understanding the component parts. This reductionist approach has had considerable success in terms of understanding essential and nonessential nutrients and their distributions, functions, and fates (Figure 1A) (1). This has effectively complemented the central dogma of biology by illuminating which gene-directed products require exogenous nutrients for function and which genetic variations can be complemented by targeted nutrient support. This success is elegantly demonstrated by management of genetic variants of phenylketonuria with therapeutic levels of tetrahydrobiopterin (2).
FIGURE 1.

A holistic approach is needed to advance research in nutrition and oxidative stress in disease. (A) Cartesian reductionism has provided details concerning oxidative stress causes and mechanisms, as well as extensive knowledge of dietary oxidants and antioxidants. (B) Omics technologies and systems biology approaches are available to study how the functional outcomes (right) of an individual interacts with the array of oxidant exposures and dietary factors (left). This interaction occurs through a redox interface in which multiple systems work together to provide a functional network to defend against and adapt to environmental challenges and resources (middle). Cys, cysteine; GSH, glutathione.
Despite such achievements in other areas, success in protecting against oxidative stress in disease has been more modest. Extensive observational and experimental research provides evidence that oxidative stress is involved in most major human diseases (3, 4). Furthermore, mechanistic studies and targeted treatments in oxidative stress models show efficacy of antioxidants in protection against chemical reactions characterized as oxidative stress (5–7). In spite of this, large-scale double-blind interventional trials in humans showed that free-radical-scavenging antioxidants had limited evidence for health benefits (8–13). Similarly, trials with the thiol antioxidant and glutathione (GSH) precursor N-acetylcysteine (NAC) provide inconsistent evidence for widespread benefit (14).
Advances in redox theory and introduction of new omics tools are beginning to address this conundrum and transform oxidative stress research. In this, oxidative stress is part of the larger scope of nutrition in systems biology and medicine (Figure 1B). Nutrition science is advancing along with other biomedical disciplines in an integrative approach toward providing personalized medicine. Pharmacogenomics has already demonstrated success in advancing patient care and treatment (15). For some drugs, genetic testing allows identification of patients who may be nonresponders or at risk of adverse outcomes (15–17). With the recognition that many complex diseases result from interacting genetic and environmental factors, gene-environment interaction studies have started to be leveraged in environmental health research to address complex disease etiologies (18, 19). By defining nutritional requirements for an individual within the context of an individual's interacting genome and exposome, science and medicine move beyond the limits of population-based guidelines. In the past, such a holistic view of nutrition for an individual has been intractable. Today omics technologies are becoming increasingly affordable to enrich phenotypic data and biomarkers of risk with details of large numbers of molecular mediators. Simultaneously, tools are being developed to describe the integrated function and dysfunction of these systems within individuals.
In the following, we start with the conclusion that a holistic approach is needed to understand nutritional impact upon oxidative stress. We finish with results from studies that use omics data and data-driven methods to understand functional network responses to nutrition and oxidative stress. In between, we use the extensive research on the GSH and related thiol antioxidant systems as a basis to summarize the advancing conceptual framework for diet and oxidative stress in which antioxidant systems are part of a redox network of proteins and small molecules with redox switches to serve as an interface between an individual and the environment to maintain internal redox organization (Figure 1B). Although such a holistic approach is not new, this perspective reveals the research needs for interacting and understudied redox players and opportunities to develop precision nutrition through leveraging advances in integrated omics and redox systems biology.
Oxidative Stress: A Disruption of Redox Signaling and Control and/or Molecular Damage
A recent review provides details on definitions and progress in oxidative stress (3). Briefly, an important role for oxidants in redox signaling was recognized over the same time frame as data accumulated from interventional trials showing that supplemental antioxidants above nutritional requirements provide little health benefit (20). With this recognition of redox signaling, the definition of oxidative stress was updated to “an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage” (21). This definition preserved the original definition of oxidative stress (22) as an adverse process, i.e., oxidative distress. With redox signaling defined as “transmission of a redox signal via an essential redox element from a source to a target,” this allows parallel use of oxidative eustress, defined as “physiological generation of oxidants,” to clearly discriminate between the adverse and beneficial aspects of oxidant production (3).
Free-radical and nonradical oxidants
Both 1-electron (free radical) and 2-electron (nonradical) transfer reactions occur in biological systems, and disruption of either can result in oxidative stress (23). Free radicals are reactive and unstable molecules leading to chain reactions of oxidation (24). Free-radical reactions are targeted by the properties of antioxidants such as vitamin E and vitamin C, which terminate the chain reactions. Although free-radical reactions have been a common target in oxidative stress research, data show that scavenging mechanisms in biological systems limit propagation under most conditions and that nonradical reactions predominate under most oxidative stress conditions (23). Nonradical oxidants include H2O2, reactive sulfur species, disulfides, and many common metabolites containing carbonyl and quinone structures (3, 25). Additionally, O2 and other chemicals can exist in activated states with electrons or electron pairs in orbitals other than the ground state. For O2, this is termed singlet oxygen (1O2) which can be formed as a consequence of chemical reactions or due to activation by visible light in the presence of photosensitizers. Activated chemicals such as 1O2 are far more reactive with biomolecules than ground-state O2 and result in oxidative stress. The number of known oxidants that cause oxidative stress (Table 1), along with their respective broad range of physical and chemical properties, is daunting. Across a lifespan, individuals are exposed regularly to dietary oxidants, glyco-oxidative stress, endoplasmic reticulum stress, proteotoxic stress, disulfide stress, photo-oxidative stress, ionizing radiation, nitrosative stress, nanoparticles, and air pollution (Table 1) (3).
TABLE 1.
Humans are commonly exposed to causes of oxidative stress and dietary antioxidants1
| Source of oxidative stress | Dietary antioxidant |
|---|---|
| Dietary oxidants: peroxides, nitrites, sulfites | Tocopherols and tocotrienols |
| Glyco-oxidative stress (AGE) | Ascorbate, GSH, ubiquinols |
| ER stress | Supporting nutrients: Se, Fe, Cu, Zn, B vitamins |
| Proteotoxic stress | Carotenoids: lutein, lycopene, zeaxanthin |
| Disulfide stress | Flavonoids: anthocyanins, phloretin |
| Photooxidative stress: ultraviolet (UV-A, UV-B), visible, infrared-A | Flavonols: keampferol, quercetin, epicatechin, epicatechin gallate |
| Ionizing radiation | Flavanones: eriodictyol, hesperetin, naringenin |
| Nitrosative stress | Flavones: luteolin |
| Nanoparticles | Isoflavonoids: daidzein, genistein |
| Air pollution: sulfur dioxide, nitrogen dioxide, ozone | Organosulfur compounds: allicin |
| Physical forces: wind, sound, heat | Phenolic acids: caffeic acid, chlorogenic acid |
| Toxic trace metals | Polyphenols: butein, curcumin, resveratrol |
| Environmental pollutants: pesticides, POPs | Stilbenes: tetrahydroxystilbene glucoside |
| — | Tannins: ellagitannins |
1Reductionist approaches are needed to identify and elucidate mechanisms of the individual elements; whereas, systems biology and network analyses are needed to understand biological responses to simultaneous exposure to multiple oxidants and antioxidants. AGE, advanced glycation end-product; ER, endoplasmic reticulum; GSH, glutathione; POP, persistent organic pollutant.
Dietary antioxidants
The number and respective characteristics of dietary antioxidants (Table 1) is similarly overwhelming. This includes antioxidant vitamins (e.g., tocopherols, tocotrienols, and ascorbate) as well as other direct antioxidants (e.g., GSH, ubiquinols) (Table 1). It also includes other nutrients (Se, Fe, Cu, Zn, and riboflavin and other B vitamins) needed to support antioxidant systems. The spectrum additionally includes thousands of dietary compounds that affect oxidative metabolism and oxidative stress, such as carotenoids (lutein, lycopene), flavonoids (anthocyanins, phloretin), flavonols (keampferol, quercetin), flavanols (catechin, epicatechin), flavanones (eriodictyol, hesperetin), flavones (luteolin), isoflavonoids (daidzein, genistein), organosulfur compounds (allicin), phenolic acids (caffeic acid, chlorogenic acid), polyphenols (curcumin, resveratrol), stilbenes (tetrahydroxystilbene glucoside), and tannins (ellagitannins) (Table 1) (26). Considering the historic use of Cartesian reductionism in nutrition and oxidative stress, one must conclude that this approach has effectively defined the relevant oxidants and antioxidants and provided useful understanding of dietary components and function. At the same time, these extensive lists, along with variability in exposure doses, metabolic pathways, and molecular interactions, force a shift away from reductionist approaches in understanding oxidative stress within a functioning organism, i.e., there are too many components to allow practical definition of variation of 1 component when this is interacting with many other varying components. The original definition of oxidative stress (22) actually captured the sense of a collective imbalance between antioxidants and oxidants, but the analytic tools were insufficient to measure the many oxidant and antioxidant components and map their interactions. New high-resolution metabolomics (HRM) methods combined with transcriptome and redox proteome analyses now provide an opportunity to fill in the details.
