

Abstract
The X and Z sex chromosomes play a disproportionately large role in intrinsic postzygotic isolation. The underlying mechanisms of this large X/Z effect are, however, still poorly understood. Here we tested whether faster rates of molecular evolution caused by more intense positive selection or genetic drift on the Z chromosome could contribute to the large Z effect in two closely related passerine birds, the Common Nightingale (Luscinia megarhynchos) and the Thrush Nightingale (L. luscinia). We found that the two species differ in patterns of molecular evolution on the Z chromosome. The Z chromosome of L. megarhynchos showed lower levels of within-species polymorphism and an excess of non-synonymous polymorphisms relative to non-synonymous substitutions. This is consistent with increased levels of genetic drift on this chromosome and may be attributed to more intense postcopulatory sexual selection acting on L. megarhynchos males as was indicated by significantly longer sperm and higher between-male variation in sperm length in L. megarhynchos compared to L. luscinia. Interestingly, analysis of interspecific gene flow on the Z chromosome revealed relatively lower levels of introgression from L. megarhynchos to L. luscinia than vice versa, indicating that the Z chromosome of L. megarhynchos accumulated more hybrid incompatibilities. Our results are consistent with the view that postcopulatory sexual selection may reduce the effective population size of the Z chromosome and thus lead to stronger genetic drift on this chromosome in birds. This can result in relatively faster accumulation of hybrid incompatibilities on the Z and thus contribute to the large Z effect.
Subject terms: Speciation, Genetic variation
Introduction
Intrinsic postzygotic isolation, i.e., sterility and inviability of interspecific hybrids, is an important reproductive barrier separating many closely related species. Unraveling evolutionary forces and genetic mechanisms responsible for its origin is thus essential for understanding the process of speciation. Intrinsic postzygotic isolation results from genetic incompatibilities between mutations that emerged after species divergence, and thus combinations of such mutations could not have been tested by natural selection (Turelli and Orr 2000). Studies using laboratory crosses, as well as natural hybrid populations have shown that mutations causing hybrid incompatibilities are more common on the homogametic sex chromosomes, X and Z, than on autosomes (e.g. Coyne and Orr 1989; Payseur et al. 2004; Storchová et al. 2004; Macholán et al. 2007; Presgraves 2008; Carling et al. 2010; Storchová et al. 2010; Lima 2014; Lavretsky et al. 2015). This phenomenon is known as the large X/Z effect and represents one of the most general rules in evolutionary biology (Coyne and Orr 2004). Mechanisms causing the large X/Z effect are, however, still not completely understood.
The large X/Z effect could be simply explained by hemizygous nature of the X and Z chromosomes leading to exposure of recessive hybrid incompatibilities in the heterogametic sex (males in organisms with XY sex chromosomes and females in organisms with ZW sex chromosomes) (True et al. 1996; Coyne and Orr 2004). However, the finding that hybrid incompatibilities accumulate faster on the homogametic sex chromosomes even when recessive autosomal incompatibilities are considered (Masly and Presgraves 2007) requires other explanations. These involve faster rates of molecular evolution on the X and Z chromosomes (faster-X/Z evolution) (Meisel and Connallon 2013), rapid co-evolutionary arms races between sex-linked segregation distorters and their suppressors (Hurst and Pomiankowski 1991; Patten 2017), and dysregulation of epigenetic modifications of the sex chromosomes during meiosis in hybrids (Presgraves 2008). Although there is some evidence for the role of sex-linked meiotic drive (Tao et al. 2001; Orr and Irving 2005; Larson et al. 2018) and disrupted meiotic sex chromosome inactivation (Good et al. 2010; Campbell et al. 2013; Bhattacharyya et al. 2013) in the large X/Z effect, a possible contribution of faster-X/Z evolution to the large X/Z effect remains unclear.
Faster-X/Z evolution is a pervasive phenomenon, which has been observed across many taxa (Meisel and Connallon 2013). It can have two causes: (1) more efficient positive selection of recessive beneficial mutations on the hemizygous X and Z chromosomes and (2) increased levels of genetic drift leading to faster fixation of mildly deleterious mutations on the X and Z. If there is equal sex ratio and males and females have same variance in reproductive success, the effective population size (Ne) of both the X and Z chromosomes is three-fourth that of the autosomes. However, more intense sexual selection on males, which leads to higher variance in male reproductive success, increases Ne of the X chromosome but decreases Ne of the Z chromosome relative to the autosomes (Vicoso and Charlesworth 2009, Mank et al. 2010a). This is because, the X chromosome spends more time in females, while the Z in males. This difference between the X and Z chromosomes may be the reason why adaptive causes (positive selection) has been mostly found for the faster-X evolution (Charlesworth et al. 1987; Khaitovich et al. 2005; Torgerson and Singh 2006; Kousathanas et al. 2014; Charlesworth et al. 2018; but see Sackton et al. 2014), while non-adaptive (genetic drift) for the faster-Z evolution (Mank et al. 2010b). In agreement with the theoretical expectations, the intensity of genetic drift on the Z chromosome has been also shown to be positively correlated with levels of promiscuity and intensity of sexual selection acting on males (Corl and Ellegren 2012, Wright et al. 2015, but see Huang and Rabosky 2015).
Here we studied a possible association between faster-Z evolution and the large Z effect in two closely related passerine birds, the Common Nightingale (L. megarhynchos) and the Thrush Nightingale (L. luscinia). These sister species diverged ~1.8 Mya (Storchová et al. 2010) and currently hybridize in a secondary contact zone spanning Central and Eastern Europe (Reifová et al. 2011a; Vokurková et al. 2013). The two species are partially isolated by hybrid female sterility (Reifová et al. 2011b; Mořkovský et al. 2018) and slightly different habitat use in sympatry (Reif et al. 2018; Sottas et al. 2018). Consistent with the large Z effect, gene flow between the species is significantly reduced on the Z chromosome compared to autosomes (Storchová et al. 2010). Although nightingales are, like most other passerines, socially monogamous, relatively strong sexual selection may occur in these species at the postcopulatory level as has been indicated by high levels of extra-pair paternity (EPP) in L. megarhynchos (Landgraf et al. 2017).
In this study, we tested whether different levels of postcopulatory sexual selection can affect patterns of molecular evolution and the rate of accumulation of hybrid incompatibilities on the Z chromosome. To do so, we performed the following analyses: First, we assessed levels of postcopulatory sexual selection in both species. In the absence of published EPP levels for L. luscinia, we used indirect methods based on analyses of sperm morphology to estimate levels of promiscuity in both species. Second, we explored levels of genetic variation and patterns of coding sequence evolution on the Z chromosome in both species. Finally, we estimated levels of interspecific gene flow along the Z chromosome in both directions. We predict that the species with higher levels of postcopulatory sexual selection will show increased levels of genetic drift and at the same time faster accumulation of hybrid incompatibilities on the Z chromosome. This should lead to asymmetrical interspecific gene flow on the Z chromosome with lower levels of gene flow from the species with higher EPP levels than in the opposite direction.
Materials and methods
Estimation of levels of postcopulatory sexual selection based on sperm traits
Levels of postcopulatory sexual selection can be estimated as the proportion of extra-pair offspring identified by the analysis of paternity (EPP; Griffith et al. 2002, Lifjeld et al. 2010). While a recent paternity study estimated the proportion of extra-pair offspring in L. megarhynchos to be 21.5% (Landgraf et al. 2017), the level of EPP in L. luscinia is unknown. It was thus necessary to use indirect methods to estimate levels of promiscuity in both species for the purposes of this study. Our first focus was on sperm length, as sperm length tends to be, across passerine species, positively associated with EPP levels (Gomendio and Roldan 2008; Kleven et al. 2009, Lifjeld et al. 2010). As an alternative measure, we used the between-male variation in sperm length (the sperm length CV index, see below). Higher variation indicates relaxed selection on sperm length and reduced levels of EPP (Calhim et al. 2007; Kleven et al. 2008; Lifjeld et al. 2010).
Sperm samples were obtained by cloacal massage (e.g., Albrecht et al. 2013), fixed in formalin, and subsequently photographed using a microscope BX51 (Olympus) with ×20 objective. We sampled 31 males of L. megarhynchos and 16 males of L. luscinia. The samples come from allopatric regions close to the contact zone, i.e., from Northeastern Poland in L. luscinia and from Southwestern Poland in L. megarhynchos. The total length of the sperm was measured using QuickPhoto Industrial software (Olympus) following standard protocol (e.g., Knief et al. 2017). As a standard, we calculated the mean length from 20 sperm cells per male (Supplementary Table 1). The sperm length CV index was calculated from the formula CV = (SD/X) × 100 × (1-1/4N), where SD is the standard deviation of mean total sperm lengths, X is the population mean sperm length, and N is the number of males measured. This formula adjusts for the variation in sample size, as the coefficient of variation tends to be deflated at low sample sizes (Sokal and Rohlf 1981). EPP levels can be estimated based on the formula SIN(0.8614–1.08826 × LOG(spermCV))2 (Lifjeld et al. 2010).
