

Abstract
Polygenic sex determination (PSD) may show variations in terms of genetic and environmental components between populations of fish species exposed/adapted to different environments. The European sea bass (Dicentrarchus labrax) is an interesting model, combining both a PSD system and a genetic subdivision into an Atlantic and a Mediterranean lineage, with genetic substructures within the Mediterranean Sea. Here, we produced experimental progeny crosses (N = 927) from broodstock sampled in four wild populations (North Atlantic, NAT; Western Mediterranean, WEM; North-Eastern Mediterranean, NEM; South-Eastern Mediterranean, SEM). We found less females than males in the progeny, both in the global dataset (32.5%) and within each paternal group (from 25.1% for NEM to 39.0% for WEM), with significant variation among populations, dams, and sires. Sex, body weight (BW), and body length (BL) showed moderate heritability (0.52 ± 0.17, 0.46 ± 0.17, 0.34 ± 0.15, respectively) and sex was genetically correlated with BW and BL, with rAsex/BW = 0.69 ± 0.12 and rA sex/BL = 0.66 ± 0.13. A weighted GWAS performed both on the global dataset and within each paternal group revealed a different genetic architecture of sex determination between Atlantic and Mediterranean populations (9 QTLs found in NAT, 7 in WEM, 5 in NEM, and 4 in SEM, with a cumulated variance explained of 27.04%, 21.87%, 15.89%, and 12.10%, respectively) and a more similar genetic architecture among geographically close populations compared to geographically distant populations, consistent with the hypothesis of a population-specific evolution of polygenic sex determination systems in different environments.
Subject terms: Evolutionary biology, Genome-wide association studies
Introduction
Sex determination is a mechanism of major evolutionary importance that exhibits a high variety of modalities, and this is especially true in fish (Mank et al. 2006; Heule et al. 2014). These modalities are generally classified as genotypic sex determination systems (or GSD, which includes male/female heterogamety and polygenic sex determination, or PSD) and environmental sex determination systems (ESD; Bull 1983).
PSD has been initially formalized by Bulmer and Bull (1982): they proposed an underlying “sex tendency” phenotype, with a polygenic determinism influencing the observed phenotype (male or female) depending on whether it lies below or beyond a fixed threshold. Under this model, any environmental or genotypic effect can equally affect the phenotype (here sex tendency) and bring its value below or beyond the threshold, therefore determining sex.
In the classical view, PSDis thought to be unstable, and should evolve either towards GSD in a fluctuating environment generating biased sex-ratios, or towards ESD if some environments increase the fitness of a specific sex (Bulmer and Bull 1982). Modeling approaches have shown that the orientation of PSD in one or the other direction depends on complex combinations of environmental variation between and within environmental patches, and on migration rates between patches (Van Dooren and Leimar 2003; Bateman and Anholt 2017). Therefore, starting with the same ancestral PSD system, we may hypothesize that there could be a population-specific evolution of the sex determination mechanism, in other words the components of the model may balance differently between sub-populations of the same species that are exposed to different environmental conditions (Vandeputte et al. 2012; Guinand et al. 2017).
In teleost fish, PSD has been well-documented in the Atlantic silverside, Menidia menidia (Conover and Heins 1987), some populations of the zebrafish, Danio rerio (Liew et al. 2012; Wilson et al. 2014), and in the European sea bass, Dicentrarchus labrax (Vandeputte et al. 2007; Palaiokostas et al. 2015). However, some authors consider that it may be more frequent than classically thought (Moore and Roberts 2013) since it is difficult to characterize.
The European sea bass (D. labrax L.) offers an interesting model to investigate the evolution of PSD. This species combines both a PSD system (Vandeputte et al. 2007; Palaiokostas et al. 2015) and a clear genetic subdivision into an Atlantic and a Mediterranean lineage, in addition to population genetic structure within the Mediterranean Sea (Naciri et al. 1999; Bahri-Sfar et al. 2000; Lemaire et al. 2005; Quéré et al. 2012; Tine et al. 2014).
Experimental evidence shows that early life rearing temperature has a strong influence on the sex-ratio of sea bass, with low rearing temperature (between 13 and 17 °C) during the first 60 days of life, favouring the production of female offspring (reviewed by Vandeputte and Piferrer 2018), which are preferred in aquaculture due to their higher growth rate (Saillant et al. 2001; Felip et al. 2006). In the natural environment, observed variation in yearly cohort sex-ratio may be indicative of natural variations in temperature influencing sex determination also in the wild populations of sea bass (Vandeputte et al. 2012).
Variation of sex-ratios between families in sea bass is consistent with the hypothesis that phenotypic sex is determined by an underlying continuous sex tendency combining the effects of polygenes and temperature (Vandeputte et al. 2007). However, at least three genome-wide significant sex determining quantitative trait loci (QTL) were recently found by Palaiokostas et al. (2015), showing that some specific loci could have a stronger effect on sex tendency, at least in some populations (in this case the western Mediterranean).
Here, we hypothesize that the sex determination system of European sea bass could have a different genetic architecture in different populations exposed to different environmental conditions (especially temperature) in the wild. Experimental progeny crosses were produced from broodstock sampled in four wild populations (corresponding to the whole distribution of natural populations, from Northern Atlantic to Eastern Mediterranean Sea). We estimated the additive genetic variation for sex tendency and the correlation between sex tendency and growth-related traits (weight and length) and performed a weighted genome-wide association study (wGWAS), both on the global dataset and within each group of paternal origin, to assess possible population-specific variations in the architecture of sex determination in sea bass.
Materials and methods
Broodstock origin, production, and rearing of experimental fish
The male broodstock used in this study belonged to four different origins, matching with most of the natural range of the species: North Atlantic (NAT), Western Mediterranean (WEM), North-Eastern Mediterranean (NEM) and South-Eastern Mediterranean (SEM). The female broodstock belonged to the WEM population. The origin and collection of the broodstock has been detailed by Vandeputte et al. (2014).