Redox Theory and Oxidative Stress Mechanisms
Advances in redox theory (27) provide a framework to address mechanisms of oxidative stress inclusive of the range of dietary oxidants and interacting antioxidant systems. Important advances resulted from studies of NADPH oxidase (NOX) family of enzymes. These are widely distributed across most cell types (28, 29) and contribute to superoxide anion radical (O2−.) production and maintenance of steady-state H2O2 concentrations in the low nanomolar range (30) (Figure 2A). Elucidation of these systems showed that in combination with well-described GSH peroxidase- and thioredoxin (TRX)-dependent peroxiredoxins, NADPH-dependent systems function in opposition to simultaneously produce and eliminate H2O2 (Figure 2A). This creates a failsafe system to maintain an oxidant tone in cells; decline in NADPH limits reactions for both oxidant generation and oxidant elimination. Thus, like the parallel NADP and NAD systems that allow anabolism and catabolism to occur simultaneously in cells (31), maintenance of steady-state pools of reductants and oxidants allows reductive and oxidative metabolism to occur simultaneously in cells. This is further elaborated in a redox control structure which regulates the redox state of protein cysteine (Cys) residues in functionally related proteins (Figure 2A) (32, 33). Importantly, this redox organizational structure allows diverse functional systems to be coordinated through redox control of thiol sulfur switches in proteins (34), and maintains relative stability of these systems against both oxidants and reductants. Redox signaling occurs within this structure by activation of NOX enzymes in specific subcellular sites with creation of localized oxidant gradients (35).
FIGURE 2.

In redox theory, NADPH plays a central role in supporting redox organization and defense against environmental threats. (A) NADPH is used by NADPH oxidases (NOX), such as NOX4, to maintain a steady-state pool of H2O2, which functions along with H2O2 from mitochondria to maintain steady-state oxidation of Cys thiols in proteins. NADPH is used by TRXRs and GR to maintain the TRX and GSH pools to counterbalance the oxidation. This redox steady state is part of a broader epiproteomic system for integration of bioenergetics and metabolism with protein structure and function. (B) NADPH is used by NOX and MPO in a respiratory burst to kill invading microorganisms. NADPH is also used by NADPH:quinone reductase 1 (NQO1) to reduce and prevent toxicity from quinones. The product quinols are conjugated and eliminated. NADPH also maintains the GSH pool to support detoxification of quinones and other electrophiles by GST. Based upon the principles of the Redox Code (27) and Redox Theory of Aging (36, 37). GPX, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione; GST, glutathione S-transferase; MPO, myeloperoxidase; NOX, NADPH oxidase; NQO1, NADPH:quinone reductase 1; NQO2, NRH:quinone reductase 2; PRX, peroxiredoxin; TRX, thioredoxin; TRXR, thioredoxin reductase.
Thiol/Disulfide Redox Systems as Measures of Oxidative Stress
The steady-state redox potential (Eh) of a molecule undergoing oxidation-reduction reactions provides a simple way to describe the tendency to donate or accept electrons. Eh is commonly expressed as an oxidation potential, given in millivolts and named in terms of the electron acceptor in the pair (e.g., GSSG for the GSH/GSSG couple); a highly oxidizing acceptor/donor couple has a more positive value, whereas a more reducing couple has a more negative value. Values are calculated from the Nernst equation, which contains a term (Eo) for a chemical's inherent tendency to accept or donate electrons at the relevant pH of the environment in which the reaction occurs, as well as a term containing the concentrations of the reduced and oxidized species in the redox couple (38). Steady-state estimates of Eh provide information about how redox-active elements are responding under different biological conditions and compartments. For example, EhGSSG for the GSH couple varies based on the cellular state. Proliferating cells are relatively reducing (EhGSSG, –260 to –230 mV) (38, 39); cells under growth arrest are more oxidized (–220 to –190 mV) (39, 40); and cells undergoing apoptosis are more oxidized (–80 to 0 mV) (41, 42).
As indicated above, these thiol/disulfide redox systems support elimination of H2O2 and other nonradical oxidants as well as maintenance of the structural and functional organization of cells through reversible sulfur-switching mechanisms (34). Of specific importance to diet and nutrition, these systems serve as part of the defensive barrier to protect tissues from the relatively oxidizing environment provided by an O2-rich atmosphere. This defensive network is further elaborated in the use of NOX2 in phagocytes to generate high levels of oxidants to kill invading microorganisms, use of GSH to eliminate reactive electrophiles and oxidants, and use of NADPH to support reduction of reactive dietary quinones for conjugation and elimination (Figure 2B).
Several nutritionally important metals, including Fe, Cu, Mn, Se and Zn, interact with redox systems both positively and negatively in oxidative stress and disease (43–45). Mn, for instance, is a critical component of the mitochondrial antioxidant enzyme superoxide dismutase-2 but also directly interacts with protein thiols and peptides and, in excess, causes oxidative stress. Nutritional and nonnutritional metals bind to many specific metal-binding proteins, such as ferritin for Fe transport and metallothionein for Cd sequestration. Additionally, many metals have less-specific interactions with protein thiols and low-molecular-weight thiols, including GSH and Cys. These interactions can have considerable importance in biological function yet are difficult to study over the usual dietary ranges because of the number and diversity of metals and the large range of thiol targets in the cysteine proteome (32).
The abundant cellular antioxidant GSH provides a useful example because this tripeptide has six possible binding sites (S, N, O) for metals, with the thiol group having the greatest affinity for metal cations (46). Metal-GSH complexes form nonenzymatically and are thermodynamically favored. GSH transporters move metal-GSH complexes across the plasma membrane (47). Thus, the redox state of the GSH system interacts with redox activity of metals, i.e., GSH levels are affected by dietary exposure to nutritional and nonnutritional metals. In turn, this can affect GSH's functions in the mobilization and delivery of metal ions within the body. These interactions are further complicated because dietary phytochemicals also bind metals. Phytic acid (48) and phytochelatins (49) have potential to broadly affect redox control through effects on absorption of trace metals. The spectrum of dietary metals, metal reactivities, and metal interactions with dietary phytochemicals adds to the complexity of oxidant and antioxidant systems described above and emphasizes the need to include nutritional and nonnutritional metals within systems biology models of nutrition and oxidative stress.
Redox Systems Biology in Nutrition
Redox systems biology is defined as the computational and mathematical modeling of redox-dependent processes in complex biological systems (50). In integrated systems, individual reactions involving electron transfer (i.e., reduction-oxidation reactions) are not separable from downstream processes dependent upon such transfer. Therefore, systems biology captures the full dynamic range of redox interactions. This includes more traditional considerations, such as reactions causing oxidative distress, to more recent concepts such as redox signaling and control (Figure 1B). Details highlighting advances in the understanding of redox organization are provided elsewhere (27, 32, 51), and additional needs to understand regional differences within tissues and subcellular structures are discussed below.
Knowledge-based assembly of redox models
Systems engineering uses bottom-up and top-down approaches to describe complex systems. The challenges to developing bottom-up redox system models with a knowledge-based assembly of component parts were recently reviewed (52). Simply, 20,000 genes are expressed into a larger number of transcripts; many of these are translated into multiple protein forms which can further undergo dozens of post-translational modifications. This amplifies the number of functional redox elements, probably into the range of hundreds of thousands in mammalian cells. To add to complexity, the proteins have differential distribution among cellular and subcellular compartments and multiple domains with large differences in propensity for interactions with lipids, other proteins, nucleic acids, metals, and small molecules. The large number of possible interactions makes knowledge-based reassembly of an individual's redox network from the component parts virtually impossible. Consequently, despite the remarkable wealth of knowledge about redox biology in mammalian systems and redox-active components in the diet, substantial barriers exist to the formulation of mechanism-based hypotheses for oxidative stress research.
