

The Ca 2+ sensor, STIM1, couples to activate Orai Ca 2+ channels in response to endoplasmic reticulum Ca 2+ depletion. New in vivo targeting approaches reveal that STIM1 mediates precise temperature sensing in skin through heat-induced regulation of Orai channels.
Cells utilize Ca2+ as a universal second messenger to activate and coordinate a wide spectrum of cellular functions. The Orai family of plasma membrane (PM) channels mediate store-operated Ca2+ entry (SOCE) that is crucial in regulating transcription, secretion, motility, and growth. Opening of these channels is controlled by the dynamic STIM proteins that function as Ca2+ sensors located in the endoplasmic reticulum (ER) membrane.1 The STIM1 protein exists as a dimer undergoing an extraordinary unfolding and activation process in response to diminished Ca2+ stored in the ER that arises after receptor-induced Ca2+ signal generation (Fig. 1a). STIM1 activation causes extension of its cytoplasmic domains and binding of the C-terminal poly-K sequence to acidic lipids in the PM, causing STIM1 to be trapped within ER-PM junctions. The unfolding of STIM1 also exposes its active STIM-Orai activating region (SOAR) to bind Orai1 channels in ER-PM junctions; as a result, Orai1 channels are trapped and opened, allowing a highly selective flow of Ca2+ ions into the cytosol.1, 2 This junctionally localized SOCE signal is critical in mediating transcriptional control, in particular, through the Ca2+-activated transcription factor, NFAT (nuclear factor of activated T-cells).2, 3 Loss-of-function mutations of either STIM1 or Orai1 are associated with severe combined immunodeficiency, and a broad spectrum of immunological, musculoskeletal, and skin disorders.4 Indeed, the STIM/Orai Ca2+ signaling pathway plays an important role in function and/or development of most cell types.
Fig. 1.
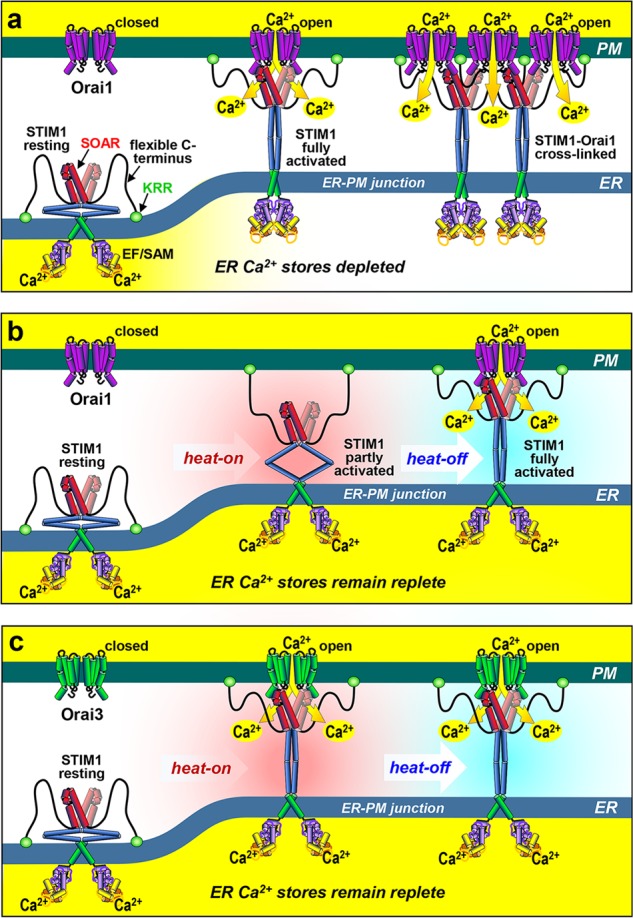
STIM-Orai coupling responses to Ca2+ store depletion and temperature change. a The resting, dimeric STIM1 Ca2+ sensor lies throughout the ER with its cytosolic STIM-Orai activating region (SOAR) occluded, and flexible C-terminus and K-rich region (KRR) folded. Luminal Ca2+ depletion causes Ca2+ dissociation and unfolding of the C-terminus, allowing PM attachment of the KRR, and exposed SOAR to tether and gate Orai1 channels. Subsequent STIM1-mediated Orai1 cross-linking enhances Ca2+ entry. b STIM1 can instead be activated by increased temperature (heat-on), independently of Ca2+ store depletion. Heat-on causes the STIM1 KRR to become PM attached, but Orai1 coupling does not occur. However, Orai1 coupling and activation does take place after subsequent cooling (heat-off). c With Orai3, heat-on alone activates STIM1 sufficiently to allow coupling and Ca2+ entry without the need for cooling, perhaps reflecting a higher energy STIM1-Orai3 interaction
An intriguing recent question is whether the activation of STIM proteins is exclusively through ER luminal Ca2+ sensing. Reports reveal that STIM1 can function as a more generalized stress sensor, responding to oxidative stress, hypoxia, and temperature change, in each case modulating the generation of Ca2+ signals via altered coupling to Orai1 channels.5 Interestingly, it was earlier reported that increased temperature causes activation of STIM1, independent of the depletion of ER luminal Ca2+.6 It was shown that heat alone activates STIM1 and its entry into ER-PM junctions, but not coupling to Orai1; subsequent