

Abstract
The nucleus accumbens is a critical integration center for reward-related circuitry and is comprised primarily of medium spiny projection neurons. The dynamic balance of excitation and inhibition onto medium spiny neurons determines the output of this structure. While nucleus accumbens excitatory synaptic plasticity is well-characterized, inhibitory synaptic plasticity mechanisms and their potential relevance to shaping motivated behaviors is poorly understood. Here we report the discovery of long-term depression of inhibitory synaptic transmission in the mouse nucleus accumbens core. This long-term depression is postsynaptically expressed, tropomyosin kinase B (TrkB) receptor-mediated, and augmented in the presence of ethanol. Our findings support the emerging view that TrkB signaling regulates inhibitory synaptic plasticity and suggest this mechanism in the nucleus accumbens as a target for ethanol modulation of reward.
Subject terms: Addiction, Neurotrophic factors, Long-term depression
Introduction
Medium spiny projection neurons (MSNs) of the nucleus accumbens (NAc) encode motivated behavior through a dynamic interplay of excitatory, inhibitory, and modulatory synaptic inputs [1–4]. Drugs of abuse such as ethanol (EtOH) alter various forms of NAc excitatory synaptic plasticity to modulate drug seeking [5]. Despite the heavy influence of GABAergic transmission on MSNs [6], which arise from several intrinsic and extrinsic sources [1], native forms of inhibitory synaptic plasticity in the NAc remain unsolved.
Gating synaptic plasticity events across the striatal complex are membrane-voltage states. MSNs exhibit bi-stable membrane potentials: the depolarized “up state” (~−60 mV) and hyperpolarized “down state” [7–9] (~−80 mV). These distinct voltage states activate various voltage-gated calcium (Ca2+) channels [10] and, as a result, enable voltage-dependent forms of synaptic plasticity [11, 12]. For instance, at inhibitory synapses onto MSNs in the dorsal striatum, distinct forms of voltage-dependent inhibitory long-term depression (iLTD) exist: up state iLTD is mediated by the endocannabinoid (eCB) 2-arachidonoylglycerol (2-AG) while down state iLTD is putatively mediated by the eCB anandamide [12]. Given that eCB-mediated iLTD represent the predominant forms of plasticity in the dorsal striatum, this raises the possibility that eCBs acting through the type 1 cannabinoid receptor, CB1, also modulate NAc synapses in a voltage-dependent manner. Accordingly, a form of up state iLTD that is eCB-mediated and induced by unitary activation of CB1-expressing fast-spiking interneurons does exist in the NAc [13]. However, the expression of CB1 receptors is much lower in the NAc than the dorsal striatum [14, 15]. This suggests that other modulatory transmitters present in the NAc, including neuropeptides and monoamines, may support NAc inhibitory synaptic plasticity [1, 3, 16].
In this study, we used whole-cell voltage-clamp recordings in acute mouse brain slices to discover a voltage-dependent iLTD onto NAc MSNs that is mediated by activation of the endogenous receptor for brain-derived neurotrophic factor (BDNF), tropomyosin receptor kinase B (TrkB). Exploring an interaction of this system with the drug of abuse EtOH, we show that NAc-iLTD is enhanced in the presence of EtOH. These findings suggest that inhibition in the NAc is under plastic control and may participate in the encoding of EtOH reward.
Methods
Experiments were carried out in accordance with the United States Public Health Service Guide for Care and Use of Laboratory Animals and approved by the University of Maryland, School of Medicine Institutional Animal Care and Use Committee.
Mice
Homozygous floxed-TrkB (Flx-TrkB) mice were maintained on a mixed C57Bl/6J × FVB/N background [17]. Drd1a-tdTomato mice were bred on a C57Bl/6J × SJL background (Jackson Laboratories). All other mice used for slice electrophysiology experiments were on a C57Bl/6J background. Animals were housed with littermates (2–5 per cage) under a regular light/dark cycle (lights on at 0700, off at 1900 h) with ad libitum access to food and water. All experiments used female and male mice that were at least 2 months old.
Acute slice preparation
Mice were deeply anesthetized with isoflurane before rapid decapitation and brain removal. 250 µm thick coronal slices were prepared in ice cold, 95% oxygen, 5% carbon dioxide (carbogen)-bubbled modified aCSF (In mM: 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose) before incubating at 32 °C for 30 min in regular aCSF (in mM: 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose). Slices were stored at room temperature for the remainder of the day.
Whole-cell voltage-clamp electrophysiology
Slices were hemisected, placed into a recording chamber, and received constant perfusion of carbogen-bubbled aCSF containing 50 µM DL-AP5 sodium salt and 5 µM NBQX disodium salt or CNQX disodium salt that was temperature controlled to 29–31 °C. MSNs located in the core region of the NAc were visually targeted using Q-Capture Pro 7 software. MSNs were voltage clamped at −60 mV using a MultiClamp 700B Amplifier (Molecular Devices) throughout the experiment unless noted. IPSCs were recorded using borosilicate glass pipettes (2–5 MΩ resistance) filled with CsCl-based internal pipette solution (in mM: 150 CsCl, 10 HEPES, 2 MgCl2, 0.3 Na-GTP, 5 QX-314, 3 Mg-ATP, and 0.2 BAPTA, osmolarity ranging from 305 to 310 mOsm, pH ~7.3). IPSCs were evoked every 20 s using a concentric bipolar stimulating electrode (World Precision Instruments) located ~100 µm away from the recording electrode. Signals were filtered at 2 kHz, digitized at 10 kHz and acquired using Clampex 10.4.1.4 software (Molecular Devices). Electrically evoked-low-frequency stimulation (eLFS; 1 Hz, 240 pulses) was delivered while holding MSNs at −80 mV (down state) after acquiring a stable baseline. To induce NAc-iLTD in the down state, the holding potential of the MSN was decreased from −60 mV to −80 mV by −4 mV increments every 4 s during the last 20 s before eLFS began. MSNs were returned to the up state (−60 mV) by reversing the protocol during the first 20 s of the post-eLFS file. If the series resistance changed more than 15% throughout the course of the experiment cells were discarded from analysis.
Viral injections
Bilateral stereotactic injections of AAV9-hSyn-cre-enhanced green fluorescent protein (eGFP; UPenn) into the NAc (A/P +1.5 mm from bregma, M/L ±0.8 from midline, D/V −3.5 from top of brain) were performed on homozygous Flx-TrkB mice [17, 18] to selectively delete TrkB from all cells in the region.
