

Abstract
Background
Astrocytes have been shown to produce several pro- and anti-inflammatory cytokines to maintain homeostasis of microenvironment in response to vast array of CNS insults. Some inflammation-related cytokines are responsible for regulating such cell events. Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine that can be inducibly expressed in the lesioned spinal cord. Unknown is whether MIF can facilitate the production of immunosuppressive factors from astrocytes to tune milieu following spinal cord injury.
Methods
Following establishment of contusion SCI rat model, correlation of PGE2 synthesis-related protein levels with that of MIF was assayed by Western blot. ELISA assay was used to detect production of PGE2, TNF-α, IL-1β, and IL-6. Immunohistochemistry was performed to observe colocalization of COX2 with GFAP- and S100β-positive astrocytes. The primary astrocytes were treated by various inhibitors to validate relevant signal pathway.
Results
The protein levels of MIF and COX2, but not of COX1, synchronously increased following spinal cord injury. Treatment of MIF inhibitor 4-IPP to the lesion sites significantly reduced the expression of COX2, mPGES-1, and as a consequence, the production of PGE2. Astrocytes responded robustly to the MIF interference, by which regulated MAPK/COX2/PGE2 signal pathway through coupling with the CD74 membrane receptor. MIF-induced production of PGE2 from astrocytes was able to suppress production of TNF-α, but boosted production of IL-1β and IL-6 in LPS-activated macrophages.
Conclusion
Collectively, these results reveal a novel function of MIF-mediated astrocytes, which fine-tune inflammatory microenvironment to maintain homeostasis. These suggest an alternative therapeutic strategy for CNS inflammation.
Electronic supplementary material
The online version of this article (10.1186/s12974-019-1468-6) contains supplementary material, which is available to authorized users.
Keywords: MIF, COX2, Spinal cord, Astrocyte, Inflammation, PGE2
Background
Spinal cord injury (SCI) always initiates a robust inflammatory response characterized by infiltration of leukocytes and synthesis of cytokines and chemokines [1, 2]. This excessive inflammation induced by SCI causes a progressive damage through aggravating the degeneration of neurons and apoptosis of oligodendrocytes [3]. The formation of inflammatory cascades in the central nervous system (CNS) is considered as a result of tightly coordinated interaction between parenchymal cells (neurons and glia), endothelia, and circulating leukocytes [2, 4]. Damage-associated molecular patterns (DAMPs) released from injured and dying cells, or from the circulation, activated the pattern-recognition receptors (PRRs) of leukocytes, microglia, and astrocytes to drive the inflammatory response in the absence of infection, thereby affect tissue functional recovery [5, 6]. Astrocytes are one of the first cell populations to sense SCI, and subsequently participate in modulation of inflammatory responses [7, 8]. On the one hand, the astrocytes express several toll-like receptors (TLR) and build up responses to innate immune triggers by releasing proinflammatory molecules that exacerbate spinal cord injuries [9, 10]. On the other hand, they can also limit the spread of inflammatory cytokines or injury-induced toxic molecules through physical glial scar and/or secretion of anti-inflammatory cytokines. For example, anti-inflammatory cytokine IL-10 can be released by human astrocyte cell lines stimulated with IFN-γ1a or combinations of proinflammatory cytokines (LPS/IFN-γ) plus IFN-γ1a [11]. Stimulation of TLR3 induces anti-inflammatory cytokines in adult human astrocytes, and observation was recapitulated by applying LPS to neonatal rats, which decreased proinflammatory responses in EAE at later age, as measured by increased IL-10 in the spinal cord [12–14]. The differential outcomes of astrocytes have been shown to tightly depend on injury modes of CNS and diversity of stimulating factors [14–16]. Obviously, astrocytes are required for the fine-tuned regulation and resolution of the inflammation and subsequent reconstruction of microenvironment homeostasis following SCI [15].
Cyclooxygenases (COX), also known as prostaglandin-endoperoxide synthases (PTGS), catalyze the reaction of arachidonic acid (AA) metabolism to prostanoids and thromboxanes [17]. COX1 and COX2 are the two main COX enzymes that participate in such catalytic process. Several bioactive lipid mediators produced in the AA metabolism have been found to play crucial roles in inflammation, thus COX1 and COX2 are widely used as targets of the nonsteroidal anti-inflammatory drugs [18]. COX1 is constitutively expressed in most tissues to maintain essential physiological functions including platelet homeostasis, gastric mucosal integrity, and neuronal function [19, 20], while COX2 is an inducible enzyme that responds to proinflammatory stimuli [17]. Since COX2 is expressed in the normal CNS, but is rare or absent in most organs, injury-induced COX2 expression has been shown to exacerbate neuronal death and neuroinflammatory responses of CNS [21, 22]. The mechanism by which COX2 modulates inflammation is poor defined, but possibly be associated with an increase in the production of prostanoids like prostaglandin E2 (PGE2) [18]. During the initial stage of inflammatory responses, PGE2 acts as a proinflammatory mediator by promotion of local vasodilatation, inflammatory edema, and fever [23]. However, it also exhibits immunosuppressive properties through contributing to the resolution phase of acute inflammation [24]. For instance, PGE2 has been shown to inhibit production of TNF-α and boost the production of IL-10 in macrophages [25]. Thus, PGE2 potentially facilitates the return of tissue microenvironment to homeostasis in the case of tissue pathology. In the CNS, COX2 is localized primarily within neurons, but it can be inducibly expressed in the astrocytes and microglia following CNS injury [17, 26, 27]. Whether COX2-induced production of PGE2 from astrocytes contributes to the microenvironment homeostasis following SCI is unclear.
Macrophage migration inhibitory factor (MIF) is a proinflammatory cytokine produced by a variety of cells and tissues, including monocytes, macrophages, T and B lymphocytes, and hepatocytes [28]. In the CNS, MIF is able to be inducibly expressed within neurons, astrocytes, and microglia, in association with neuronal apoptosis and facilitation of neuroinflammation [29–31]. Therefore, MIF is supposed to be a potential therapeutic target for several autoimmune-mediated CNS diseases such as multiple sclerosis (MS) [31]. It is interesting to note that MIF is capable of promoting the release of chemokine CCL5 from astrocytes, which preferentially attracts M2-type macrophages toward the lesion sites of spinal cord [30]. The accumulation of M2-type macrophages has been shown to be tightly coordinated with the resolution of inflammation, hinting at a function of MIF in tune the microenvironment through astrocytes. Since MIF has been shown to activate COX2/PGE2 signaling in spinal microglia, and the synthesis of PGE2 in astrocytes is also induced by proinflammatory stimuli [27, 32], it is assumed that MIF can exert the same function to induce the PGE2 production from astrocytes, which in turn regulates the activities of inflammatory cells. In the present study, we examined the relevance between expression of MIF and PGE2 following rat spinal cord contusion. We further investigated the mechanism of MIF-induced production of PGE2 in the astrocytes, as well as its effects on the inflammatory responses of macrophages. Our results have revealed that MIF can tune inflammatory microenvironment of injured spinal cord through influence of astrocytes.
Methods
Animals
Adult male Sprague–Dawley (SD) rats, weighing 180–220 g, were provided by the Center of Experimental Animals, Nantong University. All animal care, breeding, and testing procedures were approved according to the Animal Care and Use Committee of Nantong University and the Jiangsu Province Animal Care Ethics Committee. All animals were housed in individual cages in a temperature and light/dark cycle controlled environment with free access to food and water.