Transcriptome and intronic sequence data
We used transcriptome data published previously by Mořkovský et al. (2015, 2018) to explore levels of genetic variation and patterns of coding sequence evolution on the Z chromosome in both nightingale species. These data represent liver transcriptomes from eight male individuals of L. megarhynchos, seven male individuals of L. luscinia and one male individual of L. svecica obtained by 454 sequencing of normalized cDNA libraries. Individual samples were barcoded using MID adaptors and sequenced in pairs on a GS FLX instrument using Titanium chemistry. The samples of L. megarhynchos and L. luscinia come from the same close allopatric regions as samples for analyses of sperm morphology (see above). Within each species, particular individuals were sampled in different localities several kilometers apart to avoid the possible relatedness of individual birds. L. svecica represents an outgroup that diverged from the other two species ~12 Mya (Jetz et al. 2012).
In addition, we sequenced eight Z-linked intronic loci in 24 allopatric individuals of L. megarhynchos, 22 individuals of L. luscinia and one individual of L. svecica. The obtained sequences were analyzed together with previously published intronic sequences from four Z-linked and eight autosomal loci (Storchová et al. 2010). Intronic sequences were used for independent comparisons of levels of genetic variation on the Z chromosome and autosomes and for estimation of levels of gene flow between the species. Samples used for intronic sequencing in this study come from allopatric regions close to the contact zone. Intronic sequences from the previous study (Storchová et al. 2010) come one-half from the same regions (and even same individuals) and one-half from more distal allopatric regions. However, we have already previously shown that there is no significant genetic differentiation between close and more distal allopatric regions within the same species (Storchová et al. 2010), suggesting that sampling in both studies was performed within the same genetically not structured populations. Within each species, individuals were sampled in different localities several kilometers apart. All introns were sequenced by the Sanger sequencing as has been described in Storchová et al. (2010). Together the 12 Z-linked intronic loci more or less evenly cover the whole Z chromosome (Supplementary Table 2) and autosomal loci are randomly distributed on autosomes. Primer sequences used for PCR amplification and Sanger sequencing, as well as PCR conditions are provided in Supplementary Table 3.
The work with live animals was approved by the Local Ethic Committee for Scientific Experiments on Animals in Poznan, Poland (permissions no. 27/2008, 36/2010).
Transcriptome assembly and variant calling
Transcriptome sequencing resulted in 6.2 million reads and the average number of reads per individual was ~390,000. The sequences were split according to MIDs using the sfffile tool and converted to fastq. Reads were assembled into 66,939 contigs using Newbler 2.6. The contigs were then mapped onto the Zebra Finch (Taeniopygia guttata) genome (taeGut1, WUSTL v3.2.4) as has been described in Mořkovský et al. (2015, 2018) to make a reference transcriptome. Reference transcriptome and BAM files with read mapping onto the reference were used to call variants by the FreeBayes program. After removal of INDELs and low quality variants (Phred < 10), we obtained 166,962 SNPs. The average coverage per site across all samples was 62.97×. The average coverage per individual was 6.538×. We note that the sequencing error rate is more than two times lower for 454 sequencing compared to Illumina sequencing (Laehnemann et al. 2016) and thus lower coverage is sufficient for 454 sequence data. Although the variability in the average coverage slightly varied across individuals, there was no statistically significant difference in quality of data between L. megarhynchos and L. luscinia (Supplementary Figure 1). In further analyses, we used only nucleotide positions present in more than 80% individuals.
Prediction of protein-coding sequences and identification of synonymous and non-synonymous mutations
We used Exonerate program (Slater and Birney 2005) to predict protein-coding sequences in the nightingale transcriptome and to identify synonymous and non-synonymous mutations. For prediction of protein-coding sequences, we used protein sequences from the Collared Flycatcher (Ficedula albicollis) and the Zebra Finch obtained from Ensembl (Ensembl, ver. 81, July 2015). To ensure that we analyze unique protein-coding sequences and not multiple paralogs or alternative splicing variants, we excluded from the downstream analyses all predictions for which (1) one protein had multiple matches in the nightingale transcriptome of which the second best hit had score higher or equal to 90% of the best hit, (2) multiple proteins matched the same location in the nightingale transcriptome and the second best hit had score more than or equal to 90% of the best hit. We then compared predictions based on the Zebra Finch and Collared Flycatcher protein sequences and chose a better prediction according to the score provided by the Exonerate. We selected only protein-coding sequences that exhibited one-to-one orthology relationship between the Zebra Finch and the Collared Flycatcher (Ensembl Compara; ver. 81). The final dataset consisted of 4259 protein-coding sequences of which 801 were predicted based on the Zebra Finch protein sequences and 3458 based on the Collared Flycatcher protein sequences. Variation at each coding site was classified using BioPerl library (http://bioperl.org) as either synonymous or non-synonymous. Codons with multiple variants were excluded from the analysis. After this stringent filtering procedure, the quality of variants with respect to the original dataset increased. The average coverage per individual was 9.47× for L. megarhynchos and 8.47× for L. luscinia.
Genomic location of individual sequences was based on the location of given orthologous genes in the Collared Flycatcher or Zebra Finch genomes depending on which prediction was used. Nightingales and the Collared Flycatcher diverged ~15 Mya and the divergence of the Zebra Finch and nightingales occurred ~45 Mya (Jetz et al. 2012). However, large-scale synteny is observed among bird genomes separated by even greater evolutionary distances (Backström et al. 2008) and especially the Z chromosome shows strong conservation of its gene content among the different avian lineages (Nanda et al. 2008). The avian chromosomes exhibit considerable variation in size and distinction is made between macrochromosomes (including the Z chromosome) and microchromosomes, which differ in recombination rate, gene density and GC content (Ellegren 2013). When studying patterns of molecular evolution on the Z chromosome, the more appropriate comparison is thus with similar sized autosomes (1–10) than with all autosomes (Mank et al. 2010b). Therefore, we distinguished between genomic location on the Z chromosome, larger autosomes (1–10) and any autosomes (1–28). Genomic location was consistent between the two outgroup species for these three categories. All comparisons between the Z chromosome and autosomes were done separately for Z vs. all autosomes and Z vs. large autosomes.
To manipulate and analyse the transcriptome data throughout the study, we used custom shell/perl scripts, samtools/bcftools (Li et al. 2009), bedtools (Quinlan and Hall 2010), and vcftools (Danecek et al. 2011).
Population genetic analyses of intronic sequences
The obtained intronic sequences were manually edited using CodonCode Aligner software and for each locus multiple alignment was generated by ClustalW as has been described in Storchová et al. (2010). Flanking exonic sequences as well as indel polymorphisms were excluded from the analyses. Individuals and/or positions with missing data (mostly at the ends of the sequences) were eliminated from the dataset to get sequences of the same length for each locus.
Diploid sequences at each locus were separated into two haplotypes using the program PHASE, version 2.1.1 (Stephens et al. 2001) as has been described in Storchová et al. (2010). For each locus, we applied the algorithm five times with different random seeds, and we checked for consistency of results across independent runs. We obtained identical haplotypes across all runs for all loci. Basic population genetic analyses of within-species polymorphism (π, θ), between-species differentiation (FST) and divergence (Dxy) were performed with the program DnaSP (Rozas et al. 2003).
Analysis of molecular evolution on the Z chromosome and autosomes using transcriptome data
To calculate population differentiation (FST) we used Weir and Cockerham estimator (Weir and Cockerham 1984) implemented as part of vcftools software (Danecek et al. 2011). π, θ, used as a measure of within-species polymorphism (Nei 1987; Watterson 1975), and Dxy (Nei 1987) were calculated using custom scripts. The proportion of non-synonymous (dN) and synonymous (dS) substitutions and its ratio was calculated using the codeml program (Yang 2007). Statistical significance of differences in these statistics between the Z chromosome and autosomes was tested by bootstrapping procedure (n = 9999), randomly sampling genes from autosomes and the Z chromosome.
