

Abstract
Many trees form ectomycorrhizal symbiosis with fungi. During symbiosis, the tree roots supply sugar to the fungi in exchange for nitrogen, and this process is critical for the nitrogen and carbon cycles in forest ecosystems. However, the extents to which ectomycorrhizal fungi can liberate nitrogen and modify the soil organic matter and the mechanisms by which they do so remain unclear since they have lost many enzymes for litter decomposition that were present in their free-living, saprotrophic ancestors. Using time-series spectroscopy and transcriptomics, we examined the ability of two ectomycorrhizal fungi from two independently evolved ectomycorrhizal lineages to mobilize soil organic nitrogen. Both species oxidized the organic matter and accessed the organic nitrogen. The expression of those events was controlled by the availability of glucose and inorganic nitrogen. Despite those similarities, the decomposition mechanisms, including the type of genes involved as well as the patterns of their expression, differed markedly between the two species. Our results suggest that in agreement with their diverse evolutionary origins, ectomycorrhizal fungi use different decomposition mechanisms to access organic nitrogen entrapped in soil organic matter. The timing and magnitude of the expression of the decomposition activity can be controlled by the below-ground nitrogen quality and the above-ground carbon supply.
Subject terms: Microbial ecology, Biogeochemistry, Fungi
Introduction
A large portion of nitrogen (N) in forest soils is found in organic form, primarily as amides and amines, but also as heterocyclic-N molecules [1]. These N molecules are associated with polyphenols, polysaccharides, lignin residues, lipids, and other degradation products of plant and microbial origin that are present in the soil organic matter (SOM) [2]. The capacity of forest trees to assimilate organic N is limited [3]. Access to organic N sources, such as proteins or chitin, requires decomposition to make the organic N molecules accessible and, subsequently, to liberate N from those molecules. Plants are generally thought to depend on microbial decomposition to access the soil N [4]. A long-standing hypothesis proposes that the ectomycorrhizal (ECM) fungal symbionts have a key role in this process [5, 6]; however, the extent of the involvement of ECM fungi in SOM decomposition and mobilization of N compounds is debated [7, 8].
ECM fungi evolved several times from saprotrophic ancestors [9, 10]. These ancestors probably utilized diverse decomposition strategies resembling those seen in white-rot (WR) wood decayers, which use an enzymatic system for the decomposition of lignocellulose; brown-rot (BR) wood decayers, which utilize a two-step mechanism involving hydroxyl radicals (∙OH) generated by Fenton chemistry and hydrolytic enzymes [11]; and litter decomposers, which presumably use enzymatic decomposition systems similar to those of WR fungi [12]. During the transition from saprotrophic to symbiotic lifestyle, ECM fungi lost a large number of plant cell wall-degrading enzymes (PCWDEs) [9, 10]. The convergent gene losses in relation to PCWDEs seen in ECM lineages have been used as an argument against a major role of ECM fungi in SOM decomposition [8]. However, ECM lineages have lost many, but not all, genes coding for PCWDEs, with diverse types and numbers of genes related to decomposition retained across lineages [10]. The high variability of the retained PCWDE-coding genes and the diverse evolutionary backgrounds of ECM lineages suggest that ECM fungi could have also retained and adapted some features of the decomposition mechanisms to the symbiotic lifestyle [13].
To what extent ECM fungi make use of the remaining decomposition systems is not well understood. At least some ECM fungi oxidize organic matter in a SOM extract in the presence of an energy source (i.e., glucose) [13, 14]. Furthermore, ECM Cortinarius species encode ligninolytic class-II peroxidases, whose gene transcription correlates with the peroxidase activity in the boreal forest soil, supporting the hypothesis that these species may play an important role in SOM decomposition [15]. In addition, decomposition activities of ECM fungi have been inferred by ecological studies that relied on enzymatic assays detecting the activity of various hydrolytic and oxidative PCWDEs [16, 17]. However, one limitation of such studies is that the assays are performed with ECM root tips and not the mycelium colonizing the soil substrate. Several of those assays, in particular the ones probing for oxidative enzyme activity, are unspecific and do not properly capture the decomposition activity [18]. Alternative hypotheses for the role of the detected enzymes include the decomposition of dead root tips [18] and remodeling of the root cell wall during host colonization [9, 19, 20].
Additionally, the environmental cues that regulate SOM decomposition in ECM fungi are not known. Laboratory experiments revealed that the oxidative decomposition system in the ECM fungus Paxillus involutus is expressed only in the presence of an energy source (i.e., glucose) [21]. In contrast, field studies based on enzyme assays suggest that ECM fungi can produce PCWDEs and metabolize SOM when the amount of carbon (C) supplied by the host plant is low [16]. Moreover, it is not known if the expression of the decomposition system of ECM fungi and the liberation of organic N compounds are concurrent. If so, the two processes might be regulated in conjunction and by similar nutritional signals, including the availability of inorganic and organic N sources.
To address these questions, we used time-series spectroscopy and transcriptomics to analyze two species of ECM fungi with independent evolutionary histories and contrasting growth characteristics. P. involutus is characterized by a rapidly growing mycelium, and a so-called long-distance exploration type [22]. The species is nested within a paraphyletic assemblage of BR wood decayers in the Boletales [10], and oxidizes SOM using a nonenzymatic Fenton-based system [14, 23]. By contrast, Laccaria bicolor develops a slow-growing, medium-distance smooth exploration subtype mycelium [22]. It belongs in the Agaricales and probably evolved from litter-decomposing saprotrophs [10]. The L. bicolor set of enzymes presumably involved in the degradation of PCW derived polymers is larger than that of P. involutus; several of these enzymes are expressed during growth on a SOM extract [10, 13].
Materials and methods
Fungal strains and culture conditions
Cultures of P. involutus (Batsch) Fr. (ATCC 200175) and L. bicolor (Maire) P.D. Orton (S238N) were grown on a layer of glass beads in Petri dishes containing a liquid minimum Melin-Norkrans (MMN) medium for 9 and 18 d, respectively (18 °C, in the dark) [14]. The medium was then replaced with MMN medium without N to induce the formation of an N-deprived mycelium. After 24 h, the mycelium was washed with sterile water and the SOM extract was added. The extract was supplemented with glucose to a final concentration similar to that in the MMN medium [14]. The soil was collected to a depth of 10 cm after removing recently fallen and partly decomposed litter from the top, in a 61-y-old Norway spruce stand growing in an N-poor site in central Sweden (soil pH, 5.0). It mainly consists of well-decomposed humus material with occasional occurrence of mineral soil. According to [24], mycelia of ECM fungi dominate therein.
SOM was extracted using hot water [25]. Further experimental details are given in the Supplementary Information.
Chemical analyses
Total organic C, glucose, total N, and ammonium-N were determined as previously described [13]. Fourier-transform infrared (FTIR) spectra were recorded in diffuse reflectance mode on mixtures of freeze-dried samples and KBr (2% w/w) using a Vertex 80v spectrometer (Bruker Optics, Ettlingen, Germany). Each spectrum was the result of 128 consecutive scans at a resolution of 4 cm−1. KBr was used as background. The IR spectral data sets were analyzed by means of a multivariate curve resolution-alternating least squares method [26, 27]. Pyrolysis-gas chromatography/mass spectrometry was performed using a PerkinElmer Turbo-Mass/Autosystem XL with Frontier Lab Double-Shot Pyrolyser (PerkinElmer, Waltham, MA, USA). The ratio of 4-acetylguaiacol to trans-propenylguaiacol (Ox/C3-G) was used as a marker of the degree of side-chain degradation of lignin [28].