The NAT and WEM sires were reared at the IFREMER facilities of Palavas-les-Flots (France) and the sperm stripped and cryopreserved following Fauvel et al. (1998), while the sperm of SEM and NEM sires were both cryopreserved in 2005 at IOLR, Eilat, Israel (SEM sperm) and at Beymelek Lagoon, Turkey (NEM sperm) following the protocol of Sansone et al. (2002). The dams were reared at the IFREMER station of Palavas-les-Flots under natural photoperiod which was 11L:13D at the time of spawning (3 March 2014) and natural temperature, which decreased from 23.5 °C in August 2013 to 13.5 °C at the time of spawning; dams with a suitable stage of development of eggs (determined after ovarian biopsy) were hormonally injected and stripped 72 h after the injection. Artificial fertilization was performed at the IFREMER station, using a full factorial mating scheme: 15 sires per origin (60 sires in total) were crossed with 9 WEM dams. The fertilization protocol and the rearing of experimental fish were described previously by Doan et al. (2017a). Briefly, after hatching, larvae were reared in a common garden at a temperature of 16.5 °C and 25‰ salinity until 58 dph (days post-hatching); the following 7 days the temperature was gradually increased to 20 °C. Fish were then reared at a mean temperature of 21.5 °C (18.1–22.4 °C) and 30‰ salinity until 102 dph. Afterwards, fish were divided into five juvenile tanks (A–E), with a mean temperature of 22.1 °C (15.5–27.9 °C). Fish were fed using a classical hatchery feeding sequence (Artemia nauplii, Le Gouessant Marine Start and Neo Start pellets). At 180 dph, fish were individually tagged and measured for body weight (BW) and fork length (BL). At the same time, fin samples were collected for genomic DNA extraction. At 226 dph, 927 randomly chosen experimental fish were euthanized with an overdose of benzocaine, dissected, and the sex was recorded by visual observation of the gonads or using the squash technique (Menu et al. 2005) when macroscopic observation was ambiguous. The reliable identification of phenotypic sex was possible for all 927 fish. Sex was coded as a binary trait, 1 for males and 2 for females.
Genotyping, parentage assignment, and descriptive statistics
Fin clips from the 927 experimental fish, from the 60 sires and from the 9 dams were sent to LABOGENA (Jouy-en-Josas, France) for genomic DNA extraction and genotyping. Genotyping was performed with an iSelect Custom Infinium Illumina® European sea bass 3K SNP array. The design of this SNP array was done by selecting 2722 SNPs from an initial genome-wide variation map containing 2,628,725 SNPs phased into chromosome-wide haplotypes. These SNPs were discovered from 14 wild individuals from both the Atlantic and Mediterranean areas, using whole-genome sequencing as described by Duranton et al. (2018). A first filtering of the SNPs was made to remove variants closer than 80 bp from another known variant. Then, as recommended by Illumina®, we filtered the remaining SNPs to avoid A/T and C/G variants, that need a particular design (Infinium I Probe Design) involving setting up two probes instead of one. Among the remaining candidates, SNPs were chosen to cover all the chromosomes with a variable SNP density depending on the local nucleotide diversity (π), as reported by Tine et al. (2014). To do so, five π-classes were defined depending on the π estimated in non-overlapping 50 kb windows: class 1 for π < 10−3; class 2 for 10−3 < π < 2×10−3; class 3 for 2×10-3 < π < 3×10−3; class 4 for 3×10−3 < π < 4×10−3; class 5 for π > 4×10−3. Based on “best quality criterion”, one SNP was selected in class 1 windows (in interval 22.5–27.5 kb of the window), 2 SNPs in class 2 windows (in intervals 12.5–17.5 and 35-40 kb), three SNPs in class 3 windows (in intervals 5.8–10.8, 22.5–27.5, and 39–44 kb), four SNPs in class 4 windows (in intervals 3.7–8.7, 16.2–21.2, 28.7–33.7, and 41.2–46.2 kb), and five SNPs in class 5 windows (in intervals 2.5–7.5, 12.5–17.5, 22.5–27.5, 32.5–37.5, and 42.5-47.5 kb). Since nucleotide diversity is negatively correlated to the local recombination rate in sea bass (which was estimated by Tine et al. 2014), this local adjustment in the density of SNPs aimed at homogenizing the density of markers along the recombination map instead of the physical map.
Parentage assignment was performed with an exclusion-based software, VITASSIGN (Vandeputte et al. 2006), using 2722 markers and allowing 29 allelic mismatches to recover pedigree.
Proportion of individuals, males and females in the global dataset, per origin, per tank, per dam and per sire, pairwise comparisons and χ2 tests were performed in R version 3.4.3 using the packages stats (R Core Team 2017) and gmodels (Warnes et al. 2015). All P-values were adjusted for multiple testing with the Bonferroni correction method.
Principal component analysis (PCA)
To describe the overall genetic structure among the 927 individuals genotyped on the basis of genome-wide SNP data, we performed a PCA using –pca function in PLINK (Purcell et al. 2007). A two-dimension scatter plot of individuals coordinates on the two first principal components was generated, indicating the percentages of variance explained.
Heritability, genetic, and phenotypic correlations
Heritability was estimated on the entire dataset through a linear mixed sire model using the software VCE 6.0 (Groeneveld et al. 2010). The model was the following:
where yijkl is the phenotype for the studied trait (coded as a binary trait, 1 for male and 2 for female in the case of sex); oi is the fixed effect of the population of origin of the sires i; tj is the fixed effect of the rearing tank j; sk(i) is the random additive genetic effect of sire k within origin i; dl is the random effect of dam l; eijkl is the random residual.
As explained by Falconer and Mackay (1996), the sire component accounts for 1/4 of the additive genetic variance; for this reason, the heritability was estimated as h2 = 4σs2/σp2, with σs2 being the sire component of variance and σp2 the phenotypic variance. When the trait was sex, heritability on the observed (binary) scale was transformed to the value on the underlying liability scale (Dempster and Lerner 1950; Lynch and Walsh 1998) following the formula:
where is the heritability on the liability scale, is the heritability on the observed scale, p is the incidence (proportion of females) in the population and z is the value of the normal distribution density at the point where the cumulative distribution function of the normal distribution reaches incidence.
Genetic and phenotypic correlations between sex and growth-related traits (body weight and length at 180 dph) were assessed using VCE 6.0 software (Groeneveld et al. 2010) applying a three traits sire model with sex, weight, and length as variables.
Genome-wide association study (GWAS)
GWAS was performed through the BLUPf90 family of programs for mixed-model computations (Misztal et al. 2015) in order to identify possible SNPs associated with phenotypic sex.
