

Abstract
Platelet microparticles (PMPs) are closely associated with diabetic macrovascular complications. The present study aimed to investigate the effects of PMPs in diabetes on aortic vascular endothelial injury and to explore the underlying mechanisms. Peritoneal injection of streptozotocin was used to generate a diabetic rat model in vivo, and human umbilical vein endothelial cells (HUVECs) treated with PMPs were used in vitro. PMP levels in the circulation and aorta tissues were time-dependently increased in streptozotocin-induced diabetic rats at weeks 4, 8, and 12 (P < 0.05). Aspirin significantly inhibited the PMP levels at each time point (P < 0.05). In diabetic rats, the endothelial nitric oxide levels were decreased significantly combined with increased endothelial permeability. PMPs were internalized by HUVECs and primarily accumulated around the nuclei. PMPs inhibited endothelial nitric oxide levels to about 50% and caused approximately twofold increase in reactive oxygen species production. Furthermore, PMPs significantly decreased the endothelial glycocalyx area and expression levels of glypican-1 and occludin (P < 0.05). Interestingly, the PMP-induced endothelial injuries were prevented by raptor siRNA and rapamycin. In conclusion, increased PMPs levels contribute to aortic vascular endothelial injuries in diabetes through activating the mTORC1 pathway.
Keywords: platelet microparticles, diabetes, vascular endothelial cells, inflammation, mammalian target of rapamycin
Introduction
Diabetes mellitus (DM) has become a global emergency due to its rapidly rising morbidity and chronic micro- and macrovascular complications [1, 2]. Despite efforts to improve treatment, the risk of cardiovascular mortality of diabetic patients is approximately threefold higher than the general population [3]. Atherosclerosis is the prominent pathophysiological disorder of diabetic cardiovascular disease. Endothelial injury is the first step and plays a key role in early atherosclerosis [4]. Endothelial cells are the first barrier between blood flow and the vascular wall. Moreover, the endothelial glycocalyx, which is composed of glycoproteins, proteoglycans, and glycosaminoglycans, coats the surface of endothelial cells and participates in the maintenance of vascular homeostasis [5]. Accumulating evidence confirms that endothelial dysfunction, including decreased nitric oxide (NO) levels and increased endothelial permeability, often occurs in the blood vessels of diabetic patients [6, 7]. To date, the pathophysiological mechanism of endothelial injury in DM is not fully elucidated as previous studies have mainly focused on inflammation, oxidative stress, hyperglycemia, fatty acids, and insulin resistance [8]. Furthermore, the initiating factor in vascular endothelial injury in DM remains unclear.
Previous studies have demonstrated that plasma platelet microparticles (PMPs) are significantly increased in diabetes [9]. PMPs are heterogeneous vesicles (0.1–1 μm in diameter) released by activated or apoptotic platelets via outward membrane budding. As the dominant source of microparticles in the blood (70–90%), PMPs exert effects on inflammation, thrombosis, immunoregulation, and biological information transmission, primarily due to their RNA and cytoplasmic and membrane protein contents from platelets [10]. Research has increasingly suggested that PMPs are associated with atherosclerosis. For example, PMPs can be internalized by endothelial cells [11] and upregulate cytokines and intercellular adhesive molecular-1 expression, which induces the migration of leukocytes to the vessel wall [12]. Moreover, PMPs mediate inflammation by reducing NO levels which depend on nitric oxide synthase activity [13]. Accordingly, PMP levels are increased in diabetic subjects with macrovascular complications [14]. However, previous studies have tended to focus specifically on the role of PMPs in the progressive phase of atherosclerotic plaque formation and in the development of thrombosis. Thus, there is limited research that has addressed the role of PMPs on endothelial injury during the early stage of atherosclerosis in diabetes, and the potential mechanism is unclear.
Evidence has highlighted that mammalian target of rapamycin (mTOR) signaling is involved in the pathogenesis of atherosclerosis [15]. mTOR includes at least two distinct complexes, namely, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Rapamycin, an inhibitor of mTOR, selectively inhibits mTORC1 activity. mTORC1 activation mediates pro-inflammatory effects in vascular endothelial cells [16]. Moreover, circulating microparticles in the plasma of chronic lymphocytic leukemia patients can activate the mTORC1 pathway to modulate the tumor microenvironment [17]. Nevertheless, current studies have not explored the role of PMPs and the participation of the mTORC1 signal pathway in endothelial injury in diabetes.
Therefore, the present study aimed to investigate the role of PMPs in diabetic endothelial injury using human umbilical vein endothelial cells (HUVECs) and aorta tissues from streptozotocin (STZ)-induced diabetic rats as well as to explore the potential mechanisms that might be involved in activation of the mTORC1 pathway.
Materials and methods
Animal model
All animals received humane care, and the experiment protocols complied with the National Institutes of Health guidelines and were approved by the Ethics Committee of Southeast University. Male diabetes in Sprague-Dawley rats (8 weeks old and 230–240 g body weight) was induced by a single intraperitoneal injection of STZ (60 mg/kg; Sigma, St. Louis, MO, USA). Animals were purchased from Sino-British SIPPR/BK Lab Animals (Shanghai, China). Rats were confirmed to have DM if their blood glucose levels were greater than or equal to 300 mg/dL 72 h after the injection [18]. Diabetic rats were randomly assigned to the following groups (n = 30 in each group): DM, given a daily intragastric infusion of carboxymethylcellulose sodium; DM+Aspirin, given a daily intragastric infusion of 15 mg/kg aspirin (Sigma, St. Louis, MO, USA); and DM+Rapa, given a daily intraperitoneal injection of 1 mg/kg rapamycin (Sinopharm Chemical Reagent, Shanghai, China). Nondiabetic rats (n = 30) given a daily intragastric dose of carboxymethylcellulose sodium were assigned as the control group. Rats from each group (n = 10) were humanely killed at weeks 4, 8, and 12. Blood samples from the hearts were used for serum analysis, and the aortas were harvested for histological evaluations.