The McDonald–Kreitman (MK) test (McDonald and Kreitman 1991) was used to assess patterns of molecular evolution on autosomes and the Z chromosomes. MK test is based on a comparison of a number of synonymous and non-synonymous mutations within species (polymorphisms) and between species (substitutions). Under neutrality, the ratio of non-synonymous polymorphisms to non-synonymous substitutions (Pns/Dns) should be the same as ratio of synonymous polymorphisms to synonymous substitutions (Ps/Ds). A deficit of non-synonymous polymorphisms relative to non-synonymous substitutions indicates positive selection, while an excess of non-synonymous polymorphisms relative to non-synonymous substitutions is indicative of relaxed purifying selection manifested by higher frequency of slightly deleterious mutations segregating within species. Substitutions were determined either between L. megarhynchos and L. luscinia or for each species separately relative to the outgroup, L. svecica. In the latter case, sequences were additionally compared to another more distantly related outgroup—either Collared Flycatcher or Zebra Finch depending on based on which species the protein-coding sequence was predicted—to assign substitutions either to L. svecica or to L. megarhynchos/L. luscinia lineage. In our analysis, we considered only those substitutions that occurred in the lineage to L. megarhynchos/L. luscinia. The significance of MK test was assessed using χ2-test.
We also calculated the neutrality index (NI; Rand and Kann 1996), representing the odds ratio of the MK-test, NI = (Pns/Dns)/(Ps/Ds). Neutrality index less than one is consistent with positive selection, whereas values greater than one are consistent with relaxed purifying selection. We also used a new statistics proposed by Stoletzki and Eyre-Walker (2011) coined as ‘Direction of Selection’ (DoS): DoS = Dns/(Dns + Ds)–Pns/(Pns + Ps). This statistics ranges from −1 to 1. Values around 0 represent neutral evolution, negative values represent excess of non-synonymous polymorphisms suggesting relaxed purifying selection, and positive values represent evidence of positive selection. The statistical significance of DoS departure from zero was tested using a bootstrap (n = 9999). Differences between the Z chromosome and autosomes were tested by comparison of the Z to autosomes DoS difference to null distribution built by sample randomization (n = 9999).
To test whether the proportions of synonymous to non-synonymous substitutions and synonymous to non-synonymous polymorphisms differ between the Z chromosome and autosomes, we constructed a three-way contingency table and applied generalized linear model (glm) with terms for the type of mutation (synonymous or non-synonymous), type of variant (polymorphism or substitution), genomic location (autosomes or Z chromosome), and the three-way interaction between these terms. The response variable was represented by counts of variants. The three-way interaction tests for the difference in two independent MK tests—one for the autosomes and the other for the Z chromosome—and thus for the difference in patterns of molecular evolution between the Z chromosome and autosomes. To assess the significance of the three-way interaction, we compared the full model containing the interaction to a simpler model without this interaction using a Log-Likelihood ratio test (ΔLL).
Estimation of levels of interspecific gene flow on the Z chromosome
We used intronic sequences and IM program (Hey and Nielsen 2004) to estimate locus-specific migration rates for Z-linked loci. The program fits data to isolation with migration model and using Markov chain Monte Carlo simulations of genealogies it estimates several demographic parameters including migration rates in both direction for each locus. As IM assumes no recombination within loci, we determined the longest region without observed recombination for each locus using the program IMgc (Woerner et al. 2007). This program removes either sites or haplotypes to produce the most information-rich contiguous DNA sequence segment that passes the four-gamete test. Non-recombinant regions represented 92% of the length of each locus, on average, and were used as an input for the IM program. We run the program three times with identical starting conditions, with the exception of the random number seed, to assess convergence. To facilitate mixing of the Markov chains we used Metropolis coupling with 10 chains and a geometric heating scheme. All runs began with a burn-in period of 100,000 steps and were allowed to continue for 6–8 million steps. We were able to achieve adequate mixing of the Markov chains as indicated by trend line plots and effective sample size (ESS) values (for most runs, all ESS values were higher than 1000 and no ESS value was lower than 70). Independent runs converged to the same result (i.e., maximum likelihood estimates and marginal posterior probability distributions for the demographic parameters were essentially the same for all three runs).
Results
Levels of postcopulatory sexual selection based on sperm traits
Spermatozoa was significantly longer in L. megarhynchos compared to L. luscinia (272.28 ± 5.63 [SD] µm and 233.40 ± 8.47 [SD] µm, respectively; F1,45 = 354, p < 0.001). The sperm length CV index was lower in L. megarhynchos (2.08) than in L. luscinia (3.69). This implies higher levels of EPP and postcopulatory sexual selection in L. megarhynchos than in L. luscinia. The estimated proportion of extra-pair paternity offspring (sensu Lifjeld et al. 2010) would be 24% in L. megarhynchos and 6% in L. luscinia. The first estimate corresponds perfectly with the direct measure of EPP level counted as the proportion of extra-pair offspring in L. megarhynchos, which is 21.5% (Landgraf et al. 2017).
Levels of within-species polymorphism on the Z chromosome and autosomes
We first compared levels of within-species polymorphism (π, θ) between the Z chromosome and autosomes for 12 Z-linked and 8 autosomal intronic loci comprising in total 6,868 bp and 3,826 bp, respectively (Table 1, Supplementary Table 4). When comparing polymorphism between the Z chromosome and autosomes, one has to take into account (1) different effective population size of the Z chromosome, which is three-fourth that of the autosomes assuming an equal sex ratio and same variance in reproductive success in males and females, and (2) different mutation rate on the Z chromosome and autosomes (Ellegren and Fridolfsson 1997). After correction for the different effective population size of the Z chromosome, the Z/A ratio for π was 0.48 in L. megarhynchos and 0.62 in L. luscinia. The Z/A ratio for θ was 0.64 in L. megarhynchos and 0.75 in L. luscinia. To correct for differences in mutation rate, we further divided π and θ (normalized for different Ne between autosomes and the Z chromosome) by divergence (Dxy) to L. svecica which can be used as a surrogate for the mutation rate. In accordance with male-biased evolution, Dxy was on average higher on the Z chromosome compared to autosomes. Z/A ratio for π/Dxy was thus even lower: 0.30 for L. megarhynchos and 0.46 for L. luscinia. Z/A ratio for θ/Dxy was 0.42 for L. megarhynchos and 0.53 for L. luscinia. The difference in π/Dxy between the Z chromosome and autosomes was significant for L. megarhynchos (Mann–Whitney U-test, p-value = 0.0011), but not for L. luscinia (Mann–Whitney U-test, p-value = 0.0632). Similarly, the difference in θ/Dxy between the Z chromosome and autosomes was significant for L. megarhynchos (Mann–Whitney U-test, p-value = 0.0003), but not for L. luscinia (Mann–Whitney U-test, p-value = 0.0632). Values of π, θ, and Dxy for the Z chromosome and autosomes are summarized in Table 1 and for individual loci are provided in Supplementary Table 4.
Table 1.
Average levels of genetic diversity and divergence for 12 Z-linked (Z) and eight autosomal (A) intronic loci in L. megarhynchos (LM) and L. luscinia (LL)
| Chr | Lengtha | Species | N b | S c | π (%)d | θ (%)e | Dxy (%)f |
|---|---|---|---|---|---|---|---|
| Z | 6,868 | LM | 31–50 | 98 | 0.285 | 0.327 | 4.629 |
| LL | 22–50 | 161 | 0.393 | 0.556 | 4.441 | ||
| A | 3,826 | LM | 38–50 | 111 | 0.796 | 0.679 | 3.927 |
| LL | 34–50 | 176 | 0.850 | 0.994 | 3.944 |
aThe total length of intronic sequences in bp
bNumbers of analyzed haploid sequences for individual loci
cThe total number of segregating sites
dAverage number of nucleotide differences, averaged across loci
eProportion of polymorphic sites, averaged across loci
fAbsolute divergence to L. svecica measured as average pairwise divergence, averaged across loci
A similar albeit not so pronounced pattern of reduced levels of within-species polymorphism on the Z chromosome of L. megarhynchos, has been seen for transcriptome data. When different Ne and mutation rate on the Z chromosome and autosomes was taken into account as explained above, the Z/A ratio for π/Dxy was 0.76 for L. megarhynchos (p-value = 0.0199) and 0.95 for L. luscinia (p-value = 0.3925). The Z/A ratio for θ/Dxy was 0.80 for L. megarhynchos (p-value = 0.0713) and 0.90 for L. luscinia (p-value = 0.3021). Similar results were found when only large autosomes (LA) were taken into account. The Z/LA ratio for π/Dxy was 0.76 for L. megarhynchos (p-value = 0.0194) and 0.95 for L. luscinia (p-value = 0.4036). The Z/LA ratio for θ/Dxy was 0.79 for L. megarhynchos (p-value = 0.0667) and 0.90 for L. luscinia (p-value = 0.3059). Together, these results suggest that the levels of within-species polymorphism are lower on the Z chromosome than expected under neutrality, an equal sex ratio and same variance in reproductive success in males and females in L. megarhynchos, but not in L. luscinia.
