

Abstract
Recent diversity studies have revealed that microbial communities of natural environments are dominated by species-like, sequence-discrete populations. However, how stable the sequence and gene-content diversity are within these populations and especially in highly dynamic lotic habitats remain unclear. Here we quantified the dynamics of intra-population diversity in samples spanning two years and five sites in the Kalamas River (Northwest Greece). A significant positive correlation was observed between higher intra-population sequence diversity and longer persistence over time, revealing that more diverse populations tended to represent more autochthonous (vs. allochthonous) community members. Assessment of intra-population gene-content changes caused by strain replacement or gene loss over time revealed different profiles with the majority of populations exhibiting gene-content changes close to 10% of the total genes, while one population exhibited ~21% change. The variable genes were enriched in hypothetical proteins and mobile elements, and thus, were probably functionally neutral or attributable to phage predation. A few notable exceptions to this pattern were also noted such as phototrophy-related proteins in summer vs. winter populations. Taken together, these results revealed that some freshwater genomes are remarkably dynamic, even across short time and spatial scales, and have implications for the bacterial species concept and microbial source tracking.
Subject terms: Microbial ecology, Water microbiology
Introduction
During the past decade, several studies assessed microbial diversity of lotic (and other) ecosystems [1–6], focusing on the importance of hydrological, environmental, geochemical, or other factors on shaping microbial community composition. Most of these studies have used operational taxonomic units (OTUs), defined typically at the 97% nucleotide identity level of the 16S rRNA gene [2, 4], in order to identify core riverine communities and explore the dynamics of such core taxa. However, due to the high conservation of the rRNA genes, this approach can group genetically and ecologically distinct populations under the same OTU, and does not typically provide insight into the dynamics of the functional gene content [7, 8].
Assembly of metagenomic data can provide complete or nearly complete genomes of the microorganisms present in the samples analyzed. Comparison of the metagenomic reads against such genome sequences can reveal the level of genetic heterogeneity of the corresponding populations. Most notably, this analysis has revealed that sequence-discrete populations may typify natural microbial communities across habitats. Members of sequence-discrete populations are characterized by high gene-content and nucleotide sequence similarity among themselves, often—but not always— > 95% genome-aggregate Average Nucleotide Identity (ANI), and also exhibit ecological differentiation (e.g., different in-situ relative abundances) and/or lower relatedness (e.g., < 90% ANI) to close relatives [9, 10]. Such populations possess the attributes of bacterial species at a level that 16S rRNA gene sequencing cannot often assess; therefore, they represent an important unit to study. Assessing the dispersal patterns of sequence-discrete populations within a lotic system could provide higher resolution as to where populations originate and thus, aid in understanding microbial community assembly patterns and in population source tracking.
Further, quantifying gene content and sequence diversity dynamics within sequence-discrete populations may provide new insights into the frequency with which genomic adaptations are occurring in response to temporal or spatial gradients within a specific habitat, and into the molecular and ecological mechanisms that maintain (or not) sequence-discrete populations. Prokaryotic species are known to be highly dynamic, with strains of the same species frequently differing in up to ~30% of their genes [11, 12], but the rates of gene loss/gain in time scales that matter for human activities (e.g., days to years) remain elusive. Hence, by studying the diversity of sequence-discrete populations over time and space we hope to provide new insights into the species concept.
Most studies on sequence-discrete populations to date have been limited to the description of the presence/absence and relative abundance of different sequence-discrete populations in freshwater lakes [13, 14] and experimentally warmed vs. control soils [15]. Only recently, Bendall et al. [16] used sequence-discrete populations from Trout Bog Lake in order to identify population genome-wide and gene-specific sequence diversity sweeps. This study mostly focused on the investigation of different models of sequence evolution and speciation, using changes in single-nucleotide polymorphism sequences (SNPs). Bendall et al. also assessed gene-content variation of selected sequence-discrete populations based on an approach employing co-assembly of all samples. However, because co-assembly represents the “average” genome present in all samples, genes that were abundant at some but not all time points could be overlooked. These genes can be more efficiently tracked by assembling genomes from individual samples and querying them against individual metagenomes (the goal of our study).
Here we used shotgun metagenomics in order to explore the spatiotemporal dynamics of sequence-discrete populations in a highly dynamic riverine ecosystem, the Kalamas River (Northwest Greece), and quantify changes in intra-population gene-content diversity between sampling time points and seasons. Kalamas is a medium sized river that runs through NATURA 2000 protected areas but also receives urban and industrial, treated and untreated, sewage from the city of Ioannina (150,000 inhabitants) through the Lapsista ditch (Fig. 1). By assessing the read coverage patterns of the genes of 14 populations across time and space we were able to quantify gene-content diversity within these populations, and assess their origin, e.g., autochthonous freshwater vs. allochthonous populations originating from the ditch.
Fig. 1.

Kalamas river drainage basin and sampling locations. a Sampling locations along the Kalamas River. The river receives water from the adjacent drainage basin of Lake Pamvotis through an artificial ditch. Samples were collected from the ditch (KL), upstream of the influx of the ditch into the river (K0), immediately downstream of the influx location (K1), from within the river dam (K2), and estuary (K3). b Land cover of the drainage basin for each sampling location along the river. The characteristic land cover represents the immediate watershed area that drains into each sampling location, while the cumulative basin land cover represents the total drainage area (including the land areas that drain into upstream sampling points)
Materials and methods
Land cover analysis and sample processing
The basin of the Kalamas River (Fig. 1) was obtained based on the first-order mountainous water basins map provided by the Greek Ministry of Productive Reconstruction, Environment and Energy (http://geodata.gov.gr/en/organization/hydroscope), using the sub-basins dataset. (Supplementary information).
Samples were collected from five different sites (Fig. 1) at five different time points, spanning a period of 23 months (Table S1a) and exhibiting different flow rates (Table S1b). Our site selection scheme aimed to reveal the effect of the Lapsista ditch, which is expected to influence the water quality and bacterial community composition due to treated and untreated sewage it carries from human activities in the most populated city along the Kalamas River (see also land use data in Fig. 1). Therefore, in our sampling scheme, site K0 was selected as a representative pristine site with minimal influence from urban areas ~600 m upstream of the entry point of the ditch into Kalamas (Fig. 1, Table S1a), and site K1 was selected as a representative site with influence from the urban area, due to its proximity to the entry point of the ditch (~300 m downstream). The site K2 is located just before a hydroelectric dam along the river and 68.6 km downstream of the Lapsista ditch entry point. Site K2 was selected in order to assess the recovery of bacterial communities downstream of the ditch and the influence of water flow rate. Site K3, located at the estuary, was selected to evaluate how the bacterial diversity is affected by the freshwater/saltwater transition. Finally, a site within the ditch (KL), only 3.37 km downstream from Lake Pamvotis and 24.61 km before the entry point of the ditch, was chosen in order to study the bacterial communities within the ditch that presumably received the highest human impact (Fig. 1, Table S1a).