PMP extraction and flow cytometry analysis
Blood was collected into citrate-containing tubes, and platelets were removed by two subsequent centrifugations of 2000 × g for 10 min at room temperature. Microparticles were obtained by centrifuging the twofold diluted plasma at 18,000 × g for 60 min to pellet microparticles [19]. The samples were incubated with allophycocyanin (APC)-anti-Annexin V and fluorescein isothiocyanate (FITC)-anti-CD61 (BD Pharmingen, San Jose, CA, USA) for 30 min at 4 °C in the dark. For gating and counting, 0.8 μm and 3 μm beads were used, respectively. PMPs were detected as Annexin V+/CD61+ particles by a FACSCalibur cytometer (BD Biosciences). Analysis was performed using FlowJo software (Tree Star Inc., Ashland, OR, USA).
Cell culture
A HUVEC cell line (EA. hy926) was obtained from the American Type Culture Collection. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/Nutrient Mixture F-12 containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C in a humidified incubator with an atmosphere of 5% CO2 and 95% O2. HUVECs were subcultured twice a week using 0.25% trypsin-EDTA.
PMP isolation and acquisition from activated platelets in vitro
Platelet isolation was conducted as previously described [20]. Platelets were collected from acid-citrate-dextrose blood from healthy volunteers in our lab. Platelet-rich plasma (PRP) was obtained after centrifugation at 282 × g for 10 min and then again at 400 × g for 5 min at room temperature. After centrifuging PRP at 1600 × g for 5 min, the pellet was resuspended in citrate-glucose-saline (pH 6.5) buffer. After the last centrifugation at 1600 × g for 5 min, platelets were adjusted at 1 × 108/mL in modified Tyrode's buffer (MTB, pH 7.4). After a high glucose (30 mmol/L) pretreatment for 10 min, platelets were stimulated with 5 μg/mL collagen (Sigma) for 30 min at 37 °C. PMPs were resuspended in MTB for flow cytometry analysis or stored at −80 °C. The PMPs collected under these conditions were used for the in vitro studies.
Detection of PMP internalization
Washed platelets obtained from healthy volunteers were preincubated with 10 μmol/L 3,3'-dioctadecyloxacarbocyanine perchlorate (DiO) (Beyotime Biotechnology, Shanghai, China) for 30 min at 37 °C before stimulation with high glucose plus collagen to acquire DiO-labeled PMPs. The internalization of PMPs by HUVECs was assessed by confocal microscopy after incubation for 2 h. Cells were fixed in 4% paraformaldehyde for 15 min and blocked before incubation with mouse anti-CD31 (Santa Cruz Biotechnology, Dallas, TX, USA) at 4 °C overnight. Cells were then incubated with goat anti-mouse Fluor 594 secondary antibodies (Invitrogen, Carlsbad, CA, USA) at room temperature for 2 h, and nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Samples were observed under an Olympus FV1000 confocal microscope.
Small interfering RNA (siRNA) transfection
Control siRNA and raptor siRNA (GenePharma, Shanghai, China) were dissolved in Opti-Minimal Essential Medium (Opti-MEM; Gibco Life Technology, Carlsbad, CA, USA) and gently mixed with Opti-MEM-diluted Lipofectamine 2000 (Life Technology) for 20 min to form the siRNA/lipofectamine compound. HUVECs pretreated with siRNA (40 nmol/L) or preincubated with rapamycin for 10 min (10 ng/mL) were then treated with PMPs (5 × 106/mL) for 24 h and harvested for subsequent measurements.
Measurements for nitric oxide (NO), superoxide anion (O2−), and endothelial nitric oxide synthase (eNOS)
NO levels were measured using the nitrate reductase method to determine nitrite production via colorimetry. The O2− levels in homogenized aorta were detected by an assay kit, and the values are expressed as an activity unit of O2− per protein concentration. The eNOS activity in HUVECs was measured by catalyzing l-arginine and molecular oxygen to form NO using an assay kit (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
Immunofluorescence staining
Frozen sections of rat aorta were permeabilized and blocked before incubation with goat anti-glypican-1, rabbit anti-occludin (Santa Cruz Biotechnology, Dallas, TX, USA), or rabbit anti-phospho-S6K1 (p-S6K1) (Abcam, Cambridge, UK) at 4 °C overnight. Sections were then incubated with donkey anti-goat Fluor 488 or donkey anti-rabbit Fluor 555 secondary antibodies (Invitrogen, Carlsbad, CA, USA) at room temperature for 2 h, and nuclei were stained with DAPI. Samples were observed using an Olympus FV1000 confocal microscope. For cell immunofluorescence staining, HUVECs cultured in 24-well plates were fixed in 4% paraformaldehyde before permeabilization.
Endothelial glycocalyx staining
The endothelial glycocalyx was stained with wheat germ agglutinin (WGA, Sigma) as previously described [21]. Frozen sections of aorta were incubated with 5 μg/mL WGA for 2 h and were observed by laser confocal microscopy.