Establishment of contusion SCI rat model
The number of animals subjected to surgical treatment was calculated by six per experimental group in triplicate. Contusion SCI rat model was prepared as previous description [33]. Briefly, rats were anesthetized with an intraperitoneal injection of 10% chloral hydrate compound anesthetic (3 mg/kg). The fur was shaved from the surgical site and the skin was disinfected with chlorhexidine. A 15-mm midline skin incision was made to expose the vertebral column. After the spinal thoracic region was exposed by separation of dorsal muscles to the side, the spinous processes of T8–T10 vertebrae were exposed. A laminectomy was performed at vertebral level T9, exposing the dorsal cord surface with the dura remaining intact. The exposed spinal cord segment (about 3 mm in length) received a 150-kilodyne spinal contusion injury using the IH-0400 Impactor (Precision Systems and Instrumentation) injury device. The impact rod was removed immediately, and the wound was irrigated. For drug delivery, 10 μl of 80 mM COX2 inhibitor NS398 or vehicle were then slowly injected intrathecally. Muscles and incisions were sutured using silk threads. Postoperative care included butorphanol administration twice a day for a 5-day period, as well as vitamins, saline, and enrofloxacin twice a day for a 7-day period. Manual expression of bladders was performed twice a day until animals recovered spontaneous voiding.
Cell culture
Astrocytes were prepared from spinal cord of newborn Sprague–Dawley rats, 1–2 days after birth for maximum homogeneity, and the astrocytes were isolated and cultured according to previously described methods [34]. Briefly, the cells were enzymatically dissociated using 0.25% trypsin (Gibco-BRL) for 6 min at 37 °C, and the suspension was then centrifuged at 1200 rpm for 5 min and cultured in 1:1 Dulbecco’s modified Eagle’s medium:Ham’s F-12 medium supplemented with 10% fetal bovine serum (FBS), 0.224% NaHCO3, and 1% penicillin/streptomycin in the presence of 5% CO2. A monolayer of astrocytes was obtained 12–14 days after the plating. Non-astrocytes were detached from the flasks by shaking and were removed by changing the medium. Third or fourth passage cells were rendered quiescent through incubation in medium containing 0.5% FBS for 4 days prior to the experiments. Astrocyte phenotype was confirmed by cells exhibiting a characteristic morphology and positive staining for the astrocytic marker glial fibrillary acid protein (GFAP).
For the culture of macrophages in the astrocyte-conditioned medium, the isolated astrocytes were cultured in the presence of 1 μg/ml recombinant MIF with or without addition of 30 μM NS398 for 24 h. Meanwhile, astrocyte-free culture was used as negative control. The astrocyte-conditioned medium was then collected for the culture of macrophages in the presence of 1 μg/ml LPS at 24 h, respectively.
Western blot
Protein was extracted from cells with a buffer containing 1% SDS, 100 mM Tris-HCl, 1 mM PMSF, and 0.1 mM β-mercaptoethanol, following treatment with 0–2.5 μg/ml rat recombinant MIF (ProSpec) for 24 h. Alternatively, protein was extracted from 1 cm spinal segments of injured site at 0 day, 1 day, 4 days, and 1 week following contusion (n = 8 in each time point). Protein concentration of each specimen was detected by the Bradford method to maintain the same loads. Protein extracts were heat denatured at 95 °C for 5 min, electrophoretically separated on 10% SDS-PAGE, and transferred to PVDF membranes. The membranes were subjected to the reaction with a 1:1000 dilution of primary antibodies in TBS buffer at 4 °C overnight, followed by a reaction with secondary antibody conjugated with goat anti-rabbit or goat anti-mouse HRP dilution 1:1000 (Santa Cruz) at room temperature for 2 h. After the membrane was washed, the HRP activity was detected using an ECL kit. The image was scanned with a GS800 Densitometer Scanner (Bio-Rad), and the data were analyzed using PDQuest 7.2.0 software (Bio-Rad). β-actin (1:5000) was used as an internal control. Antibodies used in Western blot are MIF, COX1, cPGES (Abcam); COX2, mPGES-1, mPGES-2 (Cayman); CD74 (Biorbyt); and β-actin (Proteintech).
ELISA
Primary astrocytes were treated with 0–2.5 μg/ml rat recombinant MIF for 24 h. Cell supernatants were harvested, and cells were lysed in the buffer containing 1% SDS, 100 mM Tris-HCl, 1 mM PMSF, and 0.1 mM β-mercaptoethanol. The lysates were centrifuged at 12,000×g for 15 min. Levels of PGE2 (ARBOR ASSAYS), TNF-α, IL-1β, IL-6, IL-10, and IL-4 (MULTI SCIENCES) were assessed using the appropriate ELISA kits according to the manufacturer’s directions. Plates were read using a 96-well plate reader (Biotek Synergy2) at a 450 nm wavelength.
For determination of PGE2 and cytokines levels in the tissue samples, protein from 1 cm spinal segments of injured site at 0 day, 1 day, 4 days, and 1 week following contusion (n = 6 in each time point) was extracted with a buffer containing 1% SDS, 100 mM Tris-HCl, 1 mM PMSF, and 0.1 mM β-mercaptoethanol, respectively. The total protein of each sample was then diluted to final concentration at 0.5 μg/μl by the buffer before detection by ELISA kits.
Tissue immunohistochemistry
The vertebra segments were harvested from six experimental models of each time point, post-fixed, and sectioned. Sections were allowed to incubate with polyclonal COX2 antibody (1:100 dilution, Cayman), goat anti-IBA-1 antibody (1:200 dilution, Abcam), monoclonal mouse anti-S-100 (β-Subunit) antibody (1:1000 dilution, Sigma), or monoclonal mouse anti-human GFAP antibody (1:400 dilution, Sigma) at 4 °C for 36 h. The sections were further reacted with the FITC-labeled secondary antibody goat anti-mouse IgG (1:400 dilution, Gibco), the TRITC-labeled secondary antibody donkey anti-rabbit IgG (1:400 dilution, Gibco), or the 488-labeled secondary antibody donkey anti-goat IgG (1:400 dilution, Abcam) at 4 °C overnight, followed by observation under a confocal laser scanning microscope (Leica, Heidelberg, Germany).
Quantitative real-time polymerase chain reaction
Total RNA was prepared with Trizol (Gibco, USA) from macrophage RAW 264.7 treated with 1 μg/ml LPS for 24 h. The first-strand cDNA was synthesized using Omniscript Reverse Transcription Kit (QIAGEN) in a 20 μl reaction system containing 2 μg total RNA, 0.2 U/μl M-MLV reverse transcriptase, 0.5 mM dNTP mix, and 1 μM Oligo-dT primer. The cDNA was diluted 1:5 before use in quantitative real-time polymerase chain reaction (Q-PCR) assays. The sequence-specific primers were designed and synthesized by Invitrogen (Shanghai, China). Primer pairs for EP1: forward primer 5′-AAG CAG GCT GGC GAC GAA C-3′, reverse primer 5′-CCA ACA GGC GAT AAT GGC ACA-3′; for EP2, forward primer 5′-CTC GGA GGT CCC ACT TTT-3′, reverse primer 5′-GCG GAT TGT CTG GCA GTA-3′; for EP3, forward primer 5′-CGG TTG AGC AAT GCA AGA CA-3′, reverse primer 5′-GGT GGA GCT GGA AGC ATA GT-3′; for EP4, forward primer 5′-GAC AGC CAG CCC ACA TAC-3′, reverse primer 5′-GCG TCC TTC TCC TCC ACT-3′. Q-PCR reactions were performed in a final volume of 20 μl (1 μl cDNA template and 19 μl Q-PCR reaction buffer containing 2.5 mmol/L MgCl2, 0.2 mmol/L dNTPs, anti-sense and sense primers 0.5 μmol/L, Taqman probe 0.4 μmol/L, DNA polymerase 0.2 μl, and 1 × DNA polymerase buffer). The Rotor-Gene 5 software (Corbett Research, Rotor-Gene, Australia) was used for real-time PCR analysis. Reactions were processed using one initial denaturation cycle at 94 °C for 5 min followed by 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. Fluorescence was recorded during each annealing step. At the end of each PCR run, data were automatically analyzed by the system and amplification plots obtained. MIF full-length plasmid was used to prepare standard curves and used as a specificity control for real-time PCR. The expression levels were normalized to an endogenous β-actin. In addition, a negative control without the first-strand cDNA was also performed.