Data-driven redox network models to test nutritional interventions to protect against oxidative stress
Redox theory provides a way around this limitation to mechanistic research of oxidative stress in complex systems. Specifically, in redox theory, redox mediators, such as thioredoxins, GSH, and Cys, interact with many other redox elements and provide hubs to monitor network activity. Considerable information is available on GSH and Cys in health and disease, and we use these below as a foundation to develop a path forward to test nutritional interventions to protect against oxidative stress as a causative factor in disease development and progression. With measures of GSH and Cys systems as a foundation, integration of omics-level data with functional and phenotypic measures can, in principle, provide a powerful top-down approach for mechanistic studies of oxidative stress in humans and model systems. As described below, tools are now available to support data-driven analyses within a redox systems biology framework.
Redox hubs, compartmentalization, and interorgan redox control
Considerable detail is available concerning the steady-state redox potentials of thioredoxins, GSH/GSSG, and Cys/CySS in different subcellular compartments (53, 54). Intracellular variation of redox potentials across macromolecular structures (e.g., mitochondria, nucleus, endoplasmic reticulum) and microcompartments (e.g., spatiotemporal variation in production of redox-signaling molecules) provides means to refine models of redox signaling and control. Differences in the free concentrations of redox couples among compartments can be driven by permeability barriers, macromolecule binding, nonhomogenous distribution of redox-sensitive molecules, and heterogeneity of cell types across different tissues (27).
Redox states of intracellular compartments
Generalizations concerning redox states of subcellular compartments are largely derived from cell culture studies and must be used with caution because each exists as a dynamic steady state and is subject to variation due to specific conditions of measurement. Mitochondria are central hubs of energy production, utilizing the electron transport chain as a means of producing ATP, and using oxygen as the final electron acceptor. Mitochondrial thioredoxin and GSH systems are maintained at relatively reducing steady-state values (54), perhaps reflecting a need for powerful defenses against the range of reductive and oxidative forces among redox couples (55). Mitochondria have unique redox-related enzymes and transporters serving in this protection, such as glutaredoxin 2, which functions to catalyze reversible oxidation and glutathionylation of mitochondrial membrane proteins as well as protecting from oxidative stress and apoptosis (56).
The nuclear redox state is also relatively reducing (53, 57), with translocation of TRX1 into nuclei in response to oxidative stress. Several transcription factors (e.g., NRF2, P53, NF-κB) have redox-sensitive cysteine residues in the DNA-binding region, providing nuclear redox control in addition to cytoplasmic activation of signaling cascades in response to oxidative stress (58–61). Studies of thiol-dependent antioxidant systems in subcellular compartments of human colonic epithelial HT-29 cells following depletion of the energy precursors, glucose and glutamine, from the culture medium showed that cytosolic TRX1 and mitochondrial TRX2 were oxidized without nuclear TRX1 oxidation (62). A TRX1 substrate, redox factor-1, was also oxidized in cytosol but was reduced in nuclei, and protein S-glutathionylation was also increased in cytosol but not in nuclei. The results indicate that nuclei are better protected against oxidative stress than cytoplasm or mitochondria and suggest that energy precursors are critical to maintain thiol redox systems.
The endoplasmic reticulum has specific enzyme systems to support introduction of disulfide bonds to ensure proper folding of proteins for secretion (63). These systems maintain an oxidized state relative to the cytoplasm, and disruption by either a more reducing or oxidizing environment leads to aberrant protein folding in the secretory pathway, endoplasmic reticulum stress, and cell death. Peroxisomes, with several H2O2-producing enzymes, and lysosomes, with degradation products of endocytosis and organelle turnover, are also relatively oxidizing (54). The available data emphasize that organelles are functionally diverse and contain multiple redox systems operating in a dynamic steady state to support specialized functions.
Among the more exciting advances for mechanistic studies of oxidative stress was the development of fluorescence-based indicators, which have become a critical tool in redox biology research. However, only recently have fluorescence-based redox-sensitive protein probes been developed to allow real-time monitoring of thiol redox dynamics intracellularly. With genetic encoding, specific subcellular locations can be targeted (64). The primary redox probes in use are the redox-sensitive yellow fluorescent proteins (rxYFP) and green fluorescent proteins (roGFPs). Through fusion of probes with redox enzymes (e.g., glutaredoxin-1 or peroxidase ORP1), the probes have better dynamic response to physiologically relevant changes in GSH/GSSG or H2O2 production. In an initial demonstration of this technology, researchers were able to assess redox changes as they related to cell density, mitochondrial depolarization, and growth factor availability across nanomolar changes in GSSG on a timescale of seconds to minutes (65). Sensors are mostly limited to the glutaredoxin/GSH system, although the feasibility for other systems such as the Trx and nicotinamide nucleotide systems have been established (64). Such tools can be expected to improve the ability to monitor the responses of redox systems, and complement the relatively limited capabilities to study complex oxidative reactions under relevant conditions of diet and nutrition.
Plasma and extracellular redox state
A holistic network model for oxidative stress must address the differences of redox systems across body compartments and account for mixed effects of dietary interventions focused on reducing global oxidative stress (8–11, 13, 66). In consideration of heterogeneous compartments, organ systems communicate through the plasma, so plasma redox measures provide both a conceptual connectivity and a practical approach to link diet, oxidative stress, and human health impact. Research is available showing dietary factors such as sulfur amino acid (SAA) and glutamine intake are linked with plasma redox state (67, 68). Additionally, a noninvasive method that used magnetic resonance spectroscopy showed a relationship between dietary SAA and human midbrain glutamate (69). Parallel redox measurements of other extracellular environments, such as the gastrointestinal lumen and cerebrospinal fluid, are needed to develop redox network maps. Such maps could then be used to investigate dietary effects on oxidative stress at a redox systems level. Stable isotopic tracer studies have provided such maps for SAA turnover (70–72), and extension of this research is needed to address interorgan thiol/disulfide redox systems homeostasis.
Intraorgan redox control
Little research is available to address the extent of redox variations in different regions of organs. Higher GSH concentrations occur in rapidly growing regions of tumors and also in crypts in intestinal epithelia. GSH contents also differ among cell types in brain, lungs, bone marrow niche, and other organs, and therefore local differences can be expected based upon the cell types present. The pancreas is a unique organ for studying redox variations due to regionally distinct endocrine and exocrine functions. Redox signaling is a key component of normal insulin release from β cells, and oxidative stress is linked to β-cell dysfunction and the risk of type 2 diabetes (73, 74). The high diversity of cell types and known endocrine activities within the pancreatic islets of Langerhans provide a natural opportunity to investigate how the interplay of redox signaling affects normal and disrupted functioning. Laminar flow in capillaries creates heterogeneity in oxygenation, and therefore additional variations could occur as a consequence of differences in position of cells relative to the oxygen supply. On the other hand, relatively small differences in GSH were observed between periportal (3.6 ± 0.8 μmol/g) and pericentral hepatocytes (3.3 ± 0.8 μmol/g) (75). Thus, additional research is needed to understand the magnitude and importance of thiol/disulfide redox heterogeneity within regions of organs.
Thiol/disulfide redox control in the extracellular space was previously reviewed (76), and more recent research shows its function in maintaining the redox status of thiols in integrins, receptors, and transporters in the cell membrane (77). A relatively tight homeostatic regulation of plasma redox potentials was noted in the original description of EhGSSG and EhCySS in human plasma, where the standard deviations were only ∼10 mV in young healthy adults despite a biological redox range of ∼1000 mV from the NADPH/NADP+ couple to H2O/O2 (78). The average diurnal variations in human plasma similarly were found to be small, i.e., <10 mV over 24 h (79). Many studies have examined interorgan Cys supply, conversion of Met to Cys, and transport systems for Cys and CySS (80–82), but relatively little research is available on mechanisms providing tight regulation of plasma Cys/CySS or GSH/GSSG redox potentials.