Permeability of the aortic endothelium
To test the permeability of the aortic endothelium in vivo, harvested rat aorta tissues were rinsed in 1% lanthanum nitrate with 2% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4) for 1 h. The tissues were then washed with 1% glutaraldehyde in 0.1 mol/L cacodylate buffer 3 times and then postfixed in 1% osmium tetroxide in 0.1 mol/L cacodylate buffer for 2 h. The specimens were then dehydrated using an ethanol gradient and embedded in Araldite. Ultrathin sections were made using an ultramicrotome (Leica, Solms, Germany) and observed by transmission electron microscopy [22].
Reactive oxygen species (ROS) measurements
DCFH-DA staining (Beyotime Biotechnology) was used to determine cellular ROS levels according to the manufacturer's instructions. Briefly, confluent HUVECs in 6-well plates were incubated with 10 μmol/L DCFH-DA diluted in serum-free DMEM/F-12 for 30 min at 37 °C in the dark. Flow cytometry assays were performed at an excitation and emission of 488 nm and 525 nm, respectively, and then analyzed using FlowJo software.
Transwell assays for endothelial permeability
Endothelial permeability was evaluated by measuring FITC-conjugated dextran (70 kDa) (Sigma) across the HUVEC monolayer [23]. Briefly, cells were seeded on a transparent transwell (1.12 cm2 filter area and 0.4 μm pore diameter), grown to full confluency and then treated as indicated. FITC-dextran (5 μL of 1 mg/mL) was added to the upper compartment and incubated at 37 °C for 2 h in the dark. Samples were then harvested from the upper and bottom chambers for fluorescence detection using a multidetection microplate reader at an excitation of 485 nm and an emission of 525 nm. The results were calculated using the permeability coefficient of FITC-dextran (P) as follows: P = ([U]/t) × (1/A)× (V/[B]); where [U] is the upper chamber concentration; t is the time in seconds; A is the area of the membrane in cm2; V is the volume of the bottom chamber; and [B] is the bottom chamber concentration.
RNA preparation and real-time PCR
Total RNA from HUVECs was extracted using TRIzol (TaKaRa, Tokyo, Japan) according to the manufacturer's instructions. Complementary DNA was obtained using PrimescriptTM RT Master Mix (TaKaRa) and then amplified in a 20 μL reaction volume using an ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA, USA) and SYBR Premix Ex TaqTM (TaKaRa). Predesigned primers were purchased from InvitrogenTM Life Technology (Table 1), and β-actin served as the housekeeping gene. Data were generated from each reaction and normalized to the control group as calculated by the 2−∆∆Ct method.
Table 1.
Taqman primers for real-time PCR
| Origins | Genes | Taqman primers |
|---|---|---|
| Human | MCP-1 |
Sense 5'-TCATAGCAGCCACCTTCATTC-3' Antisense 5'-CTCTGCACTGAGATCTTCCTATTG-3' |
| Human | TNF-α |
Sense 5'-CCAGGGACCTCTCTCTAATCA-3' Antisense 5'-TCAGCTTGAGGGTTTGCTAC-3' |
| Human | IL-6 |
Sense 5'-GGAGACTTGCCTGGTGAAA-3' Antisense 5'-CTGGCTTGTTCCTCACTACTC-3' |
| Human | IL-1β |
Sense 5'-ATGGACAAGCTGAGGAAGATG-3' Antisense 5'-CCCATGTGTCGAAGAAGATAGG-3' |
| Human | β-Actin |
Sense 5'-GGACCTGACTGACTACCTCAT-3' Antisense 5'-CGTAGCACAGCTTCTCCTTAAT-3' |
Western blotting
Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then semiwet transferred onto polyvinylidene fluoride membranes. Membranes were incubated with primary antibodies overnight at 4 °C. The following primary antibodies were used: anti-glypican-1, anti-occludin (Santa Cruz Biotechnology), anti-mTOR, anti-p-mTOR, anti-4EBP1, anti-p-4EBP1, anti-S6K1, anti-p-S6K1 (Abcam), and anti-β-actin (KeyGEN BioTECH, Nanjing, China). Peroxidase-conjugated secondary antibodies were incubated with the membranes for 1 h. Detection of the bands was performed using chemiluminescence horseradish peroxidase substrate (Merck Millipore, Whitehouse Station, NJ, USA) with an ImageQuant LAS 4000 mini acquisition system.
Statistical analysis
Data are shown as the mean values ± SEM and were analyzed using SPSS 19.0 and GraphPad Prism 7.0. Data comparisons between two groups with only one treatment were analyzed using t-tests. Experiments with multiple treatment groups were assessed by one-way analysis of variance followed by Student–Newman–Keuls multiple comparison test. P < 0.05 was considered statistically significant.