Levels of between-species differentiation and divergence on the Z chromosome and autosomes
In contrast to levels of within-species polymorphism, levels of between-species differentiation were higher on the Z chromosome compared to autosomes. FST was consistently and significantly higher for Z-linked intronic loci (average FST = 0.728) than for autosomal intronic loci (average FST = 0.298) (Mann–Whitney U-test, p-value = 0.0003; Table 2). A similar contrast was found in the number of fixed differences and shared polymorphisms. In total, the 12 Z-linked loci showed 35 fixed differences and 13 shared polymorphisms. On the other hand, the 8 autosomal loci showed 0 fixed differences and 52 shared polymorphisms (Fisher exact test, p-value < 0.0001).
Table 2.
Levels of genetic differentiation between the species and estimates of migration rates between the species from IM model
| Locus | F ST | S shared a | S fixed b | Parameterc | MLEd | HPD95Loe | HPD95Hie |
|---|---|---|---|---|---|---|---|
| ADAMTS6 | 0.767 | 0 | 2 | m1 | 0.0075 | 0.0015 | 0.6075 |
| m2 | 0.0015 | 0.0015 | 0.3165 | ||||
| GHR-5.2 | 0.775 | 1 | 5 | m1 | 0.0015 | 0.0015 | 0.6135 |
| m2 | 0.0015 | 0.0015 | 0.2895 | ||||
| PPWD1 | 0.808 | 0 | 0 | m1 | 0.2025 | 0.0045 | 1.0605 |
| m2 | 0.0015 | 0.0015 | 0.3495 | ||||
| SPINZ-2 | 0.780 | 0 | 7 | m1 | 0.0015 | 0.0015 | 0.6075 |
| m2 | 0.0015 | 0.0015 | 0.2745 | ||||
| TG401 | 0.792 | 0 | 3 | m1 | 0.0045 | 0.0015 | 0.6435 |
| m2 | 0.0015 | 0.0015 | 0.3675 | ||||
| TG853 | 0.567 | 8 | 0 | m1 | 0.3615 | 0.0225 | 1.5495 |
| m2 | 0.0705 | 0.0015 | 0.6765 | ||||
| TG1015 | 0.672 | 0 | 1 | m1 | 0.0195 | 0.0015 | 0.9345 |
| m2 | 0.0045 | 0.0015 | 0.3375 | ||||
| TG1505 | 0.480 | 3 | 1 | m1 | 0.0015 | 0.0015 | 0.6975 |
| m2 | 0.0015 | 0.0015 | 0.3405 | ||||
| TG1925 | 0.834 | 0 | 9 | m1 | 0.0015 | 0.0015 | 0.6945 |
| m2 | 0.0015 | 0.0015 | 0.3465 | ||||
| TG5287 | 0.836 | 0 | 4 | m1 | 0.0015 | 0.0015 | 0.6255 |
| m2 | 0.0015 | 0.0015 | 0.2955 | ||||
| TG5729 | 0.860 | 0 | 3 | m1 | 0.0045 | 0.0015 | 0.6585 |
| m2 | 0.0045 | 0.0015 | 0.3705 | ||||
| VLDLR-7 | 0.561 | 1 | 0 | m1 | 0.2775 | 0.0015 | 1.3005 |
| m2 | 0.0015 | 0.0015 | 0.6555 |
Non-zero estimates of migration parameters are indicated in bold
aNumber of shared polymorphisms between the species
bNumber of fixed polymorphisms between the species
cMigration from L. luscinia to L. megarhynchos (m1), migration from L. megarhynchos to L. luscinia (m2)
dMaximum likelihood estimate (MLE) of migration parameters
eLower and higher 95% highest posterior density (HPD) intervals
Higher levels of FST on the Z chromosome compared to autosomes were found also for transcriptome data (Fig. 1). The average FST was 0.540 on the Z chromosome, and 0.385 on autosomes. This difference was statistically significant (Mann–Whitney U-test, p-value = 0.0223). Difference in FST between the Z chromosome and autosomes was still significant when we included in the analysis only large autosomes (FST = 0.392; Mann–Whitney U-test, p-value = 0.0325). The Z chromosome exhibited 23 fixed differences between L. megarhynchos and L. luscinia and 18 shared polymorphisms, autosomes exhibited 100 fixed differences and 665 shared polymorphisms. The ratio is clearly shifted toward higher proportion of fixed differences on the Z chromosomes as opposed to autosomes (Fisher exact test, p-value < 0.0001).
Fig. 1.
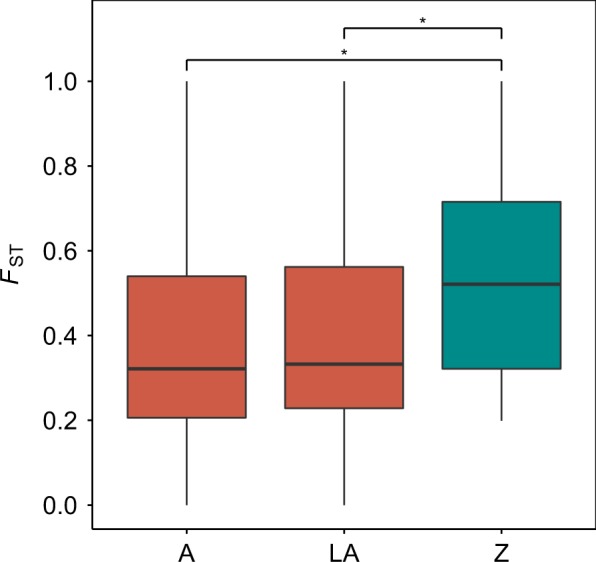
Levels of genetic differentiation (FST) between L. megarhynchos and L. luscinia on the Z chromosome and autosomes. A all autosomes, LA large autosomes, Z Z chromosome
To test whether there are higher rates of coding sequence evolution on the Z chromosome in nightingales, we compared dN, dS, and dN/dS between the two nightingale species on the Z chromosome and autosomes using transcriptome data. We found significantly higher dN (p-value = 0.0446) as well as dS (p-value = 0.0006) on the Z chromosome compared to all autosomes (Fig. 2), which is consistent with increased mutation rate on the Z chromosome (Ellegren and Fridolfsson 1997). dN/dS ratio was, however, not significantly different between the Z chromosome and autosomes (Fig. 2). The same result was found for comparison of large autosomes and the Z chromosome (dN: p-value = 0.0418; dS: p-value = 0.0007).
Fig. 2.

Estimates of dN, dS, and dN/dS between L. megarhynchos and L. luscinia on the Z chromosome and autosomes. A all autosomes, LA large autosomes, Z Z chromosome
McDonald–Kreitman test of neutral evolution
To test whether lower levels of within-species polymorphism on the Z chromosome of L. megarhynchos are driven by increased levels of positive selection or genetic drift on the Z chromosome, we conducted MK tests using transcriptome data. MK tests were performed for comparison L. megarhynchos vs. L. luscinia and for each species separately relative to the outgroup (see Materials and Methods). For each dataset, MK tests were performed separately for the Z chromosome (Z), all autosomes (A), and large autosomes (LA). Numbers of synonymous and non-synonymous polymorphisms and substitutions for each dataset are shown in Table 3. For L. megarhynchos vs. L. luscinia comparison, MK tests provided non-significant results for the Z chromosome as well as autosomes; however, the neutrality index (NI) was much higher for the Z chromosome (NIZ = 2.778) than for autosomes (NIA = 0.796, NILA = 0.813) (Table 3). DoS exhibited statistically significant negative value for the Z chromosome (DoSZ = −0.164, p-value = 0.0276) as oppose to the two groups of autosomes (DoSA = 0.041, p-value = 0.1776; DoSLA = 0.043, p-value = 0.2172; Fig. 3), suggesting that there is an excess of non-synonymous polymorphisms relative to non-synonymous substitutions associated with the Z chromosome (Table 3). Among MK tests that were done for each species separately relative to the outgroup, the only significant MK test was for the Z chromosome in L. megarhynchos (p-value = 0.001). The positive neutrality index corresponding to this MK test (NI = 2.640) revealed that the ratio of non-synonymous to synonymous mutations was higher for polymorphisms than for substitutions. Datasets for both species provided negative DoS for the Z chromosome as opposed to autosomes, however, only for L. megarhynchos the departure from zero was statistically significant (LM: DoSZ = −0.178, p-value = 0.0002; LL: DoSZ = −0.05, p-value = 0.1452; Fig. 3). The DoS for autosomes in either of the two species did not significantly differ from zero.
Table 3.
