

Abstract
We have reported that hepatitis B X-interacting protein (HBXIP, also termed LAMTOR5) can act as an oncogenic transcriptional co-activator to modulate gene expression, promoting breast cancer development. Pyruvate kinase muscle isozyme M2 (PKM2), encoded by PKM gene, has emerged as a key oncoprotein in breast cancer. Yet, the regulatory mechanism of PKM2 is still unexplored. Here, we report that HBXIP can upregulate PKM2 to accelerate proliferation of estrogen receptor positive (ER+) breast cancer. Immunohistochemistry analysis using breast cancer tissue microarray uncovered a positive association between the expression of HBXIP and PKM2. We also discovered that PKM2 expression was positively related with HBXIP expression in clinical breast cancer patients by real-time PCR assay. Interestingly, in ER+ breast cancer cells, HBXIP was capable of upregulating PKM2 expression at mRNA and protein levels in a dose-dependent manner, as well as increasing the activity of PKM promoter. Mechanistically, HBXIP could stimulate PKM promoter through binding to the −779/−579 promoter region involving co-activation of E2F transcription factor 1 (E2F1). In function, cell viability, EdU, colony formation, and xenograft tumor growth assays showed that HBXIP contributed to accelerating cell proliferation through PKM2 in ER+ breast cancer. Collectively, we conclude that HBXIP induces PKM2 through transcription factor E2F1 to facilitate ER+ breast cancer cell proliferation. We provide new evidence for the mechanism of transcription regulation of PKM2 in promotion of breast cancer progression.
Keywords: HBXIP, PKM2, E2F1, breast cancer, proliferation
Introduction
Breast cancer is a leading cause of cancer mortality among females worldwide [1]. Up to 75% of breast cancer patients express estrogen receptor (ER), which is essential for the growth and proliferation of breast cancer cell [2]. Hepatitis B X-interacting protein (HBXIP) is initially identified since its binding with hepatitis B virus X protein [3]. It has been reported that HBXIP functions as a regulator to participate in the activation of mTORC1 induced by amino acids [4]. We previously discovered that oncoprotein HBXIP was highly expressed in breast tumor and took great part in the developmental processes of breast cancer such as proliferation, migration, and abnormal glucose metabolism [5–7]. Marusawa’s team reported that HBXIP was capable of modulating cell division and apoptosis by interacting with survivin [8]. Recently, we have proven that HBXIP can promote reprogramming of glucose metabolism via reducing PDHA and SCO2, facilitating the growth of breast cancer [7]. Nevertheless, the underling mechanism by which HBXIP facilitates ER-positive (ER+) breast cancer proliferation needs to be investigated.
Pyruvate kinase is a rate-limiting glycolytic enzyme that catalyzes the last step of glycolysis [9]. Pyruvate kinase muscle isozyme M1 and M2 (PKM1 and PKM2) are two different splicing forms of an identical messenger RNA (mRNA) encoded by PKM gene [10]. PKM2 is back in the spotlight on account of its multiple roles in the promotion of cancer growth and angiogenesis [9, 11]. Numerous articles have revealed a high expression of PKM2 in breast cancer [12, 13]. Due to the low activity of PKM2 as a pyruvate kinase, the intermediate metabolites of glucose accumulate and then flow to an alternative glycolytic pathway, assisting anabolic synthesis and cancer cell growth [9]. Moreover, PKM2 can translocate to nucleus and function as a co-activator to modulate cancer-related gene expression [14, 15]. In addition, PKM2 acts as a protein kinase that directly phosphorylates proteins like STAT3, which is possibly one of the most crucial molecular signatures for accelerating the progression of cancer [16]. Nevertheless, whether PKM2 participates in HBXIP-accelerated proliferation of ER+ breast cancer still remains unclear.
Here, we attempt to go deep into the study of the relation of PKM2 and HBXIP in ER+ breast cancer. Intriguingly, we reveal that HBXIP can increase PKM2 expression via transcription factor E2F1, leading to the cell proliferation acceleration in ER+ breast cancer. Our findings unearth a novel mechanism by which HBXIP promotes the proliferation of ER+ breast cancer cells via PKM2.
Materials and methods
Patient samples
Thirty-four pairs of breast cancer and noncancerous tissues were gained from Tianjin Tumor Hospital (Tianjin, China). The Research Ethics Committee of Nankai University permitted the research procedures. The patients signed the written consent to authorize the usage of tissues for study. The patients’ information is recorded in Supplementary Table S1.