Samples were collected from November 2012 (M1) to September 2014 (M23) [Sample naming scheme: M for metagenomes; number reflects the number of months since the first sampling point, which was given the number 1]. November 2012 (M1), February (M4), and May (M7) 2013 samples were collected from three sites (K1, K2, and K3) along the Kalamas River (Fig. 1, Table S1a), and were described previously [6]. Additional samples were collected in June 2014 (M20) and September 2014 (M23) from sites KL (within the ditch), K1 (where the ditch merges with the main river), and K0 (upstream of the merging point). All water samples were immediately transferred in the lab and were processed and sequenced as previously described [[6], Supplementary information].
Metagenome sequence trimming and assembly
Illumina reads were trimmed using a Q = 20 Phred quality score cut-off using SolexaQA++ [17] and only trimmed reads longer than 50 bp were considered for further analysis. Metagenomic reads were assembled using IDBA with default settings for metagenomes [18]. Protein-coding genes were predicted from contigs longer than 500 bp using MetaGeneMark.hmm with default parameters [19]. Sequencing and assembly statistics for the metagenomic datasets are provided in Table S1b. Metagenomic datasets have been deposited in NCBI SRA under the bioproject PRJNA304352.
Population genome binning
Contigs longer than 500 bp were used for binning using MaxBin v2.1.1 with default settings [20]. In each binning run, only contigs from the assembly of an individual sample were used. CheckM and the MiGA webserver (www.microbial-genomes.org) were used to estimate completeness and contamination of each metagenome-assembled genome (MAG) based on the recovery of single-copy universal bacterial proteins [21, 22]. Recruitment plots were constructed for MAG contigs and genes using the ‘enveomics.R’ package v1.4.1 from the Enveomics Collection [23]. MAGs were manually checked for consistent coverage across contigs by assessing how many times—on average—the query contig sequence was covered by reads. Contigs with substantially different coverage than the average (> 2 fold) and those containing duplicated universal genes or having different taxonomy from the majority of the contigs based on a MyTaxa [24] scan as implemented in MiGA were removed in order to reduce contamination. Final MAGs are available through: http://enve-omics.ce.gatech.edu/data/
Abundance and gene-content diversity evaluation of recovered populations
Recruitment of metagenomic reads to the reference MAG population sequence was used to identify areas of sequence discontinuity for each MAG as described previously [10]. For most MAGs, 95% nucleotide identity, the most common threshold for the distinction of species from sequence data [25], corresponded to the area of sequence discontinuity and thus, was used as the threshold to identify reads representing the reference population and estimate the coverage of the populations (Table 1, Fig. S1). Reads with less than 95% identity were assumed to represent additional, co-occurring populations. For four populations (out of 14 evaluated in total), the area of sequence discontinuity was determined to be around 98% nucleotide identity based on visual inspection of the recruitment plots and/or steep drops in coverage (by more than 3–4 orders of magnitude), and a 98% minimum identity threshold was used in these cases (Table 1, Fig. S1.1, S1.2). Read mapping against the MAGs was performed with BLAT in competitive best-match searches [26], using all available MAGs as a reference database in order to eliminate multiple nonspecific matches of the same read(s) to different MAGs. Coverage values were normalized for dataset and genome size (Reads per Kbp of genome per Million reads, RPKM). For the linear correlation analysis, the coefficient of variation of the normalized coverage values over time was correlated with the Average Nucleotide Identity of mapped reads (ANIr) against the reference MAG genome sequence, using all mapped reads with identities above the selected nucleotide identity threshold (i.e., 95 or 98%). For samples (time points) with > 1× coverage the actual normalized coverage value was used in the correlation analysis; for samples with lower coverage a zero coverage value was used.
Table 1.
Analyzed population metagenome-assembled genomes (MAGs)
| MAG | Origina | Completeness | Contamination | N50 | CheckM /MiGA taxonomy | SDP boundary (%)b | Genes | Annotated genes |
|---|---|---|---|---|---|---|---|---|
| kal1.Rhi | K1-M1 | 99.1 | 2.7 | 32622 | Rhizobiales | 98 | 3812 | 2868 |
| kal2.Com | K1-M20 | 30.6 | 0 | 11298 | Comamonadaceae | 95 | 2145 | 2049 |
| kal3.Com | K1-M20 | 30.6 | 1.8 | 13718 | Comamonadaceae | 95 | 904 | 796 |
| kal4.Rhi | K1-M20 | 34.38 | 0 | 2622 | Rhizobiales | 95 | 2442 | 1988 |
| kal5.Flav | K0-M20 | 84.7 | 5.4 | 4132 | Flavobacteriaceae | 95 | 2210 | 2008 |
| kal6.Flav | K0-M23 | 64.00 | 0 | 2923 | Flavobacteriaceae | 95 | 2202 | 1956 |
| kal7.Gam | K0-M23 | 22.78 | 1.9 | 1763 | Gammaproteobacteria | 95 | 1046 | 947 |
| kal8.Bac | K1-M7 | 96.71 | 4.31 | 9189 | Bacilli/Exiguobacterium | 95 | 3386 | 3283 |
| kal9.Com | K1-M1 | 99.07 | 0.76 | 39707 | Comamonadaceae | 98 | 4556 | 3672 |
| kal10.Por | K1-M1 | 89.6 | 3.2 | 77881 | Porphyromonadaceae | 98 | 3127 | 2430 |
| kal11.Mol | K1-M1 | 81.46 | 3.37 | 10908 | Mollicutes | 95 | 2286 | 618 |
| kal12.Act | K1-M4 | 65.8 | 4.5 | 5022 | Actinobacteria | 95 | 1420 | 1068 |
| kal13.Act | K1-M4 | 44.1 | 1.8 | 4747 | Actinobacteria | 95 | 1613 | 1239 |
| kal14.Noc | K1-M1 | 70.3 | 2.7 | 2661 | Actinobacteria/Nocardiaceae | 98 | 5269 | 4820 |
aOrigin denotes the metagenomes from which the bin was extracted.