Owing to the genetic subdivision between Atlantic and Mediterranean sea bass lineages and the finer scale differentiation between Western and Eastern Mediterranean populations (Naciri et al. 1999; Bahri-Sfar et al. 2000; Lemaire et al. 2005; Quéré et al. 2012; Tine et al. 2014; Duranton et al. 2018), the dataset was split into four groups depending on paternal origin (NAT, WEM, NEM, and SEM) and the analyses were performed separately within each group. The origin of the sire was taken into account as a fixed effect when we performed GWAS on the global dataset.
The raw SNP dataset was quality-filtered before running the GWAS. We firstly removed eight individuals with a call rate lower than 0.9. We then applied variant filters to exclude SNPs showing either significant Mendelian distortions, a minor allele frequency (MAF) lower than 0.05, or a proportion of missing genotypes greater than 0.1. This resulted in a final dataset containing 1205 retained markers out of the total 2722 SNPs which were genotyped in 919 offsprings and 69 parents (988 individuals in total). The high number of discarded SNPs is mainly due to technical design problems, which lead to target the wrong (i.e. non-variable) base position for 50% of the markers. The mapping of the SNPs used for further analyses was reported by Doan (2017b).
For the weighted GWAS (wGWAS) the following model was applied:
where y is the vector of phenotypes, b vector of the fixed effects (intercept, tank and origin of the sires), X the incidence matrix relating phenotypes with the fixed effects, W the incidence matrix relating phenotypes with the random animal effects, u the vector of random animal effects ~N(0, Gσg2) with G being the genomic relationship matrix (VanRaden 2008), σg2 the additive genetic variance, e the vector of residuals ~N(0, I σe2) and σe2 the residual variance. The genomic relationship matrix G was established as follows:
where D is a diagonal matrix with weights for SNP effects, Z is a matrix of gene content adjusted for allele frequencies and q is a weighting factor equal to 2Σpi (1 − pi), where pi is the MAF of SNP i.
The wGWAS was implemented through an iterative process (Zhang et al. 2010, 2016; Wang et al. 2012) and using Gibbs sampling (THRGIBBS1F90) to estimate the genomic estimated breeding values (GEBVs), since this is specifically adapted to the analysis of binary traits. The following steps were performed:
(1) in the first iteration, D was first set equal to I, the identity matrix (VanRaden 2008);
(2) G was calculated as G = ZDZ′/q;
(3) Direct genomic values (DGVs) were obtained from GEBVs as DGVi = −(Σj, j≠i gijGEBVj/gii) with gij elements of the G−1 matrix (Lourenco et al. 2015), and converted to SNP effects as ai = DZ′G-1DGVi;
(4) new SNP weights over a 5 SNPs window as were calculated and normalized so that the total genetic variance remained constant;
(5) the process was then iteratively repeated from step 2 with the new D matrix.
After three rounds there was no further modification of the variance explained by the SNPs (i.e. the correlation coefficient R2 between rounds 1 and 2 was equal to 0.93, 0.97 between rounds 2 and 3, and 0.99 between rounds 3 and 4).
We calculated the regional variance explained by summing neighboring SNP variance in overlapping windows of five adjacent SNPs, suggested by Habier et al. (2011) as the more appropriate method to infer the effect of QTLs. We considered as QTLs the genomic segments that explained a proportion of genetic variance higher than 2%.
Results
Parentage assignment and descriptive statistics
Assignment to a unique parental pair was achieved for all 927 offspring. The number of fish per paternal origin varied from a minimum of 150 (NAT) to a maximum of 328 individuals (SEM). The number of offspring per sire varied from 1 to 36 and the number of offspring per dam varied from 2 to 250.
The proportion of females in the global dataset was 32.5% and was variable among groups of paternal origin (χ2 = 12.27, df = 3, P-value = 7 × 10−3), ranging from 25.1% for NEM to 39.0% for WEM (Table 1). Pairwise comparisons showed significant sex-ratio differences between NAT and NEM and between WEM and NEM (Table 1).
Table 1.
Number of individuals (N), sex-ratio, mean body weight (g) and length (mm) at 180 dph (total and separately for males and females) with the coefficient of variation expressed in percentage (CV%); the results are showed for the global dataset and separately per group of paternal origin; different letters indicate a significant difference at P ≤ 0.05
| Paternal origin | Global N = 927 | ||||
|---|---|---|---|---|---|
| NAT | WEM | NEM | SEM | ||
| N = 150 | N = 182 | N = 267 | N = 328 | ||
| Sex-ratio | |||||
| Proportion of males | 0.620a | 0.610a | 0.749a | 0.677ab | 0.675 |
| Proportion of females | 0.380a | 0.390a | 0.251b | 0.323ab | 0.325 |
| Mean BW180 dph (CV%) | 17.57 (36.4%) | 14.70 (30.3%) | 16.22 (34.5%) | 16.09 (34.7%) | 16.09 (34.7%) |
| Males | 16.00 (38.4%) | 13.02 (28.1%) | 14.90 (31.5%) | 14.19 (30.5%) | 14.48 (32.7%) |
| Females | 20.11 (29.9%) | 17.32 (25.0%) | 20.17 (30.8%) | 20.08 (28.9%) | 19.46 (29.4%) |
| Mean BL180 dph (CV%) | 113.01 (11.4%) | 108.89 (9.4%) | 111.48 (10.5%) | 110.46 (10.7%) | 110.86 (10.6%) |
| Males | 109.46 (11.4%) | 105.23 (8.9%) | 108.90 (10.0%) | 106.54 (9.4%) | 107.50 (9.9%) |
| Females | 118.79 (9.6%) | 114.62 (7.8%) | 119.18 (9.0%) | 118.68 (9.4%) | 117.85 (9.1%) |
There were neither significant differences in the proportion of males and females between the five rearing tanks (χ2 = 9.48, df = 4, P-value = 5.02 × 10−2) nor in the proportion of animals belonging to the four paternal origins between tanks (χ2 = 16.37, df = 12, P-value = 17.47 × 10−2; Table S1). On the contrary, the proportion of females in the offspring strongly differed per sire and per dam (χ2 = 124.56, df = 59, P-value = 10−6 for sires, χ2 = 34.72, df = 8, P-value = 3 × 10−6 for dams; Fig. 1). The proportion of females ranged from 0% to 100% in paternal half-sib families and from 0% to 55% in maternal half-sib families.
Fig. 1.