Immunohistochemistry staining
Immunohistochemistry (IHC) staining assay has been described previously [17]. Primary antibodies used in immunohistochemistry (IHC) staining were rabbit anti-PKM2 (Proteintech, USA), rabbit anti-HBXIP (Santa Cruz Biotechnology, USA), and rabbit anti-Ki67 antibody (Santa Cruz Biotechnology). The breast cancer tissue microarray (No. 08C14) was purchased from Aomei Biotechnology (Xi’an, China). The image of whole-tissue array was captured by the Motic digital slide system BA600 (Motic, Xiamen, China). The patient information and statistical result of tissue microarray are recorded in Supplementary Tables S2 and S3, respectively. The rule to grade the levels of staining of PKM2 and HBXIP was described previously [18, 19].
Cell lines and cell culture
The ER+ breast cancer cell lines MCF-7-HBXIP (stably transfected with the HBXIP gene [17]), MCF-7, and T47D were maintained according to ATCC protocol. Lipofectamine 2000 (Invitrogen, USA) was used in all transfections following the manufacturer’s protocol.
Plasmids and siRNA
pGL3-Basic, pcDNA3.1 (+), and pcDNA-HBXIP were saved in our lab. pRL-TK was purchased from Promega in USA. Using genomic DNA of MCF-7 cells as template, different promoter regions of human PKM gene were cloned into the pGL3-Basic vector involving KpnI/HindIII site. The primers used in this study were shown as follows: pGL-P0, sense—CGGGGTACCAACAACAACTAAAGGGACCAGGAA, anti-sense—CCCAAGCTTGACTGATGGCGTAGCCTCCTG. pGL-P1, sense—CGGGGTACCAGAAAAGTTCCAACAATACTGACTTAAAC, anti-sense—CCCAAGCTTGACTGATGGCGTAGCCTCCTG. pGL-P2, sense—CGGGGTACCGAAGGCGGCCAGGACCTC, anti-sense—CCCAAGCTTGACTGATGGCGTAGCCTCCTG. pGL-P3, sense—CGGGGTACCAGAGCCAAGAAAAGACACCCC, anti-sense—CCCAAGCTTGACTGATGGCGTAGCCTCCTG. Mutant constructs of pGL-P2: Sp1-M, sense—GGCAGGGCTACTCCGGGAGAATGCT, anti-sense—AGCATTCTCCCGGAGTAGCCCTGCC. E2F1-M, sense—TCTCCGAGATGACGCCAGAGCAGA, anti-sense—TCTGCTCTGGCGTCATCTCGGAGA. The sequences of small interfering RNAs (siRNAs) (RiboBio, Guangzhou, China) applied in this study were as follows: HBXIP, CGGAAGCGCAGUGAUGUUUdTdT; E2F1#1, CACUGACUCUGCCACCAUAGdTdT; E2F1#2, AAGUCACGCUAUGAGACCUCAdTdT; PKM2#1, CTGTGGACAGTTACCAGTCdTdT; PKM2#2, CCAUAAUCGUCCUCACCAAdTdT. Control siRNA, UUCUCCGAACGUGUCACGUdTdT.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed as described formerly [5]. The primers used to examine PKM promoter were: sense—GAAGGCGGCCAGGACCTCCAAC, anti-sense—TCGGCCCAGGCCACCCCAGCTG. Primers sets for the negative control were: sense— AGCGGGATAACCTTGAGGCTGAGG, anti-sense—CACCTCCGGCGCTGACCGACTCGG.
Reverse-transcription PCR and real-time PCR
Reverse-transcription PCR (RT-PCR) and real-time PCR assays were conducted as described previously [5]. The primers used here were shown as follows: GAPDH, sense—AACGGATTTGGTCGTATTG, anti-sense—GGAAGATGGTGATGGGATT. HBXIP, sense—CTTGGAGCAGCACTTGGAAGA, anti-sense—ATGCCATCGTGTTTCTGGATC. PKM2, sense—CCACTTGCAATTATTTGAGGAA, anti-sense—GTGAGCAGACCTGCCAGACT.