Immunohistochemistry (IHC)
To verify injection sites following slice physiology recordings, NAc slices incubated in 4% PFA at 4 °C for at least 3 days and a shortened Brain BLAQ IHC procedure was performed [19]. To visualize eGFP fluorescence chicken-anti-GFP (Abcam ab13970; 1:2000) and AlexaFluor488 donkey-anti-chicken (Jackson ImmunoResearch 703-545-155; 1:500) antibodies were used. Slides were imaged using a Nikon Eclipse 90i.
Statistical analysis
Raw IPSC amplitudes were calculated on Clampfit 10.4.1.4, averaged per minute and expressed as a percent change from baseline. The final 5 min of all cells in the experiment were averaged and compared to the 5 min baseline using a two-tailed paired t-test (GraphPad Prism 6.01). To compare between experimental groups, the final 5 min of the recordings were averaged and compared using a two-tailed unpaired t-test. All data are reported as mean ± SEM.
Drugs
All salts for aCSF, as well as sulpiride and 95% EtOH were purchased from Sigma-Aldrich. DL-AP5 was purchased from Abcam. BAPTA (MW: 1004.03) was obtained from Molecular Probes. BDNF was purchased from PeproTech and Tocris Bioscience. All other drugs were purchased from Tocris Bioscience.
Results
Nucleus accumbens inhibitory long-term depression (NAc-iLTD)
Delivering electrical low-frequency stimulation (eLFS, 1 Hz for 240 pulses, Fig. 1a) locally to the core region of the NAc while voltage-clamping MSNs in the down state (−80 mV) resulted in NAc-iLTD (Fig. 1b). To determine whether NAc-iLTD preferentially disinhibits the direct- or indirect pathways of the NAc, we induced NAc-iLTD in slices taken from the Drd1a-tdTomato reporter line of mice that express tdTomato in D1-dopamine receptor expressing MSNs (Fig. 1b). Down state NAc-iLTD occurred onto both D1-dopamine receptor expressing MSNs (Inhibitory postsynaptic current (IPSC) amplitude = 79.67 ± 8.86% of baseline, t = 2.29, df = 9 cells, p = 0.047, paired t-test) and putative D2-dopamine receptor expressing MSNs that were negative for tdTomato expression (IPSC amplitude = 75.99 ± 7.01% of baseline, t = 3.43, df = 9, p = 0.008, paired t-test, Fig. 1b). NAc-iLTD was not due to changes in series resistance over time (Fig. 1b) and there was no effect of sex on NAc-iLTD expression: NAc-iLTD amplitude in females = 84.72 ± 11.45% of baseline, n = 8, NAc-iLTD amplitude in males = 74.9 ± 6.83% of baseline, n = 7, t = 0.76, df = 13, p = 0.46, unpaired t-test).
Fig. 1.

Nucleus accumbens long-term depression of inhibitory synapses (NAc-iLTD) is tropomysin receptor kinase B (TrkB)-dependent. a Schematic representing the experimental configuration. b Left top: tdTomato expression under the Drd1a promoter. Left bottom: Representative patch onto a dopamine D1-receptor expressing medium spiny neuron (MSN). Scale bars: 125 µm top and 20 µm bottom. Right top: Electrical low-frequency stimulation (eLFS) delivered while voltage-clamping (Vhold) D1- (red) and putative D2-dopamine receptor expressing (black) MSNs in the down state (Vhold = −80 mV) induced NAc-iLTD. Right bottom: NAc-iLTD in D1-expressing MSNs and D2-expressing MSNs was not due to changes in series resistance (Rs). c Inclusion of GDP-β-S (500 µM) in the internal pipette solution did not eliminate NAc-iLTD. d Bath application of the TrkB antagonist, cyclotraxin B (2 µM) eliminated NAc-iLTD expression (red) compared to control conditions (black). e Left: Schematic of the experimental condition. Right: Expression of cre-recombinase in the NAc in floxed-TrkB mice (eGFP). Dashed line depicts the location of the NAc core. Scale bar: 250 µm. f Conditional deletion of TrkB from the NAc eliminated NAc-iLTD expression. g NAc-iLTD was stable; applying cyclotraxin B (2 µM) after NAc-iLTD induction did not return inhibitory postsynaptic current (IPSC) amplitudes to baseline. h Acute application