Number of non-synonymous (NS) and synonymous (S) substitutions (D) and polymorphisms (P) and test statistics of McDonald–Kreitman (MK) test
| Dataset | Chr | D | P | NIa | MK-test p-value | DoSb | DoS p-value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NS | S | % NS | NS | S | % NS | ||||||
| LL vs. LM | A | 27 | 73 | 27.00 | 808 | 2746 | 22.73 | 0.796 | 0.378 | 0.043 | 0.178 |
| LA | 23 | 55 | 29.49 | 703 | 2069 | 25.36 | 0.813 | 0.488 | 0.041 | 0.217 | |
| Z | 3 | 20 | 13.04 | 55 | 132 | 29.41 | 2.778 | 0.159 | −0.164 | 0.028 | |
| LL | A | 300 | 933 | 24.33 | 588 | 1846 | 24.16 | 0.991 | 0.940 | 0.002 | 0.457 |
| LA | 249 | 741 | 25.15 | 489 | 1493 | 24.67 | 0.975 | 0.810 | 0.005 | 0.386 | |
| Z | 23 | 102 | 18.40 | 36 | 118 | 23.38 | 1.353 | 0.387 | −0.05 | 0.145 | |
| LM | A | 380 | 1217 | 23.79 | 532 | 1531 | 25.79 | 1.113 | 0.179 | −0.02 | 0.085 |
| LA | 314 | 965 | 24.55 | 443 | 1225 | 26.56 | 1.111 | 0.232 | −0.02 | 0.106 | |
| Z | 28 | 142 | 16.47 | 38 | 73 | 34.23 | 2.640 | 0.001 | −0.178 | 0.000 | |
LM vs. LL is for comparison between L. megarhynchos and L. luscinia, LM and LL states for lineage-specific test for L. megarhynchos and L. luscinia lineage, respectively. Significant p-values are indicated in bold
aNeutrality index
bDirection of selection
Fig. 3.
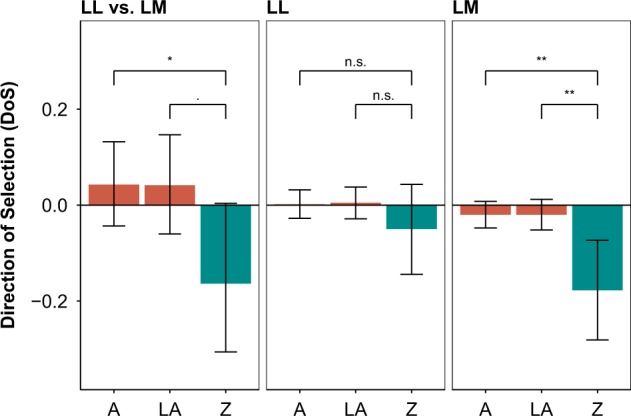
Estimates of direction of selection (DoS) between L. megarhynchos and L. luscinia (LM vs. LL) and for each species separately in comparison with the outgroup. Positive numbers of DoS indicate excess of non-synonymous substitutions, while negative numbers excess of non-synonymous polymorphisms. A all autosomes, LA large autosomes, Z Z chromosome
Importantly, when amount of synonymous and non-synonymous variation within and between species was compared between autosomes and the Z chromosome using glm models (see Materials and Methods), we found significant difference in patterns of molecular evolution between the Z chromosome and all autosomes for L. megarhynchos vs. L. luscinia comparison (ΔLL = 4.070, p-value = 0.0436; Table 4). Difference in molecular evolution between large autosomes and the Z chromosome was, however, marginally insignificant for this comparison (ΔLL = 3.791, p-value = 0.0515; Table 4). In the datasets, where the nightingale species were separately compared to the outgroup, we found statistically significant difference in patterns of molecular evolution between the Z chromosome and autosomes in L. megarhynchos (A vs. Z: ΔLL = 8.537, p-value = 0.0035; LA vs. Z: ΔLL = 8.436, p-value = 0.0037; Table 4), but not in L. luscinia (Table 4). Together, these results provide evidence for the excess of non-synonymous polymorphisms relative to non-synonymous substitutions on the Z chromosome compared to autosomes in the L. megarhynchos, but not in the L. luscinia, lineage.
Table 4.
Comparison of patterns of molecular evolution between autosomes and the Z chromosome using GLM framework
| Dataset | Comparison | ΔLL ratio | p-value |
|---|---|---|---|
| LL vs. LM | A vs. Z | 4.070 | 0.0436 |
| LA vs. Z | 3.791 | 0.0515 | |
| LL | A vs. Z | 1.021 | 0.3123 |
| LA vs. Z | 1.114 | 0.2913 | |
| LM | A vs. Z | 8.537 | 0.0035 |
| LA vs. Z | 8.436 | 0.0037 |
LM vs. LL is for comparison between L. megarhynchos and L. luscinia, LM and LL states for lineage-specific test for L. megarhynchos and L. luscinia lineage, respectively. The statistical significance of the three-way interaction between type of mutation (synonymous vs. non-synonymous), type of variant (polymorphism vs. substitution) and genomic location (autosomes vs. Z chromosome) was tested by comparison of the full model and the model without the three-way interaction using a Log-Likelihood ratio test (ΔLL). Significant p-values are indicated in bold
Patterns of interspecific gene flow along the Z chromosome
To explore how levels of interspecific gene flow varies along the Z chromosome, we fitted an IM model to sequence data from 12 Z-linked intronic loci using the IM program. This program allows estimation of migration rates separately for each locus. Estimates of migration rates are shown in Table 2 and posterior probability distributions for each locus are shown in Supplementary Figure 2. Most Z-linked loci showed zero migration rates in both directions, suggesting that interspecific gene flow is reduced along the whole Z chromosome (Table 2). Non-zero estimates of migration rates were obtained for four loci. In TG853 migration was observed in both directions, although it was higher from L. luscinia to L. megarhynchos. In other three loci (PPWD1, TG1015 and VLDLR-7) migration was observed only in one direction, from L. luscinia to L. megarhynchos. Thus, although gene flow on the Z chromosome occurs only in a few loci, it is asymmetrical and lower from L. megarhynchos to L. luscinia.
The asymmetry in levels of interspecific gene flow can be also seen on haplotype networks of the Z-linked intronic loci (Supplementary Figure 3). In all four loci with non-zero estimates of migration rates in the IM analysis, there is one or a few haplotypes evidently or very likely of L. luscinia origin introgressed in L. megarhynchos individuals (white circles surrounded by the black ones; Supplementary Figure 3). By contrast, there are no clear examples of introgression in the opposite direction.
Discussion
This study investigated a possible interconnection between intensity of postcopulatory sexual selection, patterns of molecular evolution on the Z chromosome, and the large Z effect in two nightingale species. We report the following main findings. (1) We found an indirect evidence that the two nightingale species differ markedly in levels of postcopulatory sexual selection, with L. megarhynchos showing higher levels as was indicated by significantly longer spermatozoa as well as lower between-male coefficients of variation in sperm length in this species compared to L. luscinia. (2) We found that the Z chromosome in L. megarhynchos, but not in L. luscinia, showed significantly lower levels of within-species polymorphism than would be expected under neutrality, equal sex ratio and same variance in reproductive success in males and females. At the same time the Z chromosome of L. megarhynchos showed an excess of non-synonymous polymorphisms relative to non-synonymous substitutions, suggesting increased levels of genetic drift acting on this chromosome. (3) We revealed asymmetrical introgression on the Z chromosome with lower levels from L. megarhynchos to L. luscinia than in the opposite direction. Together, these results are consistent with the prediction that postcopulatory sexual selection can significantly reduce the effective population size of the Z chromosome and thus lead to higher levels of genetic drift on this chromosome (Vicoso and Charlesworth 2009, Mank et al. 2010a). This can result in relatively faster accumulation of hybrid incompatibilities on the Z chromosome and thus contribute to the large Z effect. Below, we discuss our findings in the light of theories on the role of sex chromosomes in speciation and present alternative explanations for the observed patterns of genomic variation and gene flow.
Studies in Galloanseres and shorebirds with contrasting mating systems have shown that high levels of female promiscuity leading to more intense sexual selection on males can reduce the effective population size of the Z chromosome and thus lead to stronger genetic drift on this chromosome (Corl and Ellegren 2012; Oyler-McCance et al. 2015; Wright et al. 2015). By contrast, a recent study in passerine birds has found no correlation between levels of sexual dimorphism, which are often used as a proxy for levels of sexual selection, and Z-linked variation (Huang and Rabosky 2015). Most passerine birds, including nightingales, are socially monogamous, however, sexual selection can be quite strong in these species at the postcopulatory level due to sperm competition and cryptic female choice (Andersson 1994; Griffith et al. 2002; Albrecht et al. 2007; Webster et al. 2007). Our results showing increased levels of genetic drift on the Z chromosome in species with higher EPP levels provide the first evidence for the possible role of postcopulatory sexual selection in shaping Z-linked variation. Further studies including more species are, however, needed to assess the general importance of postcopulatory sexual selection in shaping Z-linked variation in birds as well as other organisms.
