

Abstract
Taxonomic and phylogenetic relationships of Streptococcus mitis and Streptococcus oralis have been difficult to establish biochemically and genetically. We used core-genome analyses of S. mitis and S. oralis, as well as the closely related species Streptococcus pneumoniae and Streptococcus parasanguinis, to clarify the phylogenetic relationships between S. mitis and S. oralis, as well as within subclades of S. oralis. All S. mitis (n = 67), S. oralis (n = 89), S. parasanguinis (n = 27), and 27 S. pneumoniae genome assemblies were downloaded from NCBI and reannotated. All genes were delineated into homologous clusters and maximum-likelihood phylogenies built from putatively nonrecombinant core gene sets. Population structure was determined using Bayesian genome clustering, and patristic distance was calculated between populations. Population-specific gene content was assessed using a phylogenetic-based genome-wide association approach. Streptococcus mitis and S. oralis formed distinct clades, but species mixing suggests taxonomic misassignment. Patristic distance between populations suggests that S. oralis subsp. dentisani is a distinct species, whereas S. oralis subsp. tigurinus and subsp. oralis are supported as subspecies, and that S. mitis comprises two subspecies. None of the genes within the pan-genomes of S. mitis and S. oralis could be statistically correlated with either, and the dispensable genomes showed extensive variation among isolates. These are likely important factors contributing to established overlap in biochemical characteristics for these taxa. Based on core-genome analysis, the substructure of S. oralis and S. mitis should be redefined, and species assignments within S. oralis and S. mitis should be made based on whole-genome analysis to be robust to misassignment.
Keywords: phylogenetics, Streptococcus mitis, Streptococcus oralis, core genome, pan genome
Introduction
The mitis group of streptococci comprises human-associated Streptococcus species, many of which are associated with good health, but may be opportunistic pathogens. Three closely related species of interest in this group are Streptococcusmitis and Streptococcusoralis, which are found in oral biofilms and occasionally cause systemic infections (Whatmore et al. 2000; Ip et al. 2006), and the well-known respiratory pathogen Streptococcuspneumoniae. The phylogenetic relationships of these three species have been historically difficult to distinguish, both biochemically and genetically (Kawamura et al. 1995; Kikuchi et al. 1995; Whatmore et al. 2000; Ip et al. 2006), despite a variety of methods applied to doing so. Determining the phylogenetic relationship between these species is important for establishing robust tests for clinical identification (Whatmore et al. 2000), understanding how host-associated Streptococcus species evolve (Lefébure and Stanhope 2007), and for appropriate identification from host-associated metagenome samples (Dadi et al. 2017).
Characterizing the species of mitis group streptococcal isolates is difficult, and the species have undergone several taxonomic changes. Isolates of S. mitis, S. oralis, and S. pneumoniae show phenotypic variation that makes identification of universal species-specific biochemical characteristics unlikely (Kikuchi et al. 1995; Whiley and Hardie 2009). Multilocus sequence typing (MLST) and 16S rRNA gene similarity are popular methods to identify isolates, but the two methods are not always consistent (Bishop et al. 2009; Camelo-Castillo et al. 2014; Jensen et al. 2016). The 16S rRNA gene is particularly limited in its ability to resolve species of S. mitis, S. oralis, and S. pneumoniae, because the three species share >99% sequence homology (Kawamura et al. 1995). Additionally, Streptococcus are naturally competent, and genetic recombination and horizontal gene transfer between species may confuse taxonomic placement and phylogenetic inference (Chi et al. 2007), particularly as genes used for MLST studies and the 16S rRNA gene may undergo recombination, which is not always accounted for in MLST or 16S rRNA gene phylogenies. Based on a recent restructuring of the mitis Streptococcus species, S. mitis is recognized as a homogeneous species, whereas S. oralis has three subspecies, namely dentisani, oralis, and tigurinus (Jensen et al. 2016). Initial descriptions of both S. oralis subsp. dentisani and S. oralis subsp. tigurinus characterized these as independent species closely related to S. mitis and S. oralis (Zbinden et al. 2012; Camelo-Castillo et al. 2014).
Restructuring of the mitis group’s Streptococcus species used three approaches, MLST, 16S rRNA gene homology, and whole-genome analysis (Jensen et al. 2016). However, the phylogenies of these three approaches show substantial mixing of species between the S. mitis and S. oralis clades, suggestive of taxonomic misassignment, and do not consider recombination. In addition, our core-genome phylogeny of low-passage clinical oral Streptococcus isolates (Velsko et al. 2018) showed no clear grouping of the S. mitis and S. oralis isolates within their clade (Velsko et al. 2018), in contrast to the other species in the phylogeny, which formed distinct clades. The lack of clarity in species assignments and phylogenetic relationships between S. mitis and S. oralis, as well as the inability of the popular metagenomic profiler MetaPhlAn2 (Segata et al. 2012; Truong et al. 2015) to distinguish between S. mitis/oralis/pneumoniae, led us to take a whole genome-centered approach to clarify the phylogenetic relationship between these species. We demonstrate that core-genome analysis that accounts for recombination is a preferred method of classifying S. mitis and S. oralis species.