Statistical analysis
Statistical significance of differences between groups was analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc comparisons tests with SPSS 15.0 (SPSS, Chicago, IL, USA). Normality and homoscedasticity of the data were verified before any statistical analysis using Levene’s test. Statistical significance was set at p < 0.05.
Results
Activation of COX2/PGE2 signal pathway in the astrocytes is relevant to expression of MIF following SCI of rat
To examine whether MIF expression correlates to the PGE2 production of astrocytes following SCI, we firstly assayed protein levels of MIF, COX1, and COX2, as well as the isoforms of PGE2 synthase in the lesion sites following SCI at 0 day, 1 day, 4 days, and 7 days, respectively. Results showed that MIF, COX2, and microsomal PGE synthase-1 (mPGES-1), but not of COX1, mPGES-2, and cytosolic PGE synthase (cPGES), were inducibly expressed following SCI with a peak level at 4 days (Fig. 1a–e). Accordingly, the production of PGE2 increased synchronously, as detected by ELISA (Fig. 1f). Treatment of 8 μl of 100 mM MIF inhibitor 4-IPP at the lesion sites resulted in remarkable a decrease of COX2 and mPGES-1 protein levels (Fig. 1c–e), as well as a reduction of PGE2 production (Fig. 1f), whereas COX1, mPGES-2, and cPGES were kept unaffected (Fig. 1b, d).
Fig. 1.

Determination of MIF protein levels and PGE2 production following spinal cord injury. a Western blot analysis of MIF, COX1, and COX2 protein levels following spinal cord contusion at 0 day, 1 day, 4 days, and 7 days, respectively. b–e Determination of COX1 (b), COX2 (c), mPGES-1, mPGES-2, and cPGES (d, e) protein levels for the injured cord at different time points with or without injection of 8 μl 4-IPP (100 mM) at the lesion sites. Quantities were normalized to endogenous β-actin. f Production of PGE2 was assayed by ELISA accordingly. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
Immunostaining was then performed to understand specific cell types that respond to MIF stimulation. As the roles of COX2 in neurons have been well described previously [22, 35], we therefore sought to detect its colocalization with astrocytes and microglia. Results demonstrated that COX2 expression was significantly induced in GFAP- and S100β-positive astrocytes at 4 days following SCI (Fig. 2a–d, g–j). Distinctively, expression of COX2 was moderately upregulated in IBA-1-positive microglia following SCI (Additional file 1: Figure S1). Treatment of 4-IPP attenuated the expression of COX2 in the astrocytes (Fig. 2e, f, k, l). These data indicate that injury-induced MIF is able to activate COX2/PGE2 signal pathway of astrocytes following SCI.
Fig. 2.

Colocalization of COX2 with astrocytes. a–l Immunostaining of COX2 in the cross sections of rat contused spinal cord showed colocalization with GFAP- (a–f) and S100β- (g–l) positive cells at 0 day and 4 days, with or without injection of 4-IPP, respectively. Rectangle indicates region magnified. Arrowheads indicate colocalization of COX2 with astrocytes. Scale bars, 500 μm in (a), (c), (e), (g), (i), and (k); 50 μm in (b), (d), (f), (h), (j), and (l)
MIF facilitated expression of COX2 and production of PGE2 in primary cultured astrocytes
For better insight into the function of MIF on the regulation of COX2/PGE2 in astrocytes, primary astrocytes were thus isolated and cultured with purity more than 95% (Fig. 3a). Addition of recombinant MIF protein at concentration of 0–2.5 μg/ml to the astrocytes for 24 h resulted in a dose-dependent elevation of COX2 protein levels, but not of COX1 (Fig. 3b, c). Since the types of prostanoids produced after COX activation are involved in the action of specific prostanoid isomerases [36], mPGES-1, mPGES-2, and cPGES were also determined. Remarkably, mPGES-1, rather than mPGES-2 and cPGES, was induced by MIF treatment (Fig. 3b, d). Addition of 100 μM 4-IPP to the culture in the presence of 1 μg/ml recombinant MIF efficiently blocked the effects of the stimulation (Fig. 4a–d). Correspondingly, the production of PGE2 also showed an increase in a dose-dependent manner to MIF stimulation, and addition of 4-IPP was able to attenuate such effects (Fig. 4e, f). An insight of MIF effect on COX2 expression of primary cultured microglia obtained the similar results to those of the astrocytes (Additional file 1: Figure S1). These findings indicate that MIF induces PGE2 production in astrocytes in dose-dependent manners relevant to activation of COX2 and mPGES-1.
Fig. 3.

Determination of PGE2 synthesis-related protein levels in response to recombinant MIF stimulation. a Showing isolated astrocytes stained with GFAP and Hoechst 33342 with purity more than 95%. b Western blot analysis of COX1, COX2, mPGES-1, mPGES-2, and cPGES following astrocytes treatment with 0–2.5 μg/ml recombinant MIF for 24 h. c, d Quantification data as shown in (b). Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05). Scale bars, 50 μm in (a)
Fig. 4.
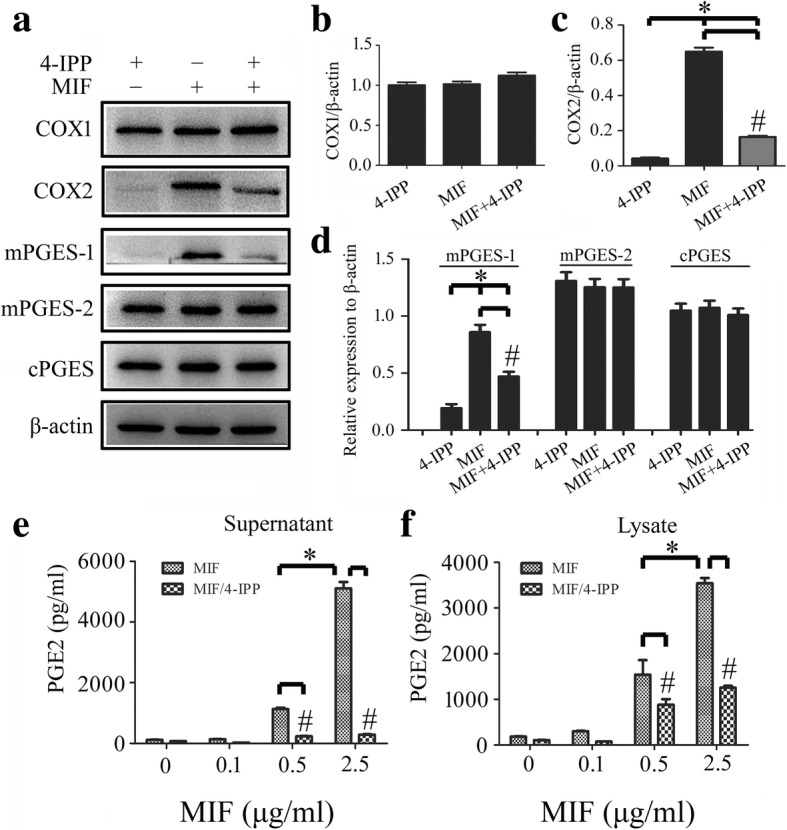
Effects of MIF inhibitor 4-IPP on the expression of PGE2 synthesis-related protein. a Western blot analysis of COX1, COX2, mPGES-1, mPGES-2, and cPGES following astrocytes treatment with 1 μg/ml recombinant MIF for 24 h in the presence of 100 μM 4-IPP. b–d Quantification data as shown in (a). e, f Cell supernatants (e) and lysates (f) were tested by ELISA for the production of PGE2, following astrocytes treatment with 0–2.5 μg/ml recombinant MIF for 24 h in the presence of 100 μM 4-IPP. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
Activation of COX2 was essential for MIF-induced production of PGE2
Now that the expression of COX2 and its downstream mPGES-1 in astrocytes was upregulated in response to stimulation of MIF, the activation of COX2 was possibly essential for the production of PGE2. Therefore, the selective COX2 inhibitor NS398 was used to examine the role of MIF in activation of COX2/PGE2 pathway. Addition of 30 μM NS398 to the culture in the presence of 1 μg/ml recombinant MIF remarkably decreased the protein levels of COX2 and mPGES-1 in astrocytes, but not of COX1, mPGES-2, and cPGES (Fig. 5a–c). Meanwhile, the production of PGE2 in response to MIF stimulation was significantly attenuated by NS398 (Fig. 5d, e). These data indicate that COX2 activation was indispensible for MIF-induced production of PGE2.