Both nightingale species are sexually monomorphic, suggesting that other traits than plumage color or other ornaments may be important for female choice in these species. Indeed, it has been found that EPP levels in nightingales correspond to song repertoire size (Landgraf et al. 2017). If song is generally more important in female choice than plumage color or feather ornaments, it could explain why no correlation has been observed between sexual dimorphism and Z-linked variation (Huang and Rabosky 2015).
Although the observed patterns of genetic variation on the Z chromosome in L. megarhynchos may be explained by increased levels of sexual selection acting on males, they might have also other causes (Sayres 2018). It has been shown that historical changes in population size can affect relative levels of genetic variation on the Z chromosome and autosomes. Specifically, reduction of population size can lead to relatively reduced Z-linked variation, whereas population growth increases Z-linked relative to autosomal variation (Pool and Nielsen 2007). Future research on demographic history of the two nightingale species will be thus needed to exclude the possible effect of demographic processes on the observed patterns of genetic variation on the Z chromosome and autosomes. Patterns of Z-linked variation could also be affected by sex-biased migration (Laporte and Charlesworth 2002). However, levels of sex-biased migration are unlikely to differ between closely related species such as nightingales and can thus hardly explain why reduced genetic variation on the Z chromosome occurs only in L. megarhynchos, but not in L. luscinia.
Although we have clearly demonstrated higher rates of genetic drift on the Z chromosome in one of the nightingale species, we found no evidence for relatively faster accumulation of non-synonymous than synonymous substitutions on the Z chromosome between the two nightingale species. This may be caused by recent divergence of the two species and thus relatively low number of substitutions. Nevertheless, the Z chromosome in nightingales was characterized by significantly higher levels of between-species differentiation (FST) as well as higher number of fixed differences when compared to autosomes, which might be the sign of faster-Z evolution in early stages of divergence (Storchová et al. 2010; Hogner et al. 2012; Irwin 2018).
We were further interested whether increased levels of genetic drift on the Z chromosome might lead to faster accumulation of hybrid incompatibilities on this chromosome and thus reduced levels of gene flow. Although our results showing lower levels of gene flow on the Z chromosome in the direction from L. megarhynchos to L. luscinia than vice versa are consistent with this prediction, they can also have alternative explanations. Asymmetry in levels of interspecific gene flow may be caused by asymmetrical premating barriers or expansion of one species into the area of other species connected with introgressive hybridization (Excoffier et al. 2008). However, in both these cases the asymmetry in levels of gene flow is expected to be same for autosomal and Z-linked loci. In nightingales, it is surprising that patterns of gene flow on the Z chromosome and autosomes are just opposite. Autosomal gene flow seems to be higher from L. megarhynchos to L. luscinia (2Nm from Lm to Ll = 0.763) than in the opposite direction (2Nm from Ll to Lm = 0.081) (Storchová et al. 2010). By contrast gene flow on the Z chromosome occurs mostly in the direction from L. luscinia to L. megarhynchos. Recently, it has been also argued that differences in effective population size between hybridizing species can lead to asymmetrical introgression as effectively neutral alleles segregating in species with low effective population size may behave as deleterious after entering population of larger effective size (Juric et al. 2016). If this occurs in nightingales, we would expect relatively low introgression from L. megarhynchos to L. luscinia on the Z chromosome but also on autosomes as L. megarhynchos has lower effective population size compared to L. luscinia (Storchová et al. 2010). We thus suggest that the most likely explanation for the asymmetrical gene flow on the Z chromosome are higher levels of genetic drift acting on the Z chromosome of L. megarhynchos, which lead to faster accumulation of hybrid incompatibilities on this chromosome in L. megarhynchos. If this is true, we would expect that the cross between L. luscinia females with L. megarhynchos males will produce less fit females than the cross in the opposite direction. Further studies involving crosses of the two nightingale species in captivity and mapping hybrid incompatibility loci should test this prediction.
Although our results suggest that stronger genetic drift on the Z chromosome could directly contribute to the large Z effect in nightingales, it is likely that the large Z effect has multiple causes in birds. Indeed, the gene flow on the Z chromosome is relatively low compared to autosomal gene flow also in the direction from L. luscinia to L. megarhynchos. In this respect, it is interesting to observe that although genetic drift seems to be the major force shaping the coding sequence variation on the Z chromosome in nightingales and Galloanseres (Wright et al. 2015), positive selection seems to reduce genetic variation on the Z chromosome in some other bird species (Reifová et al. 2016; Irwin 2018). In addition, positive selection has been shown to be responsible for the faster-Z evolution of female-biased gene expression in birds (Dean et al. 2015). It is also possible that reduced recombination rate on the Z chromosome (Z chromosome in birds recombines only in males) increases the levels of linked-selection, which might elevate the substitution rate of slightly deleterious mutations on this chromosome (Muirhead and Presgraves 2016). The further research will be needed to better understand the relative contributions of all evolutionary forces contributing to the large Z effect in nightingales and birds generally.
There has been a long-lasting discussion on the relative roles of natural selection and genetic drift in the evolution of reproductive isolation (Coyne and Orr 2004). Evidence for signatures of positive selection in several identified speciation genes in Drosophila and house mouse (Ting et al. 1998; Presgraves et al. 2003; Barbash et al. 2004; Brideau et al. 2006; Oliver et al. 2009) led to the prevailing view that positive selection is the main driver of the origin of hybrid incompatibilities both on autosomes and the sex chromosomes (Tao et al. 2001; Orr and Irving 2005). In contrast to this view, our results highlight the importance of genetic drift in accumulation of hybrid incompatibilities between the species on the Z chromosome. In this respect it is interesting to note, that hybrid incompatibilities are quite frequently observed also on mtDNA (Burton and Barreto 2012; Trier et al. 2014; Ma et al. 2016). Although evolutionary forces responsible for the origin of these incompatibilities have not been studied, effective population size of mtDNA is one-quarter that of the autosomes and mtDNA is thus also characterized by relatively high levels of genetic drift. If the genetic drift is really important driver of the origin of hybrid incompatibilities, we would expect faster origin of reproductive isolation in species with low effective population size. Further studies should test this hypothesis to assess currently neglected role of genetic drift in speciation.
Data archiving
Transcriptome sequences, demultiplexed FASTQ files, and accompanying sample identification are available from Dryad repository: doi:10.5061/dryad.2p4t3, doi:10.5061/dryad.41ng6. Intron sequences have been deposited in GenBank under accession numbers MH891170-MH891488.
Electronic supplementary material
Acknowledgements
We thank Camille Sottas, Emily R.A. Cramer and Pavel Kverek for the assistance in the field and to Kamila Opletalová for measuring spermatozoa. Three anonymous reviewers provided useful comments on earlier versions of this paper. The study was supported by grants of the Czech Science Foundation (15-10884Y, 18-14325S) and a grant of the Charles University (PRIMUS/19/SCI/08). This work was also supported by ELIXIR CZ research infrastructure project (MEYS Grant No: LM2015047) including access to computing and storage facilities.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41437-018-0161-3) contains supplementary material, which is available to authorized users.