Materials and Methods
Isolate Selection, Genome Annotation, and Clustering
All GenBank S.mitis, S.oralis, Streptococcus parasanguinis, and S.pneumoniae genome assemblies were downloaded from NCBI in May 2018. Streptococcusparasanguinis was used as an outgroup in our analyses, selected because it is the most phylogenetically distant mitis group species from S. oralis, S. mitis, and S. pneumonia (Richards et al. 2014). Twenty-seven S. pneumoniae genomes were randomly selected for inclusion in the study to match the number of S. parasanguinis genomes. An additional 22 S. mitis and 10 S. oralis assemblies, previously annotated with Prokka (Seemann 2014), were included (Velsko et al. 2018). All genomes used in this study are listed in supplementary table S1, Supplementary Material online. For consistency, the GenBank genomes were reannotated using Prokka (Seemann 2014), using a custom Streptococcus database, and the same settings as in Velsko et al. (2018).
Homologous gene clustering was done twice, first using only S. oralis and S. mitis genomes, then again using all S. mitis and S. oralis genomes as well as the 27 S. pneumoniae and S. parasanguinis genomes. Prokka-annotated amino acid fasta files were concatenated into one file, and built into a BLAST database, and then the concatenated file was searched against this database using an all-versus-all BLAST with E-value cut-off of 1e-5 and 10,000 maximum target sequences. Homologous genes among all genomes were delineated using the MCL algorithm (Brohée and van Helden 2006) as implemented in the MCLBLASTLINE pipeline (available at http://micans.org/mcl). The pipeline used Markov clustering (MCL) to assign genes to homologous clusters based on an all-versus-all BLASTP search between all pairs of protein sequences using an E-value cut-off of 1e-5. The MCL algorithm was implemented using an inflation parameter of 1.8. Simulations have shown this value to be generally robust to false positives and negatives (Dongen 2000).
Phylogenomics
A core genome of single-copy genes present in S. mitis and S. oralis, as well as in S. mitis, S. oralis, S. pneumoniae, and S. parasanguinis, was determined from the MCL clustering. A total of 180 single-copy core gene clusters were identified in S. mitis and S. oralis, whereas a total of 141 single-copy core gene clusters were identified in S. mitis, S. oralis, S. pneumoniae, and S. parasanguinis. The single-copy core gene clusters for each of the two groups were aligned using Probalign (Roshan and Livesay 2006) and checked for recombination using PhiPack (Bruen et al. 2006). Genes identified as recombinant by Phi were removed from the core gene groups. The remaining putatively nonrecombinant single-copy core gene alignments (76 in S. mitis and S. oralis, and 81 in S. mitis, S. oralis, S. pneumoniae, and S. parasanguinis) were concatenated and the concatenated alignments were used to build a core phylogeny using phyML v. 3.0 (Guindon et al. 2003) with the GTR+G substitution model. Bootstrap support was provided by generating 500 bootstrap replicates using RAxML (Stamatakis 2014).
Then, a phylogeny based on the consensus of the separate phylogenies for each gene (gene trees) in the putatively nonrecombinant core gene set was constructed using the Triple Construction Method as implemented in the program Triplec (Ewing et al. 2008) (10,000 iterations). All gene phylogenies were built using phyML v. 3.0 with the GTR+G substitution model. All phylogenies were graphed using the R package ggtree (Yu et al. 2017).
Population Delineation and Admixture Analysis
BAPS v 6 (Corander et al. 2003) was used to determine the number of genetically distinct clusters (populations) in the S. mitis/S. oralis phylogeny and in the S. mitis/S. oralis/S. pneumoniae phylogeny. Using the concatenated core gene alignments, patristic distance among populations was calculated using p-distance neighbor-joining phylogenies generated using PAUP (Swofford 2002). Population distances were calculated using the average pairwise distances among isolates in each population. For these calculations, S. parasanguinis isolates were removed from the alignments. To calculate the patristic distances in the S. mitis/S. oralis/S. pneumoniae phylogeny of BAPS populations that were present in the S. mitis/S. oralis phylogeny but not the S. mitis/S. oralis/S. pneumoniae phylogeny, we used the same isolates that were in the S. mitis/S. oralis phylogeny BAPS populations. We additionally performed admixture analysis on our core-genome alignment using BAPS v 6, with the populations as defined by our BAPS clustering results. The admixture bar plots were generated in R with ggplot2 (Wickham 2016).
Assessment of Gene Content
Gene presence/absence matrices based on the MCL gene clusters for S. mitis/S. oralis, S. mitis/S. oralis/S. pneumoniae, and S. mitis/S. oralis/S. pneumoniae/S. parasanguinis were used to build gene content dendograms. Jaccard similarity matrices were calculated from the gene presence/absence matrices using the R package vegan (Oksanen et al. 2018), and then neighbor-joining dendograms were generated from the distance matrices using the R package ape (Paradis et al. 2004). Gene content dendograms and the gene presence/absence matrix were plotted using the R package ggtree (Yu et al. 2017).
We searched for gene clusters statistically significantly associated with either the species designations or the BAPS populations in our phylogenies using treeWAS (Collins and Didelot 2018). Associations with each species or BAPS population were tested independently, with a binary metadata matrix having the species or BAPS population of interest as 1 and all other entries as 0.
16S rRNA Gene Phylogeny
All 16S rRNA gene sequences in the isolates we included in this study were identified by a BlastN search. An alignment of representative 16S rRNA gene sequences from S. oralis, S. mitis, S. parasanguinis, and S. pneumoniae was searched in Geneious (https://www.geneious.com) against a custom-built Streptococcus species database that included all RefSeq sequences we included in this study, and again against a custom-built database of Streptococcus species that we sequenced for Velsko et al. (2018). All hits with >70% coverage from S. oralis, S. mitis, S. parasanguinis, and S. pneumoniae were extracted and aligned with MAFFT (Katoh et al. 2002). For 18 isolates (11 S. mitis, 6 S. oralis, and 1 S. parasanguinis) a full 16S rRNA gene sequence could not be identified, but regions of the gene were identified on two or more contigs, and therefore these isolates were omitted from the phylogenetic analysis. Several isolates had multiple copies of the gene. A single representative copy for each isolate was selected using FABox (Villesen 2007), realigned using MAFFT, and built into a phylogeny with PhyML v3.0 using the GTR+G substitution model.