of exogenous brain-derived neurotrophic factor (BDNF, 4 nM) depressed inhibitory synaptic transmission onto MSNs while voltage-clamping MSNs at −80 mV. i eLFS delivery while voltage clamping MSNs at −80 mV depressed excitatory transmission onto MSNs (black). This excitatory LTD was not TrkB dependent (red, 2 µM cyclotraxin B). Insets: Representative traces of the first 5 min (dark) and final 5 min (light) of the experiment. Scale bars for traces: 200 pA, 200 ms. EPSC excitatory postsynaptic current; AP anterior/posterior; ac anterior commissure. Data are represented as mean ± SEM
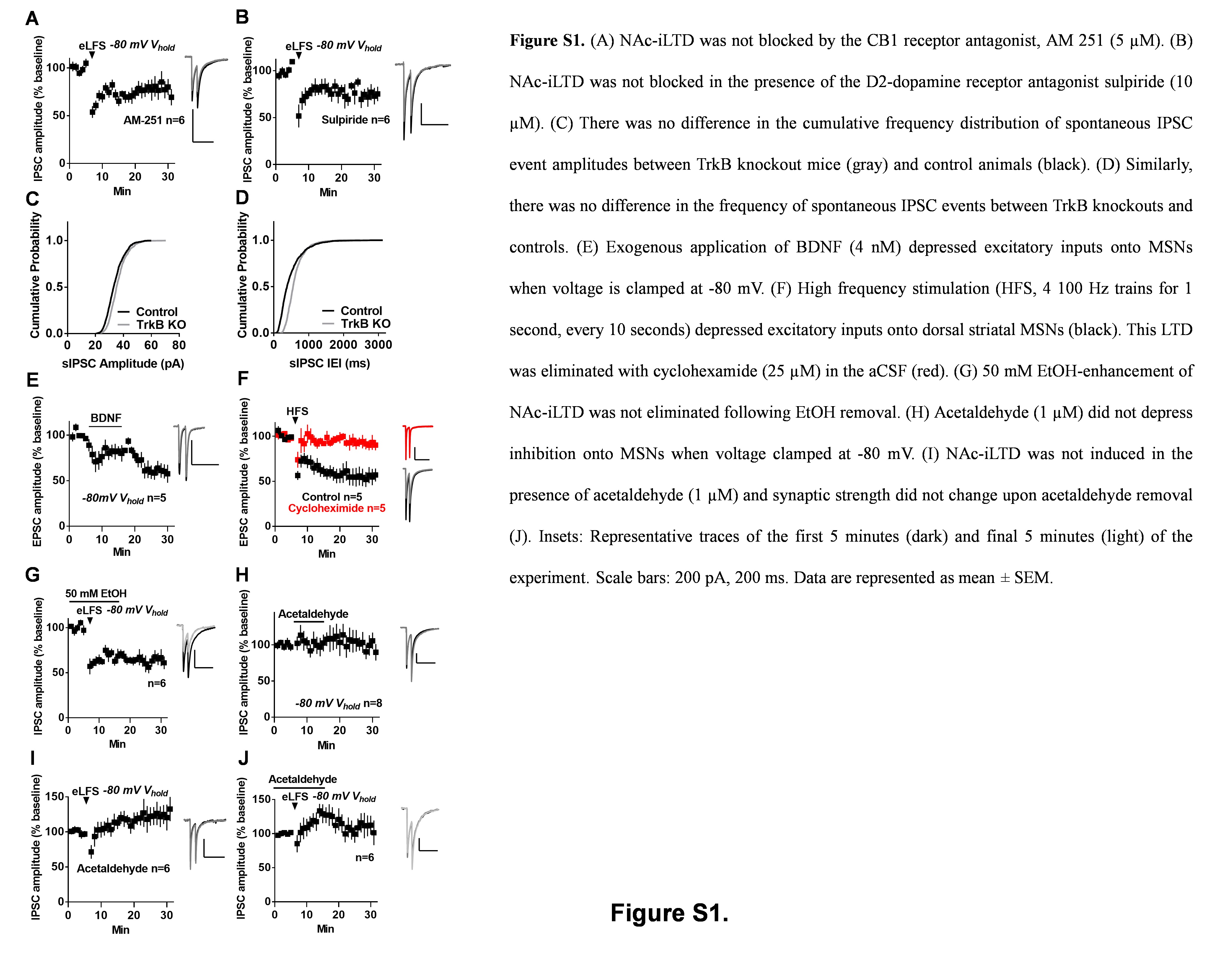
Down state iLTD in the dorsal striatum is eCB mediated [12] and, thus, we tested whether NAc-iLTD occurs through the same mechanism. Following at least 30 min of incubation in the CB1 antagonist AM251 (5 µM), eLFS was delivered. Down state NAc-iLTD expression was not abolished in the presence of AM 251 (IPSC amplitude = 77.7 ± 8.59% of baseline, t = 2.59, df = 5, p = 0.048, paired t-test, Figure S1A). As corticostriatal LTD in the dorsal striatum is D2 dopamine receptor dependent, we examined whether NAc-iLTD is also sensitive to D2 receptor blockade. Including the D2 receptor antagonist sulpiride (10 µM) in the artificial cerebrospinal fluid (aCSF) did not eliminate NAc-iLTD (IPSC amplitude in sulpiride = 74.38 ± 6.71% of baseline, t = 3.82, df = 5, p = 0.01, paired t-test, Figure S1B). To test whether NAc-iLTD requires the activity of any postsynaptic G-protein-coupled receptors (GPCR), we included the GPCR-inhibitor, GDP-β-S (500 µM) in the internal pipette solution. Inhibition of postsynaptic GPCRs did not affect NAc-iLTD expression (IPSC amplitude = 81.63 ± 6.24% of baseline, t = 2.94, df = 5, p = 0.03, paired t-test, Fig. 1c).
We hypothesized that activation of another receptor protein family, such as receptor tyrosine kinases, may be necessary for NAc-iLTD expression. We tested this by inhibiting the receptor for BDNF, TrkB, with cyclotraxin B (2 µM) in the aCSF. This manipulation blocked the expression of down state NAc-iLTD (IPSC amplitude = 107.7 ± 11.83% of baseline, t = 0.65, df = 13, p = 0.53, paired t-test, Fig. 1d). Cyclotraxin B blockade of NAc-iLTD was significantly different from control NAc-iLTD (control IPSC amplitude = 72.97 ± 6.97% of baseline, versus cyclotraxin B IPSC amplitude = 107.7 ± 11.83% of baseline, t = 2.28, df = 22, p = 0.03, unpaired t-test). Conditionally knocking out the TrkB receptor from the NAc by injecting a virus expressing cre-recombinase (cre) into a line of mice in which the TrkB gene is flanked by loxP sites (Flx-TrkB, Fig. 1e) resulted in a failure to induce NAc-iLTD (IPSC amplitude = 94.96 ± 7.87% of baseline, t = 0.64, df = 6, p = 0.55, paired t-test, Fig. 1f). Notably, there was no difference in the cumulative frequency distribution of the amplitude (Figure S1C) or frequency (Figure S1D) of spontaneous IPSC (sIPSC) events between TrkB knockout and control animals (sIPSC amplitude: Kolmogorov-Smirnov D = 0.042, p = 0.48; sIPSC frequency: Kolmogorov-Smirnov D = 0.002, p > 0.999).
The stability of LTD can be categorized into two forms: static and labile (for review, see [20]). In brief, labile LTD requires the presence of an agonist to maintain synaptic depression, while static LTD does not. To determine whether down state NAc-iLTD is static or labile, we applied cyclotraxin B onto slices after NAc-iLTD was established. GABAergic transmission remained depressed in the presence of cyclotraxin B (IPSC amplitude 25 min post induction = 66.52 ± 11.76% of baseline, IPSC amplitude with cyclotraxin B = 67.68 ± 10.51% of baseline, t = 0.10, df = 5, p = 0.92, paired t-test, Fig. 1g), demonstrating that down state NAc-iLTD is static and that downstream signaling from TrkB is likely necessary for NAc-iLTD maintenance.