References
- Albrecht T, Schnitzer J, Kreisinger J, Exnerová A, Bryja J, Munclinger P. Extrapair paternity and the opportunity for sexual selection in long-distant migratory passerines. Behav Ecol. 2007;18:477–486. [Google Scholar]
- Albrecht T, Kleven O, Kreisinger J, Laskemoen T, Omotoriogun TC, Ottosson U, Reif J, Sedláček O, Hořák D, Robertson RJ, Lifjeld JT. Sperm competition in tropical versus temperate zone birds. Proc R Soc B. 2013;280:20122434. doi: 10.1098/rspb.2012.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M. Sexual selection. Princeton, NJ: Princeton Univ. Press; 1994. [Google Scholar]
- Backström N, Fagerberg S, Ellegren H. Genomics of natural bird populations: a gene-based set of reference markers evenly spread across the avian genome. Mol Ecol. 2008;17:964–980. doi: 10.1111/j.1365-294X.2007.03551.x. [DOI] [PubMed] [Google Scholar]
- Barbash DA, Awadalla P, Tarone AM. Functional divergence caused by ancient positive selection of a Drosophila hybrid incompatibility locus. PLoS Biol. 2004;2:839–848. doi: 10.1371/journal.pbio.0020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya T, Gregorova S, Mihola O, Anger M, Sebestova J, Denny P, Simecek P, Forejt J. Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc Natl Acad Sci USA. 2013;110:E468–477. doi: 10.1073/pnas.1219126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton RS, Barreto FS. A disproportionate role for mtDNA in Dobzhansky-Muller incompatibilities? Mol Ecol. 2012;21:4942–4957. doi: 10.1111/mec.12006. [DOI] [PubMed] [Google Scholar]
- Brideau NJ, Flores HA, Wang J, Maheshwari S, Wang X, Barbash DA. Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science. 2006;314:1292–1295. doi: 10.1126/science.1133953. [DOI] [PubMed] [Google Scholar]
- Calhim S, Immler S, Birkhead TR. Postcopulatory sexual selection is associated with reduced variation in sperm morphology. PLoS ONE. 2007;2:e413. doi: 10.1371/journal.pone.0000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P, Good JM, Nachman MW. Meiotic sex chromosome inactivation is disrupted in sterile hybrid male house mice. Genetics. 2013;193:819–828. doi: 10.1534/genetics.112.148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling MD, Lovette IJ, Brumfield RT. Historical divergence and gene flow: coalescent analyses of mitochondrial, autosomal and sex-linked loci in Passerina buntings. Evolution. 2010;64:1762–1772. doi: 10.1111/j.1558-5646.2010.00954.x. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 1987;130:113–146. [Google Scholar]
- Charlesworth Brian, Campos José L., Jackson Benjamin C. Faster-X evolution: Theory and evidence from Drosophila. Molecular Ecology. 2018;27(19):3753–3771. doi: 10.1111/mec.14534. [DOI] [PubMed] [Google Scholar]
- Corl A, Ellegren H. The genomic signature of sexual selection in the genetic diversity of the sex chromosomes and autosomes. Evolution. 2012;66:2138–2149. doi: 10.1111/j.1558-5646.2012.01586.x. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Two rules of speciation. In: Otte D, Endler J, editors. Speciation and Its Consequences. Sunderland, MA: Sinauer Associates; 1989. [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sunderland, MA: Sinauer Associates; 2004. [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean R, Harrison PW, Wright AE, Zimmer F, Mank JE. Positive selection underlies faster-Z evolution of gene expression in birds. Mol Biol Evol. 2015;32:2646–2656. doi: 10.1093/molbev/msv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Fridolfsson AK. Male-driven evolution of DNA sequences in birds. Nat Genet. 1997;17:182–184. doi: 10.1038/ng1097-182. [DOI] [PubMed] [Google Scholar]
- Ellegren H. The evolutionary genomics of birds. Annu Rev Ecol Evol Syst. 2013;44:239–259. [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annu Rev Ecol Evol Syst. 2008;40:481–501. [Google Scholar]
- Gomendio M, Roldan ERS. Implications of diversity in sperm size and function for sperm competition and fertility. Int J Dev Biol. 2008;52:439–447. doi: 10.1387/ijdb.082595mg. [DOI] [PubMed] [Google Scholar]
- Good JM, Giger T, Dean MD, Nachman MW. Widespread over-expression of the X chromosome in sterile F1 hybrid mice. PLoS Genet. 2010;6:e1001148. doi: 10.1371/journal.pgen.1001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith SC, Owens IPE, Thuman KA. Extra pair paternity in birds: a review of interspecific variation and adaptive function. Mol Ecol. 2002;11:2195–2211. doi: 10.1046/j.1365-294x.2002.01613.x. [DOI] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogner S, Saether SA, Borge T, Bruvik T, Johnsen A, Saetre GP. Increased divergence but reduced variation on the Z chromosome relative to autosomes in Ficedula flycatchers: differential introgression or the faster-Z effect? Ecol Evol. 2012;2:379–396. doi: 10.1002/ece3.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Rabosky DL. Sex-linked genomic variation and its relationship to avian plumage dichromatism and sexual selection. BMC Evol Biol. 2015;15:199. doi: 10.1186/s12862-015-0480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Pomiankowski A. Causes of sex ratio bias may account for unisexual sterility in hybrids: a new explanation of Haldane’s rule and related phenomena. Genetics. 1991;128:841–858. doi: 10.1093/genetics/128.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin Darren E. Sex chromosomes and speciation in birds and other ZW systems. Molecular Ecology. 2018;27(19):3831–3851. doi: 10.1111/mec.14537. [DOI] [PubMed] [Google Scholar]
- Jetz W, Thomas GH, Joy JB, Hartmann K, Mooers AO. The global diversity of birds in space and time. Nature. 2012;491:444–448. doi: 10.1038/nature11631. [DOI] [PubMed] [Google Scholar]
- Juric I, Aeschbacher S, Coop G. The strength of selection against neanderthal introgression. PLoS Genet. 2016;12:e1006340. doi: 10.1371/journal.pgen.1006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Hellmann I, Enard W, Nowick K, Leinweber M, Franz H, Weiss G, Lachmann M, Pääbo S. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science. 2005;309:1850–1854. doi: 10.1126/science.1108296. [DOI] [PubMed] [Google Scholar]
- Kleven O, Laskemoen T, Fossøy F, Robertson RJ, Lifjeld JT. Intraspecific variation in sperm length is negatively related to sperm competition in passerine birds. Evolution. 2008;62:494–499. doi: 10.1111/j.1558-5646.2007.00287.x. [DOI] [PubMed] [Google Scholar]
- Kleven O, Fossøy F, Laskemoen T, Robertson RJ, Rudolfsen G, Lifjeld JT. Comparative evidence for the evolution of sperm swimming speed by sperm competition and female sperm storage duration in passerine birds. Evolution. 2009;63:2466–2473. doi: 10.1111/j.1558-5646.2009.00725.x. [DOI] [PubMed] [Google Scholar]
- Knief U, Forstmeier W, Pei Y, Ihle M, Wang D, Martin K, Opatová P, Albrechtová J, Wittig M, Franke A, Albrecht T, Kempenaers B. A sex-chromosome inversion causes strong overdominance for sperm traits that affect siring success. Nat Ecol Evol. 2017;1:1177–1184. doi: 10.1038/s41559-017-0236-1. [DOI] [PubMed] [Google Scholar]
- Kousathanas A, Halligan DL, Keightley PD. Faster-X adaptive protein evolution in house mice. Genetics. 2014;196:1131–1143. doi: 10.1534/genetics.113.158246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laehnemann D, Borkhardt A, McHardy AC. Denoising DNA deep sequencing data-high-throughput sequencing errors and their correction. Brief Bioinform. 2016;17:154–79. doi: 10.1093/bib/bbv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf C, Wilhelm K, Wirth J, Weiss M, Kipper S. Affairs happen–to whom? A study on extrapair paternity in common nightingales. Curr Zool. 2017;63:421–431. doi: 10.1093/cz/zox024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte V, Charlesworth B. Effective population size and population subdivision in demographically structured populations. Genetics. 2002;162:501–519. doi: 10.1093/genetics/162.1.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EL, Vanderpool D, Sarver BAJ, Callahan C, Keeble S, Provencio LP, Kessler MD, Stewart V, Nordquist E, Dean MD, Good JM (2018) The evolution of polymorphic hybrid incompatibilities in house mice. Genetics 10.1534/genetics.118.300840. [DOI] [PMC free article] [PubMed]
- Lavretsky P, Dacosta JM, Hernández-Baños BE, Engilis A, Jr, Sorenson MD, Peters JL. Speciation genomics and a role for the Z chromosome in the early stages of divergence between Mexican ducks and mallards. Mol Ecol. 2015;24:5364–5378. doi: 10.1111/mec.13402. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifjeld JT, Laskemoen T, Kleven O, Albrecht T, Robertson RJ. Sperm length variation as a predictor of extrapair paternity in passerine birds. PLoS ONE. 2010;5:e13456. doi: 10.1371/journal.pone.0013456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima TG. Higher levels of sex chromosome heteromorphism are associated with markedly stronger reproductive isolation. Nat Commun. 2014;5:4743. doi: 10.1038/ncomms5743. [DOI] [PubMed] [Google Scholar]
- Ma H, Marti Gutierrez N, Morey R, Van Dyken C, Kang E, Hayama T, Lee Y, Li Y, Tippner-Hedges R, Wolf DP, Laurent LC, Mitalipov S. Incompatibility between nuclear and mitochondrial genomes contributes to an interspecies reproductive barrier. Cell Metab. 2016;24:283–294. doi: 10.1016/j.cmet.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macholán M, Munclinger P, Šugerková M, Dufková P, Bímová B, Božíková E, Zima J, Piálek J. Genetic analysis of autosomal and X-linked markers across a mouse hybrid zone. Evolution. 2007;61:746–771. doi: 10.1111/j.1558-5646.2007.00065.x. [DOI] [PubMed] [Google Scholar]