Results
Core Phylogeny Supports Distinct Species and Subspecies
The phylogenetic relationships between S.mitis and S.oralis, as well as within S. oralis, are not well resolved, as was extensively demonstrated by Jensen et al. (2016). To investigate differences between S. mitis and S. oralis compared with well-established independent species, we built two unbiased phylogenies based on putatively nonrecombinant single-copy core genes. The first phylogeny contained S. mitis and S. oralis to investigate the relationship between and within these two species, and the second contained S. mitis/S. oralis/S. pneumoniae/S. parasanguinis. The branching patterns of both phylogenies formed two clades corresponding to current species designations, one including predominantly S. mitis, and the other including predominantly S. oralis and its subspecies (fig. 1). Streptococcus pneumoniae formed a distinct clade more closely related to S. mitis than to S. oralis, and S. parasanguinis formed a distinct clade that was distant to all others. These branching patterns reflect the phylogenetic relationships previously described between these species (Richards et al. 2014).
Fig. 1.

—Core-genome phylogenies of mitis streptococci. Core-genome phylogenies based on clustering of (A) S. mitis and S. oralis, (B) S. mitis, S. oralis, S. parasanguinis, and S. pneumoniae, with S. parasanguinis removed from the alignment to better visualize the branching patterns, and (C) S. mitis, S. oralis, S. parasanguinis, and S. pneumoniae. The BAPS populations are shown as a matrix to the right of each phylogeny. The type strains for S. oralis (NTC12261) and S. mitis (ATCC 35037) are indicated in phylogenies A and B with black horizontal lines to the left of their branches. ND, not determined.
Within the S. oralis species clade (including all S. oralis isolates with and without a subspecies designation), the subspecies dentisani, tigurinus, and oralis do not form exclusive clades. Most S. oralis subsp. dentisani isolates are in a clade with a handful of S. oralis, S. oralis subsp. tigurinus, and S. mitis (referred to here as the S. oralis subsp. dentisani clade), whereas most S. oralis subsp. tigurinus isolates are in a single clade with several S. oralis (referred to here as the S. oralis subsp. tigurinus clade), and S. oralis subsp. oralis is mixed among S. oralis (referred to here as the S. oralis/S. oralis subsp. oralis clade). Fourteen S. mitis are within the S. oralis species clade, and three S. oralis subsp. dentisani are in the S. mitis clade, suggesting that these isolates have been misidentified. Supplementary table S1, Supplementary Material online, details the current species designation for all isolates we included, as well as our suggested corrected species designation (column titled Phylogeny-Based Species). One unusual isolate, categorized as S. oralis, did not cluster with any of the other isolates, and we found that the core genes from this isolate are closely matched to S. cristatus based on a BLAST search.
To delineate the populations found within the S. mitis/S. oralis phylogeny, we ran BAPS on the S. mitis/S. oralis core gene cluster alignment and on the S. mitis/S. oralis/S. pneumoniae core gene cluster alignment. The first gave us distinct populations within the species of interest, and the second gave us distinct populations in the context of a well-defined related species, S. pneumoniae. BAPS delineated six populations within the S. mitis/S. oralis phylogeny, which correlated well with the branching patterns of the core phylogeny (fig. 1A). In the S. mitis/S. oralis clustering, the S. mitis clade separated into two populations, which we designate populations A and B, and the type strain NCTC 12261 falls into population B (fig. 1). The S. oralis species clade separated into three clusters corresponding roughly to the subspecies S. oralis subsp. dentisani, S. oralis subsp. tigurinus, and S. oralis/S. oralis subsp. oralis. In the S. mitis/S. oralis/S. pneumoniae clustering, S. mitis formed one population, S. oralis formed two populations, and S. pneumoniae formed one population (fig. 1B). The unusual S. oralis was its own population in both, further supporting that this isolate is a different species.
Admixture analysis of the BAPS populations was used to assess the extent of recombination within the remaining putatively nonrecombinant genes of our core-genome alignment. Analysis demonstrated minimal admixture within each population (supplementary fig. S1, Supplementary Material online), with the majority of isolates in each population exhibiting no evidence of admixture. Most of the admixture we did detect was between populations of the same species, that is, between S. oralis populations or between S. mitis populations, rather than between different species. Restricted gene flow may indicate different niche partitioning of these species, or other incompatibility. The presence of admixture in the core genome, albeit at low levels and in few isolates, suggests that the core genome is largely not subject to recombination.