Immunoblotting assay
Immunoblotting assay was applied by following the procedures described formerly [17]. Primary antibodies used in this study were rabbit anti-PKM2 (Proteintech), rabbit anti-E2F1 antibody (Abcam, UK), rabbit anti-HBXIP (Santa Cruz Biotechnology, USA), and mouse anti-β-actin (Sigma-Aldrich, USA).
Cell viability assay
Firstly, the indicated plasmids were transfected into MCF-7 or T47D cells for 24 h. Then, the cells were digested and plated at 1000 cells per well into 96-well plate with six replications. Subsequently, the cells were cultured for indicated time course. 10 μL of 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) reagent (Sigma) was added into each well and the cells were incubated for 4 h. Next, after the remove of supernatant, 100 μL of dimethyl sulfoxide was used to dissolve the purple crystals in each well. The result was evaluated by multi-well scanning spectrophotometer (Thermo) at an absorbance (A492 nm-A620 nm, A be Italic, 492 nm and 620 nm be subscript).
Luciferase reporter gene assay
For the assay of luciferase, the indicated plasmids were transiently transfected into the MCF-7, T47D, or MCF-7-HBXIP cells grown in 24-wells. The plasmid of pRL-TK that encodes Renilla luciferase was used to normalize the activity of firefly luciferase. Luciferase activities were measured after transfection for 24 h. By following the procedures described by manufacturer, the activity of luciferase was examined through a luciferase reporter assay system (Promega) on a luminometer (TD-20/20; Turner Designs, Sunnyvale, CA, USA).
Colony formation assay
Corresponding plasmids were transfected into MCF-7 or T47D cells for 48 h, and then the cells were trypsinized and cultured in six-well plate (500 cells per well). The medium was replaced twice a week. After growth for 14 days, cells were managed as described formerly [18]. The efficiency of colony formation was evaluated as follows: colony number/cell population number × 100%.
EdU assay
The EdU kit was bought from RiboBio of China. After transient transfection as indicated, the cells were added with 30 (μM) 5-ethynyl-2′-deoxyuridine (EdU) and maintained for 4 h. Next, 4% (w/v) formaldehyde was used to fix cells for 20 min. The rest procedures were carried out by following the manufacturer’s description. The result was measured by fluorescence microscope (Axio Imager Z1, ZEISS, Germany).
Xenograft tumor growth assay
Five- to six-week-old female BALB/c athymic nude mice (Experiment Animal Center of Peking, China) were housed and treated according to guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. MCF-7 cells pre-treated with pcDNA3.1 plasmid (or pcDNA-HBXIP plasmid, control siRNA (si-Control), PKM2 siRNA (si-PKM2#2)) were collected and suspended at 5 × 107 cells in 200 μL of phosphate-buffered saline. Then, the corresponding cells were injected into the fourth mammary fat pad (mfp) of mice (each group, n = 5). β-estradiol (sigma) dissolved in olive oil (5 mg/mL, 0.1 mL) was administered by gavage twice a week. Tumor size was measured in two dimensions with calipers twice a week, up to 5 weeks after injection. Blind measurements were carried out to avoid unconscious biases. Tumor volume (v) was calculated according to the formula: (length × width2) × 0.5. All experiments were approved by the animal care committee of Nankai University.
Statistical analysis
By using a two-tailed Student’s t test, the mean values (±SD) were compared to evaluate statistical significance. The significance was defined as P < 0.05(*), P < 0.01(**), P < 0.001(***). Student’s t test and correlation analysis were performed using Prism 6 (GraphPad, San Diego, CA, USA).