The sensitivity of NAc-iLTD to TrkB inhibition suggests that BDNF application should recapitulate synaptic depression. Applying BDNF (4 nM) while voltage-clamping MSNs in the down state depressed inhibition onto MSNs (IPSC amplitude = 75.09 ± 2.24% of baseline, t = 11.11, df = 5, p = 0.0001, paired t-test, Fig. 1h). To determine if BDNF depresses excitatory transmission onto MSNs, we applied BDNF onto NAc slices in the presence of picrotoxin (50 μM) to block GABAA-receptor-mediated currents and found that, similar to inhibitory transmission onto MSNs, BDNF depressed excitatory transmission (Excitatory postsynaptic current (EPSC) amplitude = 60.83 ± 8.43% of baseline, t = 4.64, df = 4, p = 0.0097, paired t-test, Figure S1E). Because BDNF depressed excitatory synaptic transmission in the NAc, we investigated if TrkB-dependent LTD occurs at excitatory synapses in the NAc in response to eLFS. We delivered eLFS to slices in aCSF containing picrotoxin (50 μM) and found that eLFS induced an excitatory long-term depression (eLTD; EPSC amplitude = 71.46 ± 10.64% of baseline, t = 2.68, df = 8, p = 0.028, paired t-test, Fig. 1i). However, this eLTD was not cyclotraxin B-sensitive (EPSC amplitude in cyclotraxin B = 76.86 ± 8.43% of baseline, t = 2.74, df = 10, p = 0.02, paired t test; control versus cyclotraxin B: t = 0.403, df = 18, p = 0.69, unpaired t-test, Fig. 1i).
NAc-iLTD is postsynaptically expressed
We next analyzed if NAc-iLTD is expressed pre- or postsynaptically by analyzing the cumulative frequency distribution of the amplitude and frequency of sIPSC events before and after iLTD induction. The cumulative frequency distribution of sIPSC event amplitude was significantly different than that of sIPSC amplitudes following NAc-iLTD induction (Kolmogorov-Smirnov D = 0.15, p < 0.0001, Fig. 2a, b), while the distribution of sIPSC event frequency was not (Kolmogorov-Smirnov D = 0.009, p < 0.99, Fig. 2a, b). Further, paired-pulse ratio (PPR) analysis prior to eLFS delivery was not significantly different from PPR 25 min following eLFS delivery in the down state, but was different at the 5 min time point following eLFS delivery (baseline PPR = 0.94 ± 0.05; PPR at minutes 6–10 = 1.06 ± 0.08, minutes 25–30 post iLTD induction PPR = 0.99 ± 0.06, F(1.61, 30.62) = 9.94, p = 0.001, RM one-way ANOVA, Fig. 2d). Accordingly, the coefficient of variation (CV) of IPSC amplitude before NAc-iLTD induction did not differ 25 min after NAc-iLTD induction, but did differ the first 5 min following (baseline CV = 0.18 ± 0.01, CV minutes 6–10 = 0.26 ± 0.02, CV minutes 25–30 = 0.2 ± 0.02, F(1.63, 19.56) = 10.56, p = 0.001, RM one-way ANOVA, Fig. 2e). Neither the PPR or CV of IPSC amplitude were changed 5 min or 25 min following BDNF application (PPR before BDNF = 0.83 ± 0.03, PPR minutes 6–10 = 0.8 ± 0.07, PPR minutes 25–30 after BDNF = 0.8 ± 0.08, F(1.38, 6.89) = 0.06, p = 0.88, RM one-way ANOVA, Fig. 2f; Baseline CV = 0.23 ± 0.05, CV 6–10 min post-BDNF = 0.21 ± 0.04, CV 25–30 min post-BDNF = 0.2 ± 0.04, F(1.49, 7.43) = 0.61, p = 0.52, RM one-way ANOVA, Fig. 2g). These findings suggest the mechanism underlying late-stage-NAc-iLTD is expressed postsynaptically while the short term synaptic depression immediately following eLFS is presynaptically mediated.
Fig. 2.

NAc-iLTD is postsynaptically expressed. a Representative traces of spontaneous IPSC events (sIPSC) before (black) and after NAc-iLTD induction (gray). Scale bars: 200 pA, 50 s. b The cumulative frequency distribution (CFD) of sIPSC amplitudes after NAc-iLTD induction (gray) differed from baseline (black). c The CFD of the frequency of sIPSC events before and after NAc-iLTD induction were not different. d Top: The paired-pulse ratio (PPR) before (black) and 25 min after (gray) NAc-iLTD induction were not different, but was different the first 5 min after induction (red). Open squares are individual cells; filled squares represent the mean PPR. Bottom: Representative traces at each time point. e Top: The coefficient of variation (CV) did not differ before and 25 min after LFS delivery. CV was significantly different the first 5 min following LFS delivery. Bottom: Representative traces at each time point; the red trace is the average of all the traces at that time point. f Top: PPR at minutes 6–10 and 25–30 did not differ following BDNF application. Bottom: Representative traces at each time point. g Top: CV of IPSC amplitude following BDNF application at minutes 25–30 did not differ from baseline values or minutes 6–10. Bottom: Representative traces at each time point; the red trace is the average of all the traces at that time point. h Including the MEK inhibitor, U0126 (2 µM) in the internal pipette solution blocked NAc-iLTD induction (red), compared to controls (black). i Inclusion of the PLC inhibitor, U73122 (1 µM) in the internal pipette solution did not eliminate NAc-iLTD. j Inhibiting protein synthesis with cycloheximide (25 µM) in the artificial cerebrospinal fluid (aCSF) did not alter NAc-iLTD expression. k Inhibiting dynamin-mediated endocytosis with a dynamin-inhibitory peptide (DIP, 50 µM) in the internal pipette solution blocked NAc-iLTD (red), while including a scrambled version of this peptide (DIPS, 50 µM) did not (black). Insets: Representative traces of the first 5 min (dark) and final 5 min (light) of the experiment. Scale bars: 200 pA, 200 ms, unless noted otherwise. *p < 0.05. Data are represented as mean ± SEM
TrkB signals through various downstream signaling cascades [1], one of which is the mitogen-activated protein kinase-extracellular signal-regulated kinase (MEK-ERK) pathway. To test whether MEK activity is necessary for NAc-iLTD, we included the MEK inhibitor U0126 (2 µM) in the internal pipette solution. NAc-iLTD was eliminated in the presence of U0126 (U0126 IPSC amplitude = 98.36 ± 11.71% of baseline, t = 0.14, df = 6 cells, p = 0.89, paired t-test; versus control NAc-iLTD amplitude = 68.15 ± 5.90% of baseline, t = 2.30, df = 12, p = 0.04, unpaired t-test, Fig. 2h). Testing an alternative downstream pathway recruited by TrkB signaling, we found that blocking phospholipase C (PLC) activity with 1 µM U73122 in the internal pipette solution had no effect on NAc-iLTD (IPSC amplitude = 76.45 ± 8.91% of baseline, t = 2.64, df = 6, p = 0.04, paired t-test, Fig. 2i). TrkB signaling through MEK activity typically exerts changes in plasticity through protein synthesis [1]. Thus, we tested whether NAc-iLTD requires protein synthesis by incubating NAc slices in the protein synthesis inhibitor cycloheximide (25 µM) for at least 30 min before delivering eLFS. Cycloheximide did not block NAc-iLTD, suggesting this form of plasticity does not require protein synthesis (IPSC amplitude = 77.97 ± 8.03% of baseline, t = 2.74, df = 8, p = 0.03, paired t-test, Fig. 2j), however incubating slices with cycloheximide prevented the expression of eCB-dependent corticostriatal LTD in the dorsal striatum, which does require protein synthesis [21] (Control IPSC amplitude = 54.94 ± 8.57% of baseline, t = 5.26, df = 4, p = 0.006, paired t-test; IPSC amplitude in cycloheximide = 91.72 ± 6.2% of baseline, t = 1.34, df = 4, p = 0.25, paired t-test; cycloheximide versus control: t = 3.48, df = 8, p = 0.008, unpaired t-test, Figure S1F).