The Fe K-edge XANES spectra were collected at beamline I811, MaxLab, Lund, Sweden. The X-ray absorption spectra were processed and analyzed using the software SIXPack [29]. In order to obtain the redox state of Fe, the pre-edge peak was examined as described by Wilke et al. [30]. The centroid position of this peak is indicative of the Fe redox state. The integrated area and the centroid were calculated and one-way ANOVA was used for the statistical analysis of the centroids to establish differences between treatments.
The N K-edge XANES spectra (390–430 eV) were collected at the beamline 11ID-1 (SGM), Canadian Light Source, Saskatoon, Canada. The organic matter extract was poured on indium foil and dried at 30 °C. The main peaks of the XANES spectra were assigned by comparison with spectra of model compounds found in the literature [31]. In order to semiquantitatively compare the spectra, an ordination method (principal component analysis) based on the relative abundance of π* transitions (a–d) was used to find differences in the changes of organic N speciation between fungi. Further experimental details are provided in the Supplementary Information.
Transcriptome analysis
Total RNA was isolated from the mycelium, and cDNA libraries were constructed as previously described [13]. The libraries were sequenced (RNA-Seq) using a HiSeq2000 instrument (Flow Cell v3) (Illumina Inc., San Diego, CA, USA) in single-read mode and with a read length of 50 bp (IGA Technology Services, Italy; www.igatechnology.com). Sequence reads were aligned to filtered gene models of P. involutus (ver. 1.0) [10] and L. bicolor (ver. 2.0) [9] using TopHat2 (ver. 2.0.13) [32]. Expression counts were determined using HTSeq (ver. 0.6.1) [33], and normalization factors were estimated using EDASeq (ver. 2.8.0) [34] and DESeq2 (ver. 1.14.1) [35]. Differentially expressed genes (DEGs) were identified using DESeq2, by testing for overall changes in expression during the time-course of the experiment using a likelihood ratio test, and for pairwise comparisons of (t2–t4) vs. t1 using a Wald test. P values were adjusted (padj) for multiple testing using the Benjamini-Hochberg method [36] to control the false discovery rate with a significance cutoff of 0.01. KOG and KEGG enrichment analyses of DEGs were performed using the GOseq (ver. 1.26.0) tool [37].
The main temporal gene expression profiles were identified using the FunPat procedure [38] integrating annotation information, gene selection, and clustering. The input annotation information on genes encoding enzymes in metabolic pathways [39], organic N assimilation [40, 41], CAZymes, AAs, peroxidases [13], biosynthesis of secondary metabolites [13, 42] (Tables S1 and S2), autophagy [43], and secreted proteins (Fig. S1) were organized into a hierarchy to prioritize the identification of co-expressed genes associated with the most specific function. The average log2 gene expression for each time point was determined using DESeq2; only the annotated and highly changing time-DEGs were used as input in FunPat. The FunPat-clusters were classified into 15 identified response types (cf. Fig. 4). Statistical significance of the identified response types was evaluated by a permutation test of time points on the FunPat output.
Fig. 4.

Transcriptional responses of ECM fungi to changing nutrient availability. A Main temporal gene expression profiles identified by clustering genes associated with metabolism and SOM-interaction, and differentially expressed in P. involutus (PAI, 1066 genes) and L. bicolor (LAB, 1120 genes) (Fig. S8). The main expression profiles were grouped into 15 distinct transcriptional response types (r1…r15) (panel B). The number of distinct genes within each response type category is indicated in each subpanel, with the font size proportional to the number of genes. B Identification of the transcriptional response types. Top, schematic representation of changes in glucose, organic N, ammonium-N, and SOM oxidation levels, determined by chemical analyses (cf. Figs. 1 and 2). AG active growth, ND ammonium depletion, CD glucose depletion, pCD prolonged glucose depletion. Bottom, observed patterns of qualitative changes in gene expression at the ND, CD, and pCD conditions relative to the preceding time point. These patterns were thus defining the 15 transcriptional response types. The response patterns are sorted according to the time point of expression change, prioritizing upregulation over downregulation, and positioning complementary pairs (up vs. down) next to each other. A missing box indicates a sustained expression level. C Genes clustered by virtue of their SOM-interaction annotation. Shown are the transcriptional response types that were specifically upregulated during ND (r5), upregulated during ND and CD (r7 and r9), sustained during ND and upregulated during CD and pCD (r11 and r12), and sustained during ND and downregulated during CD and pCD (r13). Functional gene categories are indicated by different colors and the boxes represent individual gene models. Genes discussed in the text are highlighted. See Dataset S1 for more detailed information
To identify genes in P. involutus and L. bicolor potentially encoding proteins that are homologs to those being upregulated in S. cerevisiae during ammonium limitation, we collected a list of 41 proteins in S. cerevisiae that are significantly up-regulated during N-starvation conditions [44, 45]. The list was used as a query to identify putative homologs in the genomes of P. involutus and L. bicolor using BLASTP search (cut-off < 4.00E−20). One-to-one orthologous proteins were predicted using the reciprocal best alignment heuristic as implemented in Proteinortho (ver. 5) using the conserved synteny option [46].
A phylogenetic analysis of the yeast amino acid transporter (YAT) family was conducted by retrieving a preliminary dataset of all gene catalog proteins carrying the PF00324 domain from eight Agaricomycotina genomes [47]. A set of characterized sequences from S. cerevisiae and Hebeloma cylindrosporum [40] the YAT family was also included. A preliminary alignment was constructed using MAFFT [48] and poorly aligned regions were removed using Jalview [49]. A phylogenetic analysis was performed using RAxML [50] at Cipres [51] under the model PROTCATWAG, with 200 bootstrap runs. Further details are given in the Supplementary Information.
Results
Changes in the C and N content and the SOM composition associated with fungal activities
P. involutus and L. bicolor were grown on SOM extracted by a hot-water method from the humic layer of a Norway spruce forest. At the start of the experiment, the extract was supplemented with glucose as an energy source. The total organic C and N content in the SOM extract decreased over time in the presence of both fungi (Fig. 1A, B, Fig. S2A). The N level in the SOM medium, in particular ammonium-N, dropped more rapidly than the C content in both cases. Nitrate was not detected in the SOM extract. The fungal biomass increased until glucose was depleted from the medium. At that moment, the biomass of P. involutus started to decline, while that of L. bicolor remained relatively constant (Fig. 1A, B).
Fig. 1.
