

Abstract
Myotoxicity is a significant factor contributing to the poor adherence and reduced effectiveness in the treatment of statins. Genetic variations and high drug plasma exposure are considered as critique causes for statin-induced myopathy (SIM). This study aims to explore the sequential influences of rosuvastatin (RST) pharmacokinetic and myopathy-related single-nucleotide polymorphisms (SNPs) on the plasma exposure to RST and its metabolites: rosuvastatin lactone (RSTL) and N-desmethyl rosuvastatin (DM-RST), and further on RST-induced myopathy. A total of 758 Chinese patients with coronary artery disease were enrolled and followed up SIM incidents for 2 years. The plasma concentrations of RST and its metabolites were determined through a validated ultra-performance liquid chromatography mass spectrometry method. Nine SNPs in six genes were genotyped by using the Sequenom MassArray iPlex platform. Results revealed that ABCG2 rs2231142 variations were highly associated with the plasma concentrations of RST, RSTL, and DM-RST (Padj < 0.01, FDR < 0.05). CYP2C9 rs1057910 significantly affected the DM-RST concentration (Padj < 0.01, FDR < 0.05). SLCO1B1 rs4149056 variant allele was significantly associated with high SIM risk (OR: 1.741, 95% CI: 1.180–2.568, P = 0.0052, FDR = 0.0468). Glycine amidinotransferase (GATM) rs9806699 was marginally associated with SIM incidents (OR: 0.617, 95% CI: 0.406–0.939, P = 0.0240, FDR = 0.0960). The plasma concentrations of RST and its metabolites were not significantly different between the SIM (n = 51) and control groups (n = 707) (all P > 0.05). In conclusion, SLCO1B1 and GATM genetic variants are potential biomarkers for predicting RST-induced myopathy, and their effects on SIM are unrelated to the high plasma exposure of RST and its metabolites.
Keywords: genetic polymorphism, rosuvastatin, metabolites, plasma concentration, myopathy
Introduction
As a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, rosuvastatin (RST) is one of the most globally popular cholesterol-reducing drugs [1]. RST can decrease the risk of cardiovascular disease during primary and secondary prevention [2]. However, RST induces a class-wide adverse myotoxicity effect, which has become the leading factor for treatment discontinuation, switching, or non-adherence [3–5]. High plasma exposure of statin or its metabolites and genetic factor play crucial roles in statin-induced myopathy (SIM) [6–8].
Patients with SIM presented significantly higher circulation exposure to atorvastatin metabolites than patients without SIM [8]. Atorvastatin lactone is one of metabolites, which has potential toxicity for skeletal muscle cells in vitro [9]. RST-5S lactone (RSTL) and N-desmethyl RST (DM-RST) are two known metabolites, and DM-RST is biotransformed by the isoenzyme CYP2C9 [10]. The effects of these two metabolites and CYP2C9 gene on RST-induced myopathy remain unclear.
Transporters play an important role in the disposition of RST owing to its low passive membrane permeability [11]. Many studies have revealed that SLCO1B1 521T>C (rs4149056) contributed to SIM [12–14]. This variant allele is also associated with the increased plasma exposure of statins [15, 16], which is considered a causative factor of SIM [7, 16, 17]. However, most studies focused on simvastatin, atorvastatin, or a combination of statins, and only few were specifically conducted for RST. Compared with that of SLCO1B1 521T>C, the variation of ABCG2 is a potentially crucial heredity factor in the pharmacokinetic of RST for East Asians because of the differences in drug property and patients’ race [18–20]. Therefore, further investigation is needed to confirm whether or not the genic variations of SLCO1B1, ABCG2, or both influence the RST-induced myotopathy in Chinese people.
Glycine amidinotransferase (GATM) gene is functionally correlated with SIM [21]. GATM G>A (rs9806699) was discovered by a genome-wide association study as a possible genetic marker for the decreased risk of SIM [21]. However, this single-nucleotide polymorphism (SNP) failed to replicate this association in an independent sample and a case–control study [22, 23]. Nonetheless, these studies were performed on Caucasians who received statins. The influence of this locus on SIM in Asians has not been reported.
In the present study, we assessed the effects of pharmacokinetic-related and GATM SNPs on the systemic exposure to RST and its metabolites, and subsequently on the RST-related myopathy in Chinese people receiving RST therapy.
Materials and methods
Ethics statement
This study was approved by the Medical Ethical Review Committee of Guangdong General Hospital and conducted in accordance with the Declaration of Helsinki. Informed consents were obtained from all participants in this study.
Study design
Nine polymorphisms were selected from five candidate genes that were potentially associated with the pharmacokinetic of RST and a functional SNP associated with SIM. These SNPs were: ABCG2 421C>A (rs2231142), ABCG2 rs2199936, ABCB1 rs1045642, SLCO1B1 521T>C (rs4149056), SLCO1B1 388A>G (rs2306283), SLCO1B1 rs4363657, SLCO1B3 rs7311358, CYP2C9 rs1057910, and GATM rs9806699.
As an index of RST and its metabolites, the Css/D (dose-adjusted steady-state plasma concentrations) was used as a dependent variable. This ratio was defined as the drug concentration per 10 mg daily dosage (ng/mL per 10 mg).
Patients
Chinese Han patients with coronary artery disease who have ingested RST for more than 1 week were prospectively recruited between January 2010 and December 2013 from Guangdong General Hospital. Patients’ baseline information, including demographics, medical history, biochemical measurements, and medication was obtained from the database of the hospital.
The exclusion criteria were as follows: (1) pretreatment with other statins in 2 weeks; (2) age ≥80 years; (3) renal insufficiency (defined as serum CREA concentration is thrice greater than the normal upper limit (345 μmol/L), renal transplantation, or dialysis); (4) liver insufficiency (defined as serum alanine aminotransferase (ALT) concentration is thrice greater the normal upper limit (120 U/L), or a cirrhosis diagnosis); (5) advanced cancer.
Blood samples were obtained from each eligible patient in the morning at 10–12 h after taking RST and collected in EDTA-coated tubes. The plasma and the blood cells were separated in 2 h by centrifugation at 3000 r/min for 10 min at 4 °C and then stored at –80 °C until usage.