Results
Increased PMP levels in the plasma of diabetic rats accelerates endothelial dysfunction in vivo
As shown in Table 2, a DM rat model was successfully generated by peritoneal injections of STZ as indicated by the presence of hyperglycemia. Aspirin had no effect on blood glucose levels in DM rats. The number of circulating PMPs was counted by flow cytometry. PMPs were indicated by both CD61- and Annexin V-positive signals. Plasma levels of PMPs were increased in a time-dependent manner in diabetic rats at weeks 4, 8, and 12, and these increases were inhibited by aspirin (Fig. 1a). To determine if PMPs were deposited in rat aortas, CD61, a protein marker of PMPs, was used to detect PMPs in aorta tissues from DM rats by Western blotting (Fig. 1b). Quantitative analysis demonstrated that there were significantly higher CD61 expression levels in the DM group than in the control group. Moreover, aspirin intervention abated the increasing trend of CD61 expression levels. The effect of aspirin on endothelial function was also assessed. Endothelial NO levels were lower in diabetic rats than that in nondiabetic rats at all time points (weeks 4, 8, and 12), and aspirin improved endothelial function (Fig. 1c). As an ROS, the aortic levels of O2− tended to be opposite to those of NO (Fig. 1d). Therefore, these findings suggested that higher levels of PMPs in DM accelerate aortic endothelial dysfunction.
Table 2.
The basic biochemical data in rats
| Time point | Group | BG (mmol/L) | BUN (mmol/L) | Creatinine (μmol/L) |
|---|---|---|---|---|
| 4W | Control | 6.7 ± 0.80 | 5.7 ± 0.4 | 61.1 ± 5.2 |
| DM | 22.90 ± 3.41* | 9.5 ± 2.7 | 54.2 ± 12.1 | |
| DM+Aspirin | 25.23 ± 3.12* | 8.2 ± 1.6 | 52 ± 5.20 | |
| 8W | Control | 6.0 ± 0.56 | 6.6 ± 0.7 | 68.2 ± 9.0 |
| DM | 33.87 ± 7.18* | 17.5 ± 6.1* | 63 ± 11.6 | |
| DM+Aspirin | 26.29 ± 5.60* | 9.6 ± 1.8 | 59.9 ± 11.7 | |
| 12W | Control | 6.2 ± 1.06 | 6.0 ± 0.6 | 59.5 ± 10.7 |
| DM | 31.47 ± 6.13* | 12.7 ± 3.4* | 61.6 ± 14.0 | |
| DM+Aspirin | 26.22 ± 2.81* | 11.5 ± 7.5* | 54.9 ± 9.4 |
DM diabetes mellitus, BG blood glucose
*P < 0.05 versus Control group
Fig. 1.

Increased PMP levels in the plasma of diabetic rats accelerate endothelial dysfunction in vivo. Normal SD rats were given a daily intragastric infusion of carboxymethylcellulose sodium (Control). Diabetic rats were given a daily intragastric infusion of carboxymethylcellulose sodium (DM) or 15 mg/kg aspirin (DM+Aspirin). a Plasma PMP levels were measured by flow cytometry as CD61+AnnexinV+ particles <1 μm in diameter. The plasma PMP levels at week 4 (4W), week 8 (8W), and week 12 (12W) for each group are represented as the mean values ± SEM (n = 8). b PMP levels in aortas were confirmed by measuring CD61 protein expression by Western blotting. The right histograms were normalized as the fold change compared to β-actin and are shown as the mean values ± SEM (n = 8). c Endothelial function was evaluated by measuring total aortic NO levels. The results represent the mean values ± SEM (n = 8). d O2- levels in the aorta were measured at three time points (week 4, week 8, and week 12) using colorimetry. The results represent the mean values ± SEM (n = 8). *P < 0.05 compared with the control group, and #P < 0.05 compared with the DM group
PMPs destroy the aortic endothelial glycocalyx and increase endothelial permeability in diabetic rats
WGA staining showed a reduction in the endothelial glycocalyx area in diabetic aortas. In addition, immunofluorescence staining demonstrated that the levels of glypican-1, a glycocalyx proteoglycan, and occludin, an intercellular tight junction protein, were both decreased in diabetic rats, and these decreases were alleviated in the aspirin group at week 12 (Fig. 2a). Western blot analysis of glypican-1 and occludin protein levels showed similar results (Fig. 2b) to those of immunofluorescence staining. In the control group, lanthanum nitrite was concentrated in the aortic lumen, but it was diffusely distributed across the endothelium to the basal lamina in diabetic rats, which indicated increased endothelial permeability. Importantly, these effects were inhibited by aspirin which decreased the plasma PMP levels (Fig. 2c). These findings suggested that PMPs contribute to endothelial damage in DM.
Fig. 2.

PMPs destroy the aortic endothelial glycocalyx and increase endothelial permeability in diabetic rats. a Immunofluorescence staining of the glycocalyx (red), glypican-1 (green), and occludin (red) in each group at week 12 as assessed by confocal microscopy. Scale bar = 20 μm (scale bar in the top frame for the glycocalyx is 5 μm). b Protein levels of glypican-1 and occludin were measured by Western blotting. Quantification of glypican-1 and occludin are normalized as the fold change compared to β-actin and represent the mean values ± SEM at the bottom (n = 8). *P < 0.05 compared with the control group, and #P < 0.05 compared with the DM group. c Aortic endothelial permeability was determined by assessing the location of lanthanum nitrite using transmission electron microscopy (scale bar = 0.5 μm; red arrows indicate lanthanum nitrate). 4W week 4, 8W week 8, 12W week 12
PMP uptake activates the mTORC1 pathway in the endothelium in vivo and in vitro
To identify the role of PMPs in endothelial injury, the uptake of PMPs by endothelial cells was evaluated in vitro. DiO-labeled PMPs primarily accumulated around the nuclei (Fig. 3a). Microparticles isolated from diabetic rats reduced the NO concentrations in the supernatants of cultured HUVECs compared with those of nondiabetic rats (Fig. 3b). In addition, under stimulation of high glucose and collagen, activated human platelet-released PMPs showed a dose-dependent effect on the NO levels released by HUVECs in vitro (Fig. 3c). These findings suggested that activated PMPs in DM cause vascular endothelial injury.