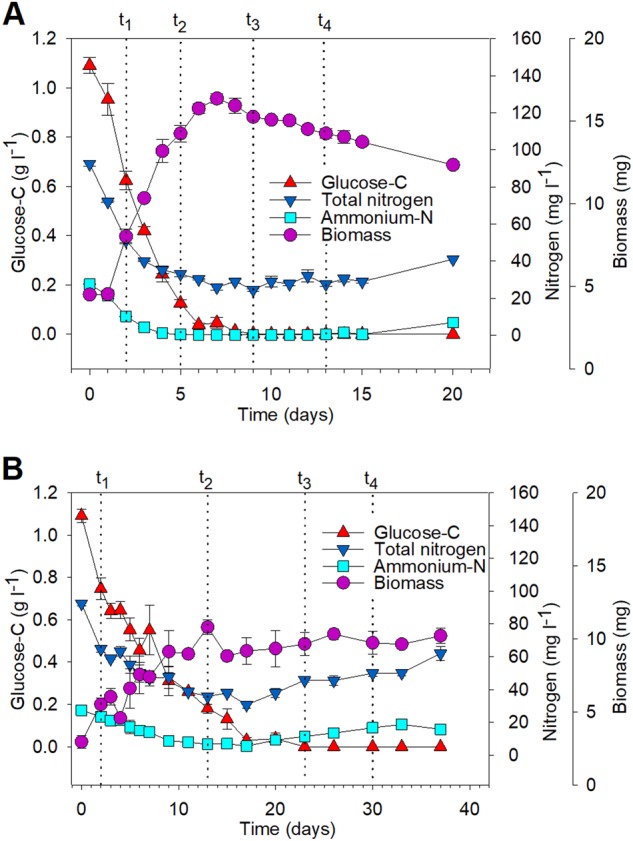
Mycelial biomass and chemical modifications of SOM extracts during decomposition by P. involutus (PAI) and L. bicolor (LAB). Dotted vertical lines indicate four time points (t1–t4) selected for spectroscopic and transcriptomic analyses. Changes in glucose-C, total N, ammonium-N, and the biomass of P. involutus (A) and L. bicolor (B) are shown (mean ± SE, n = 3). Note the different time-scales for the two fungi. Changes in the pH of the SOM extract are shown in Fig. S2B
Comparison of the FTIR spectra of the SOM extracts before and after incubation revealed a decrease of the bands associated with polysaccharides and ammonium, and an increase in the carbonyl band (C = O stretching) after the incubation (Fig. S3A, Table S3). The C = O stretching was more pronounced in the SOM extract incubated with P. involutus than the one incubated with L. bicolor (Fig. S3A). The infrared spectra of the SOM extract were resolved into four major components by the multivariate curve resolution-alternating least squares analysis, with a total explained variance equal to 99.99% (Fig. S3B). For both fungi, the first and second major resolved components decreased with time, which could be attributed to the uptake of nutrients (sugar-C and ammonium-N) from the medium. The third and fourth components increased with time, although they were not clearly resolved for L. bicolor (Fig. S3C).
Four time points were selected for a more detailed spectroscopy and transcriptome analysis (Fig. 1A, B). At t1, both glucose and ammonium were present in the SOM extract, and the fungal biomass increased. Thus, t1 represented an active growth (AG) phase. Next, t2 represented a phase when glucose was present, but ammonium was depleted from the medium (ND). At t3, glucose was not detected in the medium (<1 mg glucose–C l−1) and the mycelial growth was arrested, suggesting that the fungi were experiencing C depletion (CD). The last time point (t4) was chosen after the fungi would have experienced a prolonged C limitation (pCD).
Lignin residue oxidation, iron speciation, and changes in organic N compound content
No oxidation of lignin residues was detected at the beginning of the SOM extract incubation with either fungus (AG, t1); in contrast, increasing oxidation was detected between t1 and t2 (ND) (Fig. 2A). The oxidation continued in the presence of P. involutus, but not L. bicolor, until t3. X-ray absorption spectroscopy at the Fe K-edge revealed iron reduction in the SOM extract, from ferric (Fe3+) to ferrous (Fe2+), during the AG and ND phases (t1 and t2) of P. involutus, and the iron was present in the oxidized form during the C starvation phases (t3 and t4) (Fig. 2B, Fig. S4). No significant changes in the iron oxidation state in the SOM extract were observed in the L. bicolor culture.
Fig. 2.
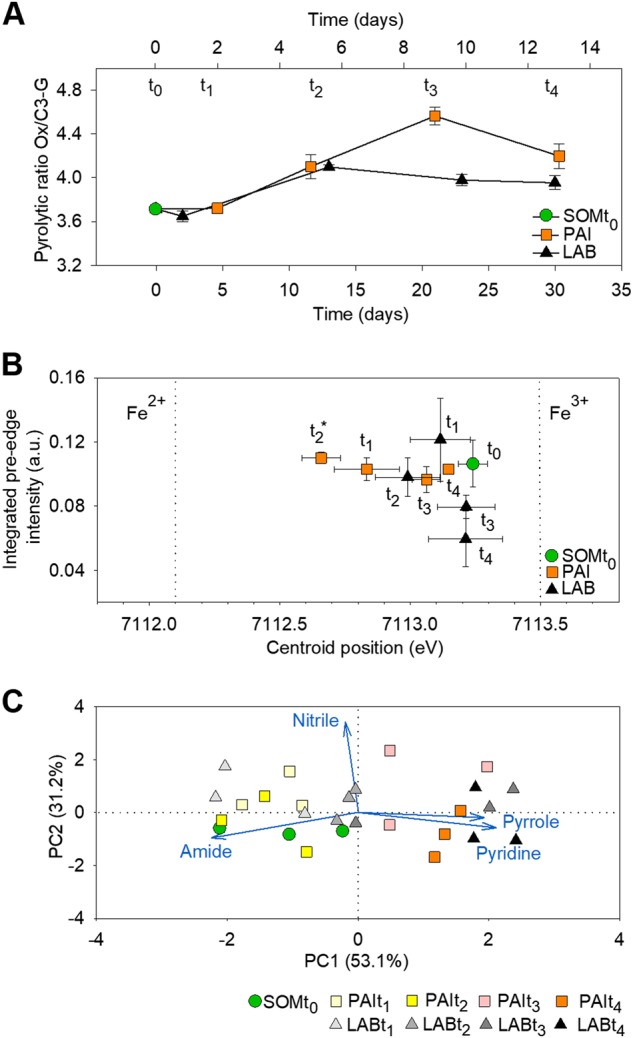
Lignin residue oxidation, iron speciation, and changes in organic N compound content. The time points t1–t4 are defined in Fig. 1. A Decomposition of lignin residues in the SOM extract, as determined by using pyrolysis-gas chromatography/mass spectrometry. The ratio of 4-acetylguaiacol to trans-propenylguaiacol (Ox/C3-G) is shown, as a marker of the degree of lignin side-chain degradation [13]. The data are corrected for the total organic C content and normalized to data for a SOM extract that had not been incubated with fungi (mean ± SE, n = 3). The top x-axis refers to the incubation time of P. involutus and the bottom x-axis refers to that of L. bicolor. B Changes in the speciation of iron in the SOM extract (mean ± SE, n = 2). Vertical lines indicate the centroid positions for ferric iron (Fe3+; at 7113.5 eV) or ferrous iron (Fe2+; at 7112.1 eV). The asterisk indicates significant difference from the initial SOM extract (SOMt0) (p < 0.05, one-way ANOVA). c The principal component analysis score and loading factor plot of the N K-edge spectra of the SOM extracts incubated with P. involutus and L. bicolor. The loading vectors were lengthened to improve the clarity of the figure
X-ray absorption spectroscopy at the N K-edge revealed that the relative abundance of various types of organic N compounds in the SOM extract (pyridine, nitrile, amide, and pyrrole) changed similarly during the incubation with P. involutus and L. bicolor, with a pronounced change between ND (t2) and CD (t3). Principal component analysis based on changes in relative abundances of these N compound types grouped SOM extracts at t1 and t2 together with the initial material, indicating low uptake of organic N by the fungi (Fig. 2C, Fig. S5, Table S4). As the incubation progressed to t3, SOM extracts incubated in the presence of P. involutus and L. bicolor were enriched in heterocyclic-N (pyridine and pyrrole) at the expense of amide, indicating uptake of amide compounds by the fungi (Fig. 2C). No separation of the samples based on the relative abundances of organic N types was observed in the PCA between t3 (CD) and t4 (pCD).
