

Abstract
Although mitochondrial genomes are typically thought of as single circular molecules, these genomes are fragmented into multiple chromosomes in many eukaryotes, raising intriguing questions about inheritance and (in)stability of mtDNA in such systems. A previous comparison of mitochondrial genomes from two different individuals of the angiosperm species Silene noctiflora found variation in the presence of entire mitochondrial chromosomes. Here, we expand on this work with a geographically diverse sampling of 25 S. noctiflora populations and the closely related species S. turkestanica and S. undulata. Using a combination of deep sequencing and PCR-based screening for the presence of 22 different mitochondrial chromosomes, we found extensive variation in the complement of chromosomes across individuals. Much of this variation could be attributed to recent chromosome loss events, suggesting that the massively expanded and fragmented mitochondrial genomes of S. noctiflora may have entered a phase of genome reduction in which they are losing entire chromosomes at a rapid rate. Sequence analysis of mitochondrial and plastid genomes revealed genealogical differences both between these organelles and within the mitochondrial genome, indicating a history of recombination. Evidence that recombination has generated novel combinations of alleles was more frequent between loci on different mitochondrial chromosomes than it was within chromosomes. Therefore, the fragmentation of mitochondrial genomes and the assortment of chromosomes during mitochondrial inheritance appears to have contributed to a history of sexual-like recombination in the mtDNA of this species.
Subject terms: Plant evolution, Phylogenetics, Evolutionary biology
Introduction
Mitochondrial genome architecture is remarkably diverse (Gray et al. 1999; Smith and Keeling 2015). Most mitochondrial genomes are represented by a single chromosome, which in some cases can even retain much of its ancestral bacterial-like architecture (Lang et al. 1997; Burger et al. 2013). But many independent eukaryotic lineages have evolved complex multichromosomal structures (Lukes et al. 2005; Shao et al. 2009; Vlcek et al. 2011; Smith et al. 2012). The evolutionary consequences of dividing an organelle genome into multiple chromosomes are not well understood and pose fundamental questions about inheritance and genome stability.
Flowering plants are particularly extreme with respect to their diverse and unusual mitochondrial DNA (mtDNA) structures (Mower et al. 2012). Angiosperm mitochondrial genomes are very large and contain recombinationally active repeat sequences, resulting in complex and dynamic structures in vivo, including low frequency alternative structures known as sublimons (Small et al. 1987; Arrieta-Montiel et al. 2009; Arrieta-Montiel and Mackenzie 2011; Gualberto and Newton 2017). However, these genome can still typically be mapped as a “master circle” structure (Sloan 2013). In contrast, multichromosomal mitochondrial genomes have been identified in at least five independent angiosperm genera: Amborella (Rice et al. 2013), Cucumis (Alverson et al. 2011), Lophophytum (Sanchez-Puerta et al. 2017), Saccharum (Shearman et al. 2016), and Silene (Sloan et al. 2012). The most dramatic examples have been found in certain Silene species in which the mitochondrial genome has expanded enormously (up to 11 Mb in size) and been fragmented into dozens of circular-mapping chromosomes (Sloan et al. 2012). In species such as S. noctiflora, many of these chromosomes share only very small repeated sequences with the rest of the genome and appear to be largely or entirely autonomous. Other chromosomes do contain long repeats (up to thousands of bp in length) and are involved in recombinational activity with repeat copies on other chromosomes. However, even in these cases, isolated chromosomes appear to be the numerically dominant form (Sloan et al. 2012).
In a recent comparison of sequenced mitochondrial genomes from two different populations of Silene noctiflora (OSR and BRP), we found that the two genomes were highly similar in sequence and structure, with the one major exception that they differed in their numbers of chromosomes (59 vs. 63) (Wu et al. 2015). Each genome contained many unique chromosomes that were absent altogether in the other, suggesting that the dominant mode of molecular evolution in this system is acting at the level of entire chromosomes. However, we could not determine whether variation in the presence of any given chromosome was the result of a recent gain in one lineage or a recent loss in the other. One hypothesis is that the differences in chromosome content are the result of an ongoing process of simple segregational loss during mitochondrial division and cell division that followed an ancestral expansion and fragmentation of the mitochondrial genome. Although most of the “missing” chromosomes contain at least some transcribed regions (Wu et al. 2015), they generally have no identifiable genes and are populated by large amounts of non-coding sequence of unrecognizable origin. Therefore, they may constitute “junk DNA” and experience little or no functional constraint that would prevent such losses.
The fragmentation of the S. noctiflora mitochondrial genome raises additional questions about whether it might facilitate independent assortment of chromosomes as a mechanism that generates novel combinations of alleles (Rand 2009; Wu et al. 2015). Although maternal inheritance is the predominant mode of mitochondrial transmission in angiosperms, evidence of low frequency paternal “leakage” of mtDNA has been observed in both natural populations and controlled crosses in many species, including some within the genus Silene (McCauley 2013). Moreover, mitochondria readily and regularly fuse with each other, allowing for intermixing of different copies of the mitochondrial genome (Arimura and Tsutsumi 2005; Chan 2006; Segui-Simarro et al. 2008), and crossovers between repeated/homologous sequences frequently occur in plant mtDNA (Arrieta-Montiel and Mackenzie 2011). Therefore, many of the ingredients for sexual-like inheritance and recombination may already be in place for plant mitochondrial genomes (Stadler and Delph 2002; Touzet and Delph 2009; McCauley 2013; Delph and Montgomery 2014; Levsen et al. 2016). For simplicity, we will refer to this process as “sexual recombination” to highlight the potential to bring together distinct mitochondrial haplotypes through biparental inheritance and to generate novel combinations of alleles, even though the process does not include meiosis or meet some formal definitions of “sex”.
In this study, we use collections from widespread natural populations of S. noctiflora and some of its closest relatives to describe its diversity in mitochondrial genome content and to address the following three questions: (1) How much variation is there in the presence/absence of entire mitochondrial chromosomes? (2) To what extent is that variation the result of recent gains vs. losses of chromosomes? (3) Is there evidence of a history of sexual recombination within and among the mitochondrial chromosomes?