Endpoints
Follow-up information was collected through inpatient and outpatient hospital visits and telephone contacts with the patients until December 2015. During each follow-up assessment (every 6 months), the participants were questioned about any uncomfortable muscle symptoms, including myalgia, weakness, stiffness, spasms, or twitches; their RST medication; and possible interference factors of SIM assessment. These data were recorded for all of the enrolled patients.
SIM was defined based on the patients’ subjective sense of muscular pain and creatine kinase (CK) elevation. The considered musculoskeletal effects were new and inexplicable muscle-related symptoms, including myalgia, weakness, stiffness, spasms, or twitches (irrespective of CK values), rhabdomyolysis, and inexplicable CK elevations of over four times than normal upper limit (irrespective of symptoms) [24, 25].
Among the 975 patients, the following were excluded: 51 patients who were older than 80 years, 10 patients who experienced renal insufficiency, 18 patients who had liver insufficiency, 41 patients whose plasma concentrations were lower than the limit of detection, and 97 patients who provided incomplete information on SIM events during follow up. Finally, 758 eligible patients were included in the analysis.
Determination of RST and its metabolites concentrations in plasma
A sensitive ultra-performance liquid chromatography–tandem mass spectrometry (UPLC–MS/MS) assay was developed and validated for the simultaneous quantification of RST, RSTL, and DM-RST in human plasma. Liquid–liquid extraction by using ethyl acetate was adopted to extract three analytes and the internal standard (carbamazepine) from 200 μL buffered plasma (adding 100 μL ammonium acetate of pH = 4.0–100 μL human plasma). The analytes were chromagraphically separated by an Acquity UPLC HSS T3 column (3.0 mm × 100 mm, 1.8 µm) with 0.1% formic acid and a gradient of 30%–85% acetonitrile at a flow rate of 0.30 mL/min. Mass detection was performed with a Waters Xevo TQ-S triple–quadrupole mass spectrometer under positive electrospray ionization mode. The responses of RST, RSTL, and DM-RST were optimized at m/z 482.1 → 258.1, m/z 464.1 → 270.1, m/z 468.0 → 258.0, respectively.
Genotyping
DNA was extracted by using a DNA automatic extractor (TGuide M16 Systems, Tiangen, China). The quality control of the DNA was based on the absorbance ratio (A260/A280: 1.8–2.0) and the concentrations were determined by using NanoDrop 2000. Nine SNPs were genotyped by using the Sequenom MassArray technology platform (Sequenom, CA, USA).
Statistical analysis
All data analyses were performed by using SAS 9.4 (SAS Institute, Cary, NC, USA). Categorical data were presented as percentages, and continuous variables were expressed as mean ± SD. The concentrations of RST, RSTL, DM-RST, serum ALT, aspartate aminotransferas (AST), triglyceride, and lipoprotein(a) levels were logarithmically transformed to normalize their distributions. Haploview 4.2 was used to determine the deviation from the Hardy–Weinberg equilibrium. Linkage disequilibrium analysis was performed online by using SHEsis (http://analysis.bio-x.cn). Linear regression analysis was adopted to evaluate the influences of the genotype and clinical baseline characteristics on the plasma exposure of RST and its two metabolites. Logistic regression analysis was used to assess the effects of the baseline features and genotypes on SIM incidents. P values <0.05 were considered statistically significant. The FDR was controlled by using the SAS PROC MULTTEST with the FDR option to correct multiple comparisons in the genetic factors analysis. P values of <0.05 were considered statistically significant, whereas FDR was controlled at 0.05.
Results
Effects of genotypes on the plasma exposure of RST and its metabolites
First, we analyzed the clinical baseline characteristics and their influences on the Css/D of RST and its metabolites. Univariate linear regression analysis revealed the following: (1) plasma AST levels and combination with angiotensin-converting enzyme inhibitors were associated with the Css/D of RST; (2) a high creatinine (CREA) level was correlated with a higher exposure to RSTL; and (3) age and heart failure could significantly affect the Css/D of DM-RST (Table S1). To exclude the effects of these clinical baseline features on the Css/D of RST, RSTL, and DM-RST, we used the factors that significantly influenced the drug concentrations in the univariate linear regression analysis as the covariates in the following analysis.
All allelic distributions in our study conformed to the Hardy–Weinberg equilibrium (P > 0.05). All of the allelic frequencies were close to the reference data of 1000G population (http://www.ncbi.nlm.nih.gov/snp/). ABCG2 rs2199936 was highly linked with rs2231142 (D′ = 0.996, r2 = 0.993). Then only the locus of ABCG2 rs2231142 was included in following analysis. The mean Css/D of RST and its metabolites were significantly higher in the subjects carrying the ABCG2 421A than in non-carriers of this allele. The effects of this allele remained significant after being adjusted by the baseline characteristics and false discovery rate (FDR) (Padj < 0.01, FDR < 0.05). The results are listed in Table 1 and Fig. 1.
Table 1.