Global transcriptional changes during SOM decomposition
The mycelial transcriptional profiles of the fungi were analyzed using RNA sequencing (Table S5). The transcriptomes of P. involutus and L. bicolor were distinct and different at the four evaluated time points (Fig. S6A). Overall, the expression of 43% (7779) of the predicted P. involutus genes and 39% (8919) of the predicted genes in L. bicolor was significantly altered over the time course of the experiment. A majority of the DEGs were most highly expressed at t1 or t4 (Table S6, Fig. S6B). Enrichment analysis revealed that many metabolic and cellular processes were overrepresented in the two fungi at t1. A small number of processes were enriched at the later time points, and the overrepresented processes exhibited time-dependent differences in the two fungi (Fig. S6C).
Gene expression induced at the onset of SOM oxidation
To identify the major transcriptional changes that occurred during the onset of SOM oxidation, the genes that were both highly upregulated (more than twofold) and highly expressed (among the 20% most highly expressed genes at t2) in pairwise comparisons of t2 and t1 samples were identified (Fig. 3). In both fungi, some of these genes were associated with N metabolism and the assimilation of organic N sources, and several shared sequence similarity with genes upregulated in Saccharomyces cerevisiae during ammonium limitation. However, the upregulated genes from the N metabolism and assimilation categories were different between P. involutus and L. bicolor. Upregulated genes in P. involutus but not in L. bicolor encoded nitrate, ammonium, and urea transporters. An upregulated amino acid permease-encoding gene of L. bicolor shared sequence similarity with several amino acid permeases of S. cerevisiae; close homologs of these S. cerevisiae genes were not found in P. involutus (Fig. S7).
Fig. 3.

Expression of P. involutus (PAI) and L. bicolor (LAB) genes at the onset of SOM oxidation. The bar graph depicts the functional categories of annotated genes (29 in P. involutus and 88 in L. bicolor) that were among the top 20% most highly expressed genes at t2, and were upregulated more than twofold in pairwise t2 vs. t1 comparisons (n = 3, padj < 0.01, Wald test). The categories CAZymes and Translation, RNA processing were identified only in L. bicolor; Intracellular trafficking was only identified in P. involutus. Detailed information on the annotated genes is given in Tables S7 and S8. Fold-changes in expression for (t2–t4) vs. t1 comparisons, presented on a log2-scale, of selected genes from the N metabolism, N assimilation, CAZymes, oxidoreductases, and cellular transport categories are shown in the panel on the right. The t2 data points depicted in black squares indicate genes that were highly upregulated (≥twofold vs. t1) and expressed (top 20% at t2). Gene names given in parentheses in the N metabolism and N assimilation categories are homologs from S. cerevisiae that are significantly upregulated during ammonium limitation (Table S9). Procedures for identifying homologs of P. involutus and L. bicolor are described in Table S10; white color indicates that no homolog was identified
The expression of genes encoding enzymes acting on carbohydrates (CAZymes) and auxiliary oxidoreductases (AAs) [52, 53] differed markedly between P. involutus and L. bicolor (Fig. 3). Genes encoding the following extracellular enzymes involved in the degradation of plant polysaccharides were highly expressed and upregulated in L. bicolor: pectin methylesterase (CE8), amylase (GH13_1), glucooligosaccharide oxidase (AA7), and two lytic polysaccharide monooxygenases (LPMOs, AA9). In addition, L. bicolor expressed several genes presumably involved in the degradation of microbe-derived material, including A–A trehalase (GH37), lysozymes (GH23 and GH24), and chitin deacetylases (CE4). Genes encoding these enzymes were either absent from the P. involutus genome or not upregulated at the onset of SOM oxidation. Further, the upregulated L. bicolor genes that encoded putative transporters were more numerous and diverse than those upregulated in P. involutus. The L. bicolor transporter genes included ones coding for the hexose import transporter LbMST1.3 [54] and several other transporters from the major facilitator superfamily (Fig. 3).
Transcriptional responses to changing nutrient availability
To characterize the major transcriptional changes that occurred over the time course of the entire experiment, genes that were coordinately expressed were identified (Fig. 4). A clustering procedure based on hierarchical functional annotations of genes was customized with the KEGG metabolism hierarchy and a novel hierarchy of genes associated with the decomposition of PCW derived polymers and assimilation of organic N (SOM-interaction genes), with a conservative statistical treatment to maximize the biological relevance (Figs. S8 and S9; Table S11). Transcriptional response type discovery was developed, and clusters of 15 types (r1… r15) were identified (Fig. 4A, B). Fourteen of these response types were found in P. involutus and 11 in L. bicolor. The largest number of genes followed the response type r10, i.e., highest expression during AG and a gradually decreasing expression over time. This expression pattern was observed for ca. 54% of the clustered genes in P. involutus (330 genes) and L. bicolor (457 genes), and was dominated by the metabolic pathway genes (Fig. S9A). Ten of the remaining response types represented genes that were upregulated during one or more time points associated with ND, CD, or pCD conditions (Fig. 4B bottom). These response types were dominated by SOM-interaction genes; functional annotations of the five most common types are shown in Fig. 4C (r5, r7, r9, r11, and r12). The functions and numbers of genes in these response types differed considerably in the two fungi: most genes in L. bicolor were upregulated in response to ND and CD/pCD; in contrast, most genes in P. involutus were only upregulated in response to CD or pCD. The temporal expression pattern associated with each response type was significant in these types as indicated by the enrichment of SOM-interaction genes for at least one of the species, or by the significantly limited metabolic genes with an expected amount of SOM-interaction genes (r5) (Fig. S9B).