Materials and methods
Silene sampling and DNA extraction
Silene noctiflora is native to Eurasia but has been widely introduced across the globe as a weedy species (McNeil, 1980). To identify variation in mitochondrial genome content across the species range of S. noctiflora, we obtained seeds from 25 geographically dispersed populations from Europe and North America (Table S1). In addition, we obtained seeds from a single collection of S. undulata ( = S. capensis), a South African species that has been identified as a close relative of S. noctiflora (Havird et al. 2017) (B. Oxelman, pers. comm.). Seeds were germinated on soil (Fafard 2SV Mix supplemented with vermiculite and perlite) in SC7 Cone-tainers (Stuewe and Sons) in February 2014. Plants were grown for two months with regular watering and fertilizer treatments under supplemental lighting (16 hr/8 hr light/dark cycle) in the Colorado State University greenhouse. To test for variation on a more local geographical scale, we also sampled leaf tissue from 19 S. noctiflora individuals collected from three sites that are within 10 km of each other in a metapopulation near Mountain Lake Biological Station in southwestern Virginia that is the subject of an annual Silene census (Fields and Taylor 2014) (Table S2). Total-cellular DNA was extracted from rosette leaf tissue with a Qiagen Plant DNeasy Kit following the manufacturer’s protocol. We also used previously extracted DNA from S. turkestanica, which is a close relative of S. noctiflora (Sloan et al. 2009; Rautenberg et al. 2012). To generate sufficient template material from the herbarium-derived S. turkestanica DNA sample, we performed whole-genome amplification with a Qiagen Repli-G Mini Kit.
Chromosome sampling and PCR presence/absence screening
To assess the variation in the presence/absence of specific mitochondrial chromosomes in Silene noctiflora, we chose a sample of 22 chromosomes, which were divided into four different groups based on our previous comparison of the mitochondrial genomes from the S. noctiflora OSR and BRP populations. These groups were selected because of a priori expectations that they might be particularly variable in chromosome presence/absence across the species. They included: (1) the five chromosomes with the highest level of nucleotide sequence divergence, (2) five chromosomes that were present in both OSR and BRP but did not contain any identifiable genes, (3) six chromosomes found in OSR but not in BRP, and (4) six chromosomes that were found BRP but not in OSR. Only four of the selected chromosomes (OSR52, OSR57, BRP45, and BRP50) contained any annotated genes, and these were all genes that were present in duplicate copies on other chromosomes and, therefore, potentially expendable. The other 18 chromosomes were “empty” with respect to functional annotations. For each of the 22 sampled chromosomes, three distantly spaced PCR primer pairs were designed using Primer3 (Untergasser et al. 2012) (Table S3).
All extracted DNA samples were quantified with a Nanodrop 2000 UV spectrophotometer (Thermo-Fisher Scientific) and diluted to a concentration of 0.5 ng/μl. For each PCR amplification, two replicates were performed to verify consistent determination of marker presence/absence. PCRs were performed in a Bio-Rad C1000 Touch Thermal Cycler in 10 μl reaction volumes containing 1 ng template DNA, 0.2 μM concentration of each primer, 0.1 mM concentration of each dNTP, 1 μl 10 × buffer, and 0.1 U Paq5000 DNA polymerase (Agilent Technologies). Amplification was achieved using 3 min of initial denaturation at 94 °C, 38 cycles of 15 s at 94 °C, 15 s annealing at 54 °C, and 30 s extension at 72 °C, followed by a final 5-min incubation at 72 °C. The reactions were screened for the presence/absence of an amplified fragment with the expected size on a 1.5% agarose gel using a 1 Kb Plus Ladder (Thermo-Fisher Scientific) as a molecular size standard.
Sampling of mitochondrial loci, Sanger sequencing, and preliminary phylogenetic analysis
To provide an initial estimate of the mitochondrial genealogy for our sample, we designed nine primer pairs (Table S4) targeting three coding regions (cox1, mttB, and nad2) and six non-coding regions from the mitochondrial genome and used them to perform PCR and Sanger sequencing. Previous comparisons of whole mitochondrial genomes from two populations (OSR and BRP) found extremely low rates of sequence polymorphisms. Therefore, the above markers were chosen to include regions known to contain single-nucleotide polymorphisms (SNPs) (Wu et al. 2015). These markers were used for sequencing all of the 25 S. noctiflora populations, as well as S. undulata and S. turkestanica (but only five of the nine markers could be amplified for S. turkestanica). PCR amplification was conducted as described above, and the resulting PCR products were purified and sent to University of Chicago Comprehensive Cancer Center DNA Sequencing Facility for Sanger sequencing.
These data were combined with existing sequence data from four additional mitochondrial coding regions (atp1, atp9, cox3, and nad9) for which there has been thorough sampling across the genus Silene, including representatives of S. noctiflora and S. turkestanica (Sloan et al. 2009; Rautenberg et al. 2012). We did not attempt to amplify and sequence these loci in all S. noctiflora populations, but we extracted the corresponding sequences from the published mitochondrial genomes of S. noctiflora OSR and BRP (Sloan et al. 2012; Wu et al. 2015) and high throughput sequencing datasets available at the time for S. noctiflora KEW 22121 and OPL, as well as S. undulata (Table S5). Because the S. undulata dataset was based on RNA-seq, all variants associated with known editing sites in Silene (Sloan et al. 2010) were removed to avoid misinterpreting sequence changes introduced by RNA editing. Finally, sequence data for all seven coding loci (but not the six non-coding loci) were obtained from published genome assemblies for the outgroups S. latifolia, S. vulgaris, and Dianthus caryophyllus (Sloan et al. 2012; Yagi et al. 2014).
Individual genes were aligned separately and concatenated using BioEdit (Hall 1999) and ClustalX v2.1 (Larkin et al. 2007). Phylogenetic trees were inferred by maximum likelihood (ML) implemented with IQ-TREE v1.66 (Nguyen et al. 2015). The ML analysis was performed with 1000 bootstrap replicates under the GTR model of nucleotide substitution.