bSDP boundary: sequence-discrete population threshold used
Each gene of a MAG was evaluated as being present or variable within a metagenomic dataset as long as the corresponding reference MAG was detected in the same dataset with at least 7× coverage in order to reduce the impact of spurious results. When population coverage is lower, genes may be missed due to sequence depth variation and not real absence resulting in a false positive result for the variable gene [27]. This coverage level (>7×) and p-value (<0.01; see also below) ensured a low number of possible false positives based on a subsampling experiment (Fig. S2). For the subsampling experiment, the variable genes of the population MAG (see below) were identified by mapping the reads of the metagenome that the MAG originated from against the MAG; the metagenome was subsequently sub-sampled in order to test the effect of different population coverage levels on gene detection (Fig. S2). Genes detected as absent in the reference metagenome (no-subsampling) and in a subsample were denoted as true positives (TP); false positives (FP) were detected as variable (absent in this case) in the subsample only. Conversely, true negatives (TN) and false negatives (FN) were defined as present (detected) in both the subsample and the reference datasets, and present in the subsample but not the reference, respectively. Accordingly, Recall (TP/[TP + FN]), Precision (TP/[TP + FP]), as well as the F1 score (2 × Precision × Recall/[Precision + Recall]) were calculated. To identify variable genes, the coverage of a gene was first calculated by mapping reads on contigs, and calculating the average coverage of all bases of each gene. The resulting distribution of gene coverage values for a MAG were fit to a log-normal distribution using the enveomics.R package v1.4.1 [23] in order to calculate the probability that a gene with zero or near zero coverage was absent from the population studied using a p = 0.01 cut-off value (null hypothesis was that the gene is present in the population); only genes with p < 0.01 were considered variable. When multiple metagenomes showed population coverage > 7× for a MAG of interest (minimum cut-off to analyze a population for gene-content diversity) only the two metagenomes with the highest abundances were used (Table S2b).
Simulations were performed to test whether or not our methodology overestimated the number of variable genes, and whether differences in coverage (abundance) of a target population could affect the ANIr values. For this, Illumina-like reads were generated in-silico from an E. coli genome (GCF_000931565.1_ASM93156v1) using Grinder (-cf 10 -md uniform 0.1 -mr 95 5 -rd 150 uniform 5), and spiked in four different metagenome-like datasets at different concentrations (coverage; 0.1, 0.2, 0.5, 1, 2, 5, and 10 × ). The first two datasets were randomly selected genomes from RefSeq excluding Escherichia coli and Shigella spp., and were used as background. The other two datasets were Kalamas metagenomes. Thus, 28 test datasets were produced. Finally BLAT and recruitment plot analysis were performed against seven reference E. coli genomes (GCF_001865295.1_ASM186529v1, GCF_003018115.1_ASM301811v1, GCF_002952895.1_ASM295289v1, GCF_002237305.1_ASM223730v1, GCF_001577325.1_ASM157732v1, GCF_002012205.1_ASM201220v1, GCF_000931565.1_ASM93156v1) to test if the gene-content and sequence diversity of the reference genome compared to the genome spiked in the datasets could be recovered reliably by the recruitment plots (Fig. S3).
Supplementary information
The supplementary information provides additional details about land use coverage, flow rates analysis, metagenome sequencing, processing of 16S rRNA gene-encoding metagenomic reads, and functional annotation and phylogenetic analysis of identified genes.
Results
Microbial taxonomic and functional diversity in Kalamas samples
The riverine bacterial community composition, as assessed by the 16S rRNA gene (16S) fragments recovered in the metagenomes, exhibited drastic changes throughout the months as well as between sites examined (Fig. S4). Only a small fraction of the community exhibited persistent abundance profiles throughout most of the samples. From a total of 6245 identified OTUs, only 199 (~0.03%) were detected in all time points examined (Fig. S5), revealing highly dynamic microbial communities. These results were, in general, consistent with our previous study [6], which surveyed microbial community-wide shifts on a subset of the available samples (November 2012 through May 2013) as opposed to individual populations, the main focus of the present study. Our previous study had shown that differences in functional diversity were much more limited compared to taxonomic differences, and were related, at least in part, to the differential flow rate of the river water and the impact of anthropogenic activities. For instance, we had previously noted the prevalence of nitrogen metabolism proteins in K1-M1, and virulence, stress response, and aromatic compounds degradation associated proteins in K2-M7 samples, presumably reflecting the effect of the treated and untreated sewage carried by the ditch. Consistent results were observed in the 2014 samples studied here (Supplementary information).
Persistent populations show higher intra-population diversity
Population binning was focused on samples from site K1 because we wanted to assess how the populations recovered from this site changed over time and space; and on K0 for assessing the persistence of populations associated with communities receiving less anthropogenic impacts. The K1 site was also the only site for which five different time points were available, allowing for higher temporal resolution. Contig binning from these samples resulted in the recovery of 14 population MAGs with > 30% estimated completeness (Average = 65.22%) and < 6% contamination (after manual curation of the MAGs; Table 1), reflecting, in part, the low sequencing effort applied (~2 Gbp/sample, on average, Table S1b) relative to the diversity of the microbial communities sampled. The high diversity of the community was also reflected in the low coverage values estimated by Nonpareil (Table S1b), a tool that estimates coverage based on read redundancy [28].
Taxonomic assignments based on phylogenetic reconstructions of the housekeeping single-copy genes for each MAG revealed that the recovered genomes represented novel bacterial taxa at the genus level of Rhizobiales, Comamonadaceae, Mollicutes, Porphyromonadaceae, Actinobacteria, Flavobacteriaceae, Gammaproteobacteria, and a novel species of the Exiguobacterium genus (Firmicutes, Bacilli) (Table 1). The 14 MAGs (Table 1) collectively recruited 2.24–22.4% of the total metagenomic reads, depending on the sample considered (Fig. S6). The largest percentage of read matches were for sample K1-M1 from which the majority of the MAGs were recovered (Table 1).