In P. involutus, the number of SOM-interaction genes upregulated in response to ND was limited and included genes that encoded oxidoreductases: laccase-like multicopper oxidase (AA1), methanol oxidase (AA3_3), and copper radical oxidase (AA5_1 (CRO_2)). Some of the genes that encode extracellular peptidases (A01, S10) and N transporters (YAT and oligopeptide transporter (OPT) families) were similarly regulated (Fig. 4C, r5, r7, and r9). Temporary upregulation in response to ND (Fig. 4C, r5) characterized genes of three coordinated N transporters: OPT, and the previously identified urea (Dur3) and nitrate transporters (NNP) (cf. Fig. 3). The cohort of P. involutus genes that were upregulated in response to CD but not ND (Fig. 4C, r12) included several PCWDE genes, i.e., endoxylanase (GH10), glucoamylase (GH15), alpha glucosidase (GH31), endoglucanase (GH5_5), and LPMO (AA9); as well as two laccases (AA1_1) and a copper radical oxidase (AA5_1 (CRO3-5)). Other upregulated genes in this category encoded chitinases (GH18 and GH18–CBM5), intracellular and extracellular peptidases, and some N transporters. Notably, a cluster of six autophagy-related (atg) genes (atg9, atg13, atg15, atg17, atg26, and Vps19) was coregulated with the CD-responsive genes, suggesting the activation of a C starvation response [43]. The analysis also revealed temporal regulation of genes involved in the biosynthesis of the Fe3+-reducing metabolite involutin (InvA5 and InvD) [23, 55]. In terms of the response type, both genes showed sustained expression during AG and ND (Fig. 4C, r13).
In L. bicolor, genes upregulated in response to ND (Fig. 4C, r5, r7, and r9) included ones that encoded enzymes putatively involved in the oxidation of organic matter, including two laccases (AA1), aryl alcohol oxidase (AA3_2), and a copper radical oxidase (AA5_1 (CRO1)), which was also among the most highly expressed and upregulated genes at t2 (Fig. 3). The cluster analysis also identified several PCWDE genes upregulated by N, including the previously identified genes for pectin methylesterase (CE8) and amylase (GH13_1) (cf. Fig. 3), and, additionally, two pectinases (GH28), a beta-glucosidase (GH3), and an alpha-glucosidase (GH31). Several genes encoding intracellular peroxidases, including haloperoxidases of the heme-thiolate family (HalPrx01, HalPrx02, HalPrx03, and HalPrx04) and a dye-decolorizing peroxidase (DyPrx01) [13] were also up-regulated. Genes that were up-regulated or with unchanged elevated expression at ND included ones that encoded proteins involved in organic N assimilation: extracellular peptidases (A01, M14, M36, and M43) and a diverse set of N transporters (DHA1, DHA2, ACS, ACT, YAT, OPT, and PTR families) (Fig. 4C).
Discussion
As demonstrated by recent studies, SOM extracted from the humic soil layer of a Norway spruce stand using hot water contains all major classes of biomolecules found in bulk SOM [13]. P. involutus and L. bicolor, when supplemented with glucose, have the capacity to decompose SOM by relying on oxidative mechanisms [13]. Here, using time-series spectroscopy and the same type of SOM extract, we show that SOM oxidation is linked to the liberation of organic N from SOM (Figs. 1 and 2). In both fungi, SOM oxidation was initiated when the readily available N source, i.e., ammonium, had been depleted. Following oxidation, organic N sources were utilized. In agreement with the findings of a previous study [21], the increased level of oxidation products declined when the fungi experienced C limitation. Therefore, despite their different evolutionary histories and foraging strategies, we observed functional convergence between the two species at the level of both SOM decomposition processes (SOM oxidation and liberation of organic N) and the regulation of SOM decomposition by similar nutritional signals, including limitation of inorganic N sources, presence of organic N source(s), and access to an energy source, e.g., glucose.
Sequential assimilation of N sources is common in fungi, with the ammonium utilized before other, less preferred, N sources [56, 57] and this was observed in our study as well. To examine whether P. involutus and L. bicolor sensed ammonium depletion, as has been observed in other fungi, we analyzed the regulation of genes homologs to ones that are upregulated during ammonium limitation in S. cerevisiae. Although most of those genes are present in the genomes of P. involutus and L. bicolor, and similar number of genes related to N-assimilation and N-metabolism were upregulated in both species, the types of upregulated genes differed markedly (Fig. 3). One gene that was upregulated in L. bicolor encoded an ammonium permease; this gene was located in a clade of fungal permeases, including Gap1 (Fig. S7). In S. cerevisiae, Gap1 is up-regulated during ammonium limitation and acts as a transceptor, i.e., both as a transporter and a receptor sensing the presence of amino acid substrates [58]. P. involutus and other related species from the Boletales examined here lack sequences in this clade. The genes upregulated during ammonium limitation in P. involutus included ones encoding inorganic N transporters and a Dur3 homolog (PiDur3) [59], which encodes a plasma membrane transporter of urea and polyamines in S. cerevisiae.
The data presented in the current study indicated that SOM oxidation by P. involutus and L. bicolor proceeded in conjunction with hydrolysis. However, the components of the decomposition mechanisms and their regulation were distinct and different in the two fungi. The data suggested that SOM decomposition by P. involutus is a two-step mechanism of oxidation and hydrolysis, controlled by N limitation and C limitation, respectively. By contrast, SOM decomposition by L. bicolor is a one-step mechanism that involves a combined activity of oxidative and hydrolytic enzymes triggered by N limitation and sustained during C limitation.
Reduced iron (Fe2+) is required for the generation of hydroxyl radicals (∙OH) in the Fenton reaction [11]. Such iron was detected in the organic matter extract after incubation with P. involutus at the onset of SOM-oxidation (t2) using X-ray absorption spectroscopy. This coincided with the expression of genes encoding enzymes involved in the biosynthesis of the Fe3+-reducing metabolite involutin, InvA5 and InvD [55]. At this stage of SOM decomposition, only a small number of genes encoding extracellular enzymes were upregulated in P. involutus. However, at later time points and during CD, P. involutus expressed a larger number of extracellular hydrolytic enzymes, including proteases, chitinases, oxidases, and glycoside hydrolases (Fig. 4C). These genes were upregulated together with several genes associated with autophagocytosis, suggesting that P. involutus was undergoing C starvation response involving mycelial autolysis [43]. At the same time, the fungal biomass declined (Fig. 1A). Although some of the upregulated genes coded for extracellular enzymes that were probably involved in the degradation and assimilation of released cellular material, others (GH10, GH15, GH31, GH5_5, and LPMOs) most likely encoded enzymes involved in the decomposition of the PCW-derived material present in the SOM extract. The above observations suggest that ∙OH generation is to some extent temporally separated from the synthesis of extracellular (both oxidative and hydrolytic) enzymes. Evidence of such a temporal separation of the production of ∙OH and proteolytic enzymes was recently presented in a study examining the protein decomposition by P. involutus [60]. A temporally separated two-stage oxidation-hydrolytic mechanism was recently shown to be utilized during wood decay by the BR fungus Postia placenta [61].