Fig. 5.
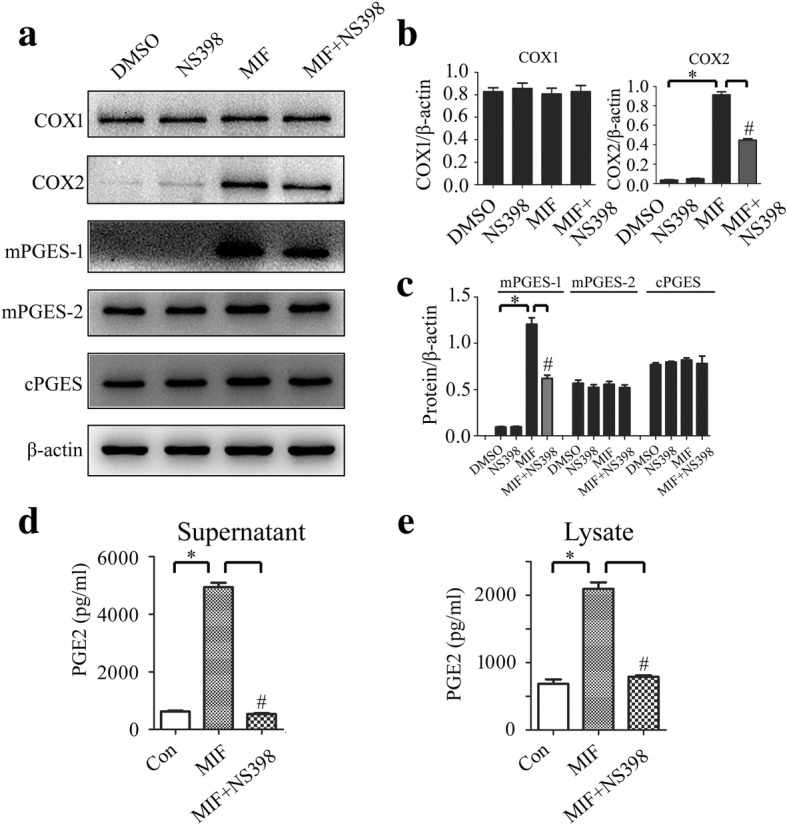
Effects of COX2 inhibitor NS398 on the MIF-induced expression of PGE2 synthesis-related protein. a Western blot analysis of COX1, COX2, mPGES-1, mPGES-2, and cPGES following astrocytes treatment with 1 μg/ml recombinant MIF for 24 h in the presence of 30 μM NS398. b, c Quantification data as shown in (a). d, e Cell supernatants (d) and lysates (e) were tested by ELISA for the production of PGE2, following astrocytes treatment with 1 μg/ml recombinant MIF for 24 h in the presence of 30 μM NS398. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
MIF activated COX2/PGE2 signal pathway through CD74 membrane receptor
Our previous studies have shown that MIF is able to interact with CD74 membrane receptor of astrocytes, which forms a receptor complex with CXCR2 and CXCR4 to elicit intracellular signaling [30]. To clarify whether this interaction can initiate the activation of COX2 and subsequent production of PGE2, the siRNA oligonucleotide (siRNA2) which showed nearly 60% knockdown efficiency of CD74 was selected to interfere expression of the receptor [30].
CD74 receptor of astrocyte was knocked down by siRNA2 for 48 h, followed by the cell treatment with 1 μg/ml recombinant MIF protein for 6 h or 12 h. Results showed that protein levels of COX2 and mPGES-1 remarkably decreased following CD74 interference, while those of COX1, mPGES-2, and cPGES exhibited no changes (Fig. 6a–c). Accordingly, production of PGE2 was significantly inhibited by CD74 siRNA following MIF stimulation for 6 h and 12 h, respectively (Fig. 6d, e). These findings indicate that MIF triggers COX2/PGE2 signaling through CD74 membrane receptor.
Fig. 6.
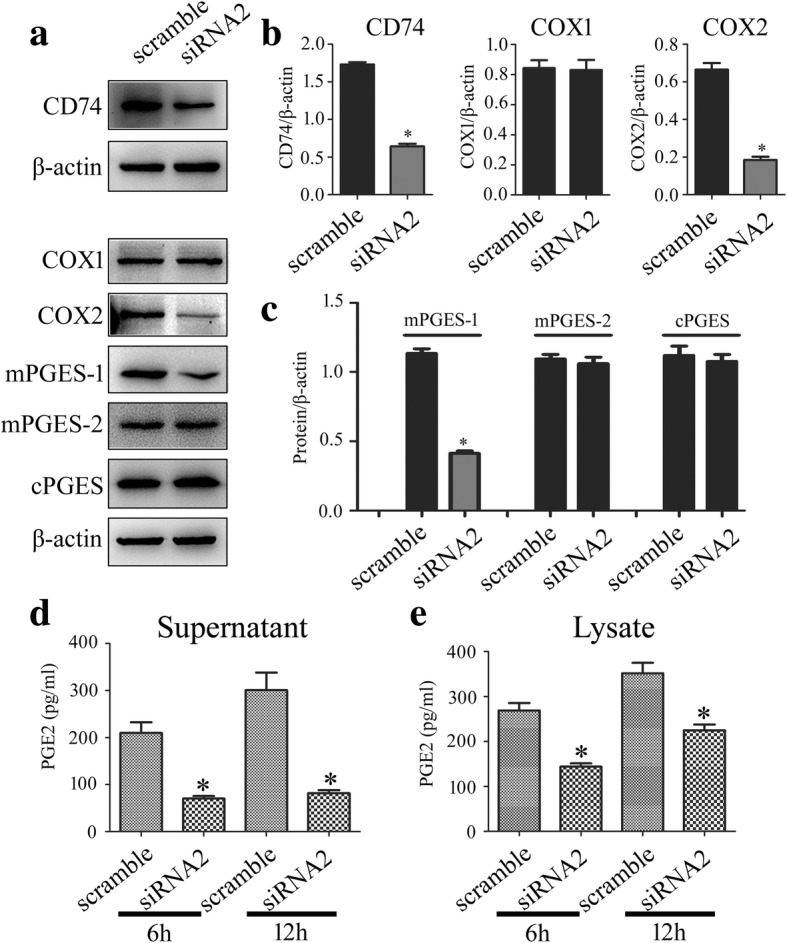
Effects of CD74 knockdown on the MIF-induced expression of PGE2 synthesis-related enzymes. a Western blot analysis of CD74, COX1, COX2, mPGES-1, mPGES-2, and cPGES following siRNA2 knockdown of CD74 receptor for 48 h, and then treated with 1 μg/ml recombinant MIF at 6 h and 12 h, respectively. A siRNA (scramble) with the same nucleotide composition as siRNA2 but which lacks sequence homology to the CD74 was also designed as negative control. b, c Quantification data as shown in (a). d, e Cell supernatants (d) and lysates (e) were tested by ELISA for the production of PGE2, following knockdown of CD74 receptor for 48 h, and then treated with 1 μg/ml recombinant MIF at 6 h and 12 h, respectively. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05)
MIF activated COX2 enzyme through MAPKs pathway
MIF has been shown to rapidly activate mitogen-activated protein kinases (MAPKs) signaling in fibroblasts, macrophages, and astrocytes [30, 37]. To unveil whether activation of COX2 by MIF was associated with MAPKs, inhibitor of JNK (SP600125), ERK (PD98059), or P38 (SB203580) was used to treat the primary astrocytes in the presence of 1 μg/ml recombinant MIF protein. Determination of PGE2 synthesis-related enzymes revealed that induction of COX2 and mPGES-1 by recombinant MIF protein was severely reduced by the inhibitor of JNK, ERK, or P38 (Fig. 7a–d). The production of PGE2 was also attenuated by these MAPK inhibitors (Fig. 7e, f). These data indicate that activation of COX2 by MIF is under regulation of MAPKs pathway.