We next asked whether the BAPS populations within S. mitis and S. oralis are supported as distinct species or subspecies. To answer this, we calculated the patristic distance between the BAPS populations (table 1) in each phylogeny. The distance between each population in the S. mitis/S. oralis phylogeny, roughly corresponding to subspecies designations, ranged from 5.8% to 7.7% (table 1, fig. 1). The distance between all population pairs in the S. mitis/S. oralis/S. pneumoniae phylogeny exceeded that of the well-established, distinct, species S. mitis versus S. pneumoniae (5.6%) (table 1, fig. 1), adding additional support that each clade is a distinct phylogenetic group. In the S. mitis/S. oralis/S. pneumoniae phylogeny, each of the populations that were delineated in the S. mitis/S. oralis phylogeny but not the S. mitis/S. oralis/S. pneumoniae phylogeny (i.e., S. oralis subsp. tigurinus, S. oralis subsp. oralis, and the two S. mitis populations) had patristic distance values of >5% (table 1). Specifically, the distance between S. oralis subsp. dentisani and S. oralis subsp. tigurinus (7.1%), and the distance between S. oralis subsp. dentisani and S. oralis/S. oralis subsp. oralis (6.9%), exceed the distance between S. pneumoniae and S. mitis, whereas the distance between S. oralis subsp. tigurinus and S. oralis/S. oralis subsp. oralis (5.3%) is less than the distance between S. pneumoniae and S. mitis. However, all of these values are less than the values between the BAPS-delineated populations, which were ∼12.5%.
Table 1.
Patristic Distance between BAPS Populations
| Phylogeny | Species/Subspecies | BAPS Pop | Patristic Distance |
|---|---|---|---|
| Streptococcus mitis/Streptococcus oralis | S. oralis/S. oralis subsp. oralis versus S. oralis subsp. tigurinus | 5 versus 2 | 0.061 |
| S. oralis/S. oralis subsp. oralis versus S. oralis subsp. dentisani | 5 versus 3 | 0.073 | |
| S. oralis subsp. tigurinus versus S. oralis subsp. dentisani | 2 versus 3 | 0.077 | |
| S. mitis population A versus S. mitis population B | 6 versus 4 | 0.058 | |
| S. mitis/S. oralis/Streptococcus pneumoniae | S. oralis/S. oralis subsp. oralis/S. oralis subsp. tigurinus versus S. oralis subsp. dentisani | 4 versus 5 | 0.069 |
| S. oralis subsp. tigurinus versus S. oralis subsp. dentisani | 4a versus 5 | 0.071 | |
| S. oralis/S. oralis subsp. oralis versus S. oralis subsp. dentisani | 4b versus 5 | 0.069 | |
| S. oralis subsp. tigurinus versus S. oralis/S. oralis subsp. oralis | 4a versus 4b | 0.053 | |
| S. oralis oralis/S. oralis subsp. oralis/S. oralis subsp. tigurinus versus S. mitis | 4 versus 2 | 0.124 | |
| S. mitis population A versus S. mitis population B | 2c versus 2d | 0.055 | |
| S. oralis subsp. dentisani versus S. mitis | 5 versus 2 | 0.125 | |
| S. oralis oralis/S. oralis subsp. oralis/S. oralis subsp. tigurinus + S. oralis subsp. dentisani versus S. mitis | 4 + 5 versus 2 | 0.124 | |
| S. oralis oralis/S. oralis subsp. oralis/S. oralis subsp. tigurinus + S. oralis subsp. dentisani versus S. pneumoniae | 4 + 5 versus 3 | 0.124 | |
| S. mitis versus S. pneumoniae | 2 versus 3 | 0.056 |
Note.—Population patristic distances are the average of pairwise comparisons among isolates in each population.
Only the isolates in population 4 corresponding to the S. oralis subsp. tigurinus population delineated in the S. mitis/S. oralis phylogeny.
Only the isolates in population 4 corresponding to the S. oralis/S. oralis subsp. oralis population delineated in the S. mitis/S. oralis phylogeny.
Only the isolates in population 2 corresponding to the S. mitis population 6 delineated in the S. mitis/S. oralis phylogeny.
Only the isolates in population 2 corresponding to the S. mitis population 4 delineated in the S. mitis/S. oralis phylogeny.
The patristic distance values we obtained, along with the BAPS populations and bootstrap values, support that the S. oralis subsp. dentisani clade is a distinct species, and we suggest adjusting this taxonomic assignment to the previously proposed S. dentisani (Camelo-Castillo et al. 2014) (although for consistency throughout this article we will continue to refer to this clade as S. oralis subsp. dentisani). Although the BAPS populations distinction for S. oralis subsp. tigurinus is lacking in the S. mitis/S. oralis/S. pneumoniae phylogeny, bootstrap values and patristic distance values support that it is a distinct group from the S. oralis/S. oralis subsp. oralis isolates in both of our phylogenies, and therefore we conclude that this group is a subspecies of S. oralis. In addition, our distance calculations support that S. mitis comprised two subspecies, which has not yet been distinguished in taxonomic naming conventions, despite previous reports in the literature (Shelburne et al. 2014). The distance between these two groups (5.5%) is just below the distance between S. pneumoniae and S. mitis, and for this reason, along with the lack of BAPS support in the S. mitis/S. oralis/S. pneumoniae phylogeny, we have designated the two S. mitis groups as subspecies rather than independent species.
Further, we investigated whether any sample metadata were correlated with the species/subspecies designations, which could indicate biases in taxonomic naming. We pulled data for the following metadata categories from the isolate GenBank files and/or the source publication for the isolates: country of origin, city of origin, isolation source, collection date, host, sequencing instrument, assembler, genome coverage, GenBank entry, complete genome, and submitter organization (supplementary table S1, Supplementary Material online). A matrix of metadata aligned with the core phylogenies showed no clear correlation between any category and the species/subspecies designations (supplementary fig. S2, Supplementary Material online).