In contrast with P. involutus, increased Fe2+ levels were not observed in the SOM extract of L. bicolor at any stage of the incubation. This suggested that the mechanism of SOM decomposition utilized by L. bicolor probably does not involve a nonenzymatic ∙OH oxidation. Furthermore, the L. bicolor biomass did not decrease and autophagy-related genes were not upregulated during C depletion, suggesting that the two fungal species respond differently to C starvation cues. Unlike in P. involutus, ND induced the expression of a number of L. bicolor genes associated with the decomposition of PCW derived polymers by saprotrophic fungi, including ones encoding LPMOs and CRO1, and genes associated with the decomposition of microbial products. Homologs of these genes were either lacking or not up-regulated in P. involutus (Fig. 3). Moreover, in L. bicolor, during C starvation, the expression of SOM decomposition genes upregulated during ND increased further, and a small set of additional genes, potentially also involved in SOM decomposition, was upregulated (Fig. 4C). The genes that responded to both ND and CD, or only to CD, included ones that are typically involved in the decomposition of PCW polymers by saprotrophic fungi, including pectin (GH28 and CE8), cutin (CE5), cellobiose (GH3), starch (GH13_1), small polysaccharides (AA7) [11], and cellulose (AA9), as well as the fungal cell wall component chitin (CE4). These genes also included ones coding for oxidative enzymes, such as DyP and HalPrx, which have been suggested to act on lignin-like compounds and participate in detoxification processes [62]. The observation that a gene of a hexose import transporter LbMST1.3 [54] was upregulated together with several PCWDE genes in L. bicolor (Fig. 3) suggests that this fungus indeed possesses some capacity to assimilate C released during SOM decomposition. Collectively, these data indicated that immediately after the onset of ND, a number of genes related to the enzymatic decomposition of PCW and microbial polymers were upregulated in L. bicolor, and continued to be upregulated during C depletion.
Studies have suggested that some of the remaining PCWDE genes seen in ECM fungi have been recruited for the modification of the PCW of the host during mycorrhizal formation [19, 20]. Here, we provide evidence that at least some PCWDE genes are upregulated in the absence of a host during N and/or glucose limitation as part of the SOM decomposition mechanisms of ectomycorrhizal fungi. This suggests that while during evolution of the ectomycorrhizal lineages the PCWD machinery got reduced [10], the remaining genes could have been either incorporated into the SOM decomposition mechanisms of ECM fungi or recruited as PCW modifying genes during mycorrhizal formation. Such a diversification of functions could happen even within a gene family. This hypothesis is supported by the fact that the endoglucanase GH5-5 gene from L. bicolor that was shown to participate in the remodeling of the PCW during mycorrhization [20] was expressed at low levels and not significantly regulated in our experiments.
The decomposition mechanisms of P. involutus and L. bicolor are distinctly different, in agreement with their diverse evolutionary origins, i.e., a BR wood decayer vs. litter-decomposing fungus [10]. In spite of these differences, the action of both mechanisms is controlled by N and C availability. This further suggests that the availability of photosynthetic products along with the type of available N in soil could act as a dual control over the decomposition activities of ECM fungi. That the host plant might actively control the decomposing activities of ECM fungi by controlling the amount of photosynthetic carbon provided to the fungus is suggested by soil microcosm experiments with Pinus sylvestris seedlings and ECM fungi including P. involutus. 14C pulse labeling of the seedlings showed that the amount of plant C allocated to the fungal mycelium was high at the early phase of colonization of litter patches, but the C flow dropped when these areas were fully colonized and the available nutrients were assimilated [63]. Alternatively, seasonality might control the decomposition activities of ECM fungi as suggested in field studies using enzyme assays [16, 64]. Further studies are needed to examine how the decomposition activities of ECM fungi are regulated when the fungi are growing in the field and in association with their plant host. Data generated from experiments in pure culture system as used in this study will provide the tools including the molecular biomarkers that could accurately predict the decomposition activities of ECM fungi in situ. Such markers must capture the action of both enzymatic and nonenzymatic (i.e., Fenton-based) reactions.
The impact of ECM fungi on soil carbon cycling remains controversial. Genomic comparisons have suggested that the decomposition potential of ECM fungi is much smaller than that of their saprotrophic ancestors, based on the gene losses for PCWDEs seen in ECM lineages [8, 10]. However, our results suggest that under nitrogen limitation ECM fungi oxidize SOM, while under nitrogen and/or carbon limitation some of the remaining PCWD related genes are upregulated. Whether these genes are used to only modify the SOM in order to further access entrapped nitrogen sources [65] or to release metabolic C is not clear, but the upregulated sugar transporter in L. bicolor suggests that some of this C can be assimilated by the mycelium. The impact of those PCWD related genes on C cycling processes might be considerable, particularly in deeper soil horizons where ECM species dominate [23]. Furthermore, ECM species might have an indirect impact on soil C cycling by affecting the availability of N. The upregulation of genes involved in the decomposition of proteins and other microbial-origin N compounds suggest that ECM fungi are able to access, decompose and assimilate organic N entrapped in SOM compounds. By doing so, ECM fungi may induce or potentiate N limitation of free-living, saprotrophic microbial decomposers, which may impede the soil C cycling and increase soil C storage [66, 67].
Supplementary information
Acknowledgments
This work was supported by the Knut and Alice Wallenberg Foundation (KAW, Nr: 2013.0073). We also acknowledge the Canadian Light Source facility, which is supported by Natural Sciences and Engineering Research Council of Canada (NSERC), National Research Council (NRC), Canadian Institutes of Health Research (CIHR), and other government agencies; and beamline I811 at the MAX-Lab, Lund University, Sweden, which is funded by the Knut and Alice Wallenberg Foundation (KAW) and the Swedish Research Council.
Author contributions
C.N., T.M.B., P.P., and A.T designed research; C.N., T.M.B., M.S. and T.J. performed research; C.N., T.M.B., D.F., J.B., C.T., P.P. and A.T. analyzed data, and C.N., T.M.B., D.F. and A.T. wrote the paper with input from all authors.
Data availability
The chemical and spectral datasets (Py-GC/MS, XANES, FTIR) generated and analyzed during the current study are available from the corresponding author on reasonable request. The RNA-Seq datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO) repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110485.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary information
The online version of this article (10.1038/s41396-018-0331-6) contains supplementary material, which is available to authorized users.