Illumina genomic DNA sequencing, read mapping, and genome-wide analysis of mitochondrial and plastid sequences
To get a more detailed assessment of the presence/absence of mitochondrial genome content and sequence variation and to obtain plastid genome sequences from the major lineages in this study, we performed Illumina sequencing of total-cellular DNA. To augment existing sequence data from the S. noctiflora OSR and BRP population, we sequenced total-cellular DNA samples from the KEW 22121, KEW 1672, and OPL populations and from S. turkestanica and S. undulata (Table S5). The KEW 22121 and KEW 1672 samples were specifically targeted because preliminary analyses based on Sanger and PCR data (see above) showed them to be genetically distinct from both OSR and BRP.
To measure sequencing coverage across the mitochondrial genome, adapter and low-quality sequences were trimmed using either Cutadapt v1.3 (Martin 2011) or Trimmomatic version 0.32 (Bolger et al. 2014). The filtered and trimmed reads were then mapped to previously published reference mitochondrial genomes from S. noctiflora OSR and BRP (Sloan et al. 2012; Wu et al. 2015) using Bowtie2 v2.2.4 with default parameters (Langmead et al. 2009). Samtools v1.3 (Li et al. 2009) was used to calculate read depth in a 1-kb sliding window analysis across the length of each chromosome. Results were visualized using the ggplot2 package (http://ggplot2.org/) in R v3.2.4 (www.r-project.org).
To generate genome-wide alignments of mitochondrial sequences for phylogenetic analysis, we individually mapped reads from our S. noctiflora, S. undulata, and S. turkestanica Illumina datasets to the S. noctiflora BRP reference genome (Wu et al. 2015), using Bowtie2. We then used ANGSD v 0.921 (http://www.popgen.dk/angsd/index.php/ANGSD) (Korneliussen et al., 2014) to extract consensus sequences from each resulting BAM file, applying a minimum read depth cutoff on 5. The resulting data matrix had many gaps because of entire missing chromosomes and the extremely heterogeneous coverage of the whole-genome-amplified S. turkestanica sample. We restricted our analysis to regions with complete taxon-sampling by extracting all sequences of at least 500 bp in length that had uninterrupted coverage for all taxa.
Whole plastid genome alignments were generated by Bowtie2 mapping of Illumina reads to the S. noctiflora OSR reference sequence (NC_016728.1) after removing one copy of its inverted repeat. Consensus sequences were extracted from individual BAM files, using CLC Genomics Workbench v7.5.1, and then aligned across their entire lengths with the FFT-NS-1 algorithm in MAFFT v7.222 (Katoh and Standley 2013).
ML phylogenetic analyses were conducted with IQ-TREE as described above. Separate analyses were conducted on the whole-plastome alignments, the concatenated mitochondrial dataset, and each of 14 mitochondrial chromosomes that included at least four parsimony-informative sites (not counting sites that only distinguished the closely related OPL and OSR samples from the rest of the taxa).
Parsimony-based reconstruction of mitochondrial chromosomal gain/loss in Silene noctiflora
To infer the history of chromosome gain/loss in S. noctiflora, a simple (Fitch) parsimony criterion was applied to reconstruct the ancestral state for the presence/absence of each individual chromosome that was surveyed with the PCR screen described above, using Mesquite v3.0 (Maddison and Maddison 2006). We used the maximum-likelihood topology based on the mitochondrial genome-wide concatenation described above as the backbone for the constraint tree in this analysis. The remaining S. noctiflora population samples were grouped with their respective clades based on the phylogenetic analysis of the Sanger sequencing dataset. This analysis was also repeated under the assumptions of Dollo parsimony, using the Count software package (Csűös 2010).
Tests of sexual recombination
We tested for a history of sexual recombination at three different levels. First, we tested for significant topological differences between phylogenies produced from our entire concatenated mitochondrial dataset and the concatenated plastid dataset by performing Shimodaira–Hasegawa (SH) and Approximately Unbiased (AU) tests with the CONSEL software package (Shimodaira and Hasegawa 2001). Second, we tested for significant topological differences among the 14 chromosomes that were selected for individual phylogenetic analyses (see above). We performed SH and AU tests for all pairwise combinations of these 14 chromosomes. All tests were performed in reciprocal fashion (using the alignment from one dataset against the optimal tree from another and vice versa), and we took the higher (less significant) p-value as our measure of significance. Multiple comparisons were controlled for by applying the Benjamini–Hochberg procedure to determine false discovery rate. Third, we applied the “four-gamete” test (Hudson and Kaplan 1985; McCauley 2013) to look for evidence of recombination between all pairs of parsimony-informative sites in the entire mitochondrial alignment, using a custom Perl script (https://github.com/dbsloan/fgt).
Digital droplet PCR validation
Unexpectedly, one chromosome (OSR chromosome 46) that had previously been found to be absent from the BRP mitochondrial genome (Wu et al. 2015), produced consistent amplification across all 25 S. noctiflora populations, including BRP. To assess whether this chromosome might be present at lower copy number in some individuals, allowing it to have escaped detection in earlier genome sequencing efforts, we performed ddPCR, using individuals from each of seven different S. noctiflora samples (BRP, OSR, MH-L, BWT-1, BWT-2, KEW 12991, and KEW 36186). We also analyzed the copy number of chromosome 37, which is present in both BRP and OSR, to serve as a comparison. For each chromosome, three pairs of ddPCR primers were designed (Table S6). We selected two mitochondrial protein-coding genes (cox1 and nad2) to serve as reference markers. All ddPCR amplifications were set up in 20-μL volumes with Bio-Rad QX200™ ddPCR™ EvaGreen Supermix, 2 μM concentration of each primer, and 1 ng of template DNA before mixing into an oil emulsion with a Bio-Rad QX200 Droplet Generator. Amplification was performed on a Bio-Rad C1000 Touch Thermal Cycler with an initial 5 min incubation at 95 °C and 40 cycles of 30 s at 95 °C and 1 min at 60 °C, followed by signal stabilization via 5 min at 4 °C and 5 min at 95 °C. The resulting droplets were read on a Bio-Rad QX200 Droplet Reader. Absolute copy numbers (per ng of total-cellular DNA) for each PCR target were calculated based on a Poisson distribution using the Bio-Rad QuantaSoft package and reported as proportions relative to the average of the cox1 and nad2 reference markers.