Because TrkB signaling can lead to dynamin-mediated GABAA-receptor endocytosis [22], we tested whether dynamin-mediated endocytosis is necessary for NAc-iLTD by including a dynamin-inhibitory peptide (DIP, 50 µM) in the internal pipette solution. Inclusion of DIP blocked NAc-iLTD, whereas a scrambled version of the peptide (DIPS, 50 µM) did not (DIP IPSC amplitude = 91.58 ± 8.82% of baseline, t = 0.95, df = 5, p = 0.38, paired t-test; DIPS IPSC amplitude = 68.17 ± 4.65% of baseline, t = 6.85, df = 5, p = 0.001, paired t-test; DIP versus DIPS internal: t = 2.35, df = 10, p = 0.04, unpaired t-test, Fig. 2k).
NAc-iLTD is voltage-state dependent
Because iLTD in the dorsal striatum is membrane-voltage-state dependent [12], we delivered eLFS while voltage-clamping MSNs in the up state (−60 mV). This did not depress GABAergic transmission onto NAc MSNs (IPSC amplitude = 87.05 ± 12.4% of baseline, t = 1.04, df = 6 cells, p = 0.34, paired t-test, Fig. 3a), suggesting NAc-iLTD is membrane-voltage dependent. Testing the voltage-dependence of BDNF-induced synaptic depression, application of BDNF while voltage-clamping MSNs in the up state did not depress inhibition onto MSNs (IPSC amplitude = 90.79 ± 5.29% of baseline, t = 1.74, df = 4, p = 0.16, paired t-test, Fig. 3b). To verify that NAc-iLTD is in fact voltage dependent and not due to a sampling error, we delivered eLFS while voltage-clamping MSNs in the up state, followed by delivery of eLFS while voltage-clamping MSNs in the down state. Inhibition onto MSNs did not depress when MSNs were held in the up state during eLFS delivery (IPSC amplitude = 91.36 ± 6.78% of baseline, t = 1.28, df = 6 cells, p = 0.25, paired t-test), but did depress when MSNs were subsequently held in the down state during eLFS (IPSC amplitude = 73.68 ± 4.66% of baseline, t = 5.65, df = 6 cells, p = 0.001, paired t-test, Fig. 3c).
Fig. 3.

NAc-iLTD membrane-voltage-state dependence is dictated by L-type voltage-gated calcium (Ca2+) channels. a eLFS delivered while voltage-clamping MSNs in the up state (Vhold = −60 mV) did not induce NAc-iLTD. b Acute application of BDNF (4 nM) while voltage clamping MSNs at −60 mV did not produce iLTD. c Down state NAc-iLTD occurred in MSNs that did not express up state NAc-iLTD. d Inclusion of the Ca2+ chelator, BAPTA (20 mM) in the internal pipette solution did not alter down state NAc-iLTD. e PPR following NAc-iLTD induction with a high BAPTA concentration is not different across the experiment. Open squares are individual cells; filled squares represent the mean PPR. f The L-type Ca2+-channel blocker, nifedipine (30–60 µM) did not alter down state NAc-iLTD. g Including BAPTA (20 mM) in the internal pipette solution unmasked the expression of up state NAc-iLTD (red), compared to controls (black). h Nifedipine (30–60 µM) also unmasked up state NAc-iLTD (red), compared to control aCSF (black). Insets: Representative traces of the first 5 min (dark) and final 5 min (light) of the experiment. Scale bars: 200 pA, 200 ms. Data are represented as mean ± SEM
The voltage-dependence of distinct forms of iLTD in the nearby dorsal striatum is gated by postsynaptic Ca2+ [12]. To test whether the voltage-dependence of NAc-iLTD is similarly Ca2+ dependent, we included a high concentration of the Ca2+ chelator BAPTA (20 mM) in the internal pipette solution. Inhibition onto MSNs was still depressed by eLFS when MSNs were held in the down state in the presence of 20 mM BAPTA (IPSC amplitude = 82.06 ± 5.48% of baseline, t = 3.28, df = 8, p = 0.01, paired t-test, Fig. 3d), suggesting that NAc-iLTD occurs in a Ca2+-independent manner. However, PPR analysis following NAc-iLTD induction with 20 mM BAPTA revealed this LTD no longer contains a presynaptic component (baseline PPR = 0.96 ± 0.09, PPR minutes 6–10 = 0.86 ± 0.05, PPR minutes 25–30 = 0.96 ± 0.11, F(1.37, 9.6) = 2.5, p = 0.14, RM one-way ANOVA, Fig. 3e). Because the down state of MSNs is beyond the activation threshold for L-type voltage-gated Ca2+ channels (VGCC) [10] and down state NAc-iLTD was insensitive to postsynaptic BAPTA loading, we predicted that the expression of down state NAc-iLTD would be unaffected in the presence of the L-type VGCC inhibitor, nifedipine. Indeed, inhibition onto MSNs was still depressed by eLFS when MSNs were held in the down state in the presence of nifedipine (30–60 µM) (IPSC amplitude = 82.16 ± 3.42% of baseline, t = 5.21, df = 4, p = 0.007, paired t-test, Fig. 3f).