cooling of cells results in robust coupling to activate the Orai1 channel, again independent of ER Ca2+ (Fig. 1b). In a recent paper in Cell Research, this temperature-sensing feature of STIM1 has been linked to physiological thermo-sensing in keratinocytes.7 Using some ingenious in vivo strategies to eliminate STIM1 and replace it with a coupling-defective mutant, the authors reveal that animals have altered preference for temperature, and that the STIM1 protein thermosensitivity may physiologically mediate precise temperature sensing in mammals.7
This new work brings together two distinct fields of study — temperature sensing and calcium signaling, providing some important new understanding on both processes. Mammals have an extraordinary ability to sense very tiny deviations (~1 °C) from normal skin temperature, in addition to the more robust sensations of noxious heat and cold. A number of channels have been implicated in temperature sensing, most notably the thermo-TRP channels.8 Among these, TRPV1 and TRPM8 are perhaps the most conclusively-determined sensors of noxious heat and cold, respectively; but others including TRPM2, TRPM3, TRPV2, TRPV3, and TRPA1 have also been implicated.9 These thermo-sensing ion channels are believed to operate within thermo-sensory neurons, particularly those of the dorsal root ganglia (DRG), nerve endings of which extend into the skin. Some of these thermo-TRP channels are also clearly involved in pain sensation, but the underlying mechanisms by which physiological thermo-regulation is mediated and relatively small temperature changes in skin are sensed, still remain enigmatic.7
Instead of DRGs, the new work from Liu et al. focuses on the keratinocytes themselves, the exposure of which on the skin lends them to be a primary site for sensing rapid and subtle temperature changes. They provide evidence that the STIM1 in keratinocytes functions as the initial warm sensor, which then activates downstream sensory neurons. Based on their earlier work on STIM1 temperature sensing,6 Liu et al. demonstrate that STIM1 acts as a mediator of optimal preference temperature (OPT) for warm preference behavior in mice.7 Thus, keratinocyte-selective in vivo deletion of STIM1 results in a small but crucially shifted OPT from 32 °C to 34 °C in mice. Neural-specific knockout of STIM1 does not give altered OPT. Interestingly, keratinocyte-specific knockin of the thermally-inactive STIM1ΔK variant (devoid of the PM lipid-anchoring domain), did not restore wild-type thermo-sensing. Earlier studies had shown that increased temperature (heat-on) drove STIM1 to unfold independently of stored Ca2+, and become PM attached within ER-PM junctions through its K-rich C-terminus (Fig. 1b), despite the observation that Orai1 coupling was prevented at this higher temperature.6 Subsequent cooling of cells allows the PM-attached STIM1 to activate Orai1 (the heat-off response) (Fig. 1b). Thus, the failure of keratinocyte-specific STIM1ΔK knockin to reverse the altered OPT in STIM1-knockout mice, provides evidence for the specific role of the intact STIM1 molecule in mediating the altered thermo-sensing. Interestingly, the warm responses of DRG neurons in the keratinocyte-specific STIM1-knockout mice, were also defective, indicating that keratinocytes are the primary thermo-sensing cells. The authors suggest that TRPA1 in DRG neurons may be the downstream transduction channel, but how keratinocytes signal to neurons is not apparent.