As individual gene phylogenies may vary (Velsko et al. 2018), we additionally generated a gene consensus phylogeny from the individual gene phylogenies of our core, putatively nonrecombinant gene clusters, to see whether the branching pattern of our core phylogeny is well supported on a gene-by-gene basis. The S. mitis clade and S. oralis clade were well supported and separated based on BAPS population assignments for both the S. oralis/S. mitis and S. oralis/S. mitis/S. parasanguinis/S. pneumoniae phylogenies (fig. 2). However, the BAPS population assignments within the S. oralis clade and those within the S. mitis clade are intermixed in the S. oralis/S. mitis phylogeny (fig. 2A), whereas they cluster distinctly in the S. oralis/S. mitis/S. parasanguinis/S. pneumoniae phylogenies (fig. 2A and B), although support values for these clades are low (fig. 2). We further found that there are no clear associations between the metadata categories and gene consensus tree branching patterns (supplementary fig. S3, Supplementary Material online).
Fig. 2.

—Gene consensus genome phylogenies of mitis streptococci. Gene consensus phylogenies based on clustering of (A) S. mitis/S. oralis, (B) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae, with S. parasanguinis removed from the alignment to better visualize the branching patterns, and (C) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae. The BAPS populations are shown as a matrix to the right of each phylogeny. ND, not determined.
Pan-Genome Content Poorly Discriminates Species/Subspecies
Several isolates of both S. mitis and S. oralis appear to be misidentified based on their placement in the core phylogeny. We investigated whether pan-genome content could explain species misassignment in S. mitis and S. oralis, which is potentially related to biochemical characterization of these species. A total of 6,610 gene clusters were delineated in the S. mitis/S. oralis clustering, whereas the S. mitis/S. oralis/S. pneumoniae/S. parasanguinis clustering delineated 7,067 total gene clusters. The average number of gene clusters in each species/subspecies group S. mitis/S. oralis clustering ranged from 1,854 to 1,893, whereas the average number in the S. mitis/S. oralis/S. pneumoniae/S. parasanguinis clustering ranged from 1,854 to 2,030 (supplementary table S3, Supplementary Material online). The average number of gene clusters in each BAPS population was similar (supplementary table S3, Supplementary Material online).
We aligned a gene presence/absence matrix with the core phylogenies and gene consensus phylogenies to visualize gene clusters that were associated with the clades. No clear patterns could be discerned within the S. mitis or S. oralis clades (fig. 3A–D); however, there was a clear pattern of gene presence/absence in S. pneumoniae and in S. parasanguinis (fig. 3C and D) that differed from both S. oralis and S. mitis. Gene content dendograms generated from gene presence/absence matrices clustered the isolates similarly to the core genome (fig. 4A–C), and neither the metadata matrix (supplementary fig. S4, Supplementary Material online) nor the gene presence/absence matrix aligned with these phylogenies revealed any groups of metadata or gene clusters associated with regions of the phylogenies (fig. 5A and B) other than gene content for S. pneumoniae and S. parasanguinis (fig. 5B). These results suggest highly variable gene content within S. mitis and S. oralis that might make biochemical characterization difficult. This appears to be the case according to tables 1 and 3 in the Streptococcus chapter of Bergey’s Manual of Systematics of Bacteria and Archaea (Whiley and Hardie 2009), which details biochemical characteristics of these species. Most of the results are identical between S. mitis and S. oralis, and those that are not are often “variable between strains.” The core phylogeny, gene consensus phylogeny, and gene content dendogram for the S. mitis/S. oralis clustering and the S. mitis/S. oralis/S. parasanguinis/S. pneumoniae clustering show similar deep branching patterns but slight variations in leaf branching patterns (supplementary figs. S5–S7, Supplementary Material online).
Fig. 3.

—Species-specific gene content of mitis streptococci. Core-genome phylogeny based on clustering of (A) S. mitis/S. oralis and (C) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae aligned with the corresponding gene-content matrix. Gene consensus phylogeny based on clustering of (B) S. mitis/S. oralis and (D) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae aligned with the corresponding gene-content matrix. Present genes are indicated in dark gray and absent genes are indicated in light gray.
Fig. 4.

—Species-specific gene content is not correlated with core or gene consensus phylogenies. Gene content trees of mitis streptococci based on clustering of (A) S. mitis/S. oralis, (B) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae, with S. parasanguinis removed from the alignment to better visualize the branching patterns, and (C) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae. The BAPS populations are shown as a matrix to the right of each phylogeny. ND, not determined.
Fig. 5.

—Species-specific gene content of mitis streptococci. Gene content tree based on clustering of (A) S. mitis/S. oralis and (B) S. mitis/S. oralis/S. parasanguinis/S. pneumoniae aligned with the corresponding gene-content matrix.
We next looked for gene clusters that were significantly associated with species designations or with our BAPS populations to check for associations that were not clear in the gene content matrices. We used treeWAS (Collins and Didelot 2018), which was developed for bacterial genome-wide association studies, where the phenotype we were investigating was either the NCBI-designated species/subspecies name or the BAPS population. There were no gene clusters significantly associated with the NCBI species designations of S. mitis or S. oralis, but there was one gene cluster significantly associated with S. oralis supsp. dentisani in the S. mitis/S. oralis cluster matrix, and two gene clusters significantly associated with S. oralis subsp. dentisani in the S. mitis/S. oralis/S. pneumoniae cluster matrix (supplementary table S3, Supplementary Material online). In contrast, there were 63 gene clusters significantly associated with BAPS cluster 4 (part of the S. mitis clade, supplementary table S3, Supplementary Material online), 55 gene clusters significantly associated with BAPS cluster 5 (S. oralis clade, supplementary table S3, Supplementary Material online), and 1 gene cluster significantly associated with BAPS cluster 6 (part of the S. mitis clade, supplementary table S3, Supplementary Material online) in the S. mitis/S. oralis cluster matrix (table 3). Similarly in the S. mitis/S. oralis/S. pneumoniae cluster matrix, there were 64 genes significantly associated with BAPS cluster 2 (the S. mitis clade, supplementary table S3, Supplementary Material online) and 66 genes significantly associated with BAPS cluster 4 (S. oralis and S. oralis subsp. tigurinus clades, supplementary table S3, Supplementary Material online). These results suggest that identification of species-specific genes may assist with correct species identification of S. oralis subsp. dentisani and S. mitis.