Among the 14 identified population genomes, five were abundant only in one out of the five time samples for the K1 site, and remained in very low abundance or below the detection limit in the other time points (Fig. 2; 5 bottom rows including kal10.Por-kal14.Noc). Those non-persistent populations were typically characterized by low intra-population sequence diversity, as reflected by the observed high ANIr values ( > 99%) in the sample in which the population was detected (no samples were mixed; Fig. 2). The remaining nine MAGs represented persistent or reoccurring populations, and were detected throughout the 5 time points spanning 23 months, exhibiting at least 10 RPKM for two time points or more (Fig. 2, kal1.Rhi-kal9.Com). The persistent populations were characterized by ANIr values ranging from 97.58 to 99.42%, with an average of 99.18%. However, in all 14 cases ANIr never dropped below 97% (Fig. 2), suggesting that all populations studied represented assemblages showing levels of intra-population sequence diversity lower than that of an average named species [25] or the Prochlorococcus marinus and SAR-11 populations in the ocean [9, 29]. For instance, ANIr of abundant Prochlocococcus populations in Tara Ocean surface water metagenomes using the exact same methodology was often 95-96%, depending on the sample considered (data not shown).
Fig. 2.

Abundance and sequence diversity of the identified bacterial populations (MAGs). The leftmost panel presents the abundance of the MAGs (rows) through time in samples (columns) from the K1 site as normalized coverage (Reads per Kbp per Million Reads, or RPKM). The middle panel shows the sequence diversity of the populations measured by ANIr for each of the time points (represented by open circles) where MAGs were detected ( > 1 RPKM). The rightmost panel denotes the potential origin of the each population based on their detection in the K0 (with drainage areas mostly representing mountainous forest regions) and KL (representative site of the urban area) sites. Presence (and thus, origin) was defined as coverage of the population by at least 100 RPKM in at least two time points from either the K0 or KL sites (M20, M23)
We further hypothesized that higher temporal persistence of MAGs (coverages > 1 × ) would be associated with higher intra-population diversity, thus, less clonal populations. The underlying assumption is that such persistent populations represent autochthonous freshwater taxa that are highly ecologically successful in the lotic habitat (Kalamas River in this case). Thus, these populations would be characterized, in general, by longer evolutionary time since the last strong intra-population diversity sweep, and larger population sizes, allowing for mutations (diversity) to accumulate. In contrast, allochthonous taxa that only recently entered the lotic environment from soil or wastewater and thus, likely underwent a strong population bottleneck event during this transition and/or were short-lived in the lotic environment, would be characterized, on average, by more clonal populations. Consistent with our hypothesis, correlation analysis of the coefficient of variation in abundance across samples (i.e., persistence) with average ANIr revealed a significant (R = 0.73, p = 0.0052) linear correlation (Fig. 3). When populations with low genome completeness ( < 70%), and thus lower quality, were removed from the analysis, this correlation became even stronger (R = 0.83, p = 0.0053) (Fig. 3). Finally, our simulations showed that ANIr values were essentially independent of the relative abundance (coverage) of the population for coverage 1–2× or higher, which was the case for all populations and samples assessed above (Fig. S3). And, the coefficient of variation (persistence) was independent of the relative abundance of the population in the metagenome; thus, the correlations reported above were not merely due to differences in relative abundances of the populations.
Fig. 3.
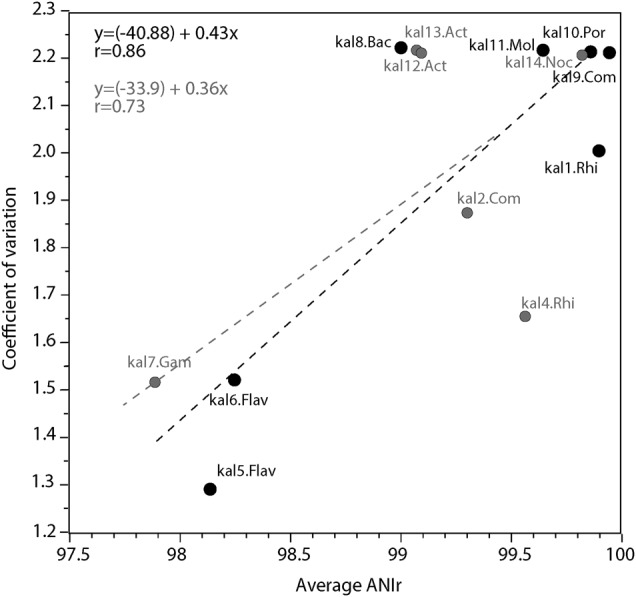
Relationship between sequence diversity and persistence through the time series of the populations studied. The coefficient of variation of the normalized relative abundance (measured by sequence coverage; y axis) shows a significant linear correlation with the estimated intra-population sequence diversity calculated as ANIr (x axis). The linear regression is shown for all the identified populations (gray) or for a selected subgroup representing bins with completeness > 70% (black)
Tracking the source of populations
To provide further support for the abovementioned conclusions, we next tested a related hypothesis, i.e., the more persistent populations should be detectable in the upstream, more pristine site (K0) since they represent more freshwater-autochthonous organisms, whereas several of the less persistent (more allochthonous and clonal) populations should be detectable only in the Lapsista ditch (KL) site. The abundance of the populations typically changed along the river, depending on the geographic distance between the samples and the season sampled (e.g., flow rate of water; Table S1b, Fig. S7), and largely agreed with our previous 16S-based findings from the same river [6]. During the first sampling in November 2012 (M1) populations that were abundant in K1 gradually decreased in abundance towards the estuary. Three months later (M4), a time at which the dam was opened and the water discharge increased (Table S1b), the identified populations exhibited similar abundances across sites, showing slight increases towards the estuary. Finally, in May (M7) some MAGs increased in K2, while others completely disappeared and reappeared in K3, and still others (Flavobacteriales) gradually increased from K1 to the estuaries. During May, water flow was relatively low compared to other time points (Table S1a), resulting in high heterogeneity between samples and probably, species sorting effects [30, 31].