Mechanisms for tight control of plasma thiol/disulfide redox potentials, despite diverse environmental exposures and oxidative stress, could be key to understanding diet, oxidative stress, and disease. Evidence is available to support 2 general models (Figure 3), one dependent upon intercellular/interorgan signaling (Figure 3A) and another dependent upon intrinsic characteristics of redox systems within cells (Figure 3B). Soon after the demonstration that GSH was transported out of liver (83), Aw et al. (84) showed that the GSH precursor, Met, inhibited hepatic GSH release. Sies and Graf (85) then showed that α-adrenergic agonists stimulated GSH release (85), and this was corroborated by studies showing that α-adrenergic agonists stimulated GSH transport in the small intestine (86, 87). Subsequent studies showed that cellular release of GSH was also stimulated by oxidized extracellular EhCySS (88). Together these results indicate that circulating factors related to plasma redox systems, as well as stress hormones, affect homeostatic mechanisms for plasma GSH (Figure 3A).
FIGURE 3.
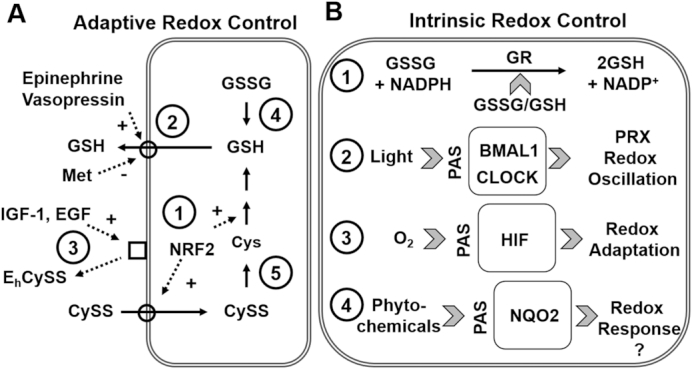
Known and hypothetical redox control systems in mammals. (A) Adaptive redox control systems have been described for 1) the NRF2 transcription factor system that controls both the initial step in GSH biosynthesis and also a major transport system for cellular uptake of CySS, an important precursor for GSH synthesis. 2) GSH efflux is stimulated by vasopressin and α-adrenergic agonists and inhibited by extracellular methionine. 3) Control of extracellular EhCySS is stimulated by growth factors IGF-1 and EGF. In principle, regulation could occur by stimulation of signaling systems for reduction of 4) GSSG and 5) CySS, but these mechanisms have not been reported. (B) Intrinsic redox control systems (hypothetical) could include endogenous control of GSSG reductase activity by GSH/GSSG and also by PAS [PER (period circadian protein), ARNT (aryl hydrocarbon receptor nuclear translocator protein), and SIM (single-minded protein)] domain proteins. 1) GR activity was reported to be stimulated by oxidative stress, suggesting that redox sensitive activation of GR could provide an intrinsic redox control mechanism. Multiple proteins with PAS domains are known to occur in mammalian cells and hypothetically could serve to autoregulate redox systems. 2) BMAL1 and CLOCK are PAS domain proteins which respond to light and serve to regulate diurnal variation of redox state of peroxiredoxins (PRX). 3) HIF is a PAS domain protein that functions to sense O2 and control adaptation to oxidative stress. 4) The PAS domain of NQO2 could serve to regulate redox responses to dietary quinones and polyphenols. Solid arrows indicate a metabolic reaction or transport of substrate. Dashed arrows indicate the agent stimulates the metabolic reaction or transport step (as marked). BMAL1, brain and muscle Arnt like protein-1; CLOCK, circadian locomotor output cycles kaput; Cys, cysteine; CySS, cystine; EGF, epidermal growth factor; Eh, redox potential; GR, glutathione reductase; GSH, glutathione; GSSG, glutathione disulfide; HIF, hypoxia-inducible factor; IGF-1, insulin-like growth factor; NQO2, NRH:quinone reductase 2; NRF2, nuclear factor (erythroid-derived 2)-like 2; PAS, Per-Arnt-Sim; PRX, peroxiredoxin.
Similar evidence exists for the Cys/CySS couple. For instance, regulation of extracellular EhCySS in cell culture was enhanced by growth factors [IGF-1, EGF (88)], and rates of thiol efflux were stimulated by high perfusate GSSG in perfused rat intestine (89) (Figure 3A). Although speculative, these results suggest that a humoral redox control system could operate to maintain systemic plasma EhGSSG and EhCySS. Such a system could account for changes in redox potential accompanying inflammation, obesity, exercise conditioning, and aging (78, 90, 91), and also be part of the CySS and GSH redox ratio (R-ratio) system ([CySS]/[GSH]) serving as a predictive biomarker of death in coronary artery disease patients (4). Such an interorgan control system would effectively parallel well-described humoral systems controlling blood glucose, blood pH, blood O2, and blood CO2. Indeed, such a control system may be essential given that EhCySS controls inflammatory signaling (77) and EhGSSG controls platelet activation (92).
If such an adaptive redox control system exists, failure of the system to control plasma redox potentials could occur as a consequence of a “redox resistance,” analogous to insulin resistance in type 2 diabetes. For instance, if NRF2 were central to this adaptive redox regulation system, decline in responsiveness of the NRF2 system with age (93) would cause oxidative stress. From this perspective, a key antioxidant strategy might be to target “redox resistance” for activation of NRF2 rather than attempting to activate NRF2 with sulforophane or other inducers. Thus, a key need is to clarify adaptive redox control mechanisms for plasma EhGSSG, EhCySS, and the associated R-ratio (4), because understanding of these mechanisms could catalyze development of new therapeutic approaches for the prevention and treatment of oxidative stress.
Redox theory also highlights alternative mechanisms for tight redox control, which depend upon the intrinsic characteristics of proteins within cells (36). Evidence for stable thiol networks has been reviewed (32, 51) and will not be further discussed here. Other redox control mechanisms depend upon intrinsic characteristics of proteins and these could be fundamental to understanding diet and oxidative stress. For instance, Per-Arnt-Sim (PAS) domain proteins are found in mammals and function to bind small molecules and regulate responses to environmental change (94) (Figure 3B). In prokaryotes, these enable responses to be made to O2, redox potential, and light (95). In mammals, PAS domain proteins are present in hypoxia, circadian, and dioxin response pathways that regulate cellular and whole-organism responses (94). Of relevance to redox control, several CLOCK-related proteins contain PAS domains, and CLOCK genes directly function in diurnal redox variation of the peroxiredoxin systems in organisms as diverse as fungus (Neurospora), fruit flies, and mice (96). Similar diurnal variation in plasma redox potential occurs in humans (27, 79). It is noteworthy that the character of diurnal variation in plasma EhGSSG and EhCySS changes with age in humans (79). Thus, it is intriguing to consider the possibility that this could be part of an intrinsic redox control system which has evolved to anticipate and respond to environmental change (94). If so, this would link to diet, nutrition, and the environment because the human N-ribosyldihydronicotinamide:quinone reductase 2 (NQO2) and human aryl hydrocarbon receptor nuclear translocator contain PAS domains. These systems interact with many phytochemicals and also with polycyclic aromatic hydrocarbons generated by cooking at high temperatures (Figure 3B). Improved understanding of these intrinsic redox control mechanisms, in addition to the adaptive control mechanisms discussed above, could catalyze development of new approaches for nutritional prevention and treatment of oxidative stress.
Detailed analysis of dietary factors in human health and disease
New HRM methods measure tens of thousands of chemicals in biological samples (97, 98), creating a possibility to quantify diverse antioxidant chemicals (e.g., see Table 1) within an individual's diet. Corresponding metabolomics and redox measures of plasma could then be used with tools such as xMWAS to connect these data (99). If performed with sequential measures, trajectory analyses would show which metabolites are linked to variations in redox parameters. Thus, nutrition science is poised to rapidly transition from targeted analysis of 1 chemical at a time to systems biology models in which complex dietary mixtures are studied. In principle, the methods will allow simultaneous analysis of components which positively and negatively affect oxidative stress and health outcomes. Such studies can build upon well-developed pharmacokinetic models to evaluate distribution, metabolism, and elimination of individual food components and also examine metabolites of the intestinal microbiome. Studies are already available in environmental health research in which computational methods were used to connect individual chemical exposures to effects on metabolism and biomarkers of health outcomes (98).