Results
Mitochondrial and plastid genealogies
An initial analysis of 13 mitochondrial markers provided evidence for four distinct, well-supported mitochondrial lineages within S. noctiflora (Figure S1). Two of these lineages correspond to the BRP and OSR backgrounds that have already been thoroughly characterized based on complete mtDNA sequences (Wu et al. 2015). Each of these two lineages is represented by multiple populations in our dataset (eight in the BRP-like group and 15 in the OSR-like group). Two additional lineages were also detected, each represented by only a single sample (KEW 1672 and KEW 22121). Despite the broad geographical sampling across Europe and North America (Table S1), the overall level of intraspecific polymorphism was extremely low, and no sequence variants were detected among populations within the BRP- or OSR-like groups, which is consistent with the general lack of mitochondrial sequence diversity in previous studies of S. noctiflora (Sloan et al. 2012; Wu et al. 2015). This mitochondrial dataset confirmed the close relationships between S. noctiflora, S. turkestanica, and S. undulata relative to the rest of the genus Silene (Sloan et al. 2009; Rautenberg et al. 2012; Havird et al. 2017). Unexpectedly, this initial phylogenetic analysis placed the S. undulata mitochondrial haplotype as nested within the small amount of observed diversity in S. noctiflora, forming a well-supported group (98% bootstrap support) with the S. noctiflora BRP-like and KEW 1672 lineages to the exclusion of the KEW 22121 and OSR-like lineages (Figure S1).
To better resolve the cytoplasmic genealogy for the major lineages within our sample, we used Illumina sequencing datasets to perform genome-wide analyses of mitochondrial and plastid sequence data (341,456-bp and 128,245-bp sequence alignments, respectively). In contrast to the much smaller Sanger dataset, these analyses both recovered trees that placed S. undulata as an outgroup to all four of the major S. noctiflora lineages (Fig. 1). However, the mitochondrial and plastid trees have conflicting topologies with regard to the major S. noctiflora lineages. Whereas the mitochondrial analysis identifies KEW 1672 as an outgroup to the rest of the S. noctiflora populations with 99% bootstrap support, the plastid dataset places it sister to BRP with 100% bootstrap support.
Fig. 1.

Phylogenetic relationships among S. turkestanica, S. undulata, and major lineages of S. noctiflora based on concatenated sequence alignments from a the mitochondrial genome and b the plastid genome. Bootstrap support values are shown for each node
The topological discrepancy between the Sanger (Figure S1) and Illumina (Fig. 1) mitochondrial datasets suggested that there was heterogeneity within the mitochondrial data, which we confirmed by analyzing sequence alignments from individual mitochondrial chromosomes. We analyzed sequence data from 14 different chromosomes (those that were present among all major lineages and had sufficient sequence variation to be phylogenetically informative). We recovered 10 different topologies, including many conflicts that were strongly supported by bootstrap resampling (Fig. 2).
Fig. 2.

Phylogenetic relationships among S. turkestanica, S. undulata, and major lineages of S. noctiflora based on concatenated sequence alignments for each of 14 different chromosomes in the mitochondrial genome. Bootstrap support values greater than 70 are shown for each node. Chromosome labels refer to the numbering in the S. noctiflora BRP mitochondrial genome
Evidence of sexual recombination in the mitochondrial genome
The conflicting tree topologies produced from phylogenetic analysis of mitochondrial vs. plastid datasets (Fig. 1), as well as the topological variation observed from different mitochondrial chromosomes (Fig. 2), suggested the potential for a history of sexual recombination. We tested for such a history at multiple genomic scales. Congruence between the mitochondrial and plastid concatenated datasets was strongly rejected by both SH (P = 0.002) and AU (P < 0.0001) tests. In addition, numerous comparisons between pairs of chromosomes revealed significant topological conflict within the mitochondrial genome. Out of 91 pairwise AU tests, 39 (42.9%) had an uncorrected p-value of <0.05, and 34 of those remained significant at a false discovery rate of 0.05 (Table S7). For the more conservative SH test, 8 of 91 comparisons were significant at an uncorrected threshold of 0.05, but they did not remain significant after corrections for multiple testing (Table S8). Overall, these phylogenetic comparisons suggested that many of the mitochondrial chromosomes do appear to have distinct genealogical histories.
“Four-gamete” tests were applied to further investigate the possibility of sexual recombination within mitochondrial genomes. When considering any pair of biallelic markers, the presence of all four possible combinations of alleles at the two loci across a set of individuals is evidence for recombination (in the absence of multiple homoplasious mutations occurring at the same site) (Hudson and Kaplan 1985; McCauley 2013). By analyzing the 183 parsimony-informative sites in our concatenated mitochondrial alignment, we found that 25.5% (4245/16,653) of pairwise comparisons contained all four possible combinations of alleles, indicating sexual recombination (Table S9). We also observed that the proportion of comparisons showing evidence of recombination was much lower for pairs of loci on the same chromosome (8.5%; 93 out of 1100 comparison) than for loci on different chromosomes (26.7%; 4152 out of 15,553).
Variation in mitochondrial chromosome presence/absence
PCR-based screening for a sample of 22 mitochondrial chromosomes revealed substantial variation in chromosome presence/absence among populations of S. noctiflora and related species (Fig. 3), especially when juxtaposed with the extremely low levels of nucleotide sequence divergence. Among the S. noctiflora populations, the patterns of chromosome presence/absence mirrored the sequence-based analysis and could be clustered into the same four lineages, with S. undulata exhibiting a fifth distinct pattern. Although the 15 S. noctiflora OSR-like mitochondrial haplotypes were all identical across the Sanger-sequenced mitochondrial markers (see above), there was variation in chromosome presence/absence within this group, with pairs of samples differing in the presence/absence of up to four chromosomes (Fig. 3). In contrast, all eight BRP-like samples had identical presence/absence profiles. Only four of the sampled mitochondrial chromosomes were detectable at all in S. turkestanica (BRP15-OSR18, BRP41-OSR45, OSR57, and BRP50), and none of those chromosomes produced positive amplification for all three markers (data not shown). The lack of amplification may indicate that mitochondrial genome content and structure is highly divergent in S. turkestanica relative to S. noctiflora/undulata. However, it may also be an artefact of the whole-genome amplification technique applied to the herbarium-derived S. turkestanica sample, which introduces a large bias in coverage and potential “drop-out” of some genomic regions.