Similar to the dorsal striatum [12], when eLFS was delivered while MSNs were held in the up state, the inclusion of 20 mM BAPTA unmasked NAc-iLTD (IPSC amplitude = 68.27 ± 4.14% of baseline, t = 7.67, df = 5, paired t-test, p = 0.0006, paired t-test; versus control internal solution: t = 2.52, df = 10, p = 0.03, unpaired t-test, Fig. 3g). Because the activation threshold of L-type VGCC is within the MSN up state membrane potential range [10], we predicted that up state NAc-iLTD would be unmasked by blocking L-type VGCC with nifedipine. Up state NAc-iLTD was unmasked in the presence of nifedipine (IPSC amplitude = 70.48 ± 10.49% of baseline, t = 2.81, df = 5, p = 0.04, paired t-test; versus control aCSF: t = 2.6, df = 9, p = 0.03, unpaired t-test, Fig. 3h).
The effect of EtOH on NAc-iLTD
Because disinhibiting the NAc may be associated with reward-related behaviors, we investigated whether EtOH interacts with NAc-iLTD. To test this, we first applied EtOH onto NAc slices at two different concentrations (a low concentration: 10 mM, and a high concentration: 50 mM) while voltage clamping MSNs in the up or down state. Delivering paired electrical stimulation pulses eliminates EtOH-induced LTD in the dorsal striatum [23]. Therefore, we delivered single electrical pulses to determine the effect of EtOH on inhibitory synaptic strength in the NAc. Acute application of 10 mM EtOH alone did not depress inhibition onto NAc MSNs when voltage clamped at either membrane-voltage state (IPSC amplitude for up state = 100.4 ± 14.11% of baseline, t = 0.03, df = 5 cells, p = 0.98, paired t-test; IPSC amplitude for down state = 92.65 ± 7.86% of baseline, t = 0.93, df = 5, p = 0.39, paired t-test, Fig. 4a). Similarly, acute application of a high dose of EtOH (50 mM) did not depress inhibition onto NAc MSNs while voltage clamped at the up or down state (IPSC amplitude for up state = 97.86 ± 2.14% of baseline, t = 0.99, df = 7, p = 0.35, paired t-test; IPSC amplitude for down state = 104.3 ± 10.1% of baseline, t = 0.43, df = 6, p = 0.68, paired t-test, Fig. 4b). However, delivering eLFS to slices in 10 mM EtOH significantly augmented the magnitude of IPSC amplitude depression (IPSC amplitude in 10 mM EtOH = 56.23 ± 7.52% of baseline, t = 5.82, df = 7, p = 0.0007, paired t-test; versus control aCSF: t = 2.22, df = 15, p = 0.04, unpaired t-test, Fig. 4c). Enhancement of NAc-iLTD similarly occurred after incubating slices in a higher concentration of EtOH (50 mM, IPSC amplitude = 50.91 ± 10.71% of baseline, t = 4.58, df = 6, p = 0.0038, paired t-test; verses control aCSF: t = 2.35, df = 13, p = 0.04, unpaired t-test, Fig. 4d). This enhanced NAc-iLTD was not eliminated upon removal of EtOH (IPSC amplitude in 50 mM EtOH = 69.69 ± 5.66% of baseline, IPSC amplitude in regular aCSF = 64.81 ± 6.81% of baseline, t = 0.83, df = 5, p = 0.44, paired t-test, Figure S1G). There was no difference in the magnitude of the enhancement of NAc-iLTD induced in 10 mM or 50 mM EtOH, suggesting this effect is not dose-dependent (Concentration: F(1, 13) = 0.72, p = 0.41, Time: F(29, 377) = 12.54, p < 0.0001, Two-way Time × Concentration mixed ANOVA). The inclusion of either concentration of EtOH in the bath eliminated the presynaptic component to NAc-iLTD (for 10 mM: baseline PPR = 0.87 ± 0.03, PPR minutes 6–10 = 0.92 ± 0.02, PPR minutes 25–30 = 0.89 ± 0.03, F(1.31, 5.24) = 0.8, p = 0.45, RM one-way ANOVA, Fig. 4e; for 50 mM: Baseline PPR = 0.97 ± 0.16, PPR minutes 6–10 = 0.91 ± 0.14, PPR minutes 25–30 = 0.99 ± 0.1, F(1.72, 6.88) = 0.24, p = 0.76, RM one-way ANOVA, Fig. 4f). Moreover, there was no effect of the animal’s sex on 50 mM EtOH-induced enhancement of NAc-iLTD (IPSC amplitude in females = 52.47 ± 7.03% of baseline, n = 8, IPSC amplitude in males = 55.91 ± 4.46% of baseline, n = 9, t = 0.42, df = 15, p = 0.68, unpaired t-test).
Fig. 4.

Ethanol (EtOH) enhances NAc-iLTD. a Acute application of 10 mM EtOH did not depress synaptic inhibition onto MSNs when voltage clamped at −60 mV (red) or −80 mV (black). b Acute application of a higher dose of EtOH (50 mM) did not depress inhibitory transmission onto MSNs when voltage clamped at either −60 mV (red) or −80 mV (black). c Incubating slices in 10 mM EtOH enhanced NAc-iLTD (red) compared to control aCSF (black). d The presence of a higher concentration of EtOH (50 mM) in the aCSF enhanced the magnitude of NAc-iLTD (red) compared to control aCSF (black). e Left: The mean PPR was not different at any time point following NAc-iLTD induction in the presence of 10 mM EtOH. Right: Representative traces for each time point. f Left: The mean PPR was not significantly different between any time point following NAc-iLTD induction in the presence of 50 mM EtOH. Right: Representative traces for each time point. g 50 mM EtOH-enhanced NAc-iLTD is eliminated in the presence of cyclotraxin B (2 µM, red). Black line (±SEM, gray) represents the 50 mM EtOH-induced enhancement of NAc-iLTD from d. h Left: The mean PPR was not significantly different between any time point following NAc-iLTD induction in the presence of 50 mM EtOH and cyclotraxin B. Right: Representative traces for each time point. Insets: Representative traces of the first 5 min (dark) and final 5 min (light) of the experiment. Scale bars: 200 pA, 200 ms. Data are represented as mean ± SEM
The enhancement of NAc-iLTD by 50 mM EtOH was eliminated in the presence of cyclotraxin B (IPSC amplitude = 105.7 ± 9.20% of baseline, t = 0.62, df = 5, p = 0.56, paired t-test, Fig. 4g) and PPR did not change throughout the course of the experiment in the presence of cyclotraxin B (baseline PPR = 1.16 ± 0.21, PPR minutes 6–10 = 1.05 ± 0.14, PPR minutes 25–30 = 1.25 ± 0.11, F(1.89, 9.45) = 1.24, p = 0.33, RM one-way ANOVA, Fig. 4h). To determine the possible involvement of the EtOH metabolite acetaldehyde in enhanced NAc-iLTD, we applied acetaldehyde (1 µM) to the slice and measured the change in IPSC amplitude. Acute application of acetaldehyde did not induce iLTD in NAc MSNs (IPSC amplitude = 97.73 ± 11.46% of baseline, t = 0.19, df = 7, p = 0.85, paired t-test, Figure S1H). Further, delivering eLFS in the presence of acetaldehyde (1 µM) did not induce NAc-iLTD (IPSC amplitude = 125.2 ± 15.55% of baseline, t = 1.62, df = 5, p = 0.17, paired t-test, Figure S1I), nor did synaptic depression occur following eLFS and subsequent acetaldehyde removal (IPSC amplitude in acetaldehyde = 122.0 ± 10.46% of baseline, IPSC amplitude in aCSF = 110.6 ± 15.06, t = 1.18, df = 7, p = 0.28, paired t-test, Figure S1J).