Fig. 3.
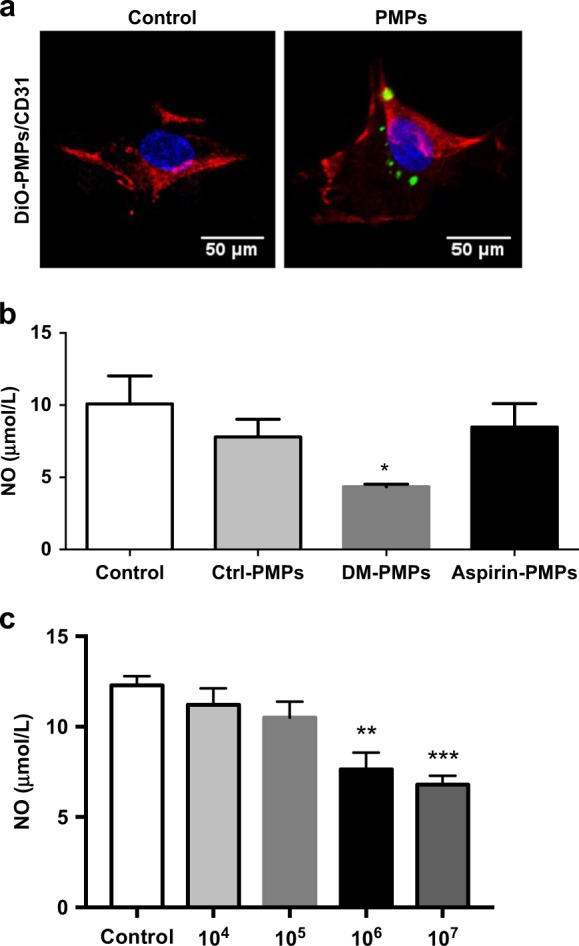
PMP uptake reduces the release of NO from endothelial cells. a Uptake of DiO-labeled PMPs (green) by HUVECs is shown using immunofluorescence. HUVECs were stained with CD31 (red), and the nuclei were labeled with DAPI (blue, scale bar = 50 μm). b Effects of isolated plasma microparticles from diabetic rats on the production of endothelial NO. Ctrl-PMPs plasma PMPs isolated from the Control group, DM-PMPs, plasma PMPs isolated from the DM group; Aspirin-PMPs, plasma PMPs isolated from the DM + Aspirin group. The results represent the mean values ± SEM (n = 8). *P < 0.05 versus Ctrl-PMPs. c Effects of PMPs (104/mL to 107/mL) obtained from activated platelets on the production of endothelial NO. The results represent the mean values ± SEM (n = 8). **P < 0.01 versus Control, ***P < 0.001 versus Control
Subsequently, the mTORC1 pathway was examined to explore the potential mechanisms of PMP-induced endothelial injury. The phosphorylation levels of the S6K1 effector protein were increased in endothelial cells both in vivo (Fig. 4a) and in vitro (Fig. 4b), and these effects were inhibited by rapamycin and raptor siRNA. Western blotting was used to measure the total and phosphorylated protein levels of mTOR, 4EBP1, and S6K1 (Fig. 4c, d). The Western blotting results were in accordance with the immunofluorescence staining results. These findings indicated that the mTORC1 pathway participates in mediating the effects of PMPs on the vascular endothelium in DM.
Fig. 4.

PMPs activate the mTORC1 pathway in the endothelium in vivo and in vitro. a, c Activation of the mTORC1 pathway in aortas of diabetic rats in vivo. Normal SD rats were given a daily intragastric infusion of carboxymethylcellulose sodium (Control). Diabetic rats were given a daily intragastric infusion of carboxymethylcellulose sodium (DM) or an intraperitoneal injection of 1 mg/kg rapamycin (DM+Rapa). a Representative pictures of immunofluorescence staining of p-S6K1 (green) in the rat aortic endothelium (scale bar = 50 μm). c Protein expression levels of p-mTOR, mTOR, p-4EBP1, 4EBP1, S6K1, and p-S6K1 in the aortas were measured by Western blotting. Quantification of the ratio of p-mTOR/mTOR, p-4EBP1/4EBP1, and p-S6K1/S6K1 in each group, and the results represent the mean values ± SEM (n = 5). *P < 0.05 compared with the control group, and #P < 0.05 compared with the DM group. b, d HUVECs were cultured for 24 h in serum-free medium (Control), in serum-free medium containing 5 × 106/mL PMPs (PMPs) or 5 × 106/mL PMPs plus 10 ng/mL rapamycin (PMPs + Rapa), or in serum-free medium containing 5 × 106/mL PMPs plus 40 nmol/L raptor siRNA (PMPs+siRaptor) or control-siRNA (PMPs+siControl). b Immunofluorescence staining of p-S6K1 (green) and nuclei (blue) in HUVECs was assessed by confocal microscopy (scale bar = 20 μm). d Protein expression levels of p-mTOR, mTOR, p-4EBP1, 4EBP1, S6K1, and p-S6K1 in HUVECs were measured by Western blotting. Quantification of the p-mTOR/mTOR, p-4EBP1/4EBP1 and p-S6K1/S6K1 ratios. The results represent the mean values ± SEM (n = 5). *P < 0.05 compared with the control group, and #P < 0.05 compared with the PMP group
PMP-activated mTORC1 pathway mediates endothelial injury
Inflammatory cytokines, such as monocyte chemotactic protein-1 (MCP-1), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-1β (IL-1β), were measured by real-time PCR. All of these inflammatory cytokines were higher in the PMP group, and these levels were blocked by rapamycin and raptor siRNA (Fig. 5a). ROS production was significantly increased in PMP-stimulated HUVECs, but it was attenuated by raptor siRNA or rapamycin (Fig. 5b). Furthermore, the decreased NO levels induced by PMPs were partly blocked by pretreating with raptor siRNA or rapamycin, and these changes were consistent with eNOS activity (Fig. 5c, d). To investigate the role of the mTORC1 pathway in PMP-induced endothelial damage, transwell assays were used to determine the permeability coefficient of FITC-dextran. As shown in Fig. 5e, PMPs significantly increased endothelial permeability compared to the control group, whereas both raptor siRNA and rapamycin suppressed the PMP-induced HUVEC permeability. Immunofluorescence staining demonstrated that PMPs decreased the endothelial glycocalyx area and the expression of glypican-1 and occludin. However, these changes were blunted by raptor siRNA and rapamycin (Fig. 5f). Protein levels of glypican-1 and occludin were also decreased in PMP-treated cells, and these effects were blocked by raptor siRNA and rapamycin (Fig. 5g). Overall, these results indicated that activation of the mTORC1 pathway contributes to the endothelial injury induced by PMPs.