Fig. 7.
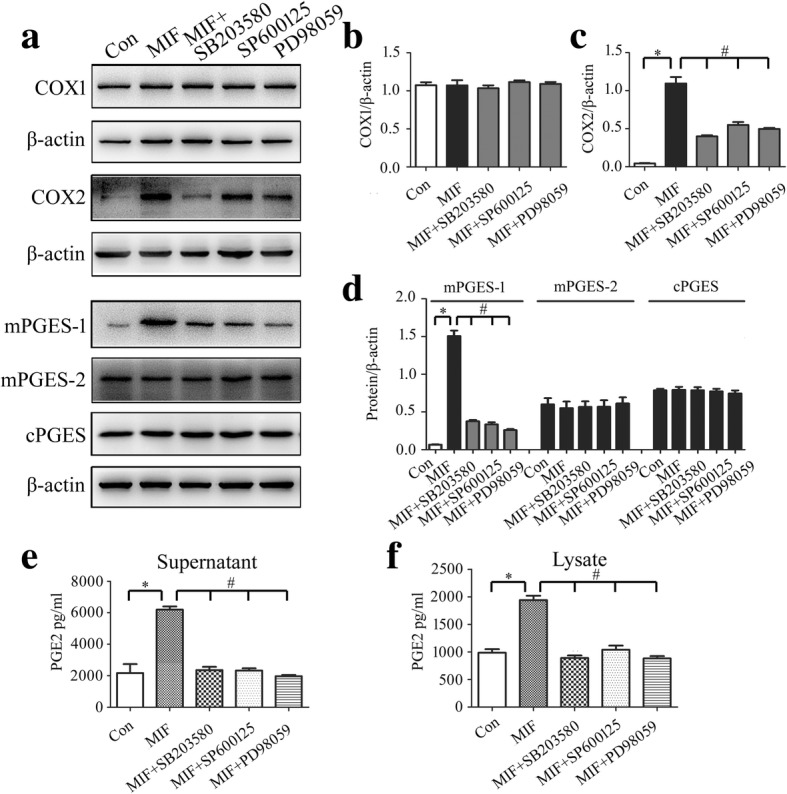
Determination of intracellular signaling associated with regulation of PGE2 production. a Western blot analysis of COX1, COX2, mPGES-1, mPGES-2, and cPGES following astrocytes treatment with 1 μg/ml recombinant MIF for 24 h in the presence of 10 μM ERK (PD98059), 10 μM P38 (SB203580), or 10 μM JNK (SP600125) inhibitor. b–d Quantification data as shown in (a). e, f Cell supernatants (e) and lysates (f) were tested by ELISA for the production of PGE2, following astrocytes treatment with 1 μg/ml recombinant MIF for 24 h in the presence of 10 μM ERK (PD98059), 10 μM P38 (SB203580), or 10 μM JNK (SP600125) inhibitor. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
PGE2 released by astrocytes exerted differential impact on the LPS-induced production of inflammatory cytokines
Several studies have shown that the production of PGE2 during inflammation negatively regulates the production of inflammatory cytokines and chemokines including TNF-α, IFN-β, CCL4, and CCL5 in immune cells [38–41]. To understand its influence on the production of injury-related inflammatory cytokines following SCI, macrophage RAW 264.7 cells were challenged with 1 μg/ml Lipopolysaccharide (LPS) for mimicking endogenous DAMPs that induce a broad-spectrum of cytokines through interaction with kinds of PRRs, with or without addition of different concentration of PGE2. Assay of cytokines by ELISA showed that PGE2 was able to suppress the production of TNF-α, but boost the production of IL-1β and IL6 in the activating macrophages in dose-dependent manner (Fig. 8a–f). Meanwhile, addition of PGE2 showed almost no influence on the production of anti-inflammatory cytokine IL-10 and IL-4, as observed from a slight increase of IL-10 protein level in the supernatant (Fig. 8g–j). Because LPS promoted the expression of EP2 receptor remarkably, the EP2-specific antagonist PF-04418948 was thus used to block PGE2 pathway (Fig. 9a). Results demonstrated that addition of 10 μM PF-04418948 could attenuate the effects of PGE2 on the production of TNF-α, IL-1β, and IL6 (Fig. 9b–d). These findings indicate that PGE2 released by astrocytes is able to change the production of proinflammatory cytokines in immune cells.
Fig. 8.

Effects of PGE2 on the production of proinflammatory and anti-inflammatory cytokines from LPS-induced macrophages. ELISA assay was used to determine TNF-α (a, b), IL-1β (c, d), IL-6 (e, f), IL-10 (g, h), or IL-4 (i, j) production in the supernatants and lysates of macrophage RAW 264.7 following treatment with 1 μg/ml LPS and 0–10 μM PGE2, respectively. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
Fig. 9.
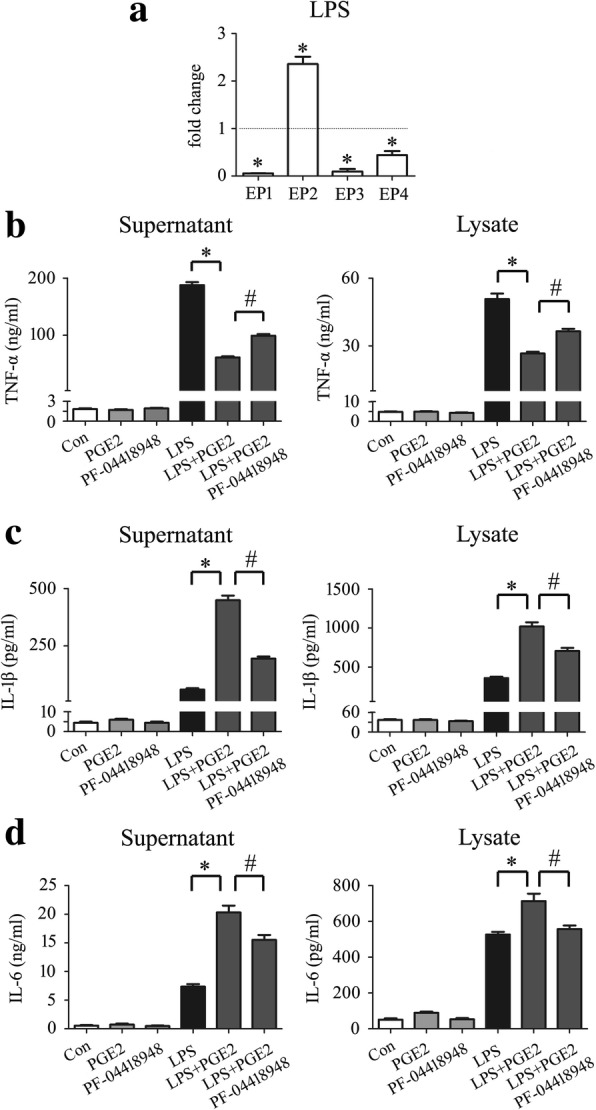
Blocking EP2 receptor attenuated PGE2-mediated function on production of inflammatory cytokines. a Expression analysis of PGE2 receptors by RT-PCR following macrophage treatment with 1 μg/ml LPS for 24 h; ELISA assay was used to determine TNF-α (b), IL-1β (c), or IL-6 (d) production in the supernatants and lysates of macrophage RAW 264.7 following treatment with 1 μg/ml LPS and 2.5 μM PGE2 in the presence or absence of 10 μM EP2 inhibitor PF-04418948. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
To further confirm that astrocyte-released PGE2 affects the production of proinflammatory cytokines in macrophages, we cultured astrocytes in the presence of 1 μg/ml recombinant MIF with or without 30 μM NS398 for 24 h. The conditioned medium (CM) from astrocytes-free, COX2 inhibition, or non-inhibition was then collected for macrophage culture in the presence of 1 μg/ml LPS at 24 h. We found that COX2-inhibited CM could increase the production of TNF-α, but significantly decreased the production of IL-1β and IL6 (Fig. 10a–f). These strengthen the conclusion that PGE2 influences production of proinflammatory cytokines in macrophages.