This research can build upon the extensive data on GSH and related systems. Considerable information is available concerning nutritional and nonnutritional dietary factors affecting plasma GSH, gastrointestinal GSH, and oxidative stress. The gastrointestinal tract has 2 sources of GSH, from bile and the diet. GSH from food plays an important role within the gastrointestinal tract (100) in increasing antioxidant capacity and protecting luminal epithelial cells from dietary reactive chemicals, including carcinogens. Quantification of GSH and total GSH reactive units (GRUs) in 142 food items showed that many foods contain GSH without a substantial amount of reactive materials, whereas other foods contain high GRU content without GSH (101, 102). The GRU includes lipid hydroperoxides formed from polyunsaturated fatty acids; dietary lipid peroxides can be absorbed via chylomicrons (103) and have consequent effects on plasma GSH when released into the bloodstream (104, 105). Studies in rodents show that supply of GSH to the intestinal lumen decreases uptake of lipid peroxides into lymph (106). Studies also show that GSH in the intestinal lumen is used to detoxify electrophiles due to the presence of GSH S-transferase in the mucus lining the intestinal epithelium (107). Effects involving the microbiome or subsequent bioactivation of polycyclic aromatic hydrocarbons and heterocyclic amines, and food processing products, such as nitrosamines (108), are also feasible. Of considerable importance for nutrition and oxidative stress research, the new HRM methods are sufficiently powerful to measure individual oxidants, electrophiles, and antioxidants within complex biological samples, and computational systems are now available to enable nutrition scientists to link dietary oxidants and antioxidants to relevant in vivo redox network responses.
Research Opportunities
Available omics technologies and integrative omics tools create an opportunity for molecular nutrition and nutritional epidemiology to achieve the goal of precision nutrition (109, 110). These tools also open a pathway forward for oxidative stress research to link functional responses to nutritional interventions (Figure 4A). In a study of Mn-induced mitochondrial oxidant production in a human neuroblastoma cell line, increasing Mn over an adequate to upper limit range (111) stimulated both H2O2 generation and O2 consumption rate. Over this range, a Redox proteome × Metabolome × Transcriptome (RMT) analysis showed that mitochondrial proteins were increasingly oxidized, adaptive changes in mitochondrial energy metabolism occurred, and transcripts for Ca2+ homeostasis, energy metabolism, and growth factor signaling were altered (52, 111) (Figure 4B). The results showed that proteomics, metabolomics, and transcriptomics data could be linked to central sites of oxidative stress that are potentially amenable to nutritional intervention (Figure 4B). Perhaps more importantly, the research showed that an integrated omics framework can support experiments to test specific nutritional interventions within a complex biological response.
FIGURE 4.

Integrative omics provides the foundation for data-driven network analysis of diet, nutrition, and oxidative stress. (A) Data-driven network analysis based on the use of the R package xMWAS (99) provides an analytical approach for visualization and identification of top correlations across multiple omics layers and nutritional or redox variables. Downstream analysis based on the use of the identified central clusters can identify functional pathways with tools such as the Kyoto Encyclopedia of Genes and Genomes. (B) Exposure of human acetyl-CoA acyltransferase 1b (Acaa1b) neuroblastoma cells to increasing nontoxic Mn concentration was used to map mitochondrial-nuclear cell signaling. Measures of redox proteomics (R), metabolomics (M), and transcriptomics (T) were combined with the use of xMWAS to provide an RMT network analysis. Results showed that metabolites of central energy metabolism correlated with oxidation of aconitase, a citric acid cycle enzyme, and metabolites and a mitochondrial translation elongation factor (TUFM) correlated with transcripts to define functional pathways linked to Mn-dependent oxidative stress. This provides an approach to study nutritional factors in complex systems. (C) In vivo study of Se supplementation in mice shows the utility of the same approach in a TMWAS to identify central response hubs in liver. In this study, 1 hub was centered on glucose transporter-2 (Glut2), with correlated TGs, CLs, PEs, and acylcarnitines. The other hub was centered on carnitine-palmitoyl transferase 2 (Cpt2) and Acaa1b, acetyl-CoA acyltransferase 1b; and correlated with bile acids, acyl-CoAs, phosphatidylcholines and acylcarnitines. Panels A and B modified with permissions from Go et al. 2018 (52). Panel C modified with permissions from Hu et al. 2018 (116). Acaa1b, acetyl-CoA acyltransferase 1b; CL, cardiolipin; Cpt2, carnitine-palmitoyl transferase 2; DHCCoA, 3α, 7α-dihydroxy-5β-cholestanoyl-CoA;Glut2, glucose transporter-2; LC, long chain; Mn, manganese; MWAS, metabolome-wide association study; PC, phosphatidylcholines; PE, phosphatidyl ethanolamine; Se, selenium; TG, triglyceride; TMWAS, transcriptome-metabolome wide association study; TrHOCCoA, 3α, 7α, 12α-trihydroxy-5β-24-oxocholestanoyl-CoA; TUFM, translation elongation factor-mitochondrial.
The value of integrated omics to understand nutrition in prevention of oxidative stress is further illustrated by effects of supplementation with the redox-active nutrient Se on oxidative stress caused by low Cd concentrations in mice (112). Low-dose Cd causes oxidative stress in mouse lung (113) and has been linked to inflammation (113, 114) and fibrosis (115). Se is an essential element for the antioxidant enzymes, thioredoxin reductases (TRXR) and GSH peroxidases. Cd caused a shift in the central hubs in an integrated transcriptome-metabolome-wide association study (TMWAS) (112). Metabolic structure collapsed around the Cd-responsive genes Zdhhc11 (protein-cysteine S-palmitoyltransferase) and Ighg1 (immunoglobulin heavy constant gamma-1), and this collapse in network structure was prevented by supplementation with Se. Although the details of these functional changes are not yet clear, the results establish feasibility to use integrated omics to evaluate nutritional interventions (112).
In vivo TMWAS data of Se supplementation in the absence of oxidative stress further indicate that integrated omics could be useful to optimize nutrition (Figure 4C) (116). Improved Se status has been associated with decreased cancer risk, and cancer chemoprevention studies have been performed in humans to test for protection (117–120). Results showed that potentially beneficial anticancer effects were offset by stimulated risk of obesity and type 2 diabetes (117, 121). In studies to determine the effects of increasing from an adequate Se intake to a 4-fold higher intake that was within an adequate intake range but below the upper limit causing toxicity, TMWAS showed 2 metabolic hubs (Figure 4C). One hub centered on the transcript for the bidirectional glucose transporter 2, Glut2, and the other centered on the transcripts for carnitine-palmitoyl transferase 2 (Cpt2) and acetyl-CoA acyltransferase (Acaa1). Correlated metabolites included acylcarnitines, triglycerides and glycerophospholipids, long-chain acyl coenzyme As, phosphatidylcholines, and sterols. The results show that integrated omics can identify functional responses to variation in an essential nutrient in vivo within the adequate intake range. In principle, this could be used to optimize intake of specific nutrients within an individual.
Together, these results illustrate the value of integrated omics to improve understanding of oxidative stress and nutritional interventions to protect against oxidative stress. Although it is not clear whether curricula in nutrition research cover concepts and tools essential to conduct integrated omics research, the potential value of such training for oxidative stress only serves to emphasize a broader utility for these approaches in all areas of nutrition. With such training will come additional demand for improved tools and databases for integrated omics research in diet and nutrition studies.
Although training and courses to analyze omics data are becoming increasingly available, additional gaps and opportunities exist to improve the databases and tools required for integrated approaches. Initiatives are underway to improve harmonization and storage of metabolomics data generated across different platforms, but this remains an ongoing challenge (122). Additionally, analysis tools for omics data have become increasingly available through open-source codes and R packages. For instance, a recently developed integrative omics package, xMWAS, is available as an app (https://kuppal.shinyapps.io/xmwas/) to enhance usability (99). However, adequate training to facilitate use of omics data and interpretation of results will continue to be a challenge with the complexity of the emerging integrative omics field (123).
Advancements in understanding redox signaling and control across unique cellular compartments, within organs, and between organs, defines the specificity with which redox processes need to be considered. With improvements in omics technology, redox sensors, and data analysis approaches, studies can now incorporate the inherent complexity of redox processes into the study design and analysis. Additionally, integrated data analyses allow incorporation of other contributing factors such as nutritional variables.
In conclusion, despite the failure of antioxidant trials to provide health benefits in humans, the recent finding that an oxidative stress biomarker, plasma CySS/GSH R-ratio, predicted death in coronary artery disease patients has renewed interest in oxidative stress as an important disease mechanism. A contemporary mechanistic view is that the broad array of causes of oxidative stress is balanced by an equally broad range of antioxidant systems; this highlights the critical nature of a holistic redox systems approach for studying nutrition and oxidative stress. Recent integrated omics research establishes the feasibility for redox network analysis in which dietary factors are systematically varied and oxidative stress markers are linked through the use of computational methods. Thus, redox systems nutrition is poised to support development of improved methods to use diet and nutrition to protect against oxidative stress and disease.