Results
PKM2 is positively related to HBXIP in clinical breast cancer patients
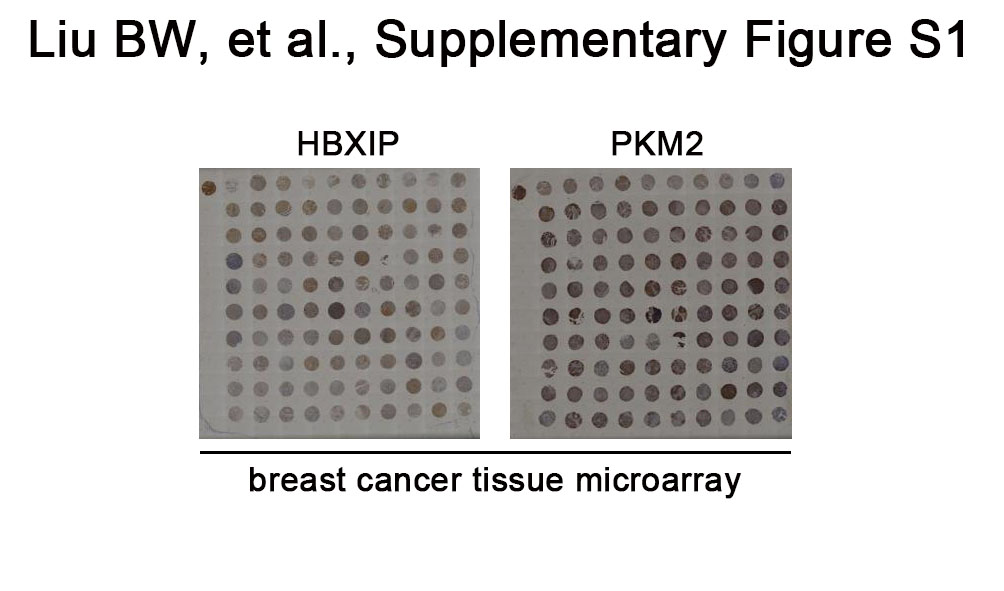
We have revealed that abundant oncoprotein HBXIP contributes to accelerated-breast cancer progression [19, 20]. Recently, a crucial function of PKM2 in cancer growth and development has been established [10]. Accordingly, we intended to explore the potential correlation between PKM2 and HBXIP in the occurrence and progression of breast cancer. Firstly, we performed IHC staining to determine the expression of PKM2 and HBXIP in human tissue microarray that contained 49 primary breast carcinomas, 13 normal tissues, and 38 metastatic lymph nodes in breast cancer. We found that both HBXIP and PKM2 were weakly stained in normal breast samples and strongly stained in breast carcinoma tissues (Fig. 1a). Moreover, we observed a positive rate of PKM2 (95.5%, 42/44) in HBXIP-positive breast carcinoma specimens, indicating that PKM2 is closely related to HBXIP in breast carcinoma patients (Pearson χ² independence test, χ² = 32.34, P < 0.001, Fig. 1a and b; Supplementary Figure S1; Supplementary Tables S2 and S3). Meanwhile, we conducted real-time PCR assay in 34 fresh breast cancer samples (30 ER+ samples and 4 ER− samples) to evaluate PKM2 and HBXIP expression (Supplementary Table S1). The data revealed an obvious positive association of the mRNA levels among HBXIP and PKM2 (Fig. 1c). Thus, we get a conclusion that the PKM2 expression is positively correlated to the HBXIP expression in clinical breast cancer patients.
Fig. 1.

PKM2 is positively related to HBXIP in clinical breast cancer patients. a IHC staining of HBXIP and PKM2 in the normal breast tissues (N) and breast carcinomas (T1, T2) from breast tissue microarray. Scale bar, 100 μm. The association between HBXIP and PKM2 expression was statistically significant, analyzed by Pearson χ² independence test, χ² = 32.34, P < 0.001. b Heatmap of the expression of HBXIP and PKM2 in above breast tissue microarray. Numbers 0, 1, or 2 represent the negative, moderate, or intense staining, respectively. c Relative mRNA levels of HBXIP and PKM2 in 34 clinical breast tumor tissues tested by real-time PCR assay. The correlation between HBXIP and PKM2 at the mRNA levels was determined by Pearson’s correlation coefficient. Statistically significant differences are indicated: ***P < 0.001; all experiments were repeated at least three times
The expression of PKM2 can be upregulated by HBXIP in ER+ breast cancer cells
Studies have demonstrated that HBXIP functions as a transcriptional co-activator to promote breast cancer by regulating the expression of different cancer-related proteins [21, 22]. Here, we intended to investigate whether PKM2 could be modulated by HBXIP in ER+ breast cancer cells. To determine HBXIP-induced effect on PKM2, we performed real-time PCR and RT-PCR assays and observed an effective elevation of mRNA level of PKM2 after HBXIP was increasingly overexpressed in MCF-7 cells (Fig. 2a, c). Oppositely, HBXIP knockdown by siRNA significantly downregulated PKM2 mRNA level in MCF-7-HBXIP cells (Fig. 2b, d). Furthermore, the similar effect of HBXIP on the protein level of PKM2 was validated by immunoblotting assay (Fig. 2e, f). Therefore, these findings indicate that the expression of PKM2 can be induced by HBXIP in ER+ breast cancer cells.