Fig. 10.

Effects of COX2 inhibitor NS398 on the production of inflammatory cytokines from macrophages in vitro or at spinal cord lesioned site. a–f ELISA assay determined production of TNF-α (a, b), IL-1β (c, d), and IL-6 (e, f) following macrophages cultured with astrocytes-conditioned medium in the presence of 1 μg/ml LPS for 24 h. The astrocytes were cultured in the presence of 1 μg/ml recombinant MIF with or without 30 μM NS398 for 24 h. g Determination of PGE2 production at lesion sites of spinal cord following injection of 10 μl 80 mM NS398 at 0 day, 1 day, 4 days, and 7 days, respectively. h, i ELISA assay determined production of TNF-α (h) and IL-1β (i) at lesion sites following injection of 10 μl 80 mM NS398 at 0 day, 1 day, 4 days, and 7 days, respectively. Experiments were performed in triplicates. Error bars represent the standard deviation (*P < 0.05; #P < 0.05)
To address the pathophysiological function of PGE2 following SCI, 10 μl of 80 mM COX2 inhibitor NS398 was injected at lesion sites. Treatment of NS398 significantly decreased PGE2 production of injured cord (Fig. 10g). However, the protein level of TNF-α increased, and contrarily, that of IL-1β decreased (Fig. 10h, i). These data indicate that COX2 inhibitor tunes the inflammatory microenvironment through blocking PGE2 production.
Discussion
Following acute CNS injury, CNS elicits a coordinated multicellular inflammatory response that involves glia, neurons, as well as various immune cells [15]. The interaction between these cells constitutes a complex regulatory network of inflammation. For instance, the resident microglia and infiltrating immune cells have been implicated in driving astrocyte-mediated inflammation, while cytokine-stimulated astrocytes also induce migration of immune cells toward lesion sites [42, 43]. It is interesting to note that injured CNS has potential to maximize the preservation of healthy tissue and restricting the spread of cytotoxic inflammation [44]. Astrocytes, the important modulator of milieu, are able to provide benefits to the CNS by phagocytosis of synapses, secretion of neurotrophins, clearance of debris, repair of the BBB, as well as formation of a scar to enclose the necrotic lesion and restrict cytotoxic inflammation [45]. Recently, the metabolites of arachidonic acid lipoxins A4 and B4 have been found to be secreted by astrocytes to promote neuroprotection from acute and chronic injury [46]. In the present study, we have presented that astrocyte-derived PGE2 is able to change the components of inflammatory microenvironment, suggesting the dynamic effects of astrocytes on inflammatory homeostasis following SCI.
MIF is constitutively expressed in a variety of immune and non-immune cells of different histogenetic origin. It shows a remarkable functional diversity ranging from activating the production of inflammatory cytokines including TNF-α, IL-1β, IL-6, and IFN-γ to inhibiting p53-mediated cell apoptosis [47]. Given such broad activities, it is not surprising that this pluripotent and pleiotropic cytokine is implicated in acute and chronic inflammatory diseases such as rheumatoid arthritis, asthma, diabetes, sepsis, cardiovascular diseases, and cancer [47–49]. During progression of cancer, MIF has the ability to change tumor microenvironment favorable for tumor aggressiveness. MIF promotes alternative macrophage differentiation that lead to formation of tumor-associated macrophages (TAMs). These TAMs are abundant in the tumor environment and act roles of immunosuppression [50]. In the central nervous system, MIF is inducibly expressed in neurons within the hypothalamus, cortex, and hippocampus to modulate nitric oxide production as well as catecholamine metabolism [51]. Also, it induces neuronal death after compression-induced spinal cord injury [29]. We have shown that MIF is capable of promoting release of chemokine CCL5 from astrocytes, which in turn primarily promote migration of M2-macrophages. In the present study, we displayed that MIF could induce PGE2 release from astrocytes to change production of inflammatory cytokines, suggesting multiple mechanisms of MIF in modulation of inflammatory microenvironment following SCI.
PGE2 has been found to play a wide range of roles in acute and chronic injury of CNS [52], such as contributing to excitotoxic and ischemic neuronal cell death [53], or neuronal protection [54, 55], depending on its interacting receptor(s). With respect to its function in modulating inflammation in the context of pathophysiological conditions, PGE2 has been demonstrated to exert both proinflammatory and immunodepressive actions. During the acute stage of the inflammatory response, PGE2 acts as a vasodilator and facilitates tissue infiltration of neutrophils [56], macrophages [57], as well as being a regulator of nociception [58]. However, mounting evidence has also shown that PGE2 modulates macrophage activation in part by suppressing the release of cytokines and/or chemokines. For example, PGE2 is a negative regulator for LPS-induced production of TNF-α in Epac/PKA-, of IFN-β in Epac/PI3K/Akt-dependent signaling in activated macrophages and during endotoxemia [40, 41]. Within the dying cells, PGE2 is induced and released to function as an inhibitory DAMP for inhibiting the expression of genes associated with inflammation, thereby limiting the cell’s immunostimulatory activities [59]. It is noteworthy that PGE2 plays an opposing role on IL-1β production from monocytes/macrophages. In primary human monocytes, PGE2 boosted LPS-induced IL-1β production [60]. While in human macrophages, PGE2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cAMP, leading to reduction in IL-1β secretion [61]. In the present study, we displayed that PGE2 attenuated LPS-induced TNF-α, but promoted IL-1β and IL-6 production in macrophage RAW 264.7 cells, suggesting its cell-specific influence on the production of inflammatory cytokines. Although PGE2-mediated effects have been found to associate with the resolution of acute inflammation [24], facilitation of tissue regeneration [62], or aggravation of the disease phenotype such as chronic inflammation or cancer [63], its contribution in the injured cord by altering the production of proinflammatory cytokines from macrophages deserves further study. Because reactive astrocytes have potential roles on promoting and restricting inflammation following CNS injury [45], PGE2-released by astrocytes might play a role in tuning the microenvironment to maintain homeostasis following SCI.
Conclusions
Injury-induced MIF activated COX2 expression of astrocytes through binding with CD74 receptor following spinal cord contusion, which in turn promoted the production of PGE2. PGE2 in turn changes the components of inflammatory microenvironment to maintain homeostasis under neuropathological conditions.
Additional file
Figure S1. Colocalization of COX2 with IBA-1-positive microglia following spinal cord contusion at 0d and 4d with or without treatment of 4-IPP, as well as examination of 4-IPP effects on COX2 expression. (PDF 187 kb)
Acknowledgements
No additional acknowledgements.