Discussion
We find that NAc-iLTD is dependent upon TrkB activation, is voltage-dependent, and is enhanced by the presence of EtOH at concentrations that correspond with physiologically relevant levels of intoxication [24]. Taken together, these results support the notion that accumbal output is under plastic control through the BDNF-TrkB signaling system and may be important for the encoding of alcohol reward (Fig. 5).
Fig. 5.
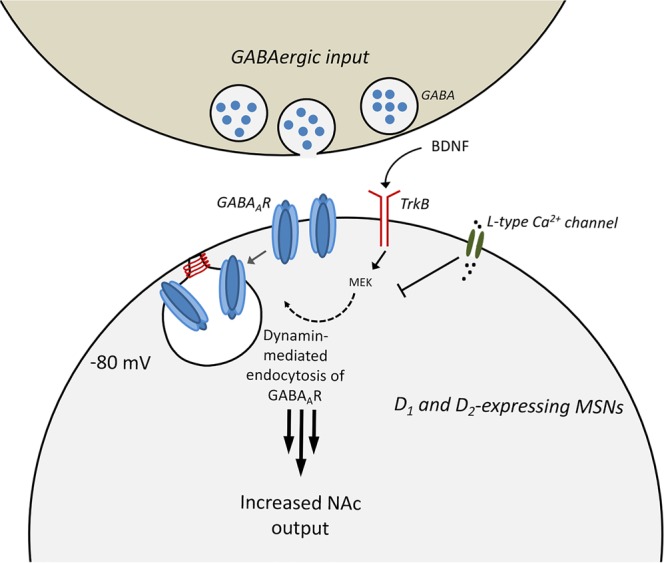
Proposed mechanism of NAc-iLTD. D1- and D2-expressing MSNs undergo NAc-iLTD through the activation of postsynaptic TrkB receptors following BDNF release. TrkB signaling enhances MEK activity, which leads to the depression of the synapse in a dynamin-dependent manner, possibly through the internalization of GABAA receptors. NAc-iLTD occurs when MSNs are in the hyperpolarized down state, and activation of the L-type Ca2+ channel can abolish this plasticity. We propose that NAc-iLTD disinhibits a subset of MSNs residing in the down state to enhance NAc output
Here, we demonstrate that inhibitory inputs onto NAc MSNs undergo iLTD. The question remains of which inhibitory input(s) onto MSNs undergo NAc-iLTD. Several candidate inhibitory inputs exist to underlie this depression, including the ventral pallidum, fast-spiking interneurons, neighboring MSNs, and midbrain dopamine neurons. As subsets of midbrain dopaminergic inputs corelease GABA or BDNF onto MSNs [25, 26], it is also possible that these neurons are the source of BDNF. However, future work is needed to determine whether midbrain- or prefrontal cortex-derived BDNF [27] mediate NAc-iLTD.
In contrast to the direct induction of iLTD by EtOH in the dorsal striatum [28], in the NAc EtOH alone does not induce synaptic depression but instead augments NAc-iLTD. EtOH differentially alters BDNF expression depending on the brain region, the dose of EtOH and the time interval following EtOH exposure [29–31]. Given that our experiments involve the acute application of EtOH to NAc slices only minutes before NAc-iLTD induction, it is unlikely that our findings of EtOH-induced enhanced depression of IPSC amplitude following NAc-iLTD induction are due to an increase in BDNF expression. It is possible however, that EtOH augments BDNF release. Alternatively, EtOH may boost signaling downstream of postsynaptic MSN TrkB activation. Consistent with this idea, Ben Hamida and colleagues (2012) demonstrated that activity of the small GTP binding protein, H-Ras increases in the NAc following acute exposure to EtOH. Further, suppression of H-Ras activation in the NAc significantly decreases EtOH consumption, suggesting H-Ras and the resulting signaling cascade are key modulators in the rewarding effects of EtOH. The Ras family of proteins is upstream of MEK and downstream of TrkB activation [1]. These findings position this family of proteins as potential permissive molecular signals underlying EtOH modulation of NAc-iLTD and a possible interventional target for the reinforcing effects of EtOH [32]. Further work is necessary to link H-Ras and NAc-iLTD.
While direct- and indirect-pathway-specific activity and plastic events are linked to reward and aversion [18, 33, 34], we did not observe pathway specificity in our slice electrophysiology experiments. This does not rule out the possibility that NAc-iLTD is induced preferentially at one pathway or another in vivo. Assuming there is no pathway specificity in vivo, this is reminiscent of the pathway non-specific, serotonin-mediated LTD at NAc excitatory synapses [16] that encodes social reward [35]. Such global disinhibition of the structure by NAc-iLTD could act in concert with pathway-specific plasticity mechanisms at excitatory synapses. At face value, the present data support the notion that global disinhibition of the NAc may encode reward and that augmentation of this mechanism by EtOH may modulate alcohol drinking behavior. As such, NAc-iLTD represents a starting point from which to devise treatment strategies for disorders of reward and aversion, such as addiction and depression.
Funding
This work was supported by the Alcoholic Beverage Medical Research Foundation/The Foundation for Alcohol Research, National Institute on Alcohol Abuse and Alcoholism grants K22AA021414, R01AA024845 (B.N.M.) and F31AA024683 (M.H.P.) and a Whitehall Foundation grant 2014-12-68 (B.N.M.).
Supplementary information
{kind=link}
Acknowledgements
The authors thank all current and previous lab members who contributed to this project.
Author contributions
M.H.P. and B.N.M. conceived experiments. M.H.P., K.E.P., P.N.M., C.M., H.Q., M.S.P., B.M.R., and B.N.M. performed experiments. M.H.P. and B.N.M. wrote the manuscript with help from K.E.P., P.N.M., C.M., H.Q., M.S.P., and B.M.R. M.H.P. and B.N.M. secured funding.