Fig. 3.

Presence/absence survey of 22 mitochondrial chromosomes from 25 different populations of S. noctiflora (Table S1) and the close relative S. undulata. For each chromosome, presence/absence was assessed with three different PCR markers in each chromosome. Dark gray shading indicates positive detection for all three markers; medium gray shading indicates positive detection for two of three markers; light gray shading indicates positive detection for only one of three markers. For three chromosomes, the analysis was performed with only two markers (BRP41-OSR45, OSR58, and BRP57). In these cases, dark gray shading indicates detection of both markers. The chromosomes are divided into four categories as described in the Methods. Note that one of the “OSR-specific” chromosomes (OSR46) was found to be present at low levels in BRP and other samples from that group (see Results). This chromosome is recorded as present, but it is also possible that it has been lost from the mitochondria and only retained as a numt
Using both simple parsimony (Fig. 4) and Dollo parsimony (Figure S2), we inferred ancestral states for the presence/absence of each sampled chromosomes and mapped them onto the topology from our mitochondrial sequence analyses. To characterize variation at a finer level of geographical resolution, we analyzed seeds from five to seven different individuals from each of three sites <10km apart) within a S. noctiflora metapopulation in southwestern Virginia near Mountain Lake Biological Station (Table S2). We found that all sampled individuals within each site had the exact same pattern of presence/absence for a sample of 10 chromosomes but that the three sites all had different patterns from each other (Figure S3). The three observed presence/absence patterns across these sites were consistent with those found for the BRP-like, OSR-like, and KEW 1672 groups that were analyzed for a larger number of chromosomes (Fig. 3). Therefore, multiple mitochondrial haplotypes occur at nearby sites in a metapopulation within the introduced range of S. noctiflora, but we did not detect evidence of variation in chromosome presence/absence at the finest scale of within-site sampling.
Fig. 4.

Parsimony-based reconstruction of ancestral States for the presence (black) or absence (white) of each of the 22 sampled chromosomes across 25 S. noctiflora samples and the close relative S. undulata. a The constraint topology used for the analysis is based on concatenated sequence data from the mitochondrial genome (see Fig. 1a), and placement of the multiple OSR-like and BRP-like samples is their respective groups in based on the phylogenetic analysis in Figure S1. The remaining panels show the presence/absence states for b the five sampled chromosomes with high sequence divergence, c the five sampled chromosomes that are conserved between BRP and OSR and have no genes, d the six sampled “OSR-specific” chromosomes, and e the six sampled “BRP-specific” chromosomes
Our inference of chromosome presence/absence was based on three independent PCR markers on each chromosome. In general, each set of three markers produced consistent results, but there were some cases in which only two of the three markers supported presence or absence (Fig. 3), raising questions about changes that have affected partial chromosomes. To address variation at this scale, we mapped Illumina sequencing reads derived from total-cellular genomic DNA against S. noctiflora reference genomes (Fig. 5 and S4-S12). Four themes emerged from this analysis. First, the mapping results confirmed predictions for the PCR-based marker analysis (e.g., the lack of coverage from KEW 22121 sequencing for BRP chromosomes 12, 13, 19, and 26; Fig. 5). Second, many “missing” chromosomes showed no coverage across their entire length with the exception of very short scattered sequences that presumably reflect the short repeats that are shared with other chromosomes (again, see chromosomes 12, 13, 19, and 26 in Fig. 5). Third, most of the other chromosomes had consistent coverage across their full length, although the observed coverage level often differed to some extent among chromosomes (e.g., chromosome 45 vs. chromosome 46 in Fig. 5). Finally, a smaller subset of chromosomes exhibited major fluctuations in coverage within the chromosome. Large regions with no coverage (e.g., chromosome 29 in Fig. 5) suggest partial chromosome absence, and large regions in which coverage abruptly jumped to higher levels suggest sequence duplications and variation in the number of copies of large repeats within the genome.
Fig. 5.

Read depth across the 63 chromosomes of the S. noctiflora BRP reference mitochondrial genome based on Illumina sequencing of total-cellular DNA from S. noctiflora KEW 22121. Coverage estimates are based on a sliding window with a window size of 1000 bp and a step size of 500 bp. The plot was generated with the ggplot2 library in R. Reference chromosomes are ordered in decreasing size from chromosome 1 (191 kb) to chromosome 63 (65 kb) on the same x-axis scale, so coverage maps end before the end of the x-axis
An unexpected result from our PCR screen was that all three markers for chromosome 46 from the OSR mitochondrial genome were detected in our BRP sample (and all other S. noctiflora samples) despite our previous finding that it was absent from the BRP genome assembly (Wu et al. 2015). One potential explanation for this discrepancy is that the chromosome was present but at an abundance that was too low to be captured in the earlier BRP sequencing and assembly dataset. Consistent with this possibility, our mapping analysis of a different sample (KEW 22121; see above) detected consistent coverage across the full length of the chromosome but at a level much lower than the other chromosomes (see chromosome 46 in Figure S4). We found further support for this interpretation by performing ddPCR, which detected the presence of this chromosome in BRP and other BRP-like samples but at a much lower relative copy number (Fig. 6). A second potential example of this phenomenon is the chromosome 40 from the BRP mitochondrial genome. Although this chromosome was not detected in the original OSR genome assembly, mapping of sequence reads from OPL (a member of the OSR-like group) produced a low but consistent level of coverage across the chromosome (Figure S5). In addition, PCR-based screening produced sporadic amplification across the OSR-like group (often with only very faint bands), including for one of the three markers in OSR (Fig. 3).
Fig. 6.