Funding
This study was supported by the National Natural Science Foundation of China (No. 31830028; 31871211; 31471011), the National Key Research and Development Program of China (2016YFC1101600; 2018YFC1105603), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AA
Arachidonic acid
- ANOVA
Analysis of variance
- COX
Cyclooxygenases
- DAMPs
Damage-associated molecular patterns
- ELISA
Enzyme-linked immunosorbent assay
- GFAP
Glial fibrillary acid protein
- LPS
Lipopolysaccharide
- MAPK
Mitogen-activated protein kinases
- MIF
Macrophage migration inhibitory factor
- mPGES-1
Microsomal PGE synthase-1
- MS
Multiple sclerosis
- PBS
Phosphate-buffered saline
- PGE2
Prostaglandin E2
- SCI
Spinal cord injury
Authors’ contributions
YoW and AG designed this work. YoW wrote the paper. YZ, YZ, and SC performed the experiments. YZ, YZ, SC, YoW, AG, ZZ, YH, YiW, ND, and TS analyzed the data. AG and YY joined discussions. All authors have approved the present version of the manuscript and have agreed to be accountable for all aspects of the work regarding questions related to the accuracy or integrity of any part of the work.
Ethics approval and consent to participate
All animal experiments were approved by the Animal Care and Use Committee of Nantong University and the Jiangsu Province Animal Care Ethics Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yuxin Zhang, Email: 603083381@qq.com.
Yue Zhou, Email: 1026054860@qq.com.
Shuxia Chen, Email: chenshuxia999@126.com.
Yuming Hu, Email: 13515215105@163.com.
Zhenjie Zhu, Email: zhuzhenjie@163.com.
Yingjie Wang, Email: wyj2010@ntu.edu.cn.
Nan Du, Email: dunan0713@sina.com.
Tiancheng Song, Email: 784064751@qq.com.
Yumin Yang, Email: yangym@ntu.edu.cn.
Aisong Guo, Email: 15851300188@163.com.
Yongjun Wang, Phone: 0086-513-85051818, Email: wyjbs@ntu.edu.cn.
References
- 1.Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- 2.Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- 3.Gadani SP, Walsh JT, Lukens JR, Kipnis J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron. 2015;87:47–62. doi: 10.1016/j.neuron.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 6.Chen KB, Uchida K, Nakajima H, Yayama T, Hirai T, Rodriguez Guerrero A, Kobayashi S, Ma WY, Liu SY, Zhu P, Baba H. High-mobility group box-1 and its receptors contribute to proinflammatory response in the acute phase of spinal cord injury in rats. Spine (Phila Pa 1976) 2011;36:2122–2129. doi: 10.1097/BRS.0b013e318203941c. [DOI] [PubMed] [Google Scholar]
- 7.Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia. 2005;49:360–374. doi: 10.1002/glia.20117. [DOI] [PubMed] [Google Scholar]
- 8.Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 10.Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, Shapiro A, Antel JP. TLR signaling tailors innate immune responses in human microglia and astrocytes. Immunol. 2005;175:4320–4330. doi: 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- 11.Mohsenzadegan M, Fayazi MR, Abdolmaleki M, Bakhshayesh M, Seif F, Mousavizadeh K. Direct immunomodulatory influence of IFN-β on human astrocytoma cells. Immunopharmacol Immunotoxicol. 2015;37:214–219. doi: 10.3109/08923973.2015.1014559. [DOI] [PubMed] [Google Scholar]
- 12.Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia. 2006;53:688–695. doi: 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- 13.Li XL, Lv J, Xi NN, Wang T, Shang XF, Xu HQ, Han Z, O'Byrne KT, Li XF, Zheng RY. Neonatal endotoxin exposure suppresses experimental autoimmune encephalomyelitis through regulating the immune cells responsivity in the central nervous system of adult rats. Biochem Biophys Res Commun. 2010;398:302–308. doi: 10.1016/j.bbrc.2010.06.086. [DOI] [PubMed] [Google Scholar]
- 14.Rothhammer V, Quintana FJ. Control of autoimmune CNS inflammation by astrocytes. Semin Immunopathol. 2015;37:625–638. doi: 10.1007/s00281-015-0515-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Raposo C, Schwartz M. Glial scar and immune cell involvement in tissue remodeling and repair following acute CNS injuries. Glia. 2014;62:1895–1904. doi: 10.1002/glia.22676. [DOI] [PubMed] [Google Scholar]
- 17.Hewett SJ, Bell SC, Hewett JA. Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacol Ther. 2006;112:335–357. doi: 10.1016/j.pharmthera.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 18.Rollason V, Samer CF, Daali Y, Desmeules JA. Prediction by pharmacogenetics of safety and efficacy of non-steroidal anti-inflammatory drugs: a review. Curr Drug Metab. 2014;15:326–343. doi: 10.2174/1389200215666140202214454. [DOI] [PubMed] [Google Scholar]
- 19.Shojo H, Borlongan CV, Mabuchi T. Genetic and histological alterations reveal key role of prostaglandin synthase and cyclooxygenase 1 and 2 in traumatic brain injury-induced Neuroinflammation in the cerebral cortex of rats exposed to moderate fluid percussion injury. Cell Transplant. 2017;26:1301–1313. doi: 10.1177/0963689717715169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U S A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, Panikashvili D, Shohami E, Jansen SA, Narayan RK, Strauss KI. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strauss KI, Marini AM. Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated cell death. J Neurotrauma. 2002;19:627–638. doi: 10.1089/089771502753754091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodríguez M, Domingo E, Municio C, Alvarez Y, Hugo E, Fernández N, Sánchez Crespo M. Polarization of the innate immune response by prostaglandin E2: a puzzle of receptors and signals. Mol Pharmacol. 2014;85:187–197. doi: 10.1124/mol.113.089573. [DOI] [PubMed] [Google Scholar]
- 24.Hellmann J, Zhang MJ, Tang Y, Rane M, Bhatnagar A, Spite M. Increased saturated fatty acids in obesity alter resolution of inflammation in part by stimulating prostaglandin production. J Immunol. 2013;191:1383–1392. doi: 10.4049/jimmunol.1203369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strassmann G, Patil-Koota V, Finkelman F, Fong M, Kambayashi T. Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. J Exp Med. 1994;180:2365–2370. doi: 10.1084/jem.180.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maślińska D, Woźniak R, Kaliszek A, Modelska I. Expression of cyclooxygenase-2 in astrocytes of human brain after global ischemia. Folia Neuropathol. 1999;37:75–79. [PubMed] [Google Scholar]
- 27.Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Pérdomo A, Márquez-Kisinousky L, Santalucia T, Planas AM. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J Biol Chem. 2012;287:6454–6468. doi: 10.1074/jbc.M111.327874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishio Y, Koda M, Hashimoto M, Kamada T, Koshizuka S, Yoshinaga K, Onodera S, Nishihira J, Okawa A, Yamazaki M. Deletion of macrophage migration inhibitory factor attenuates neuronal death and promotes functional recovery after compression-induced spinal cord injury in mice. Acta Neuropathol. 2009;117:321–328. doi: 10.1007/s00401-008-0476-x. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y, Guo W, Zhu Z, Hu Y, Wang Y, Zhang X, Wang W, Du N, Song T, Yang K, Guan Z, Wang Y, Guo A. Macrophage migration inhibitory factor facilitates production of CCL5 in astrocytes following rat spinal cord injury. J Neuroinflammation. 2018;15:253. doi: 10.1186/s12974-018-1297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cox GM, Kithcart AP, Pitt D, Guan Z, Alexander J, Williams JL, Shawler T, Dagia NM, Popovich PG, Satoskar AR, Whitacre CC. Macrophage migration inhibitory factor potentiates autoimmune-mediated neuroinflammation. J Immunol. 2013;191:1043–1054. doi: 10.4049/jimmunol.1200485. [DOI] [PubMed] [Google Scholar]