Acknowledgments
The authors’ responsibilities were as follows—all authors (KKD, Y-MG, DPJ) were responsible for the design, writing, and final content, and have read and approved the final manuscript.
Notes
This work was funded by NIDDK T32 DK007734 (KKD), NIEHS Grant R01 ES023485 (DPJ and Y-MG), R21 ES025632 (DPJ and Y-MG), P30 ES019776 (DPJ), NIH S10 OD018006 (DPJ).
Author disclosures: KKD, Y-MG, and DPJ, no conflicts of interest.
Abbreviations used: Cys, cysteine; CySS, cystine; Eh, redox potential; GR, glutathione disulfide reductase; GRU, GSH reactive unit; GSH, glutathione; GSSG, glutathione disulfide; HRM, high-resolution metabolomics; MWAS, metabolome-wide association study; NAC, N-acetylcysteine; NOX, NADPH oxidase; RMT, redox proteome × metabolome x × transcriptome; R-ratio, redox ratio; SAA, sulfur amino acid; TMWAS, transcriptome-metabolome-wide association study; TRX, thioredoxin; TRXR, thioredoxin reductase.
References
- 1. Food and Nutrition Board, Institute of Medicine. [Internet]. Available from: https://ods.od.nih.gov/Health_Information/Dietary_Reference_Intakes.aspx (accessed 9 October, 2018).
- 2. Hegge KA, Horning KK, Peitz GJ, Hegge K. Sapropterin: a new therapeutic agent for phenylketonuria. Ann Pharmacother. 2009;43(9):1466–73. [DOI] [PubMed] [Google Scholar]
- 3. Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–48. [DOI] [PubMed] [Google Scholar]
- 4. Patel RS, Ghasemzadeh N, Eapen DJ, Sher S, Arshad S, Ko YA, Veledar E, Samady H, Zafari AM, Sperling Let al.. Novel biomarker of oxidative stress is associated with risk of death in patients with coronary artery disease. Circulation. 2016;133(4):361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muscoli C, Cuzzocrea S, Riley DP, Zweier JL, Thiemermann C, Wang ZQ, Salvemini D. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. Br J Pharmacol. 2003;140(3):445–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jaffer OA, Carter AB, Sanders PN, Dibbern ME, Winters CJ, Murthy S, Ryan AJ, Rokita AG, Prasad AM, Zabner J et al.. Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-β-mediated collagen production in a murine asthma model. Am J Respir Cell Mol Biol. 2015;52(1):106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krause KH, Lambeth D, Kronke M. NOX enzymes as drug targets. Cell Mol Life Sci. 2012;69(14):2279–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brion LP, Bell EF, Raghuveer TS. Vitamin E supplementation for prevention of morbidity and mortality in preterm infants. Cochrane Database Syst Rev. 2003;4:CD003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goodman GE, Thornquist MD, Balmes J, Cullen MR, Meyskens FL Jr., Omenn GS, Valanis B, Williams JH Jr.. The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. J Natl Cancer Inst. 2004;96(23):1743–50. [DOI] [PubMed] [Google Scholar]
- 10. Lonn E, Bosch J, Yusuf S, Sheridan P, Pogue J, Arnold JM, Ross C, Arnold A, Sleight P, Probstfield J et al.. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293(11):1338–47. [DOI] [PubMed] [Google Scholar]
- 11. Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005;25(1):29–38. [DOI] [PubMed] [Google Scholar]
- 12. Omenn GS, Goodman GE, Thornquist MD, Balmes J, Cullen MR, Glass A, Keogh JP, Meyskens FL Jr., Valanis B, Williams JH Jr. et al.. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and Retinol Efficacy Trial. J Natl Cancer Inst. 1996;88(21):1550–9. [DOI] [PubMed] [Google Scholar]
- 13. Williams KJ, Fisher EA.. Oxidation, lipoproteins, and atherosclerosis: which is wrong, the antioxidants or the theory?. Curr Opin Clin Nutr Metab Care. 2005;8(2):139–46. [DOI] [PubMed] [Google Scholar]
- 14. Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-Acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther. 2008;8(12):1955–62. [DOI] [PubMed] [Google Scholar]
- 15. Meyer UA. Pharmacogenetics and adverse drug reactions. Lancet. 2000;356(9242):1667–71. [DOI] [PubMed] [Google Scholar]
- 16. Evans WE, Johnson JA.. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet. 2001;2:9–39. [DOI] [PubMed] [Google Scholar]
- 17. Evans WE, McLeod HL.. Pharmacogenomics—drug disposition, drug targets, and side effects. N Engl J Med. 2003;348(6):538–49. [DOI] [PubMed] [Google Scholar]
- 18. Aschard H, Lutz S, Maus B, Duell EJ, Fingerlin TE, Chatterjee N, Kraft P, Van Steen K. Challenges and opportunities in Genome-Wide Environmental Interaction (GWEI) studies. Hum Genet. 2012;131(10):1591–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schwartz D, Collins F.. Medicine. Environmental biology and human disease. Science. 2007;316(5825):695–6. [DOI] [PubMed] [Google Scholar]
- 20. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006;8(9–10):1865–79. [DOI] [PubMed] [Google Scholar]
- 21. Sies H, Jones DP.. Oxidative stress. In: Fink G.Encyclopedia of stress. Amsterdam: Elsevier; 2007. pp. 45–8. [Google Scholar]
- 22. Sies H. Introductory remarks. In: Sies H.Oxidative stress, London: Academic Press; 1985. pp. 1–8. [Google Scholar]
- 23. Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295(4):C849–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jones DP. Defenses against oxidative stress. In: Ross AC, Caballero B, Cousins R, Tucker KL, Ziegler TR. Modern nutrition in health and disease, Philadelphia: Lippincott, Williams, & Wilkins; 2014. [Google Scholar]
- 25. Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 5th ed. Oxford: Oxford University Press; 2015. [Google Scholar]
- 26. Zhang YJ, Gan RY, Li S, Zhou Y, Li AN, Xu DP, Li HB. Antioxidant phytochemicals for the prevention and treatment of chronic diseases. Molecules. 2015;20(12):21138–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones DP, Sies H. The redox code. Antioxid Redox Signal. 2015;23(9):734–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–9. [DOI] [PubMed] [Google Scholar]
- 29. Griendling KK. NADPH oxidases: new regulators of old functions. Antioxid Redox Signal. 2006;8(9–10):1443–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biology. 2017;11:613–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sies H. Nicotinamide nucleotide compartmentation. In: Sies H, editor. Metabolic compartmentation, New York:Academic Press; 1982. pp. 205–31. [Google Scholar]
- 32. Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med. 2015;84:227–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones DP, Go YM. Mapping the cysteine proteome: analysis of redox-sensing thiols. Curr Opin Chem Biol. 2011;15(1):103–12. [DOI] [PubMed] [Google Scholar]
- 34. Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30(11):1191–212. [DOI] [PubMed] [Google Scholar]
- 35. Brown DI, Griendling KK. NOX proteins in signal transduction. Free Radic Biol Med. 2009;47(9):1239–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Go YM, Jones DP. Redox theory of aging: implications for health and disease. Clin Sci. 2017;131(14):1669–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones DP. Redox theory of aging. Redox Biol. 2015;5:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. [DOI] [PubMed] [Google Scholar]
- 39. Kirlin WG, Cai J, Thompson SA, Diaz D, Kavanagh TJ, Jones DP. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic Biol Med. 1999;27(11–12):1208–18. [DOI] [PubMed] [Google Scholar]
- 40. Hutter DE, Till BG, Greene JJ. Redox state changes in density-dependent regulation of proliferation. Exp Cell Res. 1997;232(2):435–8. [DOI] [PubMed] [Google Scholar]
- 41. Cai J, Sun WM, Lu SC. Hormonal and cell density regulation of hepatic gamma-glutamylcysteine synthetase gene expression. Mol Pharmacol. 1995;48(2):212–8. [PubMed] [Google Scholar]
- 42. Jiang S, Moriarty SE, Grossniklaus H, Nelson KC, Jones DP, Sternberg P Jr.. Increased oxidant-induced apoptosis in cultured nondividing human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2002;43(8):2546–53. [PubMed] [Google Scholar]