Genotypes and their effects on the plasma exposure of RST and its metabolites
| Gene | SNP | Genotype | n (%) | Plasma RST concentration (ng/mL per 10 mg) | Plasma RSTL concentration (ng/mL per 10 mg) | Plasma DM-RST concentration (ng/mL per 10 mg) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Concentration | P value | P valuea | FDR | Concentration | P value | P valueb | FDR | Concentration | P value | P valuec | FDR | ||||
| ABCG2 | rs2231142 | CC | 359 (47.55) | 2.78 ± 3.59 | 1.0 × 10-4 | 1.2 × 10-4 | 0.0010 | 0.38 ± 0.49 | 1.3 × 10-5 | 1.0 × 10-5 | <0.0001 | 0.38 ± 0.63 | 4.0 × 10-6 | 6.0 × 10-6 | <0.0001 |
| AC | 324 (42.91) | 3.89 ± 4.03 | 0.50 ± 0.51 | 0.52 ± 0.67 | |||||||||||
| AA | 72 (9.54) | 4.73 ± 3.88 | 0.67 ± 0.64 | 0.62 ± 0.63 | |||||||||||
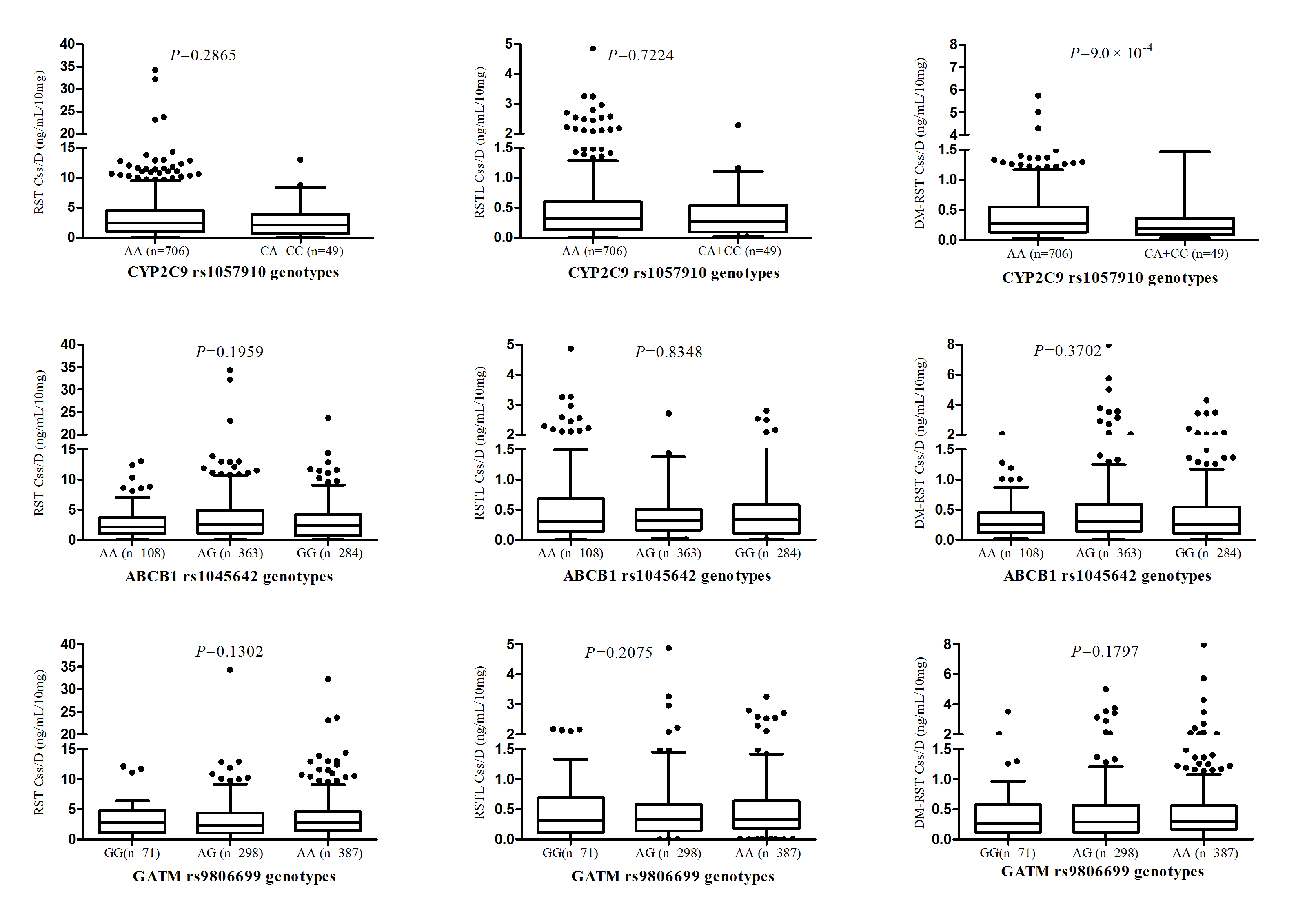
| ABCB1 | rs1045642 | AA | 108 (14.30) | 2.89 ± 2.98 | 0.1959 | 0.1471 | 0.2354 | 0.40 ± 0.42 | 0.8348 | 0.7974 | 0.8399 | 0.36 ± 0.37 | 0.3702 | 0.5224 | 0.8536 |
| AG | 363 (48.08) | 3.78 ± 4.26 | 0.48 ± 0.58 | 0.52 ± 0.77 | |||||||||||
| GG | 284 (37.62) | 3.22 ± 3.58 | 0.44 ± 0.48 | 0.43 ± 0.57 | |||||||||||
| SLCO1B1 | rs4149056 | TT | 568 (75.53) | 3.21 ± 3.77 | 0.0184 | 0.0247 | 0.0988 | 0.45 ± 0.53 | 0.3891 | 0.5621 | 0.8253 | 0.45 ± 0.58 | 0.4932 | 0.5763 | 0.8536 |
| CT | 130 (17.29) | 4.45 ± 4.39 | 0.48 ± 0.51 | 0.55 ± 0.94 | |||||||||||
| CC | 54 (7.18) | 3.59 ± 3.92 | 0.46 ± 0.44 | 0.42 ± 0.56 | |||||||||||
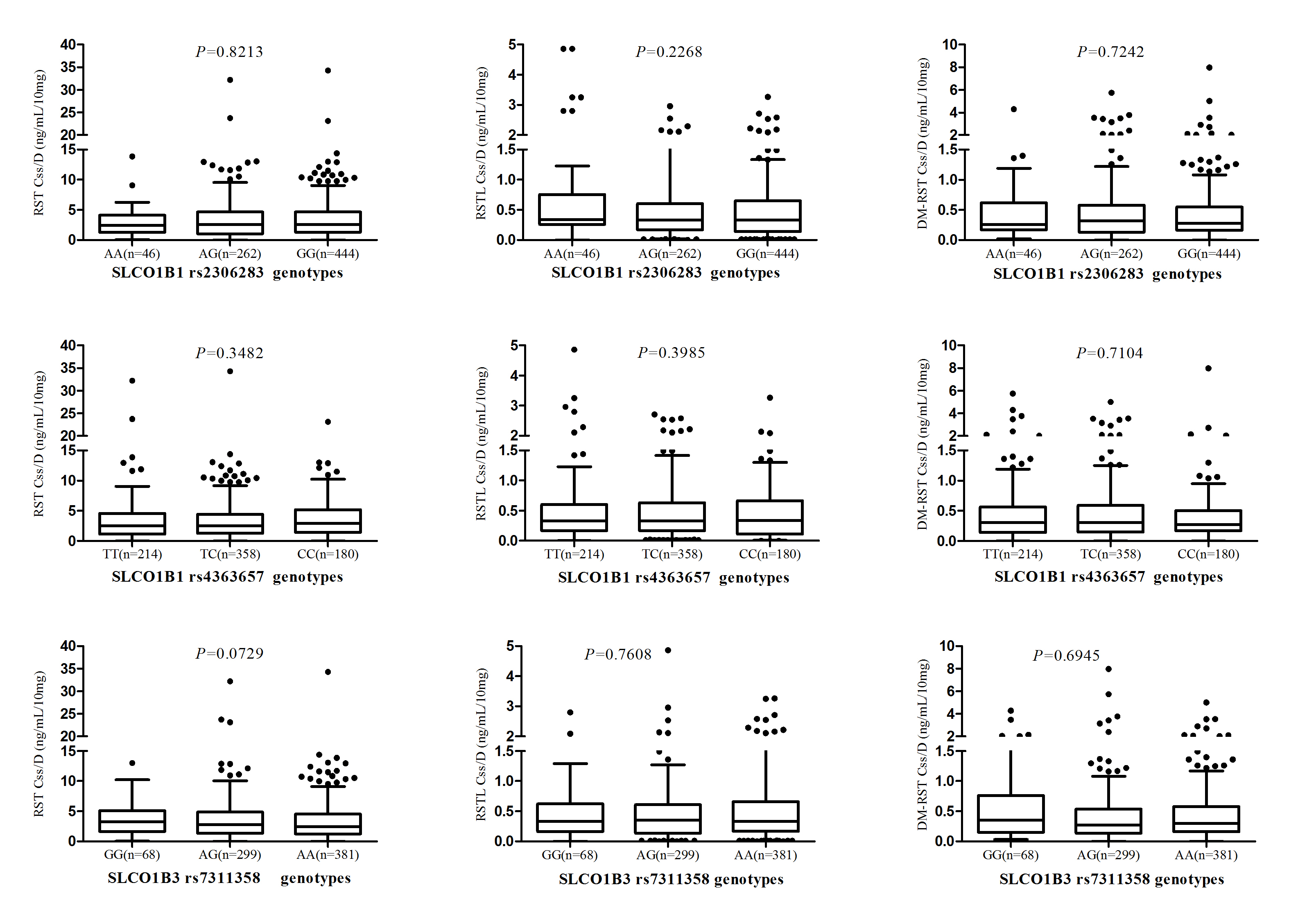
| rs2306283 | AA | 46 (6.07) | 3.71 ± 4.16 | 0.8213 | 0.8517 | 0.8517 | 0.70 ± 0.97 | 0.2268 | 0.2436 | 0.6496 | 0.54 ± 0.75 | 0.7242 | 0.8617 | 0.8617 | |
| AG | 262 (34.89) | 3.64 ± 4.06 | 0.47 ± 0.49 | 0.52 ± 0.71 | |||||||||||
| GG | 444 (59.03) | 3.57 ± 3.66 | 0.46 ± 0.47 | 0.47 ± 0.65 | |||||||||||
| rs4363657 | TT | 214 (28.46) | 3.53 ± 4.14 | 0.3482 | 0.3386 | 0.3870 | 0.52 ± 0.64 | 0.3985 | 0.4838 | 0.8253 | 0.51 ± 0.73 | 0.7104 | 0.6678 | 0.8536 | |
| TC | 358 (47.59) | 3.52 ± 3.83 | 0.47 ± 0.47 | 0.49 ± 0.61 | |||||||||||
| CC | 180 (23.95) | 3.78 ± 3.44 | 0.45 ± 0.47 | 0.45 ± 0.74 | |||||||||||