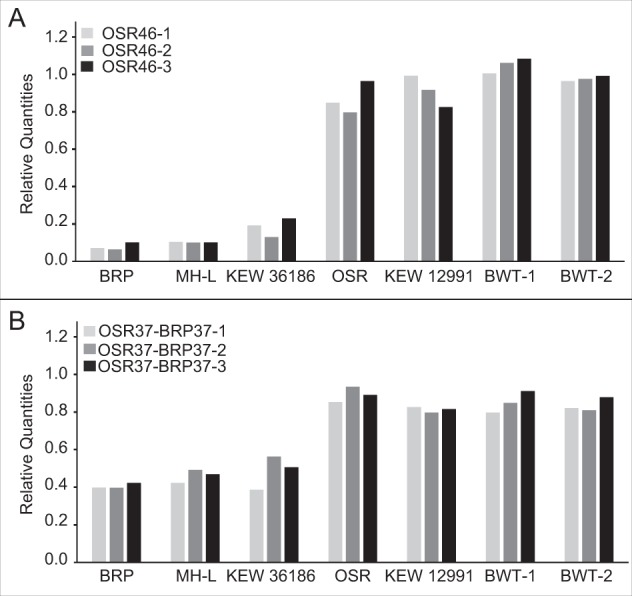
ddPCR analysis of copy number variation for two S. noctiflora mitochondrial chromosomes OSR46 (top) and OSR37-BRP37 (bottom) in seven S. noctiflora samples. Copy numbers are reported relative to two mitochondrial genes (nad2 and cox1). For each chromosome, three loci (ddPCR markers) were analyzed. The especially low copy number across all three markers for OSR46 in the BRP and BRP-like samples is consistent with the low observed sequencing coverage of this chromosome. The OSR37-BRP37 chromosome also exhibits a lower copy number than the reference markers in the BRP clade, but it more abundant than OSR46, and it was recovered in the original BRP assembly
Previous analysis of differences in copy number among chromosomes within a sample found that relative abundance does vary but across a relatively narrow range (less than two-fold) (Wu et al. 2015). Our observation of the OSR46 chromosome suggest that much wider ranges can occur. However, because our Illumina and ddPCR analyses were performed on total-cellular DNA, there is an alternative possibility that some of the sampled individuals have lost the mitochondrial chromosome but still harbor an older insertion of this mitochondrial sequence into the nuclear genome (i.e., a numt; (Hazkani-Covo et al. 2010)). Further work will be required to determine whether the low-copy sequences are indeed mitochondrial or only remnants of older nuclear insertions. More generally, rapid fluctuations in relative copy number of mitochondrial genome regions and sublimons is a common observation in plants. In angiosperms with more typical mitochondrial genomes, this phenomenon—termed substoichiometric shifting—is thought to involve crossovers between small repeats and preferential replication of alternative genome structures (Abdelnoor et al. 2003; Woloszynska 2010), often in response to stress (Shedge et al. 2010; Xu et al. 2011). It is not clear whether such processes share any underlying mechanisms with the variation in copy number that we have observed in S. noctiflora.
Discussion
Gain vs. loss of mitochondrial chromosomes
Our observations confirm and extend previous work showing remarkable intraspecific variation in chromosome content among samples that barely differ in nucleotide sequence (Wu et al. 2015). We find that many chromosomes that are present in one individual appear to be missing entirely in another, at least with respect to the tissues we have samples and subject to the limits of detection of PCR and sequencing methods. Previous analyses were unable to assess the extent to which these differences in chromosome content reflected recent gains vs. losses of chromosomes. In this study, parsimony reconstruction of ancestral states suggests variation in the timing of gains and losses (Fig. 4 and S2). These include clear cases of recent chromosome loss, in which mitochondrial markers are broadly shared across the S. noctiflora and S. undulata samples but are absent from one or a small number of lineages that are deeply nested within the group (e.g., chromosomes BRP12/OSR13, BRP56/OSR54, and BRP57; Figs. 3 and 4, and S2). One notable example is chromosome 57 in BRP, which is absent from two different highly nested parts of the tree (OSR/MH-F and KEW 1672), providing strong evidence for two independent losses of this chromosome.
This result indicates a high rate of chromosome loss and may be relevant to interpreting the presence/absence patterns of other chromosomes. For example, a simple-parsimony interpretation of chromosomes such as OSR 52 implies two independent gains of the same chromosome (Fig. 4). However, unless horizontal transfer of sequence content has occurred, we feel that this inference is less likely than a history in which the chromosome was ancestrally present at the base of the group and then lost independently in multiple lineages (as inferred under a Dollo parsimony model; Figure S2). We also note that a case such as chromosome 30 in OSR, which is only found in the KEW 22121 and OSR-like clade (Fig. 4), implies that the chromosome was not ancestrally present in S. noctiflora and was instead gained more recently at the base of that specific clade. Therefore, it is possible that the history of chromosome gain that shaped the massive, multichromosomal genomes of S. noctiflora occurred over a prolonged period that extended past the point when extant lineages within this species began to diversify. However, as above, parsimony reconstructions may be neglecting the possibility of ancestral presence followed by multiple independent losses. More generally, the evidence for conflicting genealogies within the mitochondrial genome (see below) implies that the assumption of a single mitochondrial tree may be faulty and that hybridization could have moved chromosomes between lineages. Overall, we conclude that there is strong evidence that recent chromosome losses have played a substantial role in the observed presence/absence variation but the evidence for recent gains is more ambiguous.
There are some important limitations to consider in our analysis to assess the relative contributions of chromosome gain vs. loss in variation among mitochondrial haplotypes. An obvious bias with our PCR-based screen is that the markers were all designed based on previously identified chromosomes. Therefore, they are capable of inferring the history of recent losses of chromosomes known to be shared between BRP and OSR, but they cannot detect recent lineage-specific gains of entirely novel chromosomes that are not found in either of those two genomes. This limitation also applies to our reference-based mapping of sequencing datasets. It is very possible that those samples contain additional chromosomes that are not found in either the OSR or BRP mitochondrial genomes.