- 43. Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2–3):65–87. [DOI] [PubMed] [Google Scholar]
- 44. Zhuang T, Han H, Yang Z. Iron, oxidative stress and gestational diabetes. Nutrients. 2014;6(9):3968–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3(3):205–14. [DOI] [PubMed] [Google Scholar]
- 46. Wang W, Ballatori N. Endogenous glutathione conjugates: occurrence and biological functions. Pharmacol Rev. 1998;50(3):335–56. [PubMed] [Google Scholar]
- 47. Ballatori N. Measurement of hepatobiliary transport. Curr Protoc Toxicol. 2004;Chapter 14:Unit14.5. [DOI] [PubMed] [Google Scholar]
- 48. Cheryan M. Phytic acid interactions in food systems. Crit Rev Food Sci Nutr. 1980;13(4):297–335. [DOI] [PubMed] [Google Scholar]
- 49. Cobbett C, Goldsbrough P.. Phytochelatins and metallothioneins: roles in heavy metal detoxification and homeostasis. Annu Rev Plant Biol. 2002;53:159–82. [DOI] [PubMed] [Google Scholar]
- 50. Kemp M, Go YM, Jones DP. Nonequilibrium thermodynamics of thiol/disulfide redox systems: a perspective on redox systems biology. Free Radic Biol Med. 2008;44(6):921–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Go YM, Jones DP. Thiol/disulfide redox states in signaling and sensing. Crit Rev Biochem Mol Biol. 2013;48(2):173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Go YM, Fernandes J, Hu X, Uppal K, Jones DP. Mitochondrial network responses in oxidative physiology and disease. Free Radic Biol Med. 2018;116:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu Rev Pharmacol Toxicol. 2006;46:215–34. [DOI] [PubMed] [Google Scholar]
- 54. Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780(11):1273–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jones DP, Go YM. Redox compartmentalization and cellular stress. Diabetes Obes Metab. 2010;12:(Suppl 2):116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem. 2010;285(51):39646–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lukosz M, Jakob S, Buchner N, Zschauer TC, Altschmied J, Haendeler J. Nuclear redox signaling. Antioxid Redox Signal. 2010;12(6):713–42. [DOI] [PubMed] [Google Scholar]
- 58. Galter D, Mihm S, Droge W. Distinct effects of glutathione disulphide on the nuclear transcription factor κB and the activator protein-1. Eur J Biochem. 1994;221(2):639–48. [DOI] [PubMed] [Google Scholar]
- 59. Allen RG, Tresini M. Oxidative stress and gene regulation. Free Radic Biol Med. 2000;28(3):463–99. [DOI] [PubMed] [Google Scholar]
- 60. Hainaut P, Milner J. Redox modulation of p53 conformation and sequence-specific DNA binding in vitro. Cancer Res. 1993;53(19):4469–73. [PubMed] [Google Scholar]
- 61. Bloom D, Dhakshinamoorthy S, Jaiswal AK. Site-directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decreases its capacity to upregulate antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene. 2002;21(14):2191–200. [DOI] [PubMed] [Google Scholar]
- 62. Go YM, Ziegler TR, Johnson JM, Gu L, Hansen JM, Jones DP. Selective protection of nuclear thioredoxin-1 and glutathione redox systems against oxidation during glucose and glutamine deficiency in human colonic epithelial cells. Free Radic Biol Med. 2007;42(3):363–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257(5076):1496–502. [DOI] [PubMed] [Google Scholar]
- 64. Schwarzlander M, Dick TP, Meyer AJ, Morgan B. Dissecting redox biology using fluorescent protein sensors. Antioxid Redox Signal. 2016;24(13):680–712. [DOI] [PubMed] [Google Scholar]
- 65. Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods. 2008;5(6):553–9. [DOI] [PubMed] [Google Scholar]
- 66. Scott JA, King GL. Oxidative stress and antioxidant treatment in diabetes. Ann N Y Acad Sci. 2004;1031:204–13. [DOI] [PubMed] [Google Scholar]
- 67. Borges-Santos MD, Moreto F, Pereira PC, Ming-Yu Y, Burini RC. Plasma glutathione of HIV(+) patients responded positively and differently to dietary supplementation with cysteine or glutamine. Nutrition. 2012;28(7–8):753–6. [DOI] [PubMed] [Google Scholar]
- 68. Jonas CR, Gu LH, Nkabyo YS, Mannery YO, Avissar NE, Sax HC, Jones DP, Ziegler TR. Glutamine and KGF each regulate extracellular thiol/disulfide redox and enhance proliferation in Caco-2 cells. Am J Physiol Regul Integr Comp Physiol. 2003;285(6):R1421–9. [DOI] [PubMed] [Google Scholar]
- 69. Park Y, Zhao T, Miller NG, Kim SB, Accardi CJ, Ziegler TR, Hu X, Jones DP. Sulfur amino acid-free diet results in increased glutamate in human midbrain: a pilot magnetic resonance spectroscopic study. Nutrition. 2012;28(3):235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fukagawa NK, Ajami AM, Young VR. Plasma methionine and cysteine kinetics in response to an intravenous glutathione infusion in adult humans. Am J Physiol. 1996;270(2 Pt 1):E209–14. [DOI] [PubMed] [Google Scholar]
- 71. Lyons J, Rauh-Pfeiffer A, Yu YM, Lu XM, Zurakowski D, Tompkins RG, Ajami AM, Young VR, Castillo L. Blood glutathione synthesis rates in healthy adults receiving a sulfur amino acid-free diet. Proc Natl Acad Sci U S A. 2000;97(10):5071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Raguso CA, Regan MM, Young VR. Cysteine kinetics and oxidation at different intakes of methionine and cystine in young adults. Am J Clin Nutr. 2000;71(2):491–9. [DOI] [PubMed] [Google Scholar]
- 73. Pi J, Zhang Q, Fu J, Woods CG, Hou Y, Corkey BE, Collins S, Andersen ME. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol Appl Pharmacol. 2010;244(1):77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26(10):501–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schon MR, Kauffman FC, Thurman RG. Selective depletion and measurement of glutathione in periportal and pericentral regions in the perfused rat liver. Toxicol Lett. 1988;42(3):265–72. [DOI] [PubMed] [Google Scholar]
- 76. Moriarty-Craige SE, Jones DP. Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr. 2004;24:481–509. [DOI] [PubMed] [Google Scholar]
- 77. Go YM, Park H, Koval M, Orr M, Reed M, Liang Y, Smith D, Pohl J, Jones DP. A key role for mitochondria in endothelial signaling by plasma cysteine/cystine redox potential. Free Radic Biol Med. 2010;48(2):275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radic Biol Med. 2000;28(4):625–35. [DOI] [PubMed] [Google Scholar]
- 79. Blanco RA, Ziegler TR, Carlson BA, Cheng PY, Park Y, Cotsonis GA, Accardi CJ, Jones DP. Diurnal variation in glutathione and cysteine redox states in human plasma. Am J Clin Nutr. 2007;86(4):1016–23. [DOI] [PubMed] [Google Scholar]
- 80. Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165(1):20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Go YM, Jones DP. Cysteine/cystine redox signaling in cardiovascular disease. Free Radic Biol Med. 2011;50(4):495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Stipanuk MH, Dominy JE Jr., Lee JI, Coloso RM. Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr. 2006;136(6 Suppl):S1652–9. [DOI] [PubMed] [Google Scholar]
- 83. Bartoli GM, Sies H. Reduced and oxidized glutathione efflux from liver. FEBS Lett. 1978;86(1):89–91. [DOI] [PubMed] [Google Scholar]
- 84. Aw TY, Ookhtens M, Kaplowitz N. Inhibition of glutathione efflux from isolated rat hepatocytes by methionine. J Biol Chem. 1984;259(15):9355–8. [PubMed] [Google Scholar]
- 85. Sies H, Graf P. Hepatic thiol and glutathione efflux under the influence of vasopressin, phenylephrine and adrenaline. Biochem J. 1985;226(2):545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hagen TM, Bai C, Jones DP. Stimulation of glutathione absorption in rat small intestine by alpha-adrenergic agonists. FASEB J. 1991;5(12):2721–7. [DOI] [PubMed] [Google Scholar]
- 87. Bai C, Jones DP. GSH transport and GSH-dependent detoxication in small intestine of rats exposed in vivo to hypoxia. Am J Physiol. 1996;271(4 Pt 1):G701–6. [DOI] [PubMed] [Google Scholar]
- 88. Jonas CR, Ziegler TR, Gu LH, Jones DP. Extracellular thiol/disulfide redox state affects proliferation rate in a human colon carcinoma (Caco2) cell line. Free Radic Biol Med. 2002;33(11):1499–506. [DOI] [PubMed] [Google Scholar]