Percentage of females per sire and per dam; blue, light blue, green and yellow bubbles identify the different origins of the sires, the pink ones identify the dams; the size of the bubble represents the total number of offspring per sire/dam
Female offspring were, on average, heavier and longer than males at 180 dph; this was true both for the global dataset and within groups of paternal origin (Table 1).
Principal component analysis
The PCA performed on the global dataset revealed that genotypic variance was mainly explained by paternal origin (first principal component explaining 5.9% of the variance, Fig. 2a). Furthermore, variations among dams were also detected (second axis explaining 4.1% of the variance, Fig. 2b).
Fig. 2.
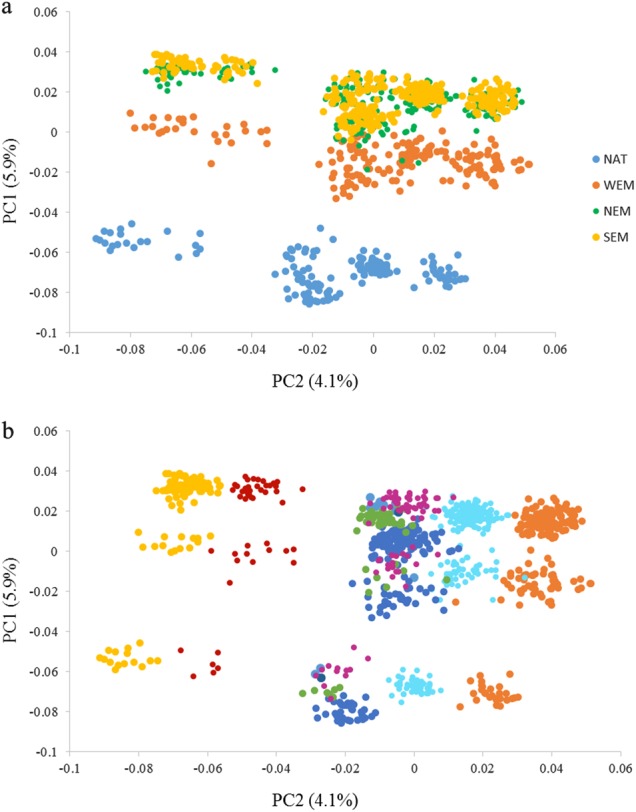
Two-dimension scatterplots showing the population stratification in the global dataset (N = 927) by paternal origin (a) and by dams (b). The first principal component was plotted against the second; the percentages of variance explained by each axis is indicated
More precisely, the first PC axis distinguished three groups c orresponding to the population of origin of the sires; NAT, Western, and Eastern Mediterranean groups were clearly separated, while the difference between North and South Eastern Mediterranean group was more subtle. This stratification explained by the population of origin of the sires was properly taken into account as a fixed effect in the models used to estimate heritability and to perform the wGWAS on the global dataset.
Heritability, genetic, and phenotypic correlations
Heritability was moderately high for all the variables (sex, BW, and body length at 180 dph, Table 2). The genetic and phenotypic correlations between sex and growth-related traits were moderately high and the genetic correlations were higher compared to the phenotypic correlations (> 0.65 vs. 0.42). The genetic and phenotypic correlations between body weight and length were close to unity (Table 2).
Table 2.
Heritability (± s.e., in bold on the diagonal) for sex (on the liability scale) and growth-related traits (body weight, BW, and body length, BL, at 180 dph), genetic (± s.e.; below the diagonal), and phenotypic correlations (above the diagonal) among traits, estimated with VCE 6.0 (Groeneveld et al. 2010)
| Trait | Sex | BW180 dph | BL180 dph |
|---|---|---|---|
| Sex | 0.52 ± 0.17 | 0.42 | 0.42 |
| BW180 dph | 0.69 ± 0.12 | 0.46 ± 0.17 | 0.97 |
| BL180 dph | 0.66 ± 0.13 | 0.99 ± 0.005 | 0.34 ± 0.15 |
Genome-wide association study (GWAS)
Results from the wGWAS performed on the global dataset identified one major group of SNPs on LG6 explaining up to 3.41% of the variance for sex. Other important groups of SNPs were detected on LG7, LG12, LG15, and LGx, explaining up to 2.73% of variance, while a minor group explaining slightly more than 2% of variance was located on LG2 (Fig. 3a and Table 3).
Fig. 3.
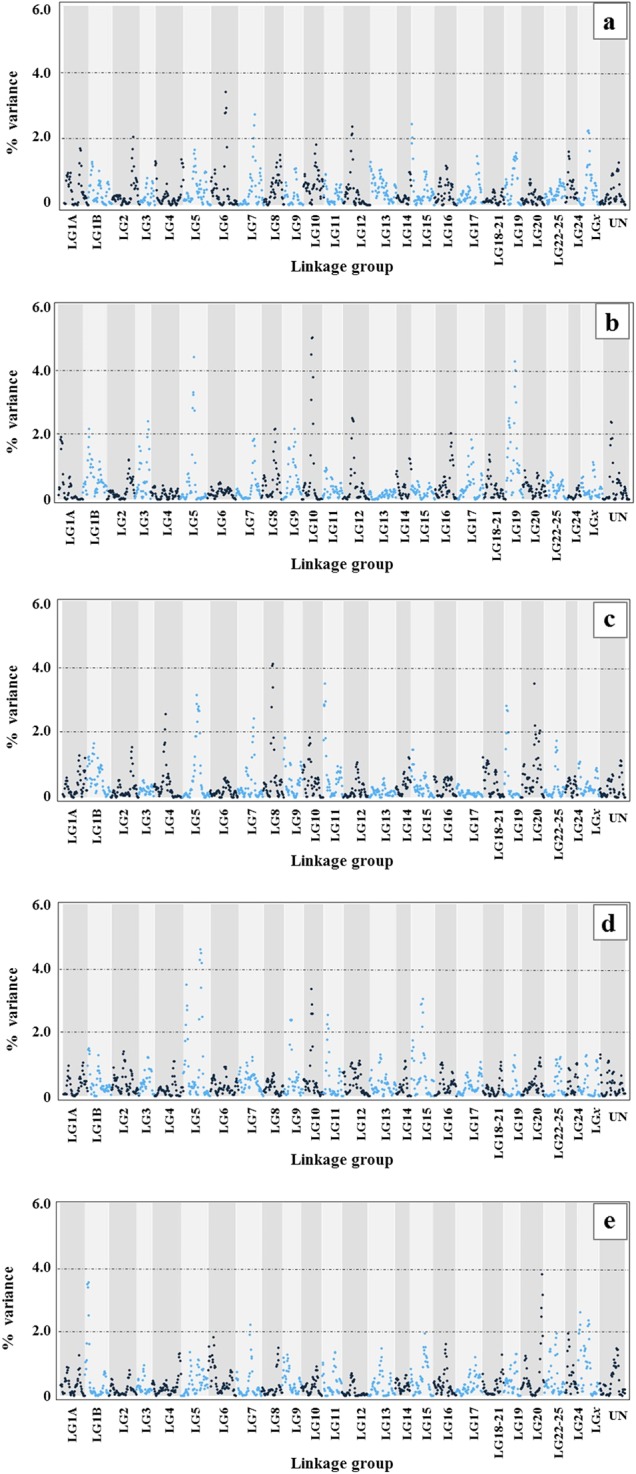
Manhattan plots showing the percentage of variance explained for sex. The results were obtained with a weighted GWAS in a sliding window of five adjacent SNPs in BLUPf90 (Misztal et al. 2015). a global dataset; b WEM × NAT; c WEM × WEM; d WEM × NEM; and e WEM × SEM
Table 3.