- Mank JE, Vicoso B, Berlin S, Charlesworth B. Effective population size and the Faster-X effect: empirical results and their interpretation. Evolution. 2010;64:663–674. doi: 10.1111/j.1558-5646.2009.00853.x. [DOI] [PubMed] [Google Scholar]
- Mank JE, Nam K, Ellegren H. Faster-Z evolution is predominantly due to genetic drift. Mol Biol Evol. 2010;27:661–670. doi: 10.1093/molbev/msp282. [DOI] [PubMed] [Google Scholar]
- Masly JP, Presgraves DC. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol. 2007;5:e243. doi: 10.1371/journal.pbio.0050243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Meisel RP, Connallon T. The faster-X effect: integrating theory and data. Trends Genet. 2013;29:537–544. doi: 10.1016/j.tig.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mořkovský L, Pačes J, Rídl J, Reifová R. Scrimer: designing primers from transcriptome data. Mol Ecol Res. 2015;15:1415–1420. doi: 10.1111/1755-0998.12403. [DOI] [PubMed] [Google Scholar]
- Mořkovský L, Janoušek V, Reif J, Rídl J, Pačes J, Choleva L, Janko K, Nachman MW, Reifová R. Genomic islands of differentiation in two songbird species reveal candidate genes for hybrid female sterility. Mol Ecol. 2018;27:949–958. doi: 10.1111/mec.14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muirhead CA, Presgraves DC. Hybrid incompatibilities, local adaptation, and the genomic distribution of natural introgression between species. Am Nat. 2016;187:249–261. doi: 10.1086/684583. [DOI] [PubMed] [Google Scholar]
- Nanda I, Schlegelmilch K, Haaf T, Schartl M, Schmid M. Synteny conservation of the Z chromosome in 14 avian species (11 families) supports a role for Z dosage in avian sex determination. Cytogenet Genome Res. 2008;122:150–156. doi: 10.1159/000163092. [DOI] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. New York, NY: Columbia Univ. Press; 1987. [Google Scholar]
- Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, Beatson SA, Lunter G, Malik HS, Ponting CP. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 2009;5:e1000753. doi: 10.1371/journal.pgen.1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA, Irving S. Segregation distortion in hybrids between the Bogota and USA subspecies of Drosophila pseudoobscura. Genetics. 2005;169:671–682. doi: 10.1534/genetics.104.033274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyler-McCance SJ, Cornman RS, Jones KL, Fike JA. Z chromosome divergence, polymorphism and relative effective population size in a genus of lekking birds. Heredity. 2015;115:452–459. doi: 10.1038/hdy.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten Manus M. Selfish X chromosomes and speciation. Molecular Ecology. 2018;27(19):3772–3782. doi: 10.1111/mec.14471. [DOI] [PubMed] [Google Scholar]
- Payseur BA, Krenz JG, Nachman MW. Differential patterns of introgression across the X chromosome in a hybrid zone between two species of house mice. Evolution. 2004;58:2064–2078. doi: 10.1111/j.0014-3820.2004.tb00490.x. [DOI] [PubMed] [Google Scholar]
- Pool JE, Nielsen R. Population size changes reshape genomic patterns of diversity. Evolution. 2007;61:3001–3006. doi: 10.1111/j.1558-5646.2007.00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC. Sex chromosomes and speciation in Drosophila. Trends Genet. 2008;24:336–343. doi: 10.1016/j.tig.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC, Balagopalan L, Abmayr SM, Orr HA. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature. 2003;423:715–719. doi: 10.1038/nature01679. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Kann LM. Excess amino acid polymorphism in mitochondrial DNA: contrasts among genes from Drosophila, mice, and humans. Mol Biol Evol. 1996;13:735–748. doi: 10.1093/oxfordjournals.molbev.a025634. [DOI] [PubMed] [Google Scholar]
- Reif J, Reifová R, Skoracka A, Kuczyński L. Competition-driven niche segregation on a landscape scale: Evidence for escaping from syntopy towards allotopy in two coexisting sibling passerine species. J Anim Ecol. 2018;87:774–789. doi: 10.1111/1365-2656.12808. [DOI] [PubMed] [Google Scholar]
- Reifová R, Reif J, Antczak M, Nachman MW. Ecological character displacement in the face of gene flow: evidence from two species of nightingales. BMC Evol Biol. 2011;11:138. doi: 10.1186/1471-2148-11-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifová R, Kverek P, Reif J. The first record of a female hybrid between the Common Nightingale (Luscinia megarhynchos) and the Thrush Nightingale (Luscinia luscinia) in nature. J Ornithol. 2011;152:1063–1068. [Google Scholar]
- Reifová R, Majerová V, Reif J, Ahola M, Lindholm A, Procházka P. Patterns of gene flow and selection across multiple species of Acrocephalus warblers: footprints of parallel selection on the Z chromosome. BMC Evol Biol. 2016;16:130. doi: 10.1186/s12862-016-0692-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sackton TB, Corbett-Detig RB, Nagaraju J, Vaishna L, Arunkumar KP, Hartl DL. Positive selection drives faster-Z evolution in silkmoths. Evolution. 2014;68:2331–2342. doi: 10.1111/evo.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayres MAW. Genetic diversity on the sex chromosomes. Genome Biol Evol. 2018;10:1064–1078. doi: 10.1093/gbe/evy039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ (1981). Biometry the principles and practice of statistics in biological research. W.H. Freeman, San Francisco
- Sottas C, Reif J, Kuczyński L, Reifová R. Interspecific competition promotes habitat and morphological divergence in a secondary contact zone between two hybridizing songbirds. J Evol Biol. 2018;31:914–923. doi: 10.1111/jeb.13275. [DOI] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. Estimation of the neutrality index. Mol Biol Evol. 2011;28:63–70. doi: 10.1093/molbev/msq249. [DOI] [PubMed] [Google Scholar]
- Storchová R, Gregorová S, Buckiová D, Kyselová V, Divina P, Forejt J. Genetic analysis of X-linked hybrid sterility in the house mouse. Mamm Genome. 2004;15:515–524. doi: 10.1007/s00335-004-2386-0. [DOI] [PubMed] [Google Scholar]
- Storchová R, Reif J, Nachman MW. Female heterogamety and speciation: reduced introgression of the Z chromosome between two species of nightingales. Evolution. 2010;64:456–471. doi: 10.1111/j.1558-5646.2009.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Hartl DL, Laurie CC. Sex-ratio segregation distortion associated with reproductive isolation in Drosophila. Proc Natl Acad Sci USA. 2001;98:13183–13188. doi: 10.1073/pnas.231478798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting CT, Tsaur SC, Wu ML, Wu CI. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science. 1998;282:1501–1504. doi: 10.1126/science.282.5393.1501. [DOI] [PubMed] [Google Scholar]
- Torgerson DG, Singh RS. Enhanced adaptive evolution of sperm-expressed genes on the mammalian X chromosome. Heredity. 2006;96:39–44. doi: 10.1038/sj.hdy.6800749. [DOI] [PubMed] [Google Scholar]
- Trier CN, Hermansen JS, Sætre GP, Bailey RI. Evidence for mito-nuclear and sex-linked reproductive barriers between the hybrid Italian sparrow and its parent species. PLoS Genet. 2014;10:e1004075. doi: 10.1371/journal.pgen.1004075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- True JR, Weir BS, Laurie CC. A genome-wide survey of hybrid incompatibility factors by the introgression of marked segments of Drosophila mauritiana chromosomes into Drosophila simulans. Genetics. 1996;142:819–837. doi: 10.1093/genetics/142.3.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli M, Orr HA. Dominance, epistasis and the genetics of postzygotic isolation. Genetics. 2000;154:1663–1679. doi: 10.1093/genetics/154.4.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. Effective population size and the faster-X effect: an extended model. Evolution. 2009;63:2413–2426. doi: 10.1111/j.1558-5646.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- Vokurková J, Petrusková T, Reifová R, Kozman A, Mořkovský L, Kipper S, Weiss M, Reif J, Dolata PT, Petrusek A. The causes and evolutionary consequences of mixed singing in two hybridizing songbird species (Luscinia spp.) PLoS ONE. 2013;8:e60172. doi: 10.1371/journal.pone.0060172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Webster MS, Tarvin KA, Tuttle EM, Pruett-Jones S. Promiscuity drives sexual selection in a socially monogamous bird. Evolution. 2007;61:2205–2211. doi: 10.1111/j.1558-5646.2007.00208.x. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Woerner AE, Cox MP, Hammer MF. Recombination-filtered genomic datasets by information maximization. Bioinformatics. 2007;23:1851–1853. doi: 10.1093/bioinformatics/btm253. [DOI] [PubMed] [Google Scholar]
- Wright AE, Harrison PW, Zimmer F, Montgomery SH, Pointer MA, Mank JE. Variation in promiscuity and sexual selection drives avian rate of Faster-Z evolution. Mol Ecol. 2015;24:1218–1235. doi: 10.1111/mec.13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: a program package for phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptome sequences, demultiplexed FASTQ files, and accompanying sample identification are available from Dryad repository: doi:10.5061/dryad.2p4t3, doi:10.5061/dryad.41ng6. Intron sequences have been deposited in GenBank under accession numbers MH891170-MH891488.