Fig. 5.

PMP-mediated endothelial injury activates the mTORC1 pathway. a The mRNA expression levels of MCP-1, TNF-α, IL-6, and IL-1β in HUVECs were measured by real-time PCR. β-Actin served as an internal control gene. Data are shown as the fold of the control by calculating the 2−∆∆Ct. b Flow cytometric analysis of ROS levels in HUVECs. The results represent the DCF fluorescence intensity. Quantification of data is shown in the right histograms. c The NO concentrations in the supernatants of HUVECs were examined in each group after PMP treatment. d eNOS activity levels in HUVECs were determined by chemiluminescence. e HUVECs were grown on transwell filters in a confluent monolayer. After treatment, FITC-dextran (70 kDa) was applied to the upper wells for the intercellular permeability assay. The results are shown as the relative permeability coefficient (fold of the control). a–e Results represent the mean values ± SEM (n = 5). *P < 0.05 compared with the control group, and #P < 0.05 compared with the PMP group. f Immunofluorescence staining of the glycocalyx (red), glypican-1 (green), and occludin (red). Nuclei are labeled with DAPI (blue). Images were captured using a confocal microscope (scale bar = 20 μm). g The protein expression levels of endothelial glypican-1 and occludin were measured by Western blotting. The histograms show the mean values ± SEM of the proteins normalized to β-Actin (n = 5). *P < 0.05 compared with the control group, and #P < 0.05 compared with the PMP group
Discussion
The present study indicated that PMPs are significantly elevated in DM rats but reduced after antiplatelet therapy with aspirin. For the first time, the present study provided evidence that increased PMPs in diabetes mediates early vascular endothelial injury via activating the mTORC1 pathway.
Increasing attention has been focused on diabetes-associated endothelial dysfunction due to its importance in early atherosclerosis. Endothelial dysfunction is characterized by failure of the vasoactive, anti-inflammatory, and anti-coagulant properties of the endothelium [24]. Inflammation [25], oxidative stress, and dyslipidemia are the main contributors of endothelial dysfunction. However, few studies have evaluated the impact of PMPs on atherosclerosis [26, 27]. The in vivo study showed that circulating PMPs were increased in diabetic rats and that aspirin, an antiplatelet agent, inhibited the increased PMP levels. Increased PMP levels in diabetes correlated to aortic endothelial injury, which was characterized by decreased NO levels, increased O2− production, decreased expression of endothelial glycocalyx, decreased expression of intercellular tight junction proteins, and increased endothelial permeability. In agreement with the present study, Giacomazzi et al. reported that antiplatelet agents inhibit the generation of PMPs in vivo [28]. One of the main manifestations in endothelial dysfunction is the decrease in endothelial NO production, and the process of NO production is dependent upon eNOS activity. Endothelial cells treated with microparticles from patients with metabolic syndrome have reduced production of NO which is associated with the inhibition of eNOS phosphorylation [29]. Based on the above results, PMPs should be considered as a new target for early intervention in diabetic atherosclerosis. Of note, in cell culture experiments, PMPs decreased endothelial NO levels via inhibiting eNOS activity. NO bioavailability depends on not only its generation but also ROS production. Elevated ROS levels are linked to diabetes and atherosclerosis. Furthermore, activated PMPs stimulate neutrophils to produce ROS [30]. The present results showed that the ROS levels in HUVECs treated with PMPs were significantly increased. In addition to impairing endothelial function, PMPs also increased endothelial permeability which primarily depended on the degree of structural injury. The endothelial glycocalyx has been considered to be a potential determinant of vascular permeability [31]. After treatment of HUVECs with PMPs, the endothelial glycocalyx was disrupted, and syndecan-1 (a core protein) expression levels were decreased. Similar changes were found in diabetic rats, but these changes were prevented when PMPs were inhibited. All these findings suggested that increased PMP levels in diabetes contribute to vascular endothelial injury.