| SLCO1B3 | rs7311358 | GG | 68 (9.09) | 4.12 ± 3.95 | 0.0729 | 0.0713 | 0.1426 | 0.52 ± 0.57 | 0.7608 | 0.8399 | 0.8399 | 0.63 ± 0.81 | 0.6945 | 0.7469 | 0.8536 |
| AG | 299 (39.97) | 3.69 ± 3.99 | 0.46 ± 0.52 | 0.47 ± 0.76 | |||||||||||
| AA | 381 (50.94) | 3.46 ± 3.69 | 0.49 ± 0.52 | 0.48 ± 0.57 | |||||||||||
| CYP2C9 | rs1057910 | AA | 706 (93.51) | 3.48 ± 3.92 | 0.2865 | 0.2225 | 0.2967 | 0.46 ± 0.53 | 0.7224 | 0.6190 | 0.8253 | 0.47 ± 0.67 | 9.0 × 10-4 | 9.0 × 10-4 | 0.0036 |
| CA + CC | 49 (6.49) | 2.79 ± 2.82 | 0.40 ± 0.44 | 0.29 ± 0.39 | |||||||||||
| GATM | rs9806699 | GG | 71 (9.39) | 3.78 ± 4.23 | 0.1302 | 0.0652 | 0.1426 | 0.54 ± 0.62 | 0.2075 | 0.1512 | 0.6048 | 0.48 ± 0.60 | 0.1797 | 0.1275 | 0.3400 |
| AG | 298 (39.42) | 3.40 ± 3.79 | 0.45 ± 0.54 | 0.48 ± 0.64 | |||||||||||
| AA | 387 (51.19) | 3.74 ± 3.84 | 0.49 ± 0.49 | 0.50 ± 0.71 | |||||||||||
ACEIs angiotensin-converting enzyme inhibitors, AST aspartate aminotransferase, CREA creatinine, DM-RST N-desmethyl rosuvastatin, GATM glycine amidinotransferase, RST rosuvastatin, RSTL rosuvastatin lactone, SNPs polymorphisms
aP value was adjusted by lnAST levels and conmination of ACEIs
bP value was adjusted by CREA levels
cP value was adjusted by age and heart failure
Fig. 1.

Effects of ABCG2 rs2231142 and SLCO1B1 rs4149056 genetic polymorphisms on the plasma exposure to RST, RSTL, and DM-RST
SLCO1B1 521T>C (rs4149056) in patients with one or two copies of the variant allele had a significantly high plasma exposure to RST, whereas the significance was not found after multiple testing (Padj = 0.0247, FDR = 0.0988), and a gene-dose effect was not observed in the three genotypes of this SNP. In addition, there were no obviously effects on the plasma concentrations of RSTL and DM-RST (Padj > 0.05, FDR > 0.05; Table 1, Fig. 1). Plasma exposure to DM-RST of patients with one and two copies of the CYP2C9 rs1057910 (CA and CC) was significantly low (Padj = 9.0 × 10–4, FDR = 0.0036). However, the difference was not statistically significant in the Css/D of RST and RSTL. The other SNPs induced no significant effects on drug exposure (Padj > 0.05, FDR > 0.05; Table 1, Figures S1 and S2).