Identification of European sea bass chromosomes with a QTL explaining more than 2% of the variance in the global dataset and in each of the four offspring groups with the same paternal origin
| 1A | 1B | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18–21 | 19 | 20 | 22–25 | 24 | x | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Global | ● | ● | ● | ● | ● | ● | ||||||||||||||||||
| NAT | ● | ● | ● | ● | ● | ● | ● | ● | ● | |||||||||||||||
| WEM | ● | ● | ● | ● | ● | ● | ● | |||||||||||||||||
| NEM | ● | ● | ● | ● | ● | |||||||||||||||||||
| SEM | ● | ● | ● | ● |
The comparisons of the Manhattan plots of the wGWAS performed separately within each group of paternal origin (Fig. 3b–e and Table 3), revealed a clear pattern of similarity between samples belonging to adjacent paternal origins. Nevertheless, taking into account genomic regions explaining at least 2% of the variance showed a very variable architecture of sex determination, with some peaks shared among populations, while others were clearly population-specific (Table 4 and Fig. 4).
Table 4.
Total number of QTL for each paternal origin (in bold on the diagonal), total number of shared QTL between origins (below the diagonal) and total number of QTL that differ between origins (above the diagonal)
| NAT | WEM | NEM | SEM | |
|---|---|---|---|---|
| NAT | 9 | 10 | 8 | 11 |
| WEM | 3 | 7 | 8 | 7 |
| NEM | 3 | 2 | 5 | 9 |
| SEM | 1 | 2 | 0 | 4 |
Fig. 4.
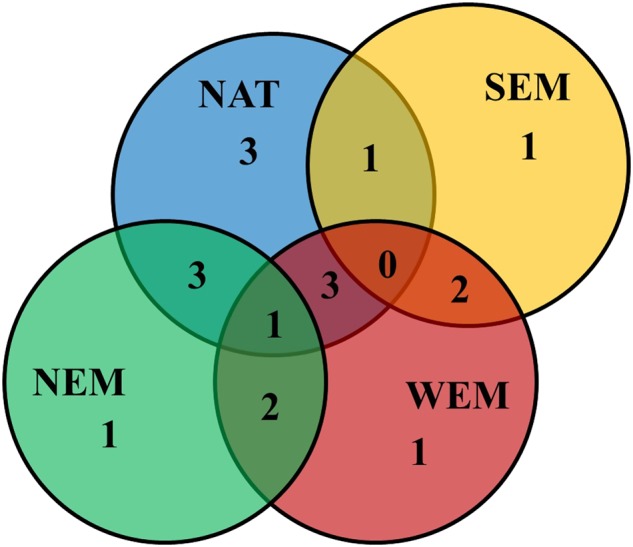
Venn diagram showing the number of QTLs for sex explaining more than 2% of the variance that were specific for each paternal group or that were shared between groups of paternal origin
A group of SNPs on LG5 explained 4.37% of variance for sex in ♀WEM × ♂NAT, 3.12% in ♀WEM × ♂WEM, and 4.6% in ♀WEM × ♂NEM, while in ♀WEM × ♂SEM this peak was not observed. Peaks shared only between ♀WEM × ♂NAT and ♀WEM × ♂WEM were identified on LG8, explaining 2.18% and 4.06% of the variance, respectively, and on LG19, explaining 4.25% and 2.80% of the variance, respectively. The crossings ♀WEM × ♂NAT and ♀WEM × ♂NEM shared two peaks on LG9 (2.17% and 2.38% of variance explained, respectively) and on LG10 (4.97% and 3.36% of variance explained, respectively).
One group of SNPs, which was shared between ♀WEM × ♂NAT and ♀WEM × ♂SEM was identified on LG1B, explaining 2.18% and 3.53%, respectively. One group of SNPs that was in common between ♀WEM × ♂WEM and ♀WEM × ♂SEM was identified on LG11 (3.48% and 2.54% of variance explained, respectively). Furthermore, ♀WEM × ♂WEM cross share two groups of SNPs with ♀WEM × ♂SEM cross, on LG7 (variance explained of 2.41% and 2.22%, respectively) and on LG20 (3.47% and 3.78% of the variance, respectively).
The ♀WEM × ♂NAT cross showed specific peaks, that were not shared with any other paternal origin, on LG3 (2.41% of variance explained), LG12 (2.51%), and LG16 (2.03%). One specific peak was found in the ♀WEM × ♂WEM cross, as well (LG4, 2.55% of variance explained).
Two groups of SNPs were identified as specific to the Eastern Mediterranean populations, one in the ♀WEM × ♂NEM cross (LG15, 3.04% of variance explained) and one in the ♀WEM × ♂SEM cross (LGx, 2.59% of variance explained).
Interestingly, we did not identify any sex QTL explaining more than 2% of the variance that was common to all populations.