16S rRNA Gene Phylogeny Does Not Follow Core-Genome Phylogeny
Finally, to investigate whether species classification by the 16S rRNA gene grouped any of the species/subspecies distinctly, we generated a phylogeny using the 16S rRNA gene in these isolates. The 16S rRNA gene phylogeny (fig. 6) shows more mixing between S. mitis and S. oralis than the core-genome phylogeny. The branching patterns form distinct clades for S. pneumoniae and S. parasanguinis similar to those seen in the core phylogeny, as well as a distinct S. mitis clade, but the branching patterns of the S. oralis clade are quite different from those of the core phylogeny. The S. oralis subspecies dentisani, oralis, and tigurinus do not form distinct clades, whereas 25 S. mitis isolates are distributed throughout the S. oralis isolates, with a distinct cluster of 12 S. mitis in the middle of the S. oralis isolates. The branching patterns of this phylogeny suggest that the species and subspecies misassignments cannot be attributed to 16S rRNA gene-based identification, with the exception of the highly divergent S. oralis isolate. Although this isolate appears to be S. cristatus based on the core genome, the 16S rRNA gene places it within the S. oralis clade, which likely explains the current species designation. We saw no clear correlations between branching patterns of the 16S rRNA gene phylogeny and any of the isolate metadata categories (supplementary fig. S8, Supplementary Material online).
Fig. 6.
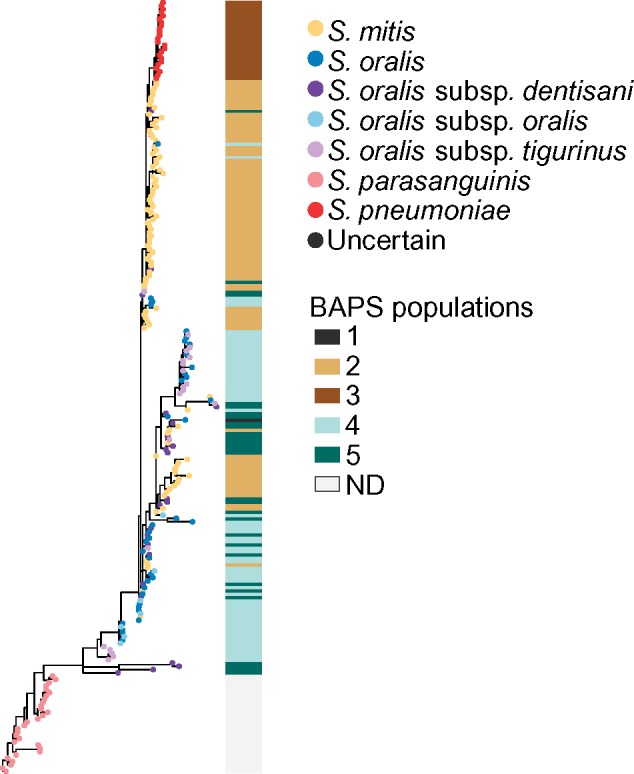
—16S rRNA gene phylogeny of mitis streptococci. ND, not determined.
Discussion
Species identification of S. mitis and S. oralis by biochemical or specific-gene-based approaches is not always reliable (Kawamura et al. 1995; Kikuchi et al. 1995; Whatmore et al. 2000; Zbinden et al. 2012; Jensen et al. 2016). Our whole genome-based approach to clarify the taxonomic relationship between S. mitis and S. oralis, and of the subspecies within S. oralis, has demonstrated that these species can be distinguished based on their core genome, and supports adjustment of species and subspecies designations as well reclassification of several apparently misidentified isolates. Our data support species/subspecies designations as follows: 1) S. oralis subsp. oralis comprised the currently designated S. oralis no subsp. and S. oralis subsp. oralis, 2) S. oralis subsp. tigurinus, 3) S. dentisani, 4) S. mitis subsp. A, and 5) S. mitis subsp. B. However, the pan-genome content of these species is highly variable, which may account for the difficulty in characterizing isolates biochemically. Different sequencing technologies have inherent biases that may impair comparison of genomes sequenced across different platforms (Kaas et al. 2014), but of the ten metadata categories we examined for this study, none appear to be related to taxonomic assignments biases or taxonomic misassignments.
Only one of the eight MLST genes used by Bishop et al. (2009) for classification of mitis streptococci was part of our core putatively nonrecombinant gene cluster list: map (methionine aminopeptidase). Five were found in duplicate in one or more strains (pfl, ppaC, pyk, rpoB, and tuf), and two were missing from a single strain (guaA and sodA), demonstrating that these genes may not be reliable for typing all sequenced isolates. This highlights an issue with noncomplete genomes that likely will continue to grow as more contig- and scaffold-level genomes are uploaded to databases such as NCBI without being completed. Namely, that not all MLST-designated genes will be found in all genomes and therefore MLST schemes may not be able to reliably type all genomes. Additionally, our 16S rRNA gene phylogeny shows that species identification by the 16S rRNA gene in the mitis streptococci may lead to taxonomic identification error. The presence of an S. mitis-specific clade in the center of a predominantly S. oralis clade reveals that the phylogenetic signal for this gene does not follow that of the core genome, and it is inappropriate to draw conclusions about phylogenetic relationships of S. mitis and S. oralis using the 16S rRNA gene alone (Velsko et al. 2018).