- 89. Dahm LJ, Jones DP. Clearance of glutathione disulfide from rat mesenteric vasculature. Toxicol Appl Pharmacol. 1994;129(2):272–82. [DOI] [PubMed] [Google Scholar]
- 90. Go YM, Jones DP. Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation. 2005;111(22):2973–80. [DOI] [PubMed] [Google Scholar]
- 91. Kelli HM, Corrigan FE 3rd, Heinl RE, Dhindsa DS, Hammadah M, Samman-Tahhan A, Sandesara P, O'Neal WT, Al Mheid I, Ko YA et al.. Relation of changes in body fat distribution to oxidative stress. Am J Cardiol. 2017;120(12):2289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Essex DW, Li M. Redox control of platelet aggregation. Biochemistry. 2003;42(1):129–36. [DOI] [PubMed] [Google Scholar]
- 93. Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, Hagen TM. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101(10):3381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. McIntosh BE, Hogenesch JB, Bradfield CA. Mammalian Per-Arnt-Sim proteins in environmental adaptation. Annu Rev Physiol. 2010;72:625–45. [DOI] [PubMed] [Google Scholar]
- 95. Taylor BL, Zhulin IB.. PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev. 1999;63(2):479–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, Xu Y, Pan M, Valekunja UK, Feeney KA et al.. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485(7399):459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Uppal K, Walker DI, Liu K, Li S, Go YM, Jones DP. Computational metabolomics: a framework for the Million Metabolome. Chem Res Toxicol. 2016;29(12):1956–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Walker DI, Uppal K, Zhang L, Vermeulen R, Smith M, Hu W, Purdue MP, Tang X, Reiss B, Kim S et al.. High-resolution metabolomics of occupational exposure to trichloroethylene. Int J Epidemiol. 2016;45(5):1517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Uppal K, Ma C, Go YM, Jones DP, Wren J. xMWAS: a data-driven integration and differential network analysis tool. Bioinformatics. 2018;34(4):701–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hagen TM, Wierzbicka GT, Bowman BB, Aw TY, Jones DP. Fate of dietary glutathione: disposition in the gastrointestinal tract. Am J Physiol. 1990;259(4 Pt 1):G530–5. [DOI] [PubMed] [Google Scholar]
- 101. Jones DP, Coates RJ, Flagg EW, Eley JW, Block G, Greenberg RS, Gunter EW, Jackson B. Glutathione in foods listed in the National Cancer Institute's Health Habits and History food frequency questionnaire. Nutr Cancer. 1992;17(1):57–75. [DOI] [PubMed] [Google Scholar]
- 102. He M, Openo K, McCullough M, Jones DP. Total equivalent of reactive chemicals in 142 human food items is highly variable within and between major food groups. J Nutr. 2004;134(5):1114–9. [DOI] [PubMed] [Google Scholar]
- 103. Staprans I, Rapp JH, Pan XM, Feingold KR. The effect of oxidized lipids in the diet on serum lipoprotein peroxides in control and diabetic rats. J Clin Invest. 1993;92(2):638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Balogh K, Weber M, Erdelyi M, Mezes M. Investigation of lipid peroxide and glutathione redox status of chicken concerning on high dietary selenium intake. Acta Biol Hung. 2007;58(3):269–79. [DOI] [PubMed] [Google Scholar]
- 105. Gupta S, Pandey R, Katyal R, Aggarwal HK, Aggarwal RP, Aggarwal SK. Lipid peroxide levels and antioxidant status in alcoholic liver disease. Indian J Clin Biochem. 2005;20(1):67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Aw TY, Williams MW, Gray L. Absorption and lymphatic transport of peroxidized lipids by rat small intestine in vivo: role of mucosal GSH. Am J Physiol. 1992;262(1 Pt 1):G99–106. [DOI] [PubMed] [Google Scholar]
- 107. Samiec PS, Dahm LJ, Jones DP. Glutathione S-transferase in mucus of rat small intestine. Toxicol Sci. 2000;54(1):52–9. [DOI] [PubMed] [Google Scholar]
- 108. Knekt P, Jarvinen R, Dich J, Hakulinen T. Risk of colorectal and other gastro-intestinal cancers after exposure to nitrate, nitrite and N-nitroso compounds: a follow-up study. Int J Cancer. 1999;80(6):852–6. [DOI] [PubMed] [Google Scholar]
- 109. Ferguson LR, De Caterina R, Gorman U, Allayee H, Kohlmeier M, Prasad C, Choi MS, Curi R, de Luis DA, Gil A et al.. Guide and position of the International Society of Nutrigenetics/Nutrigenomics on personalised nutrition: part 1—fields of precision nutrition. J Nutrigenet Nutrigenomics. 2016;9(1):12–27. [DOI] [PubMed] [Google Scholar]
- 110. Boeing H. Nutritional epidemiology: new perspectives for understanding the diet-disease relationship?. Eur J Clin Nutr. 2013;67(5):424–9. [DOI] [PubMed] [Google Scholar]
- 111. Fernandes J, Hao L, Bijli KM, Chandler JD, Orr M, Hu X, Jones DP, Go YM. From the cover: manganese stimulates mitochondrial H2O2 production in SH-SY5Y human neuroblastoma cells over physiologic as well as toxicologic range. Toxicol Sci. 2017;155(1):213–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hu X, Chandler JD, Fernandes J, Orr ML, Hao L, Uppal K, Neujahr DC, Jones DP, Go YM. Selenium supplementation prevents metabolic and transcriptomic responses to cadmium in mouse lung. Biochim Biophys Acta. 2018;1862:2417–26. doi: 10.1016/j.bbagen.2018.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chandler JD, Wongtrakool C, Banton SA, Li S, Orr ML, Barr DB, Neujahr DC, Sutliff RL, Go YM, Jones DP. Low-dose oral cadmium increases airway reactivity and lung neuronal gene expression in mice. Physiol Rep. 2016;4(13):e12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chandler JD, Hu X, Ko E, Park S, Jolyn Fernandes J, Lee Y-T, Orr ML, Hao L, Smith MR, Neujahr DC et al.. Low-dose cadmium potentiates lung inflammatory response to 2009 pandemic H1N1 influenza virus in mice. bioRxiv. 2018;346866 10.1101/346866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hu X, Fernandes J, Jones DP, Go YM. Cadmium stimulates myofibroblast differentiation and mouse lung fibrosis. Toxicology. 2017;383:50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hu X, Chandler JD, Orr ML, Hao L, Liu K, Uppal K, Go YM, Jones DP. Selenium supplementation alters hepatic energy and fatty acid metabolism in mice. J Nutr. 2018;148(5):675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA et al.. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2009;301(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Algotar AM, Stratton MS, Ahmann FR, Ranger-Moore J, Nagle RB, Thompson PA, Slate E, Hsu CH, Dalkin BL, Sindhwani P et al.. Phase 3 clinical trial investigating the effect of selenium supplementation in men at high-risk for prostate cancer. Prostate. 2013;73(3):328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Clark LC, Combs GF Jr., Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG et al.. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. JAMA. 1996;276(24):1957–63. [PubMed] [Google Scholar]
- 120. Duffield-Lillico AJ, Reid ME, Turnbull BW, Combs GF Jr., Slate EH, Fischbach LA, Marshall JR, Clark LC. Baseline characteristics and the effect of selenium supplementation on cancer incidence in a randomized clinical trial: a summary report of the Nutritional Prevention of Cancer Trial. Cancer Epidemiol Biomarkers Prev. 2002;11(7):630–9. [PubMed] [Google Scholar]
- 121. Stranges S, Marshall JR, Natarajan R, Donahue RP, Trevisan M, Combs GF, Cappuccio FP, Ceriello A, Reid ME. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: a randomized trial. Ann Intern Med. 2007;147(4):217–23. [DOI] [PubMed] [Google Scholar]
- 122. Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS et al.. Metabolomics Workbench: an international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016;44(D1):D463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Karczewski KJ, Snyder MP. Integrative omics for health and disease. Nat Rev Genet. 2018;19(5):299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]