Fig. 2.

The expression of PKM2 can be upregulated by HBXIP in ER+ breast cancer cells. a, b Real-time PCR assay of PKM2 mRNA level in MCF-7 (a) and MCF-7-HBXIP (b) cells transiently transfected with indicated plasmids or siRNA for 48 h. c, d RT-PCR assay of PKM2 mRNA level in MCF-7 (c) and MCF-7-HBXIP (d) cells transiently transfected with indicated plasmids or siRNA for 48 h. e, f Immunoblotting assay of PKM2 protein level in MCF-7 (e) and MCF-7-HBXIP (f) cells transiently transfected with indicated plasmids or siRNA for 48 h. Statistically significant differences are indicated: *P < 0.05, **P < 0.01; all experiments were repeated at least three times
HBXIP stimulates PKM promoter through binding to the −779/−579 region
To clarify the essence that HBXIP modulates PKM2, we constructed and subsequently transfected the human PKM promoter vector (−1468/−98) into the MCF-7 and MCF-7-HBXIP cells. Notably, HBXIP was capable of enhancing the activity of PKM promoter in a dose-dependent manner, demonstrated by luciferase reporter gene assay (Fig. 3a, b). Then, we further cloned a series of fragments of PKM promoter shown in Fig. 3c. The ectopic expression of HBXIP in MCF-7 cells effectively enhanced the activities of PKM promoter fragments except pGL-P3, suggesting that the region (−779/−579) in PKM promoter is the core sequence regulated by HBXIP (Fig. 3c). Furthermore, ChIP assay further revealed that the −779/−579 region of PKM promoter was detectable in the anti-HBXIP-immunoprecipitated candidates (Fig. 3d). Thus, our data support that HBXIP stimulates PKM promoter through binding to the −779/−579 region.
Fig. 3.

HBXIP stimulates PKM promoter through binding to the −779/−579 region. a, b Luciferase reporter gene assay of PKM promoter activity in MCF-7 (a) and MCF-7-HBXIP cells (b). The cells were transiently transfected with indicated plasmids or siRNA, respectively. Luciferase activities were measured after transfection for 24 h. c Activities of corresponding fragments of PKM promoter were examined by luciferase reporter gene assay in MCF-7 cells, respectively. The cells were transiently transfected with pcDNA or pcDNA-HBXIP along with indicated fragments of PKM promoter. d ChIP assay was performed to evaluate the interaction of HBXIP with the promoter region (−779/−579) of PKM in MCF-7 cells. The right panel was the quantitative data of enrichment in PKM promoter analyzed by real-time PCR and normalized against the input. An upstream region of PKM gene promoter (−1458/−1345) was used as a negative control. Statistically significant differences are indicated: *P < 0.05, **P < 0.01, ***P < 0.001; All experiments were repeated at least three times
Transcriptional factor E2F1 is responsible for HBXIP-stimulated PKM promoter
To take a step further, we analyzed the core region by the online bioinformatics tool JASPAR database (http://jaspar.binf.ku.dk/cgi-bin/jaspar_db.pl). We observed that transcription factors Sp1 and E2F1 had putative binding sites in this region. Then, we mutated the corresponding binding sites of Sp1 (termed Sp1-M) and E2F1 (termed E2F1-M) in pGL-P2 (Fig. 4a). In MCF-7 and T47D cells, the mutant of binding site of E2F1, rather than Sp1, efficiently blocked HBXIP-mediated increase of pGL-P2 activity, implying that the transcription factor E2F1 participates in HBXIP-induced stimulation of PKM promoter (Fig. 4b, c). To further confirm the role of E2F1 in HBXIP-mediated elevation of PKM2, we introduced two different siRNAs of E2F1 into MCF-7 cells and conducted luciferase reporter gene and immunoblotting assays. Notably, knockdown of E2F1 by siRNAs remarkably abolished HBXIP-induced increase of PKM promoter activity and PKM2 protein level (Fig. 4d, e). Thus, we draw a conclusion that transcriptional factor E2F1 is responsible for HBXIP-stimulated PKM promoter.
Fig. 4.