The S. mitis B6 genome has numerous insertion sequences, phage remnants, and a full set of competence genes (Denapaite et al. 2016), suggesting that it has undergone, and has high potential to continue, genetic exchange and rearrangement. It was suggested, however, that S. mitis does not undergo frequent recombination (Kilian et al. 2014), whereas S. oralis has evidence of substantial recombination (Do et al. 2009). The difference may explain the number of apparent species misassignments in the S. oralis clades compared with the S. mitis clade. Furthermore, our data, like that of Do et al. (2009), do not support that S. mitis is a recent derivative of S. pneumoniae, as has been proposed (Jensen et al. 2016). Instead, the very short branch lengths of S. pneumoniae and relatively long branch lengths of S. mitis, and placement of the S. peumoniae clade toward the tip of the S. mitis–S. pneumoniae grouping, suggest that S. pneumoniae is a recently emerged and rapidly expanding derivative of S. mitis, whereas S. mitis is an older lineage.
Our gene association tests found that the S. mitis and S. oralis populations defined by BAPS have sets of genes significantly enriched compared with the other populations, so some genes may be species-specific but not universally present in the given species. Despite clear patterns of gene presence/absence in S. pneumoniae and S. parasanguinis, treeWAS did not report any genes significantly associated with these species. Both had fewer isolates in our analyses than S. mitis and S. oralis, and hence we may have lacked statistical power. Small sample numbers could also explain the lack of genes associated with the S. oralis subsp. dentisani and subsp. tigurinus populations, as it has been reported that certain genes are enriched in S. oralis subsp. tigurinus (Diene et al. 2016). Clinical isolates of S. mitis can be found that contain the S. pneumoniae virulence factors autolysin (lytA) and pneumolysn (ply) (Whatmore et al. 2000), which highlights the potential of genetic exchange to confound gene-specific species identification.
Several studies that examined phylogenetic relationships of the species in the genus Streptococcus using different approaches have presented strong support for independent species designations of S. mitis, S. oralis, and S. pneumoniae (Thompson et al. 2013; Gao et al. 2014; Richards et al. 2014; Póntigo et al. 2015). If the species assignments of the misassigned isolates identified in this study are corrected, it should be possible to determine core single-copy marker genes that distinguish between S. mitis, S. oralis, and S. pneumoniae. This will improve resolution of metagenome classifiers that use single-copy marker genes, such as MetaPhlAn2 (Segata et al. 2012; Truong et al. 2015), to distinguish between species and even strains. Such a scheme could also be used to classify new isolates of mitis streptococci by replacing current MLST schemes (Bishop et al. 2009). However, it will need to be periodically reviewed and updated as more genomes are sequenced. With rapid advances in long-read sequencing technology such as PacBio, whole-genome sequencing has the potential to become standard practice in clinical laboratories (Rhoads and Au 2015; Ardui et al. 2018), which would improve the accuracy of taxonomic assignments.
The populations we identified within the mitis streptococci are consistent with previously recognized species and shed light on why these species have historically been difficult to distinguish. A highly variable pan genome, especially within isolates classified as S. oralis, likely contributes to the variation in biochemical characteristics used for classification prior to genetic methods. The core genomes of these species, however, reveal a distinct and well-supported phylogenetic signal. As methods for whole-genome sequencing, assembly, and annotation continue to improve and are widely adopted, particularly in clinical diagnostic laboratories, taxonomic assignment of the mitis streptococci is likely to become more straight-forward. Accurate taxonomic assignment will improve our ability to study the evolution of these species with respect to each other and their human host.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by Departmental Start-up Funds (to V.P.R.) and the NSF Research Experience for Undergraduates. CDBI 1460895 (to M.S.P.).
Data deposition: All data used in this study are previously published and were downloaded from NCBI. All accession numbers are listed in supplementary table S1, Supplementary Material online.
Reference
- Ardui S, Ameur A, Vermeesch JR, Hestand MS.. 2018. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 46(5):2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop CJ, et al. 2009. Assigning strains to bacterial species via the internet. BMC Biol. 7(1):3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohée S, van Helden J.. 2006. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7:488.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D.. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172(4):2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelo-Castillo A, Benitez-Paez A, Belda-Ferre P, Cabrera-Rubio R, Mira A.. 2014. Streptococcus dentisani sp. nov., a novel member of the mitis group. Int J Syst Evol Microbiol. 64(Pt 1):60–65. [DOI] [PubMed] [Google Scholar]
- Chi F, Nolte O, Bergmann C, Ip M, Hakenbeck R.. 2007. Crossing the barrier: evolution and spread of a major class of mosaic pbp2x in Streptococcus pneumoniae, S. mitis and S. oralis. Int J Med Microbiol. 297(7–8):503–512. [DOI] [PubMed] [Google Scholar]
- Collins C, Didelot X.. 2018. A phylogenetic method to perform genome-wide association studies in microbes that accounts for population structure and recombination. PLoS Comput Biol. 14(2):e1005958.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corander J, Waldmann P, Sillanpää MJ.. 2003. Bayesian analysis of genetic differentiation between populations. Genetics 163(1):367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadi TH, Renard BY, Wieler LH, Semmler T, Reinert K.. 2017. SLIMM: species level identification of microorganisms from metagenomes. PeerJ 5:e3138.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denapaite D, et al. 2016. Highly variable Streptococcus oralis strains are common among viridans streptococci isolated from primates. Blokesch M, editor. mSphere 1:e00041–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diene SM, François P, Zbinden A, Entenza JM, Resch G.. 2016. Comparative genomics analysis of Streptococcus tigurinus strains identifies genetic elements specifically and uniquely present in highly virulent strains. Schuch R, editor. PLoS One 11(8):e0160554–e0160517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T, et al. 2009. Population structure of Streptococcus oralis. Microbiology (Reading, Engl.). 155(8):2593–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongen S. 2000. Graph clustering by flow simulation.