Discussion
In this study, we explored the genetic basis of the sex determination system in the European sea bass by implementing GWAS genome-wide association study approach in a factorial crossing experiment. For the first time, sea bass belonging to different origins across the whole distribution range of natural populations were compared to assess variation in the genetic architecture of sex, including a comparison between the Atlantic and Mediterranean sea bass lineages. We found different QTLs underlying sex determination between Atlantic and Mediterranean populations, with a gradient of similarities from Western to Eastern Mediterranean populations, reflecting the previously documented introgression of Atlantic genes within the Mediterranean genetic background (Guinand et al. 2017; Duranton et al. 2018). This finding is consistent with the hypothesis of a population-specific evolution of PSD polygenic sex determination systems in different evolutionary lineages occupying different environments.
An important result was the increased sharing of QTLs for sex determination in adjacent populations, which could result from an ongoing admixture between two evolutionary lineages (i.e. Atlantic and Mediterranean) characterized by different genetic architectures of sex determination systems. The detected geographical gradient in the architecture, from NAT to SEM, would then reflect the level of introgression and indeed corresponds to the admixture gradient recently found in sea bass population genomic studies (Duranton et al. 2018).
The ancestral architecture of the sex determination in sea bass might have evolved differently during the 300, 000 years of divergence between Atlantic and Mediterranean lineages, explaining the origin of the variation that now has population-specific influences on sexual determination. Indeed, we did not find any linkage group common to all populations with groups of SNPs explaining more than 2% of the variance, which support the hypothesis put forward by Guinand et al. (2017) that the most important genes affecting sex may differ between sea bass populations.
♀WEM × ♂NAT cross showed some similarities compared to ♀WEM × ♂WEM and ♀WEM × ♂NEM, that have gradually reduced in ♀WEM × ♂SEM. This finding can be more likely explained by the recent history of inter-basins connectivity, since Atlantic alleles have been progressively diffused from the Western to the Eastern Mediterranean since the end of the last glacial maximum (Tine et al. 2014; Duranton et al. 2018). The resulting longitudinal gradient of admixture across the Mediterranean populations makes the WEM population (31% of Atlantic ancestry) more similar to the Atlantic than the NEM and SEM population (13% of Atlantic ancestry) in most of the genome (Duranton et al. 2018).
Therefore, a gradient in similarity of genomic architecture is expected if sex determination QTLs introgress similarly to neutral genes. We do not reject, however, the possibility that differential adaptations between Atlantic and Mediterranean environments have also contributed to the patterns we observed, although this hypothesis is difficult to distinguish from historical admixture. Finally, the presence of a biogeographical barrier to gene flow located in the Siculo-Tunisian Strait (Quignard 1978; Bahri-Sfar et al. 2000), which limits the connections between Western and Eastern Mediterranean, may explain the further reductions of similarities between Western and Eastern Mediterranean populations.
In our case, the four paternal groups are all related by the WEM dams (i.e. all the individuals have 50% of the genome coming from WEM), with the result that even the Eastern Mediterranean group contains a higher level of Atlantic ancestry than what is expected in “pure” wild ♀NEM × ♂NEM or ♀SEM × ♂SEM crosses. This leads to the conclusion that the real differences existing in nature could be even stronger than what we observed here, due to our experimental design.
We interpreted our results as differences between male origins with the implicit assumption that they mostly reflect additive QTLs effects from the sire population of origin. Still, some QTLs could be due to the dam population (WEM). This is not the preferred hypothesis as no QTLs are shared between all paternal group, although they all share the same dams. Another possibility is that some of the QTLs observed are not linked to additive genetic variation but to dominance. The higher number of QTLs in the ♀WEM × ♂NAT cross could be indicative of dominant alleles involved in sex determination (especially since some heterosis in sex-ratio has been shown by Guinand et al. (2017) when mating Atlantic and Mediterranean individuals).
Within each paternal origin, we performed a cross validation analysis of QTLs, by removing four times 25% of the offspring along the second axis of the PCA (representative of variation between dams, see Fig. 2b). Most of the QTLs were identified in all subgroups, suggesting they were linked to the sire origin studied, but several of them were absent in some of the subgroups, which may be indicative of sire origin by dam interaction, i.e. dominance variation (see Supplementary material S2).
Parental effects on phenotypic sex were clearly significant: the dams (χ2 = 34.72, df = 8, P-value = 3 × ·10−6) and sires (χ2 = 124.56, df = 59, P-value = 10−6) variation for the proportion of females in the offspring was strongly different, similar to Vandeputte et al. (2007), where both sires and dams had a similar-size effect on the sex-ratio of the progeny.
The heritability of sex tendency we estimated in the present study through a linear mixed sire model was relatively high (h2 = 0.52 ± 0.17), similar to the estimate obtained for sire heritability by Vandeputte et al. (2007) on a larger dataset consisting of individuals of Northern Atlantic origin (0.52 ± 0.13), suggesting that the influence of the genetic and the environmental components on sex-ratio variance should be roughly equivalent.
The genetic correlation between sex and growth-related traits was significant (h2 = 0.69 ± 0.12 between sex and weight, h2 = 0.66 ± 0.13 between sex and length) and higher compared to previous studies (rA between sex and size in the range of 0.23 and 0.59; Vandeputte et al. 2007; Palaiokostas et al. 2015). Overall, these results confirm the hypothesis of a strong link between genes affecting sex and growth (reviewed by Vandeputte and Piferrer 2018), with a clear sexual growth dimorphism (at the age of 180 dph, females were 34.4% heavier and 9.63% longer than males).
The sex-ratio in the global dataset was strongly skewed towards males, with a percentage of females less than half the percentage of males (32.5% versus 67.5%, respectively). This is consistent with the general observation that cultured sea bass, because of the hatchery environment (especially temperature), show an unbalanced sex-ratio in favor of males (Saillant et al. 2003; Piferrer et al. 2005), different if compared to wild-born sea bass, where in younger fish the sex-ratio seems to be balanced (Vandeputte et al. 2012).
The percentage of females was slightly higher in Atlantic/West-Med populations compared to Eastern Mediterranean populations, suggesting a possible different tendency in sex-ratio related to the origin of the individuals under aquaculture conditions. This is consistent with the study by Guinand et al. (2017), where the ♀WEM × ♂Atlantic and ♀WEM × ♂WEM crosses showed a higher mean proportion of females compared to the ♀WEM × ♂NEM and ♀WEM × ♂SEM crosses.