To assess the origin of the populations, i.e., to identify whether the abundant populations originated from the Lapsista ditch (KL) or from the upstream site (K0), we focused on the June (M20) and September 2014 (M23) samples. These were the only time points during which we sampled from all three sites: K0, K1, and KL. Further, we focused on relatively abundant populations for this analysis for more robust assessment of intra-population genetic diversity, i.e., MAGs that exhibited > 1× coverage in one of the samples K1-M20 and/or K1-M23. From the nine MAGs that were present in June or September, we were able to determine the potential origin of five. Specifically, representatives of Flavobacteriales (kal5.Flav, kal6.Flav) and Gammaproteobacteria (kal7.Gam) likely originated from the more pristine site (minimal urban impact, Fig. 1) based on their presence in both K0 and K1 sites but complete absence in KL, while kal4.Rhi (Rhizobiales) and kal8.Bac (Bacilli) originated from inside the Lapsista ditch (Fig. 2). Kal1.Rhi, most likely a member of the class Alphaproteobacteria and probably the order Rhizobiales, and kal2.Com and kal3.Com (Burkholderiales, Comamonadaceae) were not detected in either K0 or KL sites, making it challenging to determine their potential origin. We hypothesize that these populations originated from the Lapsista ditch since they were completely absent from the upstream pristine site, which could not be otherwise easily explained due to the very short distance (997 m) between the two sites (K0 and K1) and the larger distance between K1 and KL (~25 Km). Other potential sources for the latter populations, which our sampling scheme could not appropriately assess, included contamination sources within the Lapsista ditch downstream of the KL sampling site, or surface runoff or local (unaccounted) events between K0 and K1. Consistent with our hypothesis above, however, the populations that were abundant in the K0 site showed lower ANIr (i.e., more diversity) than those originating from the KL site (97.81 vs. 99.11 ANI on average).
Temporal gene-content diversity of populations
Overall, between 904 and 5269 genes were identified in each MAG, with ~25–80% of these genes annotated by a function other than hypothetical (Table 1, Table S3). We sought to quantify the variability in gene content through time for each of the identified populations. For each time point, the absence of a gene was assessed by deviation (i.e., drop) in sequencing depth (coverage) from the MAG average as described in the Materials and Methods section. Since all our MAGs were constructed from specific sampling points and sites (Table 1), we focused on genes that were variable (e.g., lost) in subsequent times points rather than those initially absent and detected (acquired) later, although the gene loss and acquisition processes likely occurred at similar rates, since genome size represents a stable property of a bacterial species -as an average of all members of the species. Overall, gene-content variability varied among the 14 populations assessed, affecting from 4.17% of the total genes in the genome (kal5.Flav) up to 21.18% (kal4.Rhi), with an average around 10% (Table S2b). Amongst these variable genes, depending on the population considered, at least 34% (average 49.01%) were either not annotated or annotated as hypothetical or conserved hypothetical (Table S2b). Amongst the variable annotated genes, 12%–34% (average 27%) were short ( < 100 amino acids), a higher fraction than that of the total annotated genes in the genome (average 14%, Table S4).
The relative abundance of sequences encoding hypothetical proteins among variable genes was higher (t-test; p < 0.008; 34–75%) than the ratio of the hypothetical genes in the genome (20–33%). Similarly, the relative abundance of mobile elements (phages, transposases, CRISPRs) among variable genes varied from 2.48% to 5.13%, exceeding in all cases their respective percentages among the total genes in the genome (t-test; p < 0.002) (Table S2b, S4). While the majority of variable genes (i.e., those encoding hypothetical proteins) were not predicted to have known functional consequences for the population (Table S4), some genes were likely related to potentially important metabolic properties such as carbohydrate utilization or adaptation to changing environmental conditions (e.g., temperature and sun radiation). For instance, for kal5.Flav and kal6.Flav, proteins related to protein biosynthesis and membrane receptors and transportation were prevalent amongst the gene categories that were variable (Table S4). Conspicuous among the cases examined, we detected the complete absence of proteins involved in photosystem II in MAG kal4.Rhi (unique in this MAG amongst all MAGs, Fig. 4), including the reaction center and light harvesting proteins, in November but not in the June samples, when sun radiation (and hence photosynthesis potential) is at its highest (Fig. 4, Table S4,S5). Similarly, in the same population, proteins involved in nitrogen limitation, and beta-carotene metabolism e.g., poly(3-hydroxyalkanoate) polymerase, also appeared only in June.
Fig. 4.

Example of intra-population gene-content variation through time. a Genome-wide coverage profiles of the identified population kal4.Rhi in two time points (M1 and M20, representing a 20 month time span). The coverage was estimated based on high-identity metagenomic reads mapping to the genes of the reference genome sequence. For each coverage profile, the absence of a particular gene (no reads mapping) is shown as a blue vertical line, the width of which is proportional to the length of the region with the variable genes. b Comparative analysis of the PSII operon of the riverine population kal4.Rhi with the PSII operon from the reference closed genome of Rhodopseudomonas palustris strain HaA2. GDR Geranylgeranyl diphosphate reductase, h.p. hypothetical protein, R.C.M/L Photosystem II Reaction center protein M/L, LHP Light harvesting protein. Note that the genomic region encoding the PSII operon was present in the population genome during the last time point but was absent during the first time point
Collectively, these results revealed that at least some of the populations were highly dynamic in terms of gene content, even within the relatively short period of time spanned by our samples. While we are currently unable to distinguish between the possible mechanisms for the gene-content variability observed, i.e., gene loss during the time sampled vs. growth of latent sub-populations with substantial gene-content differences in the river or its feeding waters, it should be noted that the intra-population sequence diversity (ANIr values) did not change significantly between the time points where gene variation was observed, and the populations were abundant ( > 7 × ) and robustly detected (as also proved from our simulation analysis) during these time points. Thus, the “same” population was detected between the time points in terms of phylogenetic affiliation or assembled (consensus) genome sequence, and it showed substantial intra-population gene-content diversity, independent of the process(es) that underlie the gene-content variation.
Only within kal2.Com and kal3.Com there were substantial differences in ANIr values ( > 0.7 percent units) observed between M1 and M20 samples (Fig. 2), and further investigation was performed in order to determine whether this was due to increased intra-population diversity or to the presence of distinct populations. Phylogenetic reconstruction among overlapping reads encoding the same marker gene (encoding the proteins FtsY, HisS, or FMT) or assembled partial genomes from each of the corresponding metagenomes provided evidence for the prevalence of different abundant genotypes between November and June, with the ANI values among the genotypes of these two populations being ~98%. (Supplementary information, Fig. S8). Thus, the most parsimonious explanation in these two cases is that the gene-content variation observed was due to sub-population or strain replacement, and these results were not artifacts of our methodology or assembly (see also below).