- Ewing GB, Ebersberger I, Schmidt HA, Haeseler von A.. 2008. Rooted triple consensus and anomalous gene trees. BMC Evol Biol. 8(1):118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X-Y, Zhi X-Y, Li H-W, Klenk H-P, Li W-J.. 2014. Comparative genomics of the bacterial genus Streptococcus illuminates evolutionary implications of species groups. Reid SD, editor. PLoS One 9:e101229–12.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O, Rannala B.. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52(5):696–704. [DOI] [PubMed] [Google Scholar]
- Ip M, et al. 2006. Use of the housekeeping genes, gdh (zwf) and gki, in multilocus sequence typing to differentiate Streptococcus pneumoniae from Streptococcus mitis and Streptococcus oralis. Diagn Microbiol Infect Dis. 56(3):321–324. [DOI] [PubMed] [Google Scholar]
- Jensen A, Scholz CFP, Kilian M.. 2016. Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int J Syst Evol Microbiol. 66:4803–4820. [DOI] [PubMed] [Google Scholar]
- Jensen A, Scholz CFP, Kilian M, Parker CT Jr, Garrity GM.. 2016. Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus Int J Syst Evol Microbiol. 66: 4803–4820. [DOI] [PubMed]
- Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O.. 2014. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. Friedrich A, editor. PLoS One 9(8):e104984.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K-I, Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30(14):3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Hou X-G, Sultana F, Miura H, Ezaki T.. 1995. Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int J Syst Bacteriol. 45(2):406–408. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Enari T, Totsuka K-I, Shimizu K.. 1995. Comparison of phenotypic characteristics, DNA-DNA hybridization results, and results with a commercial rapid biochemical and enzymatic reaction system for identification of viridans group streptococci. J Clin Microbiol. 33:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian M, Riley DR, Jensen A, Bruggemann H, Tettelin H.. 2014. Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. MBio 5(4):e01490–e01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefébure T, Stanhope MJ.. 2007. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 8(5):R71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, et al. 2018. vegan: community ecology package. R Package Version 2.4-6. Available from: https://––CRAN.R–project.org–package=vegan.
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics.20(2):289–290. [DOI] [PubMed] [Google Scholar]
- Póntigo F, Moraga M, Flores SV.. 2015. Molecular phylogeny and a taxonomic proposal for the genus Streptococcus. Genet Mol Res. 14(3):10905–10918. [DOI] [PubMed] [Google Scholar]
- Rhoads A, Au KF.. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13(5):278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards VP, et al. 2014. Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol Evol. 6:741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshan U, Livesay DR.. 2006. Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics 22(22):2715–2721. [DOI] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069.24642063 [Google Scholar]
- Segata N, et al. 2012. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 9(8):811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne SA, et al. 2014. Streptococcus mitis strains causing severe clinical disease in cancer patients. Emerg Infect Dis. 20(5):762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. 2002. PAUP*. Phylogenetic analysis using parsimony (*and other methods). Sunderland (MA: ): Sinauer Associates. [Google Scholar]
- Thompson CC, Emmel VE, Fonseca EL, Marin MA, Vicente ACP.. 2013. Streptococcal taxonomy based on genome sequence analyses. F1000Res. 2:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, et al. 2015. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 12(10):902–903. [DOI] [PubMed] [Google Scholar]
- Velsko IM, et al. 2018. Species designations belie phenotypic and genotypic heterogeneity in oral streptococci. Turnbaugh PJ, editor. mSystems 3:e00158–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villesen P. 2007. FaBox: an online toolbox for fasta sequences. Mol Ecol Notes. 7(6):965–968. [Google Scholar]
- Whatmore AM, et al. 2000. Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis, and Streptococcus mitis: characterization of ‘atypical’ pneumococci and organisms allied to S. mitis Harboring S. pneumoniae virulence factor-encoding genes. Infect Immun. 68:1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiley RA, Hardie JM.. 2009. Streptococcus In: William B. Whitman, editor. Bergeys manual of systematics of archaea and bacteria. Vol. 33. Chichester (United Kingdom: ): John Wiley & Sons, Ltd; p. 1–86. [Google Scholar]
- Wickham H. 2016. ggplot2. Cham (Switzerland): Springer International Publishing.
- Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y.. 2017. ggtree: an rpackage for visualization and annotation of phylogenetic trees with their covariates and other associated data. McInerny G, editor. Methods Ecol Evol. 8: 28–36. [Google Scholar]
- Zbinden A, et al. 2012. Streptococcus tigurinus sp. nov., isolated from blood of patients with endocarditis, meningitis and spondylodiscitis. Int J Syst Evol Microbiol. 62(Pt 12):2941–2945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.