A limitation in our study is represented by the fact that the between-populations variation of sex-ratio could be confounded by non-additive genetic effects, as NAT, NEM, and SEM broodstock were used only as sires. As previously reported by Guinand et al. (2017), sex-ratio can show non-additive components of genetic variance, and we have no possibility to disentangle additive and non-additive genetic effects in our case.
Finally, a better understanding of the genetic architecture of sex tendency in sea bass could have applications in aquaculture production. European sea bass is one of the most important marine species widely cultured in the Mediterranean areas and represents 49% of the marine Mediterranean aquaculture production (FEAP Annual report 2016). The strong bias towards males under aquaculture condition has been recognized by farmers as a problem for different reasons (lower growth rates of males compared to females, reduced flesh quality and general decrease of the commercial values of the product; Felip et al. 2006). Uncovering the population-specific sex determination system may help to produce stocks with higher proportions of females, through selective breeding and genomic selection. Moreover, the choice of broodstock coming from a specific origin could be interesting to start new breeding programs, due to the between-population differences in sex-ratio we found.
Data archiving
The dataset underlying our findings is available in the institutional public data repository (SEANOE: http://www.seanoe.org/), 10.17882/55576
Electronic supplementary material
Acknowledgements
S.F. was supported by a grant from Valle Cà Zuliani Società Agricola s.r.l. (Pila di Porto Tolle, Italy). The experimentaldata and genotyping were derived from RE-SIST project “Improvement of disease resistance of farmed fish by selective breeding” selected at the 15th “Fonds interministériel”.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41437-018-0157-z) contains supplementary material, which is available to authorized users.
References
- Bahri-Sfar L, Lemaire C, Ben Hassine OK, Bonhomme F. Fragmentation of sea bass populations in the western and eastern Mediterranean as revealed by microsatellite polymorphism. Proc R Soc Lond B Biol Sci. 2000;267:929–935. doi: 10.1098/rspb.2000.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman AW, Anholt BR. Maintenance of polygenic sex determination in a fluctuating environment: an individual-based model. J Evolut Biol. 2017;30:915–925. doi: 10.1111/jeb.13054. [DOI] [PubMed] [Google Scholar]
- Bull JJ. Evolution of sex determining mechanisms. Menlo Park, CA: Benjamin/Cummings; 1983. [Google Scholar]
- Bulmer MG, Bull JJ. Models of polygenic sex determination and sex ratio control. Evolution. 1982;36:13–26. doi: 10.1111/j.1558-5646.1982.tb05005.x. [DOI] [PubMed] [Google Scholar]
- Conover DO, Heins SW. Adaptive variation in environmental and genetic sex determination in a fish. Nature. 1987;326:496–498. doi: 10.1038/326496a0. [DOI] [PubMed] [Google Scholar]
- Dempster ER, Lerner IM. Heritability of threshold characters. Genetics. 1950;35:212–236. doi: 10.1093/genetics/35.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan QK, Vandeputte M, Chatain B, Haffray P, Vergnet A, Breuil G, et al. Genetic variation of resistance to viral nervous necrosis and genetic correlations with production traits in wild populations of the European sea bass (Dicentrarchus labrax) Aquaculture. 2017;478:1–8. doi: 10.1016/j.aquaculture.2017.05.011. [DOI] [Google Scholar]
- Doan QK (2017b). Genetic and genomic variation of resistance to viral nervous necrosis in wild populations of European seabass (Dicentrachus labrax). PhD thesis, Université de Montpellier.
- Duranton M, Allal F, Fraïsse C, Bierne N, Bonhomme F, Gagnaire PA. The origin and remolding of genomic islands of differentiation in the European sea bass. Nat Commun. 2018;9:2518. doi: 10.1038/s41467-018-04963-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics. Longman, Harlow, England
- Fauvel C, Suquet M, Dreanno C, Menu B. Cryopreservation of sea bass (Dicentrarchus labrax) spermatozoa in experimental and production simulating conditions. Aquat Living Resour. 1998;11:387–394. doi: 10.1016/S0990-7440(99)80004-7. [DOI] [Google Scholar]
- Felip A, Zanuy S, Carrillo N. Comparative analysis of growth performance and sperm motility between precocious and non-precocious males in the European sea bass (Dicentrarchus labrax, L.) Aquaculture. 2006;256:570–578. doi: 10.1016/j.aquaculture.2006.02.014. [DOI] [Google Scholar]
- Groeneveld E, Kovac M, Mielenz N (2010). VCE user’s guide and reference manual version 6.0. https://vce.tzv.fal.de/
- Guinand B, Vandeputte M, Dupont-Nivet M, Vergnet A, Haffray P, Chavanne H, et al. Metapopulation patterns of additive and nonadditive genetic variance in the sea bass (Dicentrarchus labrax) Ecol Evol. 2017;7:2777–2790. doi: 10.1002/ece3.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habier D, Fernando RL, Kizilkaya K, Garrick DJ. Extension of the bayesian alphabet for genomic selection. BMC Bioinforma. 2011;12:186. doi: 10.1186/1471-2105-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heule C, Salzburger W, Böhne A. Genetics of sexual development: an evolutionary playground for fish. Genetics. 2014;196:579–591. doi: 10.1534/genetics.114.161158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire C, Versini JJ, Bonhomme F. Maintenance of genetic differentiation across a transition zone in the sea: discordance between nuclear and cytoplasmic markers. J Evol Biol. 2005;18:70–80. doi: 10.1111/j.1420-9101.2004.00828.x. [DOI] [PubMed] [Google Scholar]
- Liew WC, Bartfai R, Lim Z, Sreenivasan R, Siegfried KR, Orieux N. Polygenic sex determination system in zebrafish. PLoS ONE. 2012;7:e34397. doi: 10.1371/journal.pone.0034397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco DAL, Tsuruta S, Fragomeni BO, Masuda Y, Aguilar I, Legarra A, Bertrand JK, et al. Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. J Anim Sci. 2015;93:2653–2662. doi: 10.2527/jas.2014-8836. [DOI] [PubMed] [Google Scholar]