Effects of plasma exposure to RST and its metabolites on SIM
Among the 758 eligible subjects, 51 patients manifested occurrences of SIM. The incidence of RST-induced myopathy was 6.73%. The clinical baseline characteristics of without SIM (control group, n = 707) and with SIM (SIM group, n = 51) were summarized in Table 2. Univariate logistic regression analyses revealed no significant differences in terms of age, sex, dosage, medical history, biochemical levels, and drug combinations between the control and SIM groups (P > 0.05). The plasma exposure of RST, RSTL, and DM-RST between control and SIM groups were 3.64 ± 4.21 vs 3.54 ± 4.75, 0.48 ± 0.54 vs 0.49 ± 0.53, 0.49 ± 0.86 vs 0.47 ± 0.71, respectively (Fig. 2). The SIM incidents were not significantly associated with the plasma concentrations of RST and its metabolites (all P > 0.05).
Table 2.
Patients’ characteristics and their effects on SIM
| Characteristics | Control group | SIM group | Univariate logistic regression | ||
|---|---|---|---|---|---|
| n (%) or mean ± SD | n (%) or mean ± SD | OR (95% CI) | P value | ||
| Demographic data | |||||
| Total no. | 707 | 51 | |||
| Age | 63.19 ± 10.33 | 61.40 ± 10.93 | 0.984 (0.958–1.011) | 0.2356 | |
| Sex | Female | 167 (23.62) | 15 (29.41) | 0.740 (0.396–1.386) | 0.3473 |
| Male | 540 (76.38) | 36 (70.59) | |||
| Dosage (mg) | 5 | 13 (1.84) | 1 (1.96) | 1.011 (0.928–1.103) | 0.7955 |
| 10 | 619 (87.55) | 44 (86.27) | |||
| 20 | 75 (10.61) | 6 (11.76) | |||
| Medical history | |||||
| Arrhythmia | No | 657 (92.93) | 49 (96.08) | 0.546 (0.129–2.311) | 0.4107 |
| Yes | 50 (7.07) | 2 (3.92) | |||
| Heart failure | No | 654 (92.50) | 51 (100) | 1.536 (0.344–6.851) | 0.5736 |
| Yes | 53 (7.50) | 0 (0) | |||
| Hypertension | No | 322 (45.54) | 24 (47.06) | 0.937 (0.530–1.657) | 0.8242 |
| Yes | 385 (54.46) | 27 (52.94) | |||
| Hyperlipidemia | No | 627(88.68) | 46 (90.20) | 0.860 (0.332–2.228) | 0.7561 |
| Yes | 80 (11.32) | 5 (9.80) | |||
| Biochemical measurements | |||||
| ALT, U/L | 32.77 ± 27.72 | 29.74 ± 18.53 | 0.995 (0.981–1.009) | 0.4669 | |
| AST, U/L | 32.47 ± 30.03 | 35.25 ± 38.19 | 1.075 (0.611–1.893) | 0.0636 | |
| CREA, μmol/L | 88.19 ± 33.26 | 83.17 ± 34.28 | 0.994 (0.984–1.005) | 0.2991 | |
| CK, U/L | 133.96 ± 239.30 | 174.21 ± 371.57 | 1.000 (1.000–1.001) | 0.2977 | |
| APOA, g/L | 1.08 ± 0.27 | 1.02 ± 0.21 | 0.418 (0.119–1.464) | 0.1724 | |
| CHOL, mmol/L | 4.45 ± 1.29 | 4.61 ± 1.48 | 1.086 (0.890–1.325) | 0.4159 | |
| CKMB, U/L | 8.03 ± 9/94 | 8.57 ± 11.29 | 1.004 (0.980–1.029) | 1.0040 | |
| HDLC, mmol/L | 1.01 ± 0.26 | 0.98 ± 0.16 | 0.563 (0.170–1.870) | 0.3484 | |
| LDLC, mmol/L | 2.73 ± 1.04 | 2.87 ± 1.25 | 1.125 (0.874–1.446) | 0.3604 | |
| Lpa, mg/L | 274.04 ± 309.39 | 246.49 ± 219.43 | 1.000 (0.999–1.001) | 0.5173 | |
| TRIG, mmol/L | 1.58 ± 1.06 | 1.63 ± 1.02 | 1.041 (0.805–1.345) | 0.7602 | |
| Medication | |||||
| β-blockers | No | 85 (12.02) | 7 (13.73) | 0.863 (0.377–1.978) | 0.1211 |
| Yes | 622 (87.98) | 44 (86.27) | |||
| ACEIs | No | 294 (41.58) | 22 (43.14) | 0.934 (0.526–1.659) | 0.8164 |
| Yes | 413 (58.42) | 29 (56.86) | |||
| CCBs | No | 506 (71.57) | 40 (78.43) | 0.693 (0.349–1.378) | 0.2960 |
| Yes | 201 (28.43) | 11(21.57) | |||
| PPIs | No | 338(47.81) | 23 (45.10) | 1.105 (0.624–1.956) | 0.7313 |
| Yes | 369 (52.19) | 28(54.90) | |||
| Clopidogrel | No | 13 (1.84) | 1 (1.96) | 0.941 (0.121–7.337) | 0.9535 |
| Yes | 694 (98.16) | 50 (98.04) | |||
| Aspirin | No | 25 (3.11) | 2 (3.92) | 0.790 (0.181–3.458) | 0.7545 |
| Yes | 682 (96.89) | 49 (96.08) | |||
| Plasma concentrations | |||||
| RST | 3.64 ± 4.21 | 3.54 ± 4.75 | 0.900 (0.789–1.027) | 0.1177 | |
| RSTL | 0.48 ± 0.54 | 0.49 ± 0.53 | 0.971 (0.799–1.178) | 0.7634 | |
| DM-RST | 0.49 ± 0.86 | 0.47 ± 0.71 | 0.889 (0.695–1.136) | 0.3469 | |
ALT alanine aminotransferase, APOA apolipoprotein a, CCBs calcium channel blockers, CHOL cholesterol, CK creatine kinase, CKMB creatine kinase MB, HDLC high-density lipoprotein cholesterol, LDLC low-density lipoprotein cholesterol, Lpa lipoprotein (a), PPIs proton pump inhibitors, TRIG triglyceride
Fig. 2.