The potential contribution of mTORC1 pathway activation was explored in PMP-mediated endothelial injury in diabetes. Previous studies have suggested that abnormal activation of the mTORC1 pathway alleviates atherosclerosis [32]. Ghosh et al. proved that circulating microparticles from chronic lymphocytic leukemia influence the tumor microenvironment via activating the mTORC1 pathway [17]. In the present study, the mTORC1 pathway was upregulated in the aortic endothelium of diabetic rats as indicated by increased phosphorylation of mTOR and its downstream targets, 4EBP1 and S6K1, confirming that excessive activation of the mTORC1 pathway contributes to endothelial injury. Furthermore, accumulating evidence has indicated that mTORC1 mediates inflammation and oxidative stress which are involved in the pathogenesis of atherosclerosis [33]. Inhibition of mTORC1 decreases atherosclerosis mediated by anti-inflammatory responses [34]. Activation of mTORC1 increases ROS formation in arterial injury and endothelial dysfunction [35]. In the present study, increased ROS levels and upregulated expression of inflammatory cytokines were observed in endothelial cells treated with PMPs. Thus, the hypothesis that mTORC1 may be involved in PMP-induced endothelial injury in diabetes was next investigated. Inhibition of the mTORC1 pathway using raptor siRNA or rapamycin prevented PMP-induced endothelial inflammation, elevated ROS levels, reduced NO levels, and increased endothelial permeability. Therefore, the present study demonstrated for the first time that activated PMPs in diabetes contribute to early endothelial injury primarily due to the activation of the mTORC1 pathway.
In conclusion, increased PMPs in diabetes contribute to early vascular endothelial injury which is mediated by activation of the mTORC1 pathway. These findings suggested that inhibiting circulating PMP levels by aspirin and targeting the mTORC1 pathway may be beneficial for alleviating endothelial injury and preventing the progression of early atherosclerosis in diabetes.
Acknowledgements
The authors would like to thank Dr. Ze-hui Hong for his kind assistance during the animal experiments. This work was supported by the National Natural Science Foundation of China (81470957), the Jiangsu Province Six Talent Peaks Project (2015-WSN-002), the Project for Jiangsu Provincial Medical Talent (ZDRCA2016077), the Fundamental Research Funds for the Central Universities (KYCX18-0182, KYCX17-0169, KYZZ15-0061), the Jiangsu Province Ordinary University Graduate Research Innovation Project (SJZZ16-004), and the Jiangsu Province Social Development Project (BE2018744).
Author contributions
G-hW performed the in vivo and in vitro experiments, analyzed data, and wrote the manuscript. K-lM designed the research, analyzed data, and wrote the manuscript. YZ performed the in vivo experiments and analyzed data. Z-bH, LL, JL, P-pC and C-cL performed the in vivo and in vitro experiments. X-zR and B-cL analyzed and interpreted the data. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
References
- 1.Gao HX, Regier EE, Close KL. International Diabetes Federation World Diabetes Congress 2015. J Diabetes. 2016;8:300–2. doi: 10.1111/1753-0407.12377. [DOI] [PubMed] [Google Scholar]
- 2.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–88. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 3.Lind M, Svensson AM, Kosiborod M, Gudbjornsdottir S, Pivodic A, Wedel H, et al. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med. 2014;371:1972–82. doi: 10.1056/NEJMoa1408214. [DOI] [PubMed] [Google Scholar]
- 4.Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595–601. doi: 10.1253/circj.CJ-08-1169. [DOI] [PubMed] [Google Scholar]
- 5.Li L, Bonventre JV. Endothelial glycocalyx: not just a sugar coat. Am J Respir Crit Care Med. 2016;194:390–3. doi: 10.1164/rccm.201603-0624ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nathan DM, Lachin J, Cleary P, Orchard T, Brillon DJ, Backlund JY, et al. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med. 2003;348:2294–303. doi: 10.1056/NEJMoa022314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradley TJ, Slorach C, Mahmud FH, Dunger DB, Deanfield J, Deda L, et al. Early changes in cardiovascular structure and function in adolescents with type 1 diabetes. Cardiovasc Diabetol. 2016;15:31. doi: 10.1186/s12933-016-0351-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang L, Mittwede PN, Clemmer JS. Glucose homeostasis and cardiovascular alterations in diabetes. Compr Physiol. 2015;5:1815–39. doi: 10.1002/cphy.c150001. [DOI] [PubMed] [Google Scholar]
- 9.Sabatier F, Darmon P, Hugel B, Combes V, Sanmarco M, Velut JG, et al. Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes. 2002;51:2840–5. doi: 10.2337/diabetes.51.9.2840. [DOI] [PubMed] [Google Scholar]
- 10.Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580–3. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dasgupta SK, Le A, Chavakis T, Rumbaut RE, Thiagarajan P. Developmental endothelial locus-1 (Del-1) mediates clearance of platelet microparticles by the endothelium. Circulation. 2012;125:1664–72. doi: 10.1161/CIRCULATIONAHA.111.068833. [DOI] [PubMed] [Google Scholar]