Conflict of interest
The authors declare no competing interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41386-019-0341-8).
References
- 1.Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56(Suppl 1):73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultz W. Reward functions of the basal ganglia. J Neural Transm. 2016;123:679–93. doi: 10.1007/s00702-016-1510-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grueter BA, Rothwell PE, Malenka RC. Integrating synaptic plasticity and striatal circuit function in addiction. Curr Opin Neurobiol. 2012;22:545–51. doi: 10.1016/j.conb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–63. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nisenbaum ES, Berger TW. Functionally distinct subpopulations of striatal neurons are differentially regulated by GABAergic and dopaminergic inputs--I. In vivo analysis. Neuroscience. 1992;48:561–78. doi: 10.1016/0306-4522(92)90402-N. [DOI] [PubMed] [Google Scholar]
- 7.Kerr JN, Plenz D. Action potential timing determines dendritic calcium during striatal up-states. J Neurosci. 2004;24:877–85. doi: 10.1523/JNEUROSCI.4475-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15(5 Pt 1):3622–39. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson CJ, Kawaguchi Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci. 1996;16:2397–410. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter AG, Sabatini BL. State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron. 2004;44:483–93. doi: 10.1016/j.neuron.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–45. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mathur BN, Tanahira C, Tamamaki N, Lovinger DM. Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nat Neurosci. 2013;16:1275–83. doi: 10.1038/nn.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright WJ, Schluter OM, Dong Y. A feedforward inhibitory circuit mediated by CB1-expressing fast-spiking interneurons in the nucleus accumbens. Neuropsychopharmacology. 2017;42:1146–56. doi: 10.1038/npp.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matyas F, Yanovsky Y, Mackie K, Kelsch W, Misgeld U, Freund TF. Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience. 2006;137:337–61. doi: 10.1016/j.neuroscience.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Davis MI, Crittenden JR, Feng AY, Kupferschmidt DA, Naydenov A, Stella N, et al. The cannabinoid-1 receptor is abundantly expressed in striatal striosomes and striosome-dendron bouquets of the substantia nigra. PLoS ONE. 2018;13:e0191436. doi: 10.1371/journal.pone.0191436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathur BN, Capik NA, Alvarez VA, Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–11. doi: 10.1523/JNEUROSCI.6250-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luikart BW, Nef S, Shipman T, Parada LF. In vivo role of truncated trkb receptors during sensory ganglion neurogenesis. Neuroscience. 2003;117:847–58. doi: 10.1016/S0306-4522(02)00719-4. [DOI] [PubMed] [Google Scholar]
- 18.Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–90. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kupferschmidt DA, Cody PA, Lovinger DM, Davis MI. Brain BLAQ: post-hoc thick-section histochemistry for localizing optogenetic constructs in neurons and their distal terminals. Front Neuroanat. 2015;9:6. doi: 10.3389/fnana.2015.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atwood BK, Lovinger DM, Mathur BN. Presynaptic long-term depression mediated by Gi/o-coupled receptors. Trends Neurosci. 2014;37:663–73. doi: 10.1016/j.tins.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin HH, Davis MI, Ronesi JA, Lovinger DM. The role of protein synthesis in striatal long-term depression. J Neurosci. 2006;26:11811–20. doi: 10.1523/JNEUROSCI.3196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hewitt SA, Bains JS. Brain-derived neurotrophic factor silences GABA synapses onto hypothalamic neuroendocrine cells through a postsynaptic dynamin-mediated mechanism. J Neurophysiol. 2006;95:2193–8. doi: 10.1152/jn.01135.2005. [DOI] [PubMed] [Google Scholar]
- 23.Patton MH, Padgett KE, McKeon PN, Lu SG, Abrams TW, Mathur BN. An Aplysia-like synaptic switch for rapid protection against ethanol-induced synaptic inhibition in a mammalian habit circuit. Neuropharmacology. 2019;144:1–8. doi: 10.1016/j.neuropharm.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovinger DM, Roberto M. Synaptic effects induced by alcohol. Curr Top Behav Neurosci. 2013;13:31–86. doi: 10.1007/978-3-642-28720-6_143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tritsch NX, Oh WJ, Gu C, Sabatini BL. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife. 2014;3:e01936. doi: 10.7554/eLife.01936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80:469–78. doi: 10.1016/j.biopsych.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–60. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 28.Patton MH, Roberts BM, Lovinger DM, Mathur BN. Ethanol disinhibits dorsolateral striatal medium spiny neurons through activation of a presynaptic delta opioid receptor. Neuropsychopharmacology. 2016;41:1831–40. doi: 10.1038/npp.2015.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeanblanc J, He DY, Carnicella S, Kharazia V, Janak PH, Ron D. Endogenous BDNF in the dorsolateral striatum gates alcohol drinking. J Neurosci. 2009;29:13494–502. doi: 10.1523/JNEUROSCI.2243-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orru A, Caffino L, Moro F, Cassina C, Giannotti G, Di Clemente A, et al. Contingent and non-contingent recreational-like exposure to ethanol alters BDNF expression and signaling in the cortico-accumbal network differently. Psychopharmacology. 2016;233:3149–60. doi: 10.1007/s00213-016-4358-y. [DOI] [PubMed] [Google Scholar]
- 31.Raivio N, Tiraboschi E, Saarikoski ST, Castren E, Kiianmaa K. Brain-derived neurotrophic factor expression after acute administration of ethanol. Eur J Pharmacol. 2012;687:9–13. doi: 10.1016/j.ejphar.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 32.Ben Hamida S, Neasta J, Lasek AW, Kharazia V, Zou M, Carnicella S, et al. The small G protein H-Ras in the mesolimbic system is a molecular gateway to alcohol-seeking and excessive drinking behaviors. J Neurosci. 2012;32:15849–58. doi: 10.1523/JNEUROSCI.2846-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kravitz AV, Kreitzer AC. Striatal mechanisms underlying movement, reinforcement, and punishment. Physiology. 2012;27:167–77. doi: 10.1152/physiol.00004.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferguson SM, Eskenazi D, Ishikawa M, Wanat MJ, Phillips PE, Dong Y, et al. Transient neuronal inhibition reveals opposing roles of indirect and direct pathways in sensitization. Nat Neurosci. 2011;14:22–24. doi: 10.1038/nn.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolen G, Darvishzadeh A, Huang KW, Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–84. doi: 10.1038/nature12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.