Comparisons of plasma exposure to RST, RSTL, and DM-RST between control and SIM groups
Effects of genetic variations on RST-induced myopathy
Among the nine SNPs in six genes, we observed two polymorphisms: SLCO1B1 rs4149056 and GATM rs9806699, which were associated with SIM incidents (Table 3). Carriers of the C allele of SLCO1B1 521T>C (rs4149056) had a high risk of SIM (odds ratio (OR): 1.741, 95% confidence interval (CI): 1.180–2.568, P = 0.0052, FDR = 0.0416). The GATM rs9806699 mutant allele indicated a marginally protective effect on SIM (OR: 0.617, 95% CI: 0.406–0.939, P = 0.024, FDR = 0.0960). The other polymorphisms, including the SNPs that were significantly relevant to the pharmacokinetics of RST, i.e., ABCG2 421C>A (rs2231142) and CYP2C9 rs1057910, did not induce significant effects on SIM.
Table 3.
Genotypes and their effects on RST-induced myopathy
| Gene | SNP | Genotype | Control group | SIM group | Univariate logistic regression | FDR | |
|---|---|---|---|---|---|---|---|
| Number of carriers (%) | Number of carriers (%) | OR (95% CI) | P value | ||||
| ABCG2 | rs2231142 | CC | 329 (46.73) | 30 (58.82) | 0.707 (0.444–1.127) | 0.1447 | 0.3859 |
| AC | 307 (43.61) | 17 (33.33) | |||||
| AA | 68 (9.66) | 4 (7.84) | |||||
| ABCB1 | rs1045642 | AA | 95 (13.53) | 11 (21.57) | 0.885 (0.585–1.337) | 0.5611 | 0.7459 |
| AG | 343 (48.86) | 20 (39.22) | |||||
| GG | 264 (37.61) | 20(39.22) | |||||
| SLCO1B1 | rs4149056 | TT | 537 (76.60) | 31 (60.78) | 1.741 (1.180–2.568) | 0.0052 | 0.0416 |
| CT | 118 (16.83) | 12 (23.53) | |||||
| CC | 46 (6.56) | 8 (15.69) | |||||
| rs2306283 | AA | 42 (5.99) | 4 (7.84) | 1.015 (0.610–1.691) | 0.9531 | 0.9531 | |
| AG | 245 (34.95) | 17 (33.33) | |||||
| GG | 414 (59.06) | 30 (58.82) | |||||
| rs4363657 | TT | 200 (28.53) | 14 (27.45) | 0.906 (0.590–1.392) | 0.6527 | 0.7459 | |
| TC | 331 (47.22) | 27 (52.94) | |||||
| CC | 170 (24.25) | 10 (19.61) | |||||
| SLCO1B3 | rs7311358 | GG | 63 (9.04) | 5 (9.80) | 1.201 (0.733–1.967) | 0.4676 | 0.7459 |
| AG | 282 (40.46) | 17 (33.33) | |||||
| AA | 352 (50.50) | 29 (56.86) | |||||
| CYP2C9 | rs1057910 | AA | 657 (93.32) | 49 (96.08) | 0.581 (0.137–2.466) | 0.4617 | 0.7459 |
| CA + CC | 47 (6.68) | 2 (3.92) | |||||
| GATM | rs9806699 | GG | 59 (8.37) | 12 (23.53) | 0.617 (0.406–0.939) | 0.0240 | 0.0960 |
| AG | 281 (39.86) | 17 (33.33) | |||||
| AA | 365 (51.77) | 22 (43.14) | |||||
SIM statin-induced myopathy
Furthermore, we analyzed the myopathy risk of carriers or no-carriers with risk alleles of both SLCO1B1 rs4149056 (C allele) and GATM rs9806699 (G allele). Results showed that SIM events occurred in nine patients among 249 individuals without any above risk allele of SIM. SIM incidence was 3.61%. However, 8 subjects had SIM in 76 patients with both risk alleles (rs4149056 CC or CT and rs9806699 GG or AG). SIM incidence was 10.53% and its risk was significantly higher (OR: 3.137, 95% CI: 1.166–8.441, P = 0.024) compared to patients without risk alleles of SIM. Only five patients simultaneously carried CC genotype of rs4149056 and GG genotype of rs9806699. Of them, SIM events occurred in one patient (OR: 6.667, 95% CI: 0.675–65.841, P = 0.104). The SIM incidence was 20.0%. No significant difference was observed because of small size sample. Results were also listed in Table 4.
Table 4.
SIM incident of patients with or without risk alleles
| SLCO1B1 rs4149056 | GATM rs9806699 | Number of carriers | SIM incident (%) | Univariate logistic regression | |
|---|---|---|---|---|---|
| OR (95% CI) | P value | ||||
| CC + CT | GG + AG | 76 | 8 (10.53) | 3.137 (1.166–8.441) | 0.024 |
| CC | GG | 5 | 1 (20.00) | 6.667 (0.675–65.841) | 0.104 |
| TT | AA | 249 | 9 (3.61) | ||
Discussion
Our study revealed that RST-induced myopathy was significantly affected by SLCO1B1 521T>C (rs4149056) and marginally affected by GATM (rs9806699). However, their influences on SIM were not associated with the high systemic exposure of RST and its metabolites. Our study identified for the first time the relationship between RST metabolites and SIM incidents, and clarified the effect of GATM on SIM in Asians.
The prevalence of SIM ranges from 7% to 29% in patients treated with statins in clinical practice [26]. In our study, the incidence of RST-induced myopathy was 6.73%. This value was similar to previous reports that SIM occurred in 2.5%–10% of subjects receiving 5–80 mg/day of oral RST in randomized controlled trials [27]. The physiological mechanism of SIM has yet to be elucidated. Our results indicated that RST-induced myopathy was not correlated with the high plasma concentrations of RST and its metabolites. This finding was consistent with a previous report that SIM was present in plasma concentrations of statins at an acceptable normal range [28].