- 12.Lukasik M, Rozalski M, Luzak B, Michalak M, Ambrosius W, Watala C, et al. Enhanced platelet-derived microparticle formation is associated with carotid atherosclerosis in convalescent stroke patients. Platelets. 2013;24:63–70. doi: 10.3109/09537104.2011.654292. [DOI] [PubMed] [Google Scholar]
- 13.Priou P, Gagnadoux F, Tesse A, Mastronardi ML, Agouni A, Meslier N, et al. Endothelial dysfunction and circulating microparticles from patients with obstructive sleep apnea. Am J Pathol. 2010;177:974–83. doi: 10.2353/ajpath.2010.091252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santilli F, Marchisio M, Lanuti P, Boccatonda A, Miscia S, Davi G. Microparticles as new markers of cardiovascular risk in diabetes and beyond. Thromb Haemost. 2016;116:220–34. doi: 10.1160/TH16-03-0176. [DOI] [PubMed] [Google Scholar]
- 15.Martinet W, De Loof H, De Meyer GR. mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis. 2014;233:601–7. doi: 10.1016/j.atherosclerosis.2014.01.040. [DOI] [PubMed] [Google Scholar]
- 16.Hassanian SM, Dinarvand P, Smith SA, Rezaie AR. Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J Thromb Haemost. 2015;13:860–71. doi: 10.1111/jth.12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010;115:1755–64. doi: 10.1182/blood-2009-09-242719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talpur N, Echard B, Ingram C, Bagchi D, Preuss H. Effects of a novel formulation of essential oils on glucose-insulin metabolism in diabetic and hypertensive rats: a pilot study. Diabetes Obes Metab. 2005;7:193–9. doi: 10.1111/j.1463-1326.2004.00386.x. [DOI] [PubMed] [Google Scholar]
- 19.Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. 2016;166:88–101. doi: 10.1016/j.cell.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duchez AC, Boudreau LH, Naika GS, Bollinger J, Belleannee C, Cloutier N, et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12-lipoxygenase and secreted phospholipase A2-IIA. Proc Natl Acad Sci USA. 2015;112:E3564–73. doi: 10.1073/pnas.1507905112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang H, Sun L, Huang Y, Wang Z, Zhao P, Fan Y, et al. Regional specific adaptation of the endothelial glycocalyx dimension in tail-suspended rats. Pflug Arch. 2015;467:1291–301. doi: 10.1007/s00424-014-1568-1. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Piontek J, Wolburg H, Piehl C, Liss M, Otten C, et al. Establishment of a neuroepithelial barrier by Claudin5a is essential for zebrafish brain ventricular lumen expansion. Proc Natl Acad Sci USA. 2010;107:1425–30. doi: 10.1073/pnas.0911996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Li X, Huang L, Jiang S, Tu F, Zhang X, et al. HSPA12B inhibits lipopolysaccharide-induced inflammatory response in human umbilical vein endothelial cells. J Cell Mol Med. 2015;19:544–54. doi: 10.1111/jcmm.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 25.Feng L, Matsumoto C, Schwartz A, Schmidt AM, Stern DM, Pile-Spellman J. Chronic vascular inflammation in patients with type 2 diabetes: endothelial biopsy and RT-PCR analysis. Diabetes Care. 2005;28:379–84. doi: 10.2337/diacare.28.2.379. [DOI] [PubMed] [Google Scholar]
- 26.Tomaniak M, Gasecka A, Filipiak KJ. Cell-derived microvesicles in cardiovascular diseases and antiplatelet therapy monitoring - a lesson for future trials? Current evidence, recent progresses and perspectives of clinical application. Int J Cardiol. 2017;226:93–102. doi: 10.1016/j.ijcard.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Chen LM, Liu ML. Microvesicles and diabetic complications--novel mediators, potential biomarkers and therapeutic targets. Acta Pharmacol Sin. 2014;35:433–43. doi: 10.1038/aps.2013.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giacomazzi A, Degan M, Calabria S, Meneguzzi A, Minuz P. Antiplatelet agents inhibit the generation of platelet-derived microparticles. Front Pharmacol. 2016;7:314. doi: 10.3389/fphar.2016.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet-Busson C, Leftheriotis G, et al. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol. 2008;173:1210–9. doi: 10.2353/ajpath.2008.080228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maugeri N, Rovere-Querini P, Baldini M, Baldissera E, Sabbadini MG, Bianchi ME, et al. Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: a candidate for microvessel injury in sytemic sclerosis. Antioxid Redox Signal. 2014;20:1060–74. doi: 10.1089/ars.2013.5298. [DOI] [PubMed] [Google Scholar]
- 31.Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–67. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- 32.Ma KL, Liu J, Wang CX, Ni J, Zhang Y, Wu Y, et al. Activation of mTOR modulates SREBP-2 to induce foam cell formation through increased retinoblastoma protein phosphorylation. Cardiovasc Res. 2013;100:450–60. doi: 10.1093/cvr/cvt203. [DOI] [PubMed] [Google Scholar]
- 33.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 34.Ai D, Jiang H, Westerterp M, Murphy AJ, Wang M, Ganda A, et al. Disruption of mammalian target of rapamycin complex 1 in macrophages decreases chemokine gene expression and atherosclerosis. Circ Res. 2014;114:1576–84. doi: 10.1161/CIRCRESAHA.114.302313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarz JB, Langwieser N, Langwieser NN, Bek MJ, Seidl S, Eckstein HH, et al. Novel role of the CXC chemokine receptor 3 in inflammatory response to arterial injury: involvement of mTORC1. Circ Res. 2009;104:189–200. doi: 10.1161/CIRCRESAHA.108.182683. [DOI] [PubMed] [Google Scholar]