Transcriptional factor E2F1 is responsible for HBXIP-stimulated PKM promoter. a Nucleotide mutation diagram of binding sites of Sp1 and E2F1 in pGL-P2. b, c Luciferase reporter gene assay of PKM promoter activities in MCF-7 (b) and T47D cells (c). The cells were transiently transfected with pcDNA or pcDNA-HBXIP along with pGL-P2 (WT) or those constructs with mutated binding sites of Sp1 (Sp1-M) and E2F1 (E2F1-M). d, e The activity of PKM promoter (d) and the protein level of PKM2 (e) were separately examined by luciferase reporter gene assay and immunoblotting assay in MCF-7 cells. The cells were transiently transfected with indicated plasmids or siRNAs. Statistically significant differences are indicated: **P < 0.01, ***P < 0.001; all experiments were repeated at least three times. NS, not significant
HBXIP-elevated PKM2 contributes to accelerated cell proliferation in ER+ breast cancer
Accumulating evidence shows that PKM2 actively participates in the facilitation of breast cancer proliferation [9, 23]. Thus, we wondered whether HBXIP accelerated ER+ breast cancer progression through PKM2. Cell viability assay in MCF-7 and T47D cells showed that silence of PKM2 by two different siRNAs efficiently blocked HBXIP-enhanced cell proliferation in ER+ breast cancer (Fig. 5a, b). Observation in EdU assay was consistent with the above result (Fig. 5c, d). Immunoblotting assay revealed a higher interference efficiency of siPKM2#2 compared with siPKM2#1 (Fig. 5e). Moreover, we conducted colony formation assay in MCF-7 and T47D cells. As expected, the colony formation efficiency obviously increased upon the overexpression of HBXIP (Fig. 6). However, the addition of siPKM2#2 dose-dependently abolished HBXIP-provoked increase of colony formation efficiency (Fig. 6).
Fig. 5.

HBXIP-elevated PKM2 contributes to accelerated cell proliferation in ER+ breast cancer. a, b Cell viability assay in MCF-7 (a) and T47D (B) cells transiently transfected with indicated plasmids or siRNAs. c, d EdU assay in MCF-7 cells transiently transfected with indicated plasmids or siRNAs. Scale bar, 50 μm. e Interference efficiency of PKM2 siRNAs validated by immunoblotting assay in MCF-7 cells. Statistically significant differences are indicated: **P < 0.01, ***P < 0.001; all experiments were repeated at least three times
Fig. 6.

HBXIP-elevated PKM2 contributes to accelerated cell proliferation in ER+ breast cancer. a, b Colony photograph (a) and colony forming efficiency (b) in MCF-7 cells after transiently transfected with the indicated plasmids or siRNA. c, d Colony photograph (c) and colony forming efficiency (d) in T47D cells after transiently transfected with the indicated plasmids or siRNA. Statistically significant differences are indicated: *P < 0.05, **P < 0.01; all experiments were repeated at least three times
To further validate the effect of PKM2 on HBXIP-mediated proliferation, we performed mfp xenograft tumor growth assay in nude mice. The data showed that elevation of HBXIP significantly enchanced the volume and weight of tumors (Fig. 7a–c). However, silence of PKM2 effectively abrogated HBXIP-mediated tumor growth (Fig. 7a–c). In addition, IHC staining was conducted to evaluate the expression of Ki67, a proliferation marker, in tumors. HBXIP overexpression promoted Ki67 expression while silence of PKM2 could abolish HBXIP-induced enhancement of Ki67 (Fig. 7d, e). Taken together, our data reveal that HBXIP enhances cell proliferation via PKM2 in ER+ breast cancer.
Fig. 7.

HBXIP-elevated PKM2 contributes to accelerated cell proliferation in ER+ breast cancer. a, b Imaging (a) and growth curve (b) of the xenograft tumors derived from MCF-7 cells with indicated treatment (each group, n = 5). c Weight of the xenograft tumors derived from MCF-7 cells. d, e Statistics of Ki67 positive cells (d) and Ki67 staining by IHC assay (e) of the xenograft tumors derived from MCF-7 cells. The blank control group was treated with PBS instead of Ki67 antibody. Scale bar, 50 μm. Statistically significant differences are indicated: **P < 0.01, ***P < 0.001; all experiments were repeated at least three times
Discussion
ER+ breast cancer is a common subtype of breast cancer, threatening the health of women around the world [2]. Therefore, it is urgent to clarify the development mechanism of ER+ breast cancer. In recent years, our findings have established a crucial role of HBXIP on the occurrence and progression of breast cancer. HBXIP provokes breast cancer proliferation by modulating the expression of multiple genes [6, 24, 25]. Evidence demonstrated that HBXIP could induce glucose metabolism reprogramming to accelerate cell proliferation [7]. PKM2, as a pyruvate kinase, has achieved the most attention due to its impact on cellular glucose metabolism in facilitation of cancer progression [26]. Furthermore, the non-canonical nuclear function of PKM2 also contributes to the promotion of cancers [12]. The abundant expression of PKM2 in cancers is unveiled by numerous reports [16, 26]. However, the regulation of PKM2 expression in ER+ breast cancer is yet to be revealed. Thus, we speculate that HBXIP might modulate PKM2 expression in ER+ breast cancer, involving cancer cell growth.