References
- 1.Leinweber P, Kruse J, Baum C, Arcand M, Knight JD, Farrell R, et al. Advances in understanding organic nitrogen chemitry in soils using state-of-the-art analytical techniques. In: Sparks DL, et al., editors. Advances in agronomy. San Diego, CA, USA: Academic Press; 2013. pp. 83–151. [Google Scholar]
- 2.Simpson AJ, Song G, Smith E, Lam B, Novotny EH, Hayes MHB. Unraveling the structural components of soil humin by use of solution-state nuclear magnetic resonance spectroscopy. Environ Sci Technol. 2007;41:876–83. doi: 10.1021/es061576c. [DOI] [PubMed] [Google Scholar]
- 3.Näsholm T, Persson J. Plant acquisition of organic nitrogen in boreal forests. Physiol Plant. 2001;111:419–26. doi: 10.1034/j.1399-3054.2001.1110401.x. [DOI] [PubMed] [Google Scholar]
- 4.Schimel JP, Bennett J. Nitrogen mineralization: challenges of a changing paradigm. Ecology. 2004;85:591–602. doi: 10.1890/03-8002. [DOI] [Google Scholar]
- 5.Frank B. Die Bedeutung der Mykorhiza Pilze für die gemeine Kiefer. Forstwiss Cent. 1894;16:185–90. [Google Scholar]
- 6.Read DJ, Leake JR, Perez-Moreno J. Mycorrhizal fungi as drivers of ecosystem processes in heathland and boreal forest biomes. Can J Bot. 2004;82:1243–63. doi: 10.1139/b04-123. [DOI] [Google Scholar]
- 7.Lindahl BD, Tunlid A. Ectomycorrhizal fungi—potential organic matter decomposers, yet not saprophytes. New Phytol. 2015;205:1443–7. doi: 10.1111/nph.13201. [DOI] [PubMed] [Google Scholar]
- 8.Pellitier PM, Zak DR. Ectomycorrhizal fungi and the enzymatic liberation of nitrogen from soil organic matter: why evolutionary history matters. New Phytol. 2018;217:68–73. doi: 10.1111/nph.14598. [DOI] [PubMed] [Google Scholar]
- 9.Martin F, Aerts A, Ahrén D, Brun A, Danchin EG, Duchaussoy F, et al. The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008;452:88–92. doi: 10.1038/nature06556. [DOI] [PubMed] [Google Scholar]
- 10.Kohler A, Kuo A, Nagy LG, Morin E, Barry KW, Buscot F, et al. Convergent losses of decay mechanisms and rapid turnover of symbiosis genes in mycorrhizal mutualists. Nat Genet. 2015;47:410–5. doi: 10.1038/ng.3223. [DOI] [PubMed] [Google Scholar]
- 11.Hatakka A, Hammel KE. Fungal biodegradation of lignocellulose. In: Hofrichter M, editor. The mycota vol. X industrial applications. Berlin, Germany: Springer-Verlag; 2010. pp. 319–40. [Google Scholar]
- 12.Morin E, Kohler A, Baker AR, Foulongne-Oriol M, Lombard V, Nagy LG, et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc Natl Acad Sci USA. 2012;109:17501–6. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah F, Nicolás C, Bentzer J, Ellström M, Smits M, Rineau F, et al. Ectomycorrhizal fungi decompose soil organic matter using oxidative mechanisms adapted from saprotrophic ancestors. New Phytol. 2016;209:1705–19. doi: 10.1111/nph.13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rineau F, Roth D, Shah F, Smits M, Johansson T, Canbäck B, et al. The ectomycorrhizal fungus Paxillus involutus converts organic matter in plant litter using a trimmed brown-rot mechanism involving Fenton chemistry. Environ Microbiol. 2012;14:1477–87. doi: 10.1111/j.1462-2920.2012.02736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bödeker IT, Clemmensen KE, de Boer W, Martin F, Olson Aring, Lindahl B. Ectomycorrhizal Cortinarius species participate in enzymatic oxidation of humus in northern forest ecosystems. New Phytol. 2014;203:245–56. doi: 10.1111/nph.12791. [DOI] [PubMed] [Google Scholar]
- 16.Courty PE, Bréda N, Garbaye J. Relation between oak tree phenology and the secretion of organic matter degrading enzymes by Lactarius quietus ectomycorrhizas before and during bud break. Soil Biol Biochem. 2007;39:655–1663. doi: 10.1016/j.soilbio.2007.01.017. [DOI] [Google Scholar]
- 17.Phillips LA, Ward V, Jones MD. Ectomycorrhizal fungi contribute to soil organic matter cycling in sub-boreal forests. ISME J. 2014;8:699–713. doi: 10.1038/ismej.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldrian P. Ectomycorrhizal fungi and their enzymes in soils: is there enough evidence for their role as facultative soil saprotrophs? Oecologia. 2009;161:657–60. doi: 10.1007/s00442-009-1433-7. [DOI] [PubMed] [Google Scholar]
- 19.Veneault-Fourrey C, Commun C, Kohler A, Morin E, Balestrini R, Plett J, et al. Genomic and transcriptomic analysis of Laccaria bicolor CAZome reveals insights into polysaccharides remodelling during symbiosis establishment. Fungal Genet Biol. 2014;72:168–81. doi: 10.1016/j.fgb.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Zhang F, Anasontzis GE, Labourel A, Champion C, Haon M, Kemppainen M, et al. The ectomycorrhizal basidiomycete Laccaria bicolor releases a secreted beta-1,4 endoglucanase that plays a key role in symbiosis development. New Phytol. 2018 doi: 10.1111/nph.15113. [DOI] [PubMed] [Google Scholar]
- 21.Rineau F, Shah F, Smits MM, Persson P, Johansson T, Carleer R, et al. Carbon availability triggers the decomposition of plant litter and assimilation of nitrogen by an ectomycorrhizal fungus. ISME J. 2013;7:2010–22. doi: 10.1038/ismej.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agerer R. Exploration types of ectomycorrhizae. A proposal to classify ectomycorrhizal systems according to their patterns of differentiation and putative ecological importance. Mycorrhiza. 2001;11:107–14. doi: 10.1007/s005720100108. [DOI] [Google Scholar]
- 23.Shah F, Schwenk D, Nicolás C, Persson P, Hoffmesier D, Tunlid A. Involutin is an Fe3+ reductant secreted by the ectomycorrhizal fungus Paxillus involutus during Fenton-based decomposition of organic matter. Appl Environ Microbiol. 2015;81:8427–33. doi: 10.1128/AEM.02312-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindahl BD, Ihrmark K, Boberg J, Trumbore SE, Högberg P, Stenlid J, et al. Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol. 2007;173:611–20. doi: 10.1111/j.1469-8137.2006.01936.x. [DOI] [PubMed] [Google Scholar]
- 25.Davidson EA, Galloway LF, Strand MK. Assessing available carbon: comparison of techniques across selected forest soils. Commun Soil Sci Plant Anal. 1987;18:45–65. doi: 10.1080/00103628709367802. [DOI] [Google Scholar]
- 26.Jaumot J, Gargallo R, de Juan A, Tauler RA. Graphical user friendly interface for MCR-ALS: a new tool for multivariate curve resolution in MATLAB. Chemom Intell Lab Syst. 2005;76:101–10. doi: 10.1016/j.chemolab.2004.12.007. [DOI] [Google Scholar]
- 27.Felten J, Hall H, Jaumot J, Tauler R, de Juan A, Gorzsá A. Vibrational spectroscopic image analysis of biological material using multivariate curve resolution-alternating least squares (MCR-ALS) Nat Protoc. 2015;10:217–40. doi: 10.1038/nprot.2015.008. [DOI] [PubMed] [Google Scholar]
- 28.Buurman P, Jongman AG, Nierop KGJ. Comparison of Michigan and Dutch podzolized soils: organic matter characterization by micromorphology and pyrolysis-GC/MS. Soil Sci Soc Am J. 2008;72:1344–56. doi: 10.2136/sssaj2007.0352. [DOI] [Google Scholar]