For all SNPs in our study, the loci that significantly affected SIM were inconsistent with that of the drug plasma concentrations. SLCO1B1 521T>C was significantly correlated with RST-induced myopathy, although this SNP was not significantly associated with the drug concentration after correction for multiple testing (FDR > 0.05). Previous studies have demonstrated the hydrophilic property of RST and hepatic bile acid uptake transporter were involved in the disposition of RST [29], which diminished the role of SLCO1B1 in the pharmacokinetic of RST [30]. The polymorphisms of ABCG2 421C>A (rs2231142) played important roles in the systemic exposure to both RST and its metabolites in our study (Padj < 0.01, FDR < 0.05). Moreover, the mutation frequency of ABCG2 421C>A was obviously higher in East Asians (allele frequency ~35%) than that in Caucasians (14%) [20, 31]. Thus, the variation of ABCG2 was likely to be the primary genetic determinant of the pharmacokinetic difference of RST in Chinese population. This result is consistent with previous studies on Chinese people [18, 19, 32]. However, variant of ABCG2 were not associated with elevated SIM risk.
Pharmacokinetic conditions result in statin accumulation in the muscle, while the molecular determinants of SIM development is statin distribution into myocytes and exerting its negative function in myocytes. Maybe they are not pharmacokinetic-related transporters (OATP1B1 (organic anion-transporting polypeptide 1B1, encoded by SLCO1B1) or BCRP2 (breast cancer resistant protein 2, encoded by ABCG2)) primarily involving in transferring RST into/out myocytes. Study have indicated that OATP2B1 and multidrug resistance protein (MRP)1, MRP4, and MRP5 played roles for statins transporting into myocytes [33].
A decreased risk was observed on patients with GATM rs9806699 G>A in our study. This correlation was replicated for the first time since the study of Mangravite et al [21–23]. As a functional gene, GATM was related to the energy metabolism of skeletal muscles. It encodes GATM, which is responsible for the synthesis of creatine. Creatine is a major source of energy in skeletal muscles. The A allele of GATM rs9806699 leads to a greater decrease in GATM RNA expression [21]. This effect of GATM on SIM found in our study should be validated by future studies on a large sample.
Our study has two limitations. First, we did not measure the exact plasma exposure to RST and its metabolites at the time of myotoxicity, and we did not conduct serial monitoring during the follow up. The drug concentration was considered to remain at a steady-state level. Second, we did not detect the CK level in the follow-up period. However, the international expert workshop on SIM proposed that the elevation of serum CK might be not a necessary sign of SIM; any of the SIM relevant symptoms with or without an elevation of CK were considered as SIM [24, 25].
Conclusions
The variant allele of SLCO1B1 521T>C (rs4149056) and GATM rs9806699 were potential biomarkers for predicting RST-induced myopathy, and their influences on SIM were unrelated to the high plasma exposure of RST and its metabolites. These findings offer further insight into the mechanism underlying RST-induced myopathy.
Electronic supplementary material
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the National Key R&D Program (No. 2017YFC0909301, 2017YFC0909302, 2017YFC0909303, 2016YFC0905003), the National Natural Science Foundation of China (No. 81673514, 81373486), the Science and Technology Development Projects of Guangdong Province, China (No. 2016B090918114), and the Science and Technology Development Projects of Guangzhou, Guangdong, China (201510010236, 201604020096).
Author contributions
In this project, X.B. and B.Z. contributed to the study design, experiment performance, follow up, and manuscript writing. S.-L.Z. and M.H. contributed to study design and management, manuscript revision. P.W., G.-l.W., J.-l.L., and H.-s.S. were involved in patient recruitment and draft revision. D.-s.W., X.-z.L., and Y.-b.L. performed the sample collection, follow up, and data analysis.
Competing interests
The authors declare no competing interests.
Contributor Information
Min Huang, Phone: 8620-39943034-806, Email: huangmin@mail.sysu.edu.cn.
Shi-long Zhong, Phone: +8620-83827812-51157, Email: zhongsl@hotmail.com.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0013-y) contains supplementary material, which is available to authorized users.
References
- 1.Ioannidis JP. More than a billion people taking statins?: Potential implications of the new cardiovascular guidelines. JAMA. 2014;311:463–4. doi: 10.1001/jama.2013.284657. [DOI] [PubMed] [Google Scholar]
- 2.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment ofblood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63:2889–934. doi: 10.1016/j.jacc.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Glueck CJ, Aregawi D, Agloria M, Khalil Q, Winiarska M, Munjal J, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther. 2006;28:933–42. doi: 10.1016/j.clinthera.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Wei MY, Ito MK, Cohen JD, Brinton EA, Jacobson TA. Predictors of statin adherence, switching, and discontinuation in the USAGE survey: understanding the use of statins in America and gaps in patient education. J Clin Lipidol. 2013;7:472–83. doi: 10.1016/j.jacl.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Serban MC, Colantonio LD, Manthripragada AD, Monda KL, Bittner VA, Banach M, et al. Statin intolerance and risk of coronary heart events and all-cause mortality following myocardial infarction. J Am Coll Cardiol. 2017;69:1386–95. doi: 10.1016/j.jacc.2016.12.036. [DOI] [PubMed] [Google Scholar]
- 6.Ghatak A, Faheem O, Thompson PD. The genetics of statin-induced myo-pathy. Atherosclerosis. 2010;210:337–43. doi: 10.1016/j.atherosclerosis.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 7.Canestaro WJ, Austin MA, Thummel KE. Genetic factors affecting statin concentrations and subsequent myopathy: a HuGENet systematic review. Genet Med. 2014;16:810–9. doi: 10.1038/gim.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hermann M, Bogsrud MP, Molden E, Asberg A, Mohebi BU, Ose L, et al. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin Pharmacol Ther. 2006;79:532–39. doi: 10.1016/j.clpt.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Skottheim IB, Gedde-Dahl A, Hejazifar S, Hoel K, Asberg A. Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur J Pharm Sci. 2008;33:317–25. doi: 10.1016/j.ejps.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 10.White CM. A review of the pharmacologic and pharmacokinetic aspects of rosuvastatin. J Clin Pharmacol. 2002;42:963–70. doi: 10.1177/009127002401102876. [DOI] [PubMed] [Google Scholar]