Discussion
In a previous study of the Kalamas River [6], we showed that hydrological and climatic factors (Table S1) largely account for the highly dynamic changes in microbial community composition in the river, and hypothesized about the allochthonous origin of several of the community members sampled. Analysis of additional sites and time points at a finer level, i.e., that of individual sequence-discrete populations, as part of the present study confirmed and expanded upon these previous findings (Fig. 2; and Supplementary information).
In particular, several of the sequence-discrete populations studied, but not all, were present at the same sampling point even after a time period of 23 months, showed similar intra-population sequence diversity (ANIr values) between sampling points, and were sequence-discrete compared to co-occurring populations (Fig. 2). Most of these populations were not clonal but showed substantial intra-population sequence diversity and were highly dynamic in terms of gene-content, which could change substantially even within a few months or between the seasons of a year (Table S2b, S4). This level of gene-content variation is comparable to what has been revealed by comparison of isolate genomes of the same named bacterial species, e.g., 5–10% gene-content difference, on average, for genomes showing > 98% ANI [32, 33]. Yet, the evolutionary time represented by isolates is much longer than that sampled here, revealing that bacterial species may be even more dynamic functionally than previously thought. Despite the highly dynamic gene content and regardless of the underlying mechanisms for it (e.g., gene loss or strain/genotype replacement), the populations remained discrete compared to other microbial community members, re-occurred, and were traceable over time and space, consistent with the interpretation that these populations represent species-like units. These results also agreed with those reported previously on other ecosystems that the ANIr values of the majority of the populations of a microbial community did not change over time or space [9, 10, 14, 34]. If these populations were equated to species, then our results would also favor an ecological definition of bacterial species.
One possible explanation for the temporal gene-content variability observed is strain/sub-population replacement. That is latent sub-populations, which showed substantial gene-content differences but did not differentiate enough in terms of sequence diversity among themselves to be detectable by our methods [e.g., to provide uneven recruitment plots or different ANIr values [9]], differentially grew in abundance between our sampling points within the river, or -most likely due to the short retention time of the river- in the feeding waters. The alternative explanation or mechanism is that of real deletion (or acquisition) of genes from the genome; for instance, through intra-genomic recombination. Analysis of recently divergent Vibrio cyclitrophicus genomes, showing > 98% ANI among themselves similar to members of the sequence-discrete population studied here, has indicated that genes can recombine and sweep through the population very rapidly [12]. Obtaining genomes of individual cells of the populations over time will be necessary, however, to distinguish between the two distinct scenarios in the future.
Further, the level of intra-population diversity (e.g., ANIr) was positively correlated with the coefficient of variation of the abundance of the population across samples (Fig. 3), revealing that persistent populations exhibit higher intra-population sequence variation. While these results are clearly preliminary and more populations and data points should be examined before more robust conclusions can emerge, it is important to note that the correlation became stronger when low quality genomes were removed from the analysis, indicating that the correlation might be even stronger with more, high-quality genomes and samples available. Thus, our findings indicated that examining sequence-discrete populations and their level of intra-population ANIr, might represent a promising approach to determine endemic/autochthonous (e.g., higher intra-population diversity) vs. allochthonous (e.g., typically lower intra-population diversity) species in such a dynamic lotic system like those found in the Kalamas River. Tracking of sequence-discrete populations supported this conclusion, at least in part. For example, (presumptive) freshwater-allochthonous or more clonal populations tended to originate from the Lapsista ditch (kal4.Rhi, kal8.Bac; Fig. 2) while freshwater-autochthonous populations were typically detected in the upstream more pristine site (kal5.Flav, kal6.Flav, kal8.Gam; Fig. 2).
In contrast, our own previous study using the origin of the 16S rRNA gene to study the same riverine system for the same purposes [6] met with mixed success due to the high sequence conservation of 16S rRNA gene sequences (see also below). In particular, the origin of the population cannot often be determined with confidence based on detection of the organism(s) that provides the best 16S rRNA gene match from a particular location because similar matches are provided by multiple organisms that thrive in different habitats and have highly similar 16S rRNA gene sequences [6]. Further, similar, yet distinct populations can be grouped under the same 16S-based OTU (97% identity cut-off) [8] as was the case for the two Comamonadaceae populations kal2.Com and kal3.Com. Even though the specific 16S sequence of each of the two Comamonadaceae populations was not assembled together with the MAG, all 16S reads that were assignable to this family (supplementary information) showed > 98% identity to each other (i.e., belonged to the same OTU). Thus, the 16S rRNA gene sequences would not be adequate for assessing the abundance, presence and origin of these two Comamonadaceae populations. These limitations were much less pronounced, if existing at all, at the sequence-discrete individual population level.
While most temporal gene-content differences were dominated by hypothetical proteins, indicating that they were likely functionally neutral for the populations, a few exceptions were also noted. Most notably, the Rhizobiales population (kal4.Rhi) was characterized by the absence of proteins involved in photosystem II (PSII), including reaction center and light harvesting proteins, which were present only in June but not in November samples (Fig. 4, Table S4). The presence of these genes was accompanied at all time points by genes encoding the different subunits of NADH quinone oxidoreductase responsible for the reversed translocation of electron between NAD+ and NADH; the latter genes were still detectable in the genome when PSII-related genes were lost (Fig. 4, Table S4). The proton translocating NADH quinone oxidoreductase, also known as complex I, is widespread across bacteria with different metabolic lifestyles such as obligate aerobes, strict anaerobes, lithotrophs, anoxic phototrophs, and methylotrophs [35]. In the case of phototrophs, the presence of complex I, along with the PSII reaction center, is required for solar energy conservation since it creates a proton motive force in order to synthesize NADH from quinols. However, in the absence of the PSII apparatus, the complex I proteins could be used in another energy-generating pathway by transferring electrons from NADH to quinones. Rhopseudomonas palustris, although phylogenetically distant to kal4.Rhi, is an example of such a metabolically versatile bacterium that can switch between aerobic and anaerobic respiration and anaerobic phototrophy in the presence of complex I proteins and PSII [36]. Comparisons between the PSII operon of R. palustris HaA2 strain and kal4.Rhi PSII operon revealed amino-acid identities ranging from 37% to 67%, while operon organization was slightly different mainly regarding the genes adjacent to genes related to reaction center (Fig. 4). Collectively, the changes in gene content in kal4.Rhi between M1 and M20 imply the presence of multiple genotypes of genetically similar populations that thrive in different environmental conditions such as light intensity in this case. These different genotypes could be the result of horizontal gene transfer that has been shown to be common in purple photosynthetic bacteria and especially in proteins responsible for photosynthetic reaction centers M and L of PSII [37], which represent the same functions as the variable genes identified in our case (Fig. 4). We find it remarkable that distinct genotypes, with such substantial metabolic and energy-generating differences, could be members of the same population and thrive in different seasons, like the kal4.Rhi genotypes appear to do. Similar results have been recently observed based on genome sequencing of Erythrobacter sp. isolates, where closely related isolates (ANI > 98%) did not share PSII genes [38]. Therefore, our analysis indicated that this pattern might not be uncommon in the environment. Whether these PSII genes were phage-encoded, similar to psbA genes in cyanophages [39], remains unclear, although kal4.Rhi variable genes were highly enriched in mobile elements related to transposases and phage integrases (4.65% vs. 3.08%), compared to the total genes in the MAG,.