To survey the potential association of PKM2 and HBXIP, we performed IHC staining using human tissue microarray and discovered a positive correlation between PKM2 and HBXIP. Meanwhile, we determined the mRNA levels of them in clinical samples including 30 ER+ and 4 ER− breast cancer tissues. The data also manifested a positive association between HBXIP and PKM2. This finding suggests that PKM2 may participate in HBXIP-associated ER+ breast cancer development. Next, we attempted to enucleate the regulation of PKM2 by HBXIP in ER+ breast cancer. Not unexpectedly, HBXIP enhanced PKM2 expression at the levels of mRNA and protein. Furthermore, HBXIP could stimulate the promoter activity of PKM, which might involve the co-activation function of HBXIP in breast cancer. Tracking the details of HBXIP-mediated upregulation of PKM2, we cloned different fragments of PKM promoter and found that the region (−779/−579) in PKM promoter was the main region for HBXIP co-activation. We have demonstrated that HBXIP can co-activate transcription factor E2F1 to modulate the expressions of different genes [22, 24]. Dai et al. [27] has revealed that there is a close connection between the higher expression of E2F1 and the poorer prognosis in breast cancer patient. Here, we reported for the first time that E2F1 was capable of stimulating the transcription of PKM gene. Moreover, E2F1 was in charge of HBXIP-activated PKM2. As to the function, our data proved that PKM2 was responsible for HBXIP-mediated cell proliferation in ER+ breast cancer in vitro and in vivo. Hence, we get the conclusion that HBXIP facilitates cellular proliferation in ER+ breast cancer through PKM2.
To summarize, we uncover that HBXIP drives cell proliferation via elevating PKM2 in ER+ breast cancer, concerning co-activating transcription factor E2F1. Our findings unearth a significant mechanism and afford a molecular basis for curing HBXIP or PKM2-associated breast cancer.
Electronic supplementary material
{kind=link}
Acknowledgements
This work was supported by the grants of National Basic Research Program of China (973 Program, No. 2015CB553905), National Natural Science Foundation of China (Nos. 81372186 and 31670771), the Fundamental Research Funds for the Central Universities, and Project of Prevention and Control of Key Chronic Non Infectious Diseases (No. 2016YFC1303401).
Author contributions
B.-w.L., T.-j.W., and L.-l.L. designed the research methods, performed the experiments, and prepared the manuscript. B.-w.L. analyzed the data. L.Z., Y.-x.L., J.-y.F., Y.W., F.-f.X., Q.-s.Z., and M.-z.B. participated in the experiments. L.-h.Y. and W.-y.Z. conceived the projects, designed, and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Wei-ying Zhang, Email: zhwybao@nankai.edu.cn.
Li-hong Ye, Email: yelihong@nankai.edu.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0015-9) contains supplementary material, which is available to authorized users.