- Lynch M, Walsh B (1998) Genetics and analysis of quantitative traits. Sinauer Associates, Sunderland, MA
- Mank JE, Promislow DE, Avise JC. Evolution of alternative sex-determining mechanisms in teleost fishes. Biol J Linn Soc. 2006;87:83–93. doi: 10.1111/j.1095-8312.2006.00558.x. [DOI] [Google Scholar]
- Menu B, Peruzzi S, Vergnet A, Vidal MO, Chatain B. A shortcut method for sexing juvenile European sea bass, Dicentrarchus labrax L. Aquacult Res. 2005;36:41–44. doi: 10.1111/j.1365-2109.2004.01181.x. [DOI] [Google Scholar]
- Misztal I, Tsuruta S, Lourenco D, Aguilar I, Legarra A, Vitezica Z. Manual for BLUPF90 family of programs. Athens, USA: University of Georgia; 2015. [Google Scholar]
- Moore EC, Roberts RB. Polygenic sex determination. Curr Biol. 2013;23:R510–R512. doi: 10.1016/j.cub.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Naciri M, Lemaire C, Borsa P, Bonhomme F. Genetic study of the Atlantic/Mediterranean transition in sea bass (Dicentrarchus labrax) J Hered. 1999;90:591–596. doi: 10.1093/jhered/90.6.591. [DOI] [Google Scholar]
- Palaiokostas C, Bekaert M, Taggart JB, Gharbi K, McAndrew BJ, Chatain B, et al. A new SNP-based vision of the genetics of sex determination in European sea bass (Dicentrachus labrax) Genet Sel Evol. 2015;47:68. doi: 10.1186/s12711-015-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piferrer F, Blazquez M, Navarro L, Gonzalez A. Genetic, endocrine, and environmental components of sex determination and differentiation in the European sea bass (Dicentrarchus labrax L.) Gen Comp Endocrinol. 2005;142:102–110. doi: 10.1016/j.ygcen.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quéré N, Desmarais E, Tsigenopoulos CS, Belkhir K, Bonhomme F, Guinand B. Gene flow at major transitional areas in sea bass (Dicentrarchus labrax) and the possible emergence of a hybrid swarm. Ecol Evol. 2012;2:3061–3078. doi: 10.1002/ece3.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignard JP. La Méditerranée: creuset ichtyologique. Boll Zool. 1978;45:23–26. doi: 10.1080/11250007809440264. [DOI] [Google Scholar]
- R Core Team (2017) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria https://www.R-project.org/
- Saillant E, Fostier A, Menu B, Haffray P, Chatain B. Sexual growth dimorphism in sea bass Dicentrarchus labrax. Aquaculture. 2001;202:371–387. doi: 10.1016/S0044-8486(01)00786-4. [DOI] [Google Scholar]
- Saillant E, Fostier A, Haffray P, Menu B, Laureau S, Thimonier J, et al. Effects of rearing density, size grading and parental factors on sex ratios of the sea bass (Dicentrarchus labrax L.) in intensive aquaculture. Aquaculture. 2003;221:183–206. doi: 10.1016/S0044-8486(02)00539-2. [DOI] [Google Scholar]
- Sansone G, Fabbrocini A, Ieropoli S, Langellotti AL, Occidente M, Matassino D. Effects of extender composition, cooling rate, and freezing on the motility of sea bass (Dicentrarchus labrax L.) spermatozoa after thawing. Cryobiology. 2002;44:229–239. doi: 10.1016/S0011-2240(02)00026-3. [DOI] [PubMed] [Google Scholar]
- Tine M, Kuhl H, Gagnaire PA, Louro B, Desmarais E, Martins RST, et al. The European sea bass genome and its variation provide insight into adaptation to euryhalinity and marine speciation. Nat Commun. 2014;5:5770. doi: 10.1038/ncomms6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dooren TJM, Leimar O. The evolution of environmental and genetic sex determination in fluctuating environments. Evolution. 2003;57:2667–2677. doi: 10.1111/j.0014-3820.2003.tb01511.x. [DOI] [PubMed] [Google Scholar]
- VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- Vandeputte M, Mauger S, Dupont-Nivet M. An evaluation of allowing for mismatches as a way to manage genotyping errors in parentage assignment by exclusion. Mol Ecol Notes. 2006;6:265–267. doi: 10.1111/j.1471-8286.2005.01167.x. [DOI] [Google Scholar]
- Vandeputte M, Dupont-Nivet M, Chavanne H, Chatain B. A polygenic hypothesis for sex determination in the European sea bass Dicentrarchus labrax. Genetics. 2007;176:1049–1057. doi: 10.1534/genetics.107.072140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeputte M, Quillet E, Chatain B. Are sex ratios in wild European sea bass (Dicentrarchus labrax) populations biased? Aquat Living Resour. 2012;25:77–81. doi: 10.1051/alr/2012002. [DOI] [Google Scholar]
- Vandeputte M, Garouste R, Dupont-Nivet M, Haffray P, Vergnet A, Chavanne H, et al. Multi-site evaluation of the rearing performances of 5 wild populations of European sea bass (Dicentrarchus labrax) Aquaculture. 2014;424-425:239–248. doi: 10.1016/j.aquaculture.2014.01.005. [DOI] [Google Scholar]
- Vandeputte M, Piferrer F (2018). Genetic and environmental components of sex determination in the European sea bass (Dicentrarchus labrax). In: Wang HP, Piferrer F, Chen SL (eds) Sex control in aquaculture: theory and practice. Wiley-Blackwell Hoboken, NJ, USA (in press).
- Wang H, Misztal I, Aguilar I, Legarra A, Muir W. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet Res. 2012;94:73–83. doi: 10.1017/S0016672312000274. [DOI] [PubMed] [Google Scholar]
- Warnes GR, Bolker B, Lumley T, Johnson RC (2015). gmodels: various R programming tools for model fitting. Available at: https://CRAN.R-project.org/package=gmodels.
- Wilson CA, High SK, McCluskey BM, Amores A, Yan Y, Titus TA, et al. Wild sex in zebrafish: loss of the natural sex determinant in domesticated strains. Genetics. 2014;198:1291–1308. doi: 10.1534/genetics.114.169284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lourenco D, Aguilar I, Legarra A, Misztal I. Weighting strategies for single-step genomic BLUP: an iterative approach for accurate calculation of GEBV and GWAS. Front Genet. 2016;7:151. doi: 10.3389/fgene.2016.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu J, Ding X, Bijma P, de Koning D, Zhang Q. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS ONE. 2010;5:e12648. doi: 10.1371/journal.pone.0012648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset underlying our findings is available in the institutional public data repository (SEANOE: http://www.seanoe.org/), 10.17882/55576