- 29.Webb SM. SIXpack: a graphical user interface for XAS analysis using IFEFFIT. Phys Scr. 2005;T115:1011–4. doi: 10.1238/Physica.Topical.115a01011. [DOI] [Google Scholar]
- 30.Wilke M, Farges F, Petit PE, Brown GE, Jr, Martin F. Oxidation state and coordination of Fe in minerals: an Fe K-XANES spectroscopic study. Am Mineral. 2001;86:714–30. doi: 10.2138/am-2001-5-612. [DOI] [Google Scholar]
- 31.Leinweber P, Kruse J, Walley FL, Gillespie A, Eckhardt KU, Blyth RI, et al. Nitrogen K-edge XANES—an overview of reference compounds used to identify unknown organic nitrogen in environmental samples. J Synchrotron Radiat. 2007;14:500–11. doi: 10.1107/S0909049507042513. [DOI] [PubMed] [Google Scholar]
- 32.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Risso D, Schwartz K, Sherlock G, Dudoit S. GC-content normalization for RNA-Seq data. BMC Bioinforma. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JR Stat Soc Ser B Methods. 1995;57:289–300. [Google Scholar]
- 37.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanavia T, Finotello F, Di CB. FunPat: function-based pattern analysis on RNA-seq time series data. BMC Genom. 2015;16:S2. doi: 10.1186/1471-2164-16-S6-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–31. doi: 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Lucic E, Fourrey C, Kohler A, Martin F, Chalot M, Brun-Jacob A. A gene repertoire for nitrogen transporters in Laccaria bicolor. New Phytol. 2008;180:343–64. doi: 10.1111/j.1469-8137.2008.02580.x. [DOI] [PubMed] [Google Scholar]
- 41.Shah F, Rineau F, Canbäck B, Johansson T, Tunlid A. The molecular components of the extracellular protein-degradation pathways of the ectomycorrhizal fungus Paxillus involutus. New Phytol. 2013;200:875–87. doi: 10.1111/nph.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, et al. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–43. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellström M, Shah F, Johansson T, Ahrén D, Persson P, Tunlid A. The carbon starvation response of the ectomycorrhizal fungus Paxillus involutus. FEMS Microbiol Ecol. 2015;91:fiv027. doi: 10.1093/femsec/fiv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boer VM, de Winde JH, Pronk JT, Piper MD. The genome-wide transcriptional responses of Saccharomyces cerevisiae grown on glucose in aerobic chemostat cultures limited for carbon, nitrogen, phosphorus, or sulfur. J Biol Chem. 2003;278:3265–74. doi: 10.1074/jbc.M209759200. [DOI] [PubMed] [Google Scholar]
- 45.Boer VM, Tai SL, Vuralhan Z, Arifin Y, Walsh MC, Piper MD, et al. Transcriptional responses of Saccharomyces cerevisiae to preferred and nonpreferred nitrogen sources in glucose-limited chemostat cultures. FEMS Yeast Res. 2007;7:604–20. doi: 10.1111/j.1567-1364.2007.00220.x. [DOI] [PubMed] [Google Scholar]
- 46.Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinforma. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014;42:D699–704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller MA, Pfieffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE); New Orleans, LA, USA; 2010, p. 1–8.
- 52.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6:1–14. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez MF, Dietz S, Grunze N, Bloschies J, Weiss M, Nehls U. The sugar porter gene family of Laccaria bicolor: function in ectomycorrhizal symbiosis and soil-growing hyphae. New Phytol. 2008;180:365–78. doi: 10.1111/j.1469-8137.2008.02539.x. [DOI] [PubMed] [Google Scholar]
- 55.Braesel J, Gotze S, Shah F, Heine D, Tauber J, Hertweck C, et al. Three redundant synthetases secure redox-active pigment production in the Basidiomycete Paxillus involutus. Chem Biol. 2015;22:1325–34. doi: 10.1016/j.chembiol.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 56.Magasanik B, Kaiser CA. Nitrogen regulation in Saccharomyces cerevisiae. Gene. 2002;290:1–18. doi: 10.1016/S0378-1119(02)00558-9. [DOI] [PubMed] [Google Scholar]
- 57.Marzluf GA. Genetic regulation of nitrogen metabolism in the fungi. Microbiol Mol Biol Rev. 1997;61:17–32. doi: 10.1128/mmbr.61.1.17-32.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van DP, Brown NA, Goldman GH, Rutherford J, Xue C, Van Zeebroeck G. Nutrient sensing at the plasma membrane of fungal cells. Microbiol Spectr. 2017;5:10–2016. doi: 10.1128/microbiolspec.FUNK-0031-2016. [DOI] [PubMed] [Google Scholar]
- 59.Morel M, Jacob C, Fitz M, Wipf D, Chalot M, Brun A. Characterization and regulation of PiDur3, a permease involved in the acquisition of urea by the ectomycorrhizal fungus Paxillus involutus. Fungal Genet Biol. 2008;45:912–21. doi: 10.1016/j.fgb.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Op De Beeck M, Troein C, Peterson C, Persson P, Tunlid A. Fenton reaction facilitates organic nitrogen acquisition by an ectomycorrhizal fungus. New Phytol. 2018;218:335–43. doi: 10.1111/nph.14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Presley GN, Hammel KE, Ryu JS, Menke JR, Figueroa M, et al. Localizing gene regulation reveals a staggered wood decay mechanism for the brown rot fungus Postia placenta. Proc Natl Acad Sci USA. 2016;113:10968–73. doi: 10.1073/pnas.1608454113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hofrichter M, Ullrich R, Pecyna MJ, Liers C, Lundell T. New and classic families of secreted fungal heme peroxidases. Appl Microbiol Biotechnol. 2010;87:871–97. doi: 10.1007/s00253-010-2633-0. [DOI] [PubMed] [Google Scholar]
- 63.Leake JR, Donnelly DP, Saunders EM, Boddy L, Read DJ. Rates and quantities of carbon flux to ectomycorrhizal mycelium following 14C pulse labeling of Pinus sylvestris seedlings: effects of litter patches and interaction with a wood-decomposer fungus. Tree Physiol. 2001;21:71–82. doi: 10.1093/treephys/21.2-3.71. [DOI] [PubMed] [Google Scholar]
- 64.Courty PE, Franc A, Garbaye J. Temporal and functional pattern of secreted enzyme activities in an ectomycorrhizal community. Soil Biol Biochem. 2010;42:2022–5. doi: 10.1016/j.soilbio.2010.07.014. [DOI] [Google Scholar]
- 65.Kieloaho AJ, Pihlatie M, Dominguez Carrasco M, Kanerva S, Parshintsev J, Riekkola ML, et al. Stimulation of soil organic nitrogen pool: the effect of plant and soil organic matter degrading enzymes. Soil Biol Biochem. 2016;96:97–106. doi: 10.1016/j.soilbio.2016.01.013. [DOI] [Google Scholar]
- 66.Averill C, Hawkes CV. Ectomycorrhizal fungi slow soil carbon cycling. Ecol Lett. 2016;19:937–47. doi: 10.1111/ele.12631. [DOI] [PubMed] [Google Scholar]
- 67.Fernandez CW, Kennedy PG. Revisiting the “Gadgil effect”: do interguild fungal interactions control carbon cycling in forest soils? New Phytol. 2016;209:1382–94. doi: 10.1111/nph.13648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The chemical and spectral datasets (Py-GC/MS, XANES, FTIR) generated and analyzed during the current study are available from the corresponding author on reasonable request. The RNA-Seq datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO) repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110485.