References
- 1.Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7:659–672. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- 2.Bhatt S, Stender JD, Joshi S, Wu G, Katzenellenbogen BS. OCT-4: a novel estrogen receptor-alpha collaborator that promotes tamoxifen resistance in breast cancer cells. Oncogene. 2016;35:5722–5734. doi: 10.1038/onc.2016.105. [DOI] [PubMed] [Google Scholar]
- 3.Melegari M, Scaglioni PP, Wands JR. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J Virol. 1998;72:1737–1743. doi: 10.1128/jvi.72.3.1737-1743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–1208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu S, Li L, Zhang Y, Zhang Y, Zhao Y, You X, et al. The oncoprotein HBXIP uses two pathways to up-regulate S100A4 in promotion of growth and migration of breast cancer cells. J Biol Chem. 2012;287:30228–30239. doi: 10.1074/jbc.M112.343947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi H, Li Y, Feng G, Li L, Fang R, Wang Z, et al. The oncoprotein HBXIP up-regulates FGF4 through activating transcriptional factor Sp1 to promote the migration of breast cancer cells. Biochem Biophys Res Commun. 2016;471:89–94. doi: 10.1016/j.bbrc.2016.01.174. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Zhang W, You X, Liu Y, Li Y, Wang Z, et al. The oncoprotein HBXIP promotes glucose metabolism reprogram-ming via downregulating SCO2 and PDHA1 in breast cancer. Oncotarget. 2015;6:27199–27213. doi: 10.18632/oncotarget.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, et al. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–2740. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris I, McCracken S, Mak TW. PKM2: a gatekeeper between growth and survival. Cell Res. 2012;22:447–449. doi: 10.1038/cr.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Q, Liu LZ, Yin Y, He J, Li Q, Qian X, et al. Regulatory circuit of PKM2/NF-kappaB/miR-148a/152-modulated tumor angiogenesis and cancer progression. Oncogene. 2015;34:5482–5493. doi: 10.1038/onc.2015.6. [DOI] [PubMed] [Google Scholar]
- 12.Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, et al. PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett. 2016;11:1980–1986. doi: 10.3892/ol.2016.4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin Y, Lv F, Liu F, Guo X, Fan Y, Gu F, et al. High expression of pyruvate kinase M2 is associated with chemosensitivity to epirubicin and 5-fluorouracil in breast cancer. J Cancer. 2015;6:1130–1139. doi: 10.7150/jca.12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2:551–556. doi: 10.18632/oncotarget.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu N, Zhang J, Cui W, Kong G, Zhang S, Yue L, et al. miR-520b regulates migration of breast cancer cells by targeting hepatitis B X-interacting protein and interleukin-8. J Biol Chem. 2011;286:13714–13722. doi: 10.1074/jbc.M110.204131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu T, Cao R, Li S, Fu M, Ren L, Chen W, et al. MiR-130b plays an oncogenic role by repressing PTEN expression in esophageal squamous cell carcinoma cells. BMC Cancer. 2015;15:29. doi: 10.1186/s12885-015-1031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, et al. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene. 2016;35:4048–4057. doi: 10.1038/onc.2015.476. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Wang Z, Shi H, Li H, Li L, Fang R, et al. HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-Myc. Cancer Res. 2016;76:293–304. doi: 10.1158/0008-5472.CAN-14-3607. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Li H, Zhang Y, Li L, Fang R, Li Y, et al. Oncoprotein HBXIP modulates abnormal lipid metabolism and growth of breast cancer cells by activating the LXRs/SREBP-1c/FAS signaling cascade. Cancer Res. 2016;76:4696–4707. doi: 10.1158/0008-5472.CAN-15-1734. [DOI] [PubMed] [Google Scholar]
- 22.Xu F, You X, Liu F, Shen X, Yao Y, Ye L, et al. The oncoprotein HBXIP up-regulates Skp2 via activating transcription factor E2F1 to promote proliferation of breast cancer cells. Cancer Lett. 2013;333:124–132. doi: 10.1016/j.canlet.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Lv L, Xu YP, Zhao D, Li FL, Wang W, Sasaki N, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol Cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Cui M, Cai X, Sun B, Liu F, Zhang X, et al. The oncoprotein HBXIP up-regulates SCG3 through modulating E2F1 and miR-509-3p in hepatoma cells. Cancer Lett. 2014;352:169–178. doi: 10.1016/j.canlet.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Cai X, Zhang S, Cui M, Liu F, Sun B, et al. HBXIP up-regulates ACSL1 through activating transcriptional factor Sp1 in breast cancer. Biochem Biophys Res Commun. 2017;484:565–571. doi: 10.1016/j.bbrc.2017.01.126. [DOI] [PubMed] [Google Scholar]
- 26.Alves-Filho JC, Palsson-McDermott EM. Pyruvate kinase M2: a potential target for regulating inflammation. Front Immunol. 2016;7:145. doi: 10.3389/fimmu.2016.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai H, van’t Veer L, Lamb J, He YD, Mao M, Fine BM, et al. A cell proliferation signature is a marker ofextremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005;65:4059–4066. doi: 10.1158/0008-5472.CAN-04-3953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.