- 11.Yoshikado T, Toshimoto K, Nakada T, Ikejiri K, Kusuhara H, Maeda K, et al. Comparison of methods for estimating unbound intracellular-to-medium concentration ratios in rat and human hepatocytes using statins. Drug Metab Dispos. 2017;45:779–89. doi: 10.1124/dmd.116.074823. [DOI] [PubMed] [Google Scholar]
- 12.Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359:789–99. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 13.Carr DF, O'Meara H, Jorgensen AL, Campbell J, Hobbs M, McCann G, et al. SLCO1B1 genetic variant associated with statin-induced myopathy: a proof-of-concept study using the clinical practice research datalink. Clin Pharmacol Ther. 2013;94:695–701. doi: 10.1038/clpt.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrari M, Guasti L, Maresca A, Mirabile M, Contini S, Grandi AM, et al. Association between statin-induced creatine kinase elevation and genetic polymorphisms in SLCO1B1, ABCB1 and ABCG2. Eur J Clin Pharmacol. 2014;70:539–47. doi: 10.1007/s00228-014-1661-6. [DOI] [PubMed] [Google Scholar]
- 15.de Keyser CE, Peters BJ, Becker ML, Visser LE, Uitterlinden AG, Klungel OH, et al. The SLCO1B1 c.521T>C polymorphism is associated with dose decrease or switching during statin therapy in the Rotterdam Study. Pharmacogenet Genomics. 2014;24:43–51. doi: 10.1097/FPC.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 16.Lee HH, Ho RH. Interindividual and interethnic variability in drug disposition: polymorphisms in organic anion transporting polypeptide 1B1 (OATP1B1; SLCO1B1) Br J Clin Pharmacol. 2017;83:1176–84. doi: 10.1111/bcp.13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mo L, He J, Yue Q, Dong B, Huang X. Increased dosage of cyclosporine induces myopathy with increased seru creatine kinase in an elderly patient on chronic statin therapy. J Clin Pharm Ther. 2015;40:245–48. doi: 10.1111/jcpt.12240. [DOI] [PubMed] [Google Scholar]
- 18.Lee HK, Hu M, Lui S, Ho CS, Wong CK, Tomlinson B. Effects of polymorphisms in ABCG2, SLCO1B1, SLC10A1 and CYP2C9/19 on plasma concentrations of rosuvastatin and lipid response in Chinese patients. Pharmacogenomics. 2013;14:1283–94. doi: 10.2217/pgs.13.115. [DOI] [PubMed] [Google Scholar]
- 19.Wan Z, Wang G, Li T, Xu B, Pei Q, Peng Y, et al. Marked alteration of rosuvastatin pharmacokinetics in healthy chinese with ABCG2 34G>A and 421C>A homozygote or compound heterozygote. J Pharmacol Exp Ther. 2015;354:310–5. doi: 10.1124/jpet.115.225045. [DOI] [PubMed] [Google Scholar]
- 20.Giacomini KM, Balimane PV, Cho SK, Eadon M, Edeki T, Hillgren KM, et al. International Transporter Consortium commentary on clinically important transporter polymorphisms. Clin Pharmacol Ther. 2013;94:23–26. doi: 10.1038/clpt.2013.12. [DOI] [PubMed] [Google Scholar]
- 21.Mangravite LM, Engelhardt BE, Medina MW, Smith JD, Brown CD, Chasman DI, et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature. 2013;502:377–80. doi: 10.1038/nature12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Floyd JS, Bis JC, Brody JA, Heckbert SR, Rice K, Psaty BM. GATM locus does not replicate in rhabdomyolysis study. Nature. 2014;513:E1–3. doi: 10.1038/nature13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luzum JA, Kitzmiller JP, Isackson PJ, Ma C, Medina MW, Dauki AM, et al. GATM polymorphism associated with the risk for statin-induced myopathy does not replicate in case-control analysis of 715 dyslipidemic individuals. Cell Metab. 2015;21:622–7. doi: 10.1016/j.cmet.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.du Souich P, Roederer G, Dufour R. Myotoxicity of statins: mechanism of action. Pharmacol Ther. 2017;175:1–16. doi: 10.1016/j.pharmthera.2017.02.029. [DOI] [PubMed] [Google Scholar]
- 25.Alfirevic A, Neely D, Armitage J, Chinoy H, Cooper RG, Laaksonen R, et al. Phenotype standardization for statin-induced myotoxicity. Clin Pharmacol Ther. 2014;96:470–6. doi: 10.1038/clpt.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muntean DM, Thompson PD, Catapano AL, Stasiolek M, Fabis J, Muntner P, et al. Statin-associated myopathy and the quest for biomarkers: can we effectively predict statin-associated muscle symptoms? Drug Discov Today. 2017;22:85–96. doi: 10.1016/j.drudis.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Kostapanos MS, Milionis HJ, Elisaf MS. Rosuvastatin-associated adverse effects and drug-drug interactions in the clinical setting of dyslipidemia. Am J Cardiovasc Drugs. 2010;10:11–28. doi: 10.2165/13168600-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 28.Teichholz LE. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2003;138:1008–9. doi: 10.7326/0003-4819-138-12-200306170-00023. [DOI] [PubMed] [Google Scholar]
- 29.Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–806. doi: 10.1053/j.gastro.2006.02.034. [DOI] [PubMed] [Google Scholar]
- 30.Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvas-tatin. Clin Pharmacol Ther. 2007;82:726–33. doi: 10.1038/sj.clpt.6100220. [DOI] [PubMed] [Google Scholar]
- 31.Zamber CP, Lamba JK, Yasuda K, Farnum J, Thummel K, Schuetz JD, et al. Natural allelic variants of breast cancer resistance protein (BCRP) and their relationship to BCRP expression in human intestine. Pharma-cogenetics. 2003;13:19–28. doi: 10.1097/00008571-200301000-00004. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Q, Ruan ZR, Yuan H, Xu DH, Zeng S. ABCB1 gene polymorphisms, ABCB1 haplotypes and ABCG2 c.421c>A are determinants of inter-subject variability in rosuvastatin pharmacokinetics. Pharmazie. 2013;68:129–34. [PubMed] [Google Scholar]
- 33.Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, Schwarz UI, Lemke CJ, Leake BF, et al. Human skeletal muscle drug transporters determine local exposureand toxicityofstatins. Circ Res. 2010;106:297–306. doi: 10.1161/CIRCRESAHA.109.203596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.