- 32.Wang F, Wu H, Xu S, Guo X, Yang J, Shen X. Macrophage migration inhibitory factor activates cyclooxygenase 2-prostaglandin E2 in cultured spinal microglia. Neurosci Res. 2011;71:210–218. doi: 10.1016/j.neures.2011.07.1821. [DOI] [PubMed] [Google Scholar]
- 33.Chehrehasa F, Cobcroft M, Young YW, Mackay-Sim A, Goss B. An acute growth factor treatment that preserves function after spinal cord contusion injury. J Neurotrauma. 2014;31:1807–1813. doi: 10.1089/neu.2013.3294. [DOI] [PubMed] [Google Scholar]
- 34.Liu J, Du L. PERK pathway is involved in oxygen-glucose-serum deprivation-induced NF-kB activation via ROS generation in spinal cord astrocytes. Biochem Biophys Res Commun. 2015;467:197–203. doi: 10.1016/j.bbrc.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 35.Ghilardi JR, Svensson CI, Rogers SD, Yaksh TL, Mantyh PW. Constitutive spinal cyclooxygenase-2 participates in the initiation of tissue injury-induced hyperalgesia. J Neurosci. 2004;2:2727–2732. doi: 10.1523/JNEUROSCI.5054-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johann S, Kampmann E, Denecke B, Arnold S, Kipp M, Mey J, Beyer C. Expression of enzymes involved in the prostanoid metabolism by cortical astrocytes after LPS-induced inflammation. J Mol Neurosci. 2008;34:177–185. doi: 10.1007/s12031-007-9028-4. [DOI] [PubMed] [Google Scholar]
- 37.Lue H, Kapurniotu A, Fingerle-Rowson G, Roger T, Leng L, Thiele M, Calandra T, Bucala R, Bernhagen J. Rapid and transient activation of the ERK MAPK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on JAB1/CSN5 and Src kinase activity. Cell Signal. 2006;18:688–703. doi: 10.1016/j.cellsig.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Qian X, Zhang J, Liu J. Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway. J Biol Chem. 2011;286:2111–2120. doi: 10.1074/jbc.M110.154971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jing H, Vassiliou E, Ganea D. Prostaglandin E2 inhibits production of the inflammatory chemokines CCL3 and CCL4 in dendritic cells. J Leukoc Biol. 2003;74:868–879. doi: 10.1189/jlb.0303116. [DOI] [PubMed] [Google Scholar]
- 40.Kunkel SL, Wiggins RC, Chensue SW, Larrick J. Regulation of macrophage tumor necrosis factor production by prostaglandin E2. Biochem Biophys Res Commun. 1986;137:404–410. doi: 10.1016/0006-291X(86)91224-6. [DOI] [PubMed] [Google Scholar]
- 41.Xu XJ, Reichner JS, Mastrofrancesco B, Henry WL, Jr, Albina JE. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol. 2008;180:2125–2131. doi: 10.4049/jimmunol.180.4.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams A, Piaton G, Lubetzki C. Astrocytes—friends or foes in multiple sclerosis? Glia. 2007;55:1300–1312. doi: 10.1002/glia.20546. [DOI] [PubMed] [Google Scholar]
- 44.Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Livne-Bar I, Wei J, Liu HH, Alqawlaq S, Won GJ, Tuccitto A, Gronert K, Flanagan JG, Sivak JM. Astrocyte-derived lipoxins A4 and B4 promote neuroprotection from acute and chronic injury. J Clin Invest. 2017;127:4403–4414. doi: 10.1172/JCI77398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schindler L, Dickerhof N, Hampton MB, Bernhagen J. Post-translational regulation of macrophage migration inhibitory factor: basis for functional fine-tuning. Redox Biol. 2018;15:135–142. doi: 10.1016/j.redox.2017.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morand EF, Leech M, Bernhagen J. MIF: a new cytokine link between rheumatoid arthritis and atherosclerosis. Nat Rev Drug Discov. 2006;5:399–410. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 50.Yaddanapudi K, Putty K, Rendon BE, Lamont GJ, Faughn JD, Satoskar A, Lasnik A, Eaton JW, Mitchell RA. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J Immunol. 2013;190:2984–2993. doi: 10.4049/jimmunol.1201650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fingerle-Rowson GR, Bucala R. Neuroendocrine properties of macrophage migration inhibitory factor (MIF) Immunol Cell Biol. 2001;79:368–375. doi: 10.1046/j.1440-1711.2001.01024.x. [DOI] [PubMed] [Google Scholar]
- 52.Tan Y, Yu L, Zhang C, Chen K, Lu J, Tan L. miRNA-146a attenuates inflammation in an in vitro spinal cord injury model via inhibition of TLR4 signaling. Exp Ther Med. 2018;16:3703–3709. doi: 10.3892/etm.2018.6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimamura M, Zhou P, Casolla B, Qian L, Capone C, Kurinami H, Iadecola C, Anrather J. Prostaglandin E2 type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab. 2013;33:1207–1214. doi: 10.1038/jcbfm.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akaike A, Kaneko S, Tamura Y, Nakata N, Shiomi H, Ushikubi F, Narumiya S. Prostaglandin E2 protects cultured cortical neurons against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res. 1994;663:237–243. doi: 10.1016/0006-8993(94)91268-8. [DOI] [PubMed] [Google Scholar]
- 56.Desouza IA, Franco-Penteado CF, Camargo EA, Lima CS, Teixeira SA, Muscará MN, De Nucci G, Antunes E. Inflammatory mechanisms underlying the rat pulmonary neutrophil influx induced by airway exposure to staphylococcal enterotoxin type a. Br J Pharmacol. 2005;146:781–791. doi: 10.1038/sj.bjp.0706393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tajima T, Murata T, Aritake K, Urade Y, Hirai H, Nakamura M, Ozaki H, Hori M. Lipopolysaccharide induces macrophage migration via prostaglandin D(2) and prostaglandin E(2) J Pharmacol Exp Ther. 2008;326:493–501. doi: 10.1124/jpet.108.137992. [DOI] [PubMed] [Google Scholar]
- 58.Natura G, Bär KJ, Eitner A, Boettger MK, Richter F, Hensellek S, Ebersberger A, Leuchtweis J, Maruyama T, Hofmann GO, Halbhuber KJ, Schaible HG. Neuronal prostaglandin E2 receptor subtype EP3 mediates antinociception during inflammation. Proc Natl Acad Sci U S A. 2013;110:13648–13653. doi: 10.1073/pnas.1300820110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hangai S, Ao T, Kimura Y, Matsuki K, Kawamura T, Negishi H, Nishio J, Kodama T, Taniguchi T, Yanai H. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci U S A. 2016;113:3844–3849. doi: 10.1073/pnas.1602023113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zasłona Z, Pålsson-McDermott EM, Menon D, Haneklaus M, Flis E, Prendeville H, Corcoran SE, Peters-Golden M, O'Neill LAJ. The induction of pro-IL-1β by lipopolysaccharide requires endogenous prostaglandin E2 production. J Immunol. 2017;198:3558–3564. doi: 10.4049/jimmunol.1602072. [DOI] [PubMed] [Google Scholar]
- 61.Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, Alsaaty S, Park YH, Kastner DL, Chae JJ, Shelhamer JH. Prostaglandin E2 inhibits NLRP3 Inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages. J Immunol. 2015;194:5472–5487. doi: 10.4049/jimmunol.1401343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kvirkvelia N, McMenamin M, Chaudhary K, Bartoli M, Madaio MP. Prostaglandin E2 promotes cellular recovery from established nephrotoxic serum nephritis in mice, prosurvival, and regenerative effects on glomerular cells. Am J Physiol Ren Physiol. 2013;304:F463–F470. doi: 10.1152/ajprenal.00575.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013;35:123–137. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Colocalization of COX2 with IBA-1-positive microglia following spinal cord contusion at 0d and 4d with or without treatment of 4-IPP, as well as examination of 4-IPP effects on COX2 expression. (PDF 187 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.