Sexual recombination in multichromosomal mitochondrial genomes
Our analysis uncovered evidence of sexual recombination within the mitochondrial genome during the history of diversification of S. noctiflora. Detecting this phenomenon required a large, genome-wide dataset. Indeed, earlier analysis of our much smaller 13-marker dataset did not identify any evidence for recombination (Wu and Sloan 2018). There are multiple factors that might contribute to difficulties in detecting mitochondrial recombination in S. noctiflora and necessitate such a large-scale analysis. First, because of the very low levels of nucleotide polymorphism, many recombination events might occur between identical sequences and therefore be impossible to detect. In our concatenated alignment of 341,456 bp, only 356 sites (0.1%) were segregating within S. noctiflora. Breeding system and modes of mitochondrial inheritance also contribute to the opportunity for biparental sexual reproduction to occur. A small number of crosses have been performed in S. noctiflora to track mtDNA inheritance, and they did not identify any paternal inheritance (Sloan et al. 2012). However, this does not preclude rare bouts of paternal transmission or low levels of paternal leakage, as has been identified in some other Silene species (McCauley 2013). In addition, S. noctiflora has a high rate of self-fertilization (Davis and Delph 2005), such that inheritance of mtDNA through both pollen and ovule may only rarely bring together two distinct haplotypes. Given these limitations, it is striking that these genome-wide signatures of recombination were detectable.
We and others have speculated that the fragmentation of mitochondrial genomes into multiple chromosomes may facilitate sexual recombination between physically unlinked loci (Rand 2009; Wu et al. 2015). Our results support this hypothesis because we found evidence of sexual recombination to be much more extensive between chromosomes than it is within chromosomes. Therefore, like in nuclear genomes where chromosomes freely recombine via the process of independent assortment, the fragmentation of mitochondrial genomes in S. noctiflora may have accelerated the generation of novel genotypic combinations. Given the diverse eukaryotic lineages in which multichromosomal mitochondrial genomes have evolved (Lukes et al. 2005; Shao et al. 2009; Vlcek et al. 2011; Smith et al. 2012), there are numerous future opportunities to assess how commonly this mechanism facilitates recombination in mitochondrial genomes.
Data archiving
The raw genomic sequence reads generated in this study were deposited in the NCBI SRA under the BioProject PRJNA450445.
Electronic supplementary material
Acknowledgements
We thank Lucas Dominguez, Jessica Hurley, and Cody Kalous for their help with PCR screening. We also thank Bengt Oxelman for suggesting S. undulata as a likely close relative of S. noctiflora and Mark Simmons for helpful discussion about ancestral state reconstruction. Suggestions from three anonymous reviewers greatly improved the manuscript and motivated us to obtain much more powerful datasets for detection of recombination. We are grateful to the Kew Millennium Seed Bank, Helena Štorchová, Mitra Menon, and especially Michael Hood for providing/collecting seeds. Justin Havird and Joel Sharbrough provided insightful comments on an earlier version of this manuscript. This work was supported by National Institutes of Health (NIGMS R01 GM118046) and start-up funding from Colorado State University. The Illumina sequencing facility at University of Colorado is supported by Cancer Center Support Grant P30-CA046934 from the National Cancer Institute.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41437-018-0153-3) contains supplementary material, which is available to authorized users.
References
- Abdelnoor RV, Yule R, Elo A, Christensen AC, Meyer-Gauen G, Mackenzie SA. Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc Natl Acad Sci USA. 2003;100:5968 LP–5965973. doi: 10.1073/pnas.1037651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson AJ, Rice DW, Dickinson S, Barry K, Palmer JD. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell. 2011;23:2499–2513. doi: 10.1105/tpc.111.087189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura S, Tsutsumi N. Plant mitochondrial fission and fusion. Plant Biotechnol. 2005;22:415–418. doi: 10.5511/plantbiotechnology.22.415. [DOI] [PubMed] [Google Scholar]
- Arrieta-Montiel MP, Mackenzie SA (2011). Plant Mitochondrial Genomes and Recombination BT - Plant Mitochondria. In: Kempken F (ed) Springer New York: New York, NY, pp 65–82.
- Arrieta-Montiel MP, Shedge V, Davila J, Christensen AC, Mackenzie SA. Diversity of the Arabidopsis mitochondrial genome occurs via nuclear-controlled recombination activity. Genetics. 2009;183:1261–1268. doi: 10.1534/genetics.109.108514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Forget L, Lang BF. Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol Evol. 2013;5:418–438. doi: 10.1093/gbe/evt008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Csűös M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- Davis SL, Delph LF. Prior selfing and gynomonoecy in Silene noctiflora L. (Caryophyllaceae): opportunities for enhanced outcrossing and reproductive assurance. Int J Plant Sci. 2005;166:475–480. doi: 10.1086/428630. [DOI] [Google Scholar]
- Delph LF, Montgomery BR. The evolutionary dynamics of gynodioecy in Lobelia. Int J Plant Sci. 2014;175:383–391. doi: 10.1086/675572. [DOI] [Google Scholar]
- Fields PD, Taylor DR. Determinants of genetic structure in a nonequilibrium metapopulation of the plant Silene latifolia. PLoS ONE. 2014;9:e104575. doi: 10.1371/journal.pone.0104575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- Gualberto JM, Newton KJ. Plant mitochondrial genomes: dynamics and mechanisms of mutation. Annu Rev Plant Biol. 2017;68:225–252. doi: 10.1146/annurev-arplant-043015-112232. [DOI] [PubMed] [Google Scholar]
- Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Havird JC, Trapp P, Miller CM, Bazos I, Sloan DB. Causes and consequences of rapidly evolving mtDNA in a plant lineage. Genome Biol Evol. 2017;9:323–336. doi: 10.1093/gbe/evx010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics. 1985;111:147–164. doi: 10.1093/genetics/111.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinforma. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, Burger G, O’Kelly CJ, Cedergren R, Golding GB, Lemieux C, et al. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387:493–497. doi: 10.1038/387493a0. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Levsen N, Bergero R, Charlesworth D, Wolff K. Frequent, geographically structured heteroplasmy in the mitochondria of a flowering plant, ribwort plantain (Plantago lanceolata) Hered (Edinb) 2016;117:1–7. doi: 10.1038/hdy.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinforma. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukes J, Hashimi H, Zikova A. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet. 2005;48:277–299. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- Maddison WP, Maddison DR (2006). Mesquite: a modular system for evolutionary analysis. http://mesquiteproject.org. Accessed 28 June 2018.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- McCauley DE. Paternal leakage, heteroplasmy, and the evolution of plant mitochondrial genomes. New Phytol. 2013;200:966–977. doi: 10.1111/nph.12431. [DOI] [PubMed] [Google Scholar]
- McNeil J. The biology of Canadian weeks. 46. Silene noctiflora L. Can J Plant Sci. 1980;60:43–1253. [Google Scholar]
- Mower J, Sloan D, Alverson A (2012). Plant Mitochondrial Genome Diversity: The Genomics Revolution. In: Wendel JF, Greilhuber J, Dolezel J, Leitch IJ (eds) Plant Genome Diversity 1 SE - 9, Springer Vienna, pp 123–144.
- Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM. ‘Why genomes in pieces?’ revisited: sucking lice do their own thing in mtDNA circle game. Genome Res. 2009;19:700–702. doi: 10.1101/gr.091132.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautenberg A, Sloan DB, Aldén V, Oxelman B. Phylogenetic relationships of Silene multinervia and Silene Section Conoimorpha (Caryophyllaceae) Syst Bot. 2012;37:226–237. doi: 10.1600/036364412X616792. [DOI] [Google Scholar]
- Rice DW, Alverson AJ, Richardson AO, Young GJ, Sanchez-Puerta MV, Munzinger J, et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science. 2013;342:1468–1473. doi: 10.1126/science.1246275. [DOI] [PubMed] [Google Scholar]
- Sanchez-Puerta MV, Garcia LE, Wohlfeiler J, Ceriotti LF. Unparalleled replacement of native mitochondrial genes by foreign homologs in a holoparasitic plant. New Phytol. 2017;214:376–387. doi: 10.1111/nph.14361. [DOI] [PubMed] [Google Scholar]
- Segui-Simarro JM, Coronado MJ, Staehelin LA. The mitochondrial cycle of Arabidopsis shoot apical meristem and leaf primordium meristematic cells is defined by a perinuclear tentaculate/cage-like mitochondrion. Plant Physiol. 2008;148:1380–1393. doi: 10.1104/pp.108.126953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19:904–912. doi: 10.1101/gr.083188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman JR, Sonthirod C, Naktang C, Pootakham W, Yoocha T, Sangsrakru D, et al. The two chromosomes of the mitochondrial genome of a sugarcane cultivar: assembly and recombination analysis using long PacBio reads. Sci Rep. 2016;6:31533. doi: 10.1038/srep31533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedge V, Davila J, Arrieta-Montiel MP, Mohammed S, Mackenzie SA. Extensive rearrangement of the arabidopsis mitochondrial genome elicits cellular conditions for thermotolerance. Plant Physiol. 2010;152:1960–1970. doi: 10.1104/pp.109.152827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- Sloan DB. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013;200:978–985. doi: 10.1111/nph.12395. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Alverson AJ, Chuckalovcak JP, Wu M, McCauley DE, Palmer JD, et al. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012;10:e1001241. doi: 10.1371/journal.pbio.1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, MacQueen AH, Alverson AJ, Palmer JD, Taylor DR. Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: selection vs. retroprocessing as the driving force. Genet. 2010;185:1369–1380. doi: 10.1534/genetics.110.118000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Oxelman B, Rautenberg A, Taylor DR. Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae. BMC Evol Biol. 2009;9:260. doi: 10.1186/1471-2148-9-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Isaac PG, Leaver CJ. Stoichiometric differences in DNA molecules containing the atpA gene suggest mechanisms for the generation of mitochondrial genome diversity in maize. EMBO J. 1987;6:865–869. doi: 10.1002/j.1460-2075.1987.tb04832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Kayal E, Yanagihara AA, Collins AG, Pirro S, Keeling PJ. First complete mitochondrial genome sequence from a box jellyfish reveals a highly fragmented linear architecture and insights into telomere evolution. Genome Biol Evol. 2012;4:52–58. doi: 10.1093/gbe/evr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ. Mitochondrial and plastid genome architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci USA. 2015;112:10177–10184. doi: 10.1073/pnas.1422049112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler T, Delph LF. Ancient mitochondrial haplotypes and evidence for intragenic recombination in a gynodioecious plant. Proc Natl Acad Sci USA. 2002;99:11730–11735. doi: 10.1073/pnas.182267799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzet P, Delph LF. The effect of breeding system on polymorphism in mitochondrial genes of Silene. Genetics. 2009;181:631–644. doi: 10.1534/genetics.108.092411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlcek C, Marande W, Teijeiro S, Lukes J, Burger G. Systematically fragmented genes in a multipartite mitochondrial genome. Nucleic Acids Res. 2011;39:979–988. doi: 10.1093/nar/gkq883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woloszynska M. Heteroplasmy and stoichiometric complexity of plant mitochondrial genomes—though this be madness, yet there’s method in’t. J Exp Bot. 2010;61:657–671. doi: 10.1093/jxb/erp361. [DOI] [PubMed] [Google Scholar]
- Wu Z, Cuthbert JM, Taylor DR, Sloan DB. The massive mitochondrial genome of the angiosperm Silene noctiflora is evolving by gain or loss of entire chromosomes. Proc Natl Acad Sci USA. 2015;112:10185–10191. doi: 10.1073/pnas.1421397112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Sloan DB (2018). Intraspecific polymorphism for the presence and absence of entire mitochondrial chromosomes. bioRxiv. [DOI] [PMC free article] [PubMed]
- Wu Z, Stone JD, Štorchová H, Sloan DB. High transcript abundance, RNA editing, and small RNAs in intergenic regions within the massive mitochondrial genome of the angiosperm Silene noctiflora. BMC Genom. 2015;16:938. doi: 10.1186/s12864-015-2155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YZ, Arrieta-Montiel MP, Virdi KS, de Paula WBM, Widhalm JR, Basset GJ, et al. MutS HOMOLOG1 is a nucleoid protein that alters mitochondrial and plastid properties and plant response to high light. Plant Cell. 2011;23:3428–3441. doi: 10.1105/tpc.111.089136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi M, Kosugi S, Hirakawa H, Ohmiya A, Tanase K, Harada T, et al. Sequence analysis of the genome of carnation (Dianthus caryophyllus L.) DNA Res. 2014;21:231–241. doi: 10.1093/dnares/dst053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw genomic sequence reads generated in this study were deposited in the NCBI SRA under the BioProject PRJNA450445.