Mechanistically, the function of the STIM/Orai Ca2+ signaling pathway in heat sensing is fascinating. Liu et al. show clearly that the ability of STIM1 to mediate temperature sensing requires its coupling to Orai channels. Thus, the weaker Orai-associating STIM2 homologue does not mediate heat responses, nor does STIM1 devoid of its Orai-interacting SOAR domain.7 Intriguingly, while Orai1 mediates only the STIM1-induced heat-off Ca2+ response, Orai3 is shown to also mediate STIM1-induced Ca2+ entry in response to heat-on.7 This may reflect a higher STIM1 affinity for Orai3 compared to Orai1. Thus, we hypothesize that while the heat-on-induced partially-activated STIM1 is unable to interact with Orai1 (Fig. 1b), a stronger interaction with Orai3 may pull STIM1 into the fully-activated configuration, even during heating (Fig. 1c). With both Orai1 and Orai3, the complex is functional during the heat-off response. Whether the heat-activated STIM1 protein induces the same coupling to Orai1 as store depletion, is unclear. Heat-induced Orai1 activation is less effective than that by store depletion,6, 7 thus STIM1 may not be optimally activated by heat. STIM1 coupling to Orai1 involves both binding and cross-linking of channels,10 and we hypothesize that the partial heat-activation of STIM 1 may not be sufficient to induce the cross-linking process and therefore results in suboptimal Orai1 channel activation (Fig. 1a–c).
The studies provide a new paradigm in thermo-sensing which, until now, has been attributed to channel molecules themselves. The new work reveals that a powerful ER-located channel regulator, STIM1, can be a primary thermo-sensor, an effect that depends on STIM1’s ability to reach across ER-PM junctions to the PM. STIM1 knockout reveals that STIM1 has no role in noxious heat-sensing which is likely mediated by neuronal thermo-TRPs. It seems that STIM1 in keratinocytes tunes the skin’s reference temperature to affect subtle thermo-sensing.
Conflict of interest
The authors declare no competing interests.
Contributor Information
Yandong Zhou, Email: zhouyd@psu.ed.
Donald L. Gill, Email: dongill@psu.edu
References
- 1.Soboloff J, Rothberg BS, Madesh M, Gill DL. Nat. Rev. Mol. Cell Biol. 2012;13:549–565. doi: 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prakriya M, Lewis RS. Physiol. Rev. 2015;95:1383–1436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shim AH, Tirado-Lee L, Prakriya M. J. Mol. Biol. 2015;427:77–93. doi: 10.1016/j.jmb.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lacruz RS, Feske S. Ann. N. Y Acad. Sci. 2015;1356:45–79. doi: 10.1111/nyas.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soboloff J, Madesh M, Gill DL. Nat. Chem. Biol. 2011;7:488–492. doi: 10.1038/nchembio.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao B, Coste B, Mathur J, Patapoutian A. Nat. Chem. Biol. 2011;7:351–358. doi: 10.1038/nchembio.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, et al. Cell Res. 2019;29:95–109. doi: 10.1038/s41422-018-0129-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castillo K, Diaz-Franulic I, Canan J, Gonzalez-Nilo F, Latorre R. Phys. Biol. 2018;15:021001. doi: 10.1088/1478-3975/aa9a6f. [DOI] [PubMed] [Google Scholar]
- 9.Palkar R, Lippoldt EK, McKemy DD. Curr. Opin. Neurobiol. 2015;34:14–19. doi: 10.1016/j.conb.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y. Proc. Natl. Acad. Sci. USA. 2018;115:E3398–E3407. doi: 10.1073/pnas.1720810115. [DOI] [PMC free article] [PubMed] [Google Scholar]