Mobile elements and hypothetical proteins were enriched amongst variable genes, by two fold or more, compared to the total genes in the genome for all populations studied. These results corroborate with previous findings showing that the majority of genes that differ between closely related strains involve hypothetical proteins and mobile elements that typically represent ephemeral invaders of the genome [40], and are shorter in length on average [41].
While it is possible that some of the intra-population gene-content diversity observed was likely the result of technical limitations such as mis-assembly or chimeric reference MAG genome sequences used in the recruitment plots, we believe that the effects of these factors on our conclusions were small, in general, e.g., likely affecting fewer than 20% of the genes detected as variable. Firstly, all genome sequences used as references to represent a population were recovered from a single sample, and were not the result of co-assembly, which is more likely to combine distinct populations into one and therefore obscure sample-specific genetic diversity. Further, the same reference genome sequences (recovered from a single sample) were assessed against reads from all samples, without mixing reads from different samples, which circumvented any sample-specific biases or sequencing artifacts affecting genome recovery. All populations that were used for gene-content analysis had at least 7× coverage in the corresponding samples, which ensured that most of the genes present in the genome were sequenced at enough depth to be recovered by metagenomic reads (Supplementary information). This was also consistent with the simulations and subsampling analyses performed here. In particular, we tested the robustness of our methodology to detect variable gene by spiking reads from an E. coli genome into an in-silico synthesized (mock) dataset or a Kalamas metagenome and used another E. coli genome, with known gene-content differences compared to the former E. coli, as reference sequence in recruitment plots. The genes of the reference identified as variable in the metagenomes by our approach matched closely ( < 10% difference) the expected gene-content differences between the two genomes as long as the coverage of the reference genome by the spiked in reads were 1× or more (lower coverage level provided for more variable results; see Fig. S3). Similar analysis but with Kalamas metagenomes that were sub-sampled in order to provide different levels of coverage (abundances) of the target population showed that the number of false positive or false negative variable genes remained 20% or less of total positives/negatives when coverage of the population was 7× or higher (Fig. S2). Therefore, our methodology likely did not overestimate the number of genes that were predicted as variable between different time points. Finally, visual inspection of assembly and read recruitment plots for the populations studied did not reveal any inconsistencies that could be attributed to sequencing and library creation artifacts such as consistently low coverage of high or low G + C% regions for the genome (Fig.S1). It is also important to note that our approach assessed only gene absence, not gene gain, in order to avoid complications related to horizontal acquisition of genes in specific samples occurring since the onset of our sampling or the presence of recently transferred genes in non-target populations (which would provide false positive matching reads).
Overall, we were able to identify the potential origin of different populations from different sites based on the level of intra-population ANIr values and their correlation to relative abundance, and to follow specific populations along the river. These results showed that the approach outlined here would be more reliable and robust, although more time consuming and demanding in terms of bioinformatics analysis, for microbial source tracking compared to alternative approaches such as PCR analysis of specific genetic markers. If more sampling points and high-quality genome sequences become available in the near future, so that a more comprehensive catalog of autochthonous (freshwater) vs. allochthonous organisms is created, high-resolution, robust microbial source tracking can be realized.
Electronic supplementary material
Acknowledgements
This research was supported, in part, by the US National Science Foundation (award DEB 1241046 to KTK), the European Union (European Regional Development Fund- ERDF), and Greek national funds through the Operational Program “THESSALY- MAINLAND GREECE AND EPIRUS-2007-2013” of the National Strategic Reference Framework (NSRF 2007–2013) (2012–2015). We are grateful to the Management Body of Straits and Estuaries of the Rivers Acheron and Kalamas and to Theofilos Vanikiotis for providing helpful information on the sampling area as well as sampling facilities, and to an anonymous reviewer for providing useful suggestions regarding the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0307-6) contains supplementary material, which is available to authorized users.
References
- 1.Ghai R, Rodriguez-Valera F, McMahon KD, Toyama D, Rinke R, Cristina Souza de Oliveira T, et al. Metagenomics of the water column in the pristine upper course of the Amazon river. PLoS ONE. 2011;6:e23785. doi: 10.1371/journal.pone.0023785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Read DS, Gweon HS, Bowes MJ, Newbold LK, Field D, Bailey MJ, et al. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 2015;9:516–26. doi: 10.1038/ismej.2014.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Portillo MC, Anderson SP, Fierer N. Temporal variability in the diversity and composition of stream bacterioplankton communities. Environ Microbiol. 2012;14:2417–28. doi: 10.1111/j.1462-2920.2012.02785.x. [DOI] [PubMed] [Google Scholar]
- 4.Savio D, Sinclair L, Ijaz UZ, Parajka J, Reischer GH, Stadler P, et al. Bacterial diversity along a 2600 km river continuum. Environ Microbiol. 2015;17:4994–5007. doi: 10.1111/1462-2920.12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Rossum T, Peabody MA, Uyaguari-Diaz MI, Cronin KI, Chan M, Slobodan JR, et al. Year-long metagenomic study of river microbiomes across land use and water quality. Front Microbiol. 2015;16:1405. doi: 10.3389/fmicb.2015.01405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meziti A, Tsementzi D, Ar. Kormas K, Karayanni H, Konstantinidis KT. Anthropogenic effects on bacterial diversity and function along a river-to-estuary gradient in Northwest Greece revealed by metagenomics. Environ Microbiol. 2016;18:4640–52. doi: 10.1111/1462-2920.13303. [DOI] [PubMed] [Google Scholar]
- 7.Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, et al. Opinion: re-evaluating prokaryotic species. Nat Rev Microbiol. 2005;3:733–9. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez RLM, Castro JC, Kyrpides NC, Cole JR, Tiedje JM, Konstantinidis KT. How much do rRNA gene surveys underestimate extant bacterial diversity? Appl Environ Microbiol. 2018;84:e00014–8. doi: 10.1128/AEM.00014-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konstantinidis KT, DeLong EF. Genomic patterns of recombination, clonal divergence and environment in marine microbial populations. ISME J. 2008;2:1052–65. doi: 10.1038/ismej.2008.62. [DOI] [PubMed] [Google Scholar]
- 10.Caro-Quintero A, Konstantinidis KT. Bacterial species may exist, metagenomics reveal. Environ Microbiol. 2012;14:347–55. doi: 10.1111/j.1462-2920.2011.02668.x. [DOI] [PubMed] [Google Scholar]
- 11.Schneider D, Duperchy E, Depeyrot J, Coursange E, Lenski RE, Blot M. Genomic comparisons among Escherichia coli strains B, K-12, and O157:H7 using IS elements as molecular markers. BMC Microbiol. 2002;2:18. doi: 10.1186/1471-2180-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szabó G, et al. Population genomics of early events in the ecological differentiation of bacteria. Science. 2012;336:48–51. doi: 10.1126/science.1218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh S, Caro-Quintero A, Tsementzi D, DeLeon-Rodriguez N, Luo C, Poretsky R, et al. Metagenomic insights into the evolution, function, and complexity of the planktonic microbial community of Lake Lanier, a temperate freshwater ecosystem. Appl Environ Microbiol. 2011;77:6000–11. doi: 10.1128/AEM.00107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsementzi D, Poretsky R, Rodriguez-R LM, Luo C, Konstantinidis KT. Evaluation of metatranscriptomic protocols and application to the study of freshwater microbial communities. Environ Microbiol Rep. 2014;6:640–55. doi: 10.1111/1758-2229.12180. [DOI] [PubMed] [Google Scholar]
- 15.Johnston ER, Rodriguez-R LM, Luo C, Yuan MM, Wu L, He Z, et al. Metagenomics reveals pervasive bacterial populations and reduced community diversity across the Alaska Tundra ecosystem. Front Microbiol. 2016;7:579. doi: 10.3389/fmicb.2016.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bendall ML, Stevens SL, Chan LK, Malfatti S, Schwientek P, Tremblay J, et al. Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. ISME J. 2016;10:1589–601. doi: 10.1038/ismej.2015.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox MP, Peterson DA, Biggs PJ. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinforma. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 19.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucl Acids Res. 2010;38:e132. doi: 10.1093/nar/gkq275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu YW, Tang YH, Tringe SG, Simmons BA, Singer SW. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2:26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-R LM, Gunturu S, Harvey WT, Rosselló-Mora R, Tiedje JM, Cole JR, et al. The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018;46:W282–8. doi: 10.1093/nar/gky467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016;4:e1900v1. [Google Scholar]
- 24.Luo C, Rodriguez-R LM, Konstantinidis KT. MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 2014;42:e73. doi: 10.1093/nar/gku169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 26.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo C, Tsementzi D, Kyrpides NC, Konstantinidis KT. Individual genome assembly from complex community short-read metagenomic datasets. ISME J. 2012;6:898–901. doi: 10.1038/ismej.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez-R LM, Konstantinidis KT. Nonpareil: a redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics. 2014;30:629–35. doi: 10.1093/bioinformatics/btt584. [DOI] [PubMed] [Google Scholar]
- 29.Tsementzi D, Wu J, Deutsch S, Nath S, Rodriguez-R LM, Burns AS, et al. SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature. 2016;536:179–83. doi: 10.1038/nature19068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crump BC, Amaral-Zettler LA, Kling GW. Microbial diversity in arctic freshwaters is structured by inoculation of microbes from soils. ISME J. 2012;6:1629–39. doi: 10.1038/ismej.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeglin LH. Stream microbial diversity in response to environmental changes: review and synthesis of existing research. Front Microbiol. 2015;18:454. doi: 10.3389/fmicb.2015.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci USA. 2005;102:2567–72. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, et al. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43:6761–71. doi: 10.1093/nar/gkv657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia SL, Stevens SLR, Crary B, Martinez-Garcia M, Stepanauskas R, Woyke T, et al. Contrasting patterns of genome-level diversity across distinct co-occurring bacterial populations. ISME J. 2018;12:742–55. doi: 10.1038/s41396-017-0001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spero MA, Aylward FO, Currie CR, Donohue TJ. Phylogenomic analysis and predicted physiological role of the proton-translocating NADH:quinone oxidoreductase (Complex I) across bacteria. mBio. 2015;6:e00389–15. doi: 10.1128/mBio.00389-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larimer FW, Chain P, Hauser L, Lamerdin J, Malfatti S, Do L, et al. Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat Biotech. 2004;22:55–61. doi: 10.1038/nbt923. [DOI] [PubMed] [Google Scholar]
- 37.Nagashima KVP, Hiraishi A, Shimada K, Matsuura K. Horizontal transfer of genes coding for the photosynthetic reaction centers of purple bacteria. J Mol Evol. 1997;45:131–6. doi: 10.1007/PL00006212. [DOI] [PubMed] [Google Scholar]
- 38.Zheng Q, Lin W, Liu Y, Chen C, Jiao N. A comparison of 14 erythrobacter genomes provides insights into the genomic divergence and scattered distribution of phototrophs. Front Microbiol. 2016;7:984. doi: 10.3389/fmicb.2016.00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006;4:e234. doi: 10.1371/journal.pbio.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Konstantinidis KT, Ramette A, Tiedje JM. The bacterial species definition in the genomic era. Philos Trans R Soc Lond B Biol Sci. 2006;361:1929–40. doi: 10.1098/rstb.2006.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akhter S, Aziz RK, Edwards RA. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012;40:e126. doi: 